

Abstract
Biological membranes constitute a critical component in all living cells. In addition to providing a conducive environment to a wide range of cellular processes, including transport and signaling, mounting evidence has established active participation of specific lipids in modulating membrane protein function through various mechanisms. Understanding lipid-protein interactions underlying these mechanisms at a sufficiently high resolution has proven extremely challenging, partly due to the semi-fluid nature of the membrane. In order to address this challenge computationally, multiple methods have been developed, including an alternative membrane representation termed HMMM (highly mobile membrane mimetic) in which lateral lipid diffusion has been significantly enhanced without compromising atomic details. The model allows for efficient sampling of lipid-protein interactions at atomic resolution, thereby significantly enhancing the effectiveness of molecular dynamics simulations in capturing membrane-associated phenomena. In this review, after providing an overview of HMMM model development, we will describe briefly successful application of the model to study a variety of membrane processes, including lipid-dependent binding and insertion of peripheral proteins, the mechanism of phospholipid insertion into lipid bilayers, and characterization of optimal tilt angle of transmembrane helices. We conclude with practical recommendations for proper usage of the model in simulation studies of membrane processes.
Keywords: Cellular membrane, lipid bilayer, membrane proteins, peripheral proteins, molecular dynamics simulation
Introduction
Biological membranes are an essential component of all living cells (Gennis, 1989; Mouritsen, 2005). The cellular membrane is a complex and dynamic environment with a large diversity of lipids that shape its characteristics (van Meer et al, 2008; van Meer and de Kroon, 2011; Ingólfsson et al, 2014). It has been estimated that several hundred distinct types of lipids populate the typical mammalian plasma membrane (Sampaio et al, 2011; Klose et al, 2013), influencing membrane fluidity, geometry, pressure, and surface charge (Klose et al, 2013).
Approximately 26% of the human proteome is known to be functional only within membranes (Fagerberg et al, 2010) and these membrane proteins account for an estimated 60% of current drug targets (Yildirim et al, 2007). Membranes, and the proteins embedded in the membrane, are essential to life. They actively establish and maintain electrochemical gradients, and further exploit these gradients to perform meaningful transport of nutrients and waste into and out of the cell. Membrane proteins are also indispensable to cellular signaling, as they provide the coupling mechanism between the inside and outside of the cell (Grecco et al, 2011). Peripheral membrane proteins, which contain structurally varying lipid-binding globular domains that recognize specific membrane components (Lemmon, 2008), associate with the membrane surface and are involved in signaling, trafficking, and regulating cell structure.
The membrane itself can also be used as a signal. By modulating the lipid composition through leaflet dependent (van Meer et al, 2008; van Meer and de Kroon, 2011) exposure of anionic phospholipids (e.g. phosphatidylserine (PS)) or biosynthesis of particular rare lipid species, such as phosphatidylinositides or cardiolipin, the activity of peripheral (Lemmon, 2008) and transmembrane (Andersen and Keoppe, II, 2007) proteins can be drastically altered. Thus, the importance of the membrane and its dynamic character in cellular homeostasis cannot be overstated.
Despite the importance of the membrane and membrane proteins to the maintenance and survival of the cell, the inherent fluid nature of the lipid bilayer (Singer and Nicolson, 1972; Engelman, 2005) makes detailed experimental studies on the specific roles of lipids in membrane proteins extremely challenging. This problem is compounded for peripheral membrane proteins. Due to the reversible nature of the interaction between peripheral proteins and the membrane, crystallizing the membrane-bound structure of peripheral proteins is exceedingly difficult. While techniques such as SAXS (Denisov et al, 2004; Mosbaek et al, 2010; Elliott et al, 2010), EPR (Lin et al, 1998; Kohout et al, 2003), NMR (Goult et al, 2009), FRET (McCallum et al, 1997; Kohout et al, 2003), and mutagenesis studies can help determine the approximate binding face of peripheral proteins, more structural information is needed to reveal molecular details critical to membrane binding of peripheral proteins and the role of lipids within this binding.
Molecular dynamics (MD) simulations offer sufficient spatial and temporal resolutions to capture specific lipid-protein interactions and have been extensively used for this purpose (Ohkubo and Tajkhorshid, 2008; Lindahl and Sansom, 2008; Ayton and Voth, 2009; Lai et al, 2010; Khalid and Bond, 2013; Bucher et al, 2013; Braun et al, 2014). Due to the slow dynamics of lipids (Klauda et al, 2006; Wohlert and Edholm, 2006) on time scales accessible to conventional atomistic MD simulations, the membrane is effectively static, preventing one from sufficiently describing the lipid motion necessary for lipid mixing and adequate sampling of lipid-protein interactions. Alternative computational approaches have been devised to circumvent the problem of slow lipid dynamics, including coarse-grained (Shelley et al, 2001; Marrink et al, 2004; Izvekov and Voth, 2005; Marrink et al, 2007; Ayton et al, 2010; Marrink and Tieleman, 2013) and implicit (Feig and Brooks III, 2004; Im and Brooks III, 2005; Chen et al, 2006; Bu et al, 2007; Mondal et al, 2010) membrane models. These approaches, which sacrifice atomistic detail for accelerated dynamics, are of limited utility in resolving lipid-specific interactions vital to membrane protein function.
In order to circumvent the aforementioned technical issues precipitated by slow lipid diffusion, a recently developed atomic membrane model, termed the Highly Mobile Membrane Mimetic (HMMM) (Arcario et al, 2011; Ohkubo et al, 2012), provides a suitable alternative for studying protein-lipid interactions. The HMMM model first fully developed by Ohkubo et al (2012) is constructed using a combination of surfactant-like short-tailed lipids, representing in full atomic detail the lipid head groups that are often the main protein-interacting elements, with a liquid representation of the membrane core (Fig. 1). The HMMM representation significantly accelerates lateral lipid diffusion and enhances lipid-protein sampling (Arcario et al, 2011; Ohkubo et al, 2012). A particular strength of this membrane representation is that it affords multiple simulations of spontaneous protein interactions with the membrane in less time than is required to simulate the biased binding of a peripheral protein in a conventional simulation (Ohkubo et al, 2012; Baylon et al, 2013; Vermaas and Tajkhorshid, 2014a; Arcario and Tajkhorshid, 2014), providing for efficient and enhanced sampling of protein-lipid interactions. By design, the kinetics of membrane-associated processes are accelerated by the HMMM model due to faster lipid diffusion. As far as thermodynamical properties are concerned, the energetics associated with protein-membrane interactions is adequately captured for the interfacial and surface regions of lipid bilayers, but significant deviations are evident in the core of the membrane due to the fluid nature and polarity of the solvent used to replace the bulk of lipid tails (Pogorelov et al, 2014).
Fig. 1.

Comparison of a full-tail and HMMM membrane representations pictorially illustrated by gradual transformation of a POPC membrane (left) into an HMMM membrane (right). The hydrophobic core of the membrane (yellow) is replaced by an organic solvent in the HMMM membrane, while the headgroup and short tails of the lipids (typically first five carbons) are preserved.
The HMMM model has been successfully applied to simulation studies of several membrane-associated protein systems(Ohkubo et al, 2012; Baylon et al, 2013; Vermaas and Tajkhorshid, 2014a; Blanchard et al, 2014; Arcario and Tajkhorshid, 2014; Wu and Schulten, 2014; Rhéault et al, 2015). We begin this review by describing the development trajectory of the HMMM model, followed by a set of its recent applications to a wide variety of membrane-associated phenomena including: phospholipid insertion into membrane (Vermaas and Tajkhorshid, 2014b), binding and insertion of peripheral proteins such as cytochrome P450 (Baylon et al, 2013), hemoglobin N (Rhéault et al, 2015), talin (Arcario and Tajkhorshid, 2014), synaptotagmin I (Wu and Schulten, 2014), and α-synuclein (Vermaas and Tajkhorshid, 2014a), and studies of pre-fusion configuration of synaptobrevin transmembrane helix in a lipid bilayer (Blanchard et al, 2014). We conclude with outlining the best practices for utilizing the HMMM model and an outlook on its future developments.
Development Trajectory of the HMMM
The initial development of the HMMM was driven by a need to study membrane-binding processes and other phenomena at the membrane surface that were inaccessible to MD simulations. The test case that accompanied the initial development of the HMMM is the GLA domain, the membrane-binding domain of the vitamin K dependent clotting factors. The GLA domain is named for its characteristic vitamin K-dependent γ-carboxyglutamate (Gla) residues that tightly coordinate multiple Ca2+ ions. The coordinated Ca2+ ions then serve two functions: they play a structural role, maintaining the “active” form of the GLA domain, as well as an adhesive role, interacting with anionic phospholipid headgroups, including phosphatidylserine (PS) (Ohkubo and Tajkhorshid, 2008). Despite the established importance of PS lipids in the process, it was unclear which moieties of the GLA domain interacted with these lipids and to what degree the protein inserted in the membrane; while Nelsestuen (1999) proposed the N-terminal ω-loop and K32 of the GLA domain made interfacial contact with the membrane, Falls et al (2001) and Mizuno et al (2001) suggested that only residues in the hydrophobic “keel” inserted into the membrane. Given the overly simplistic view of the membrane going into these postulated models, they turned out to only partially capture the interactions involved in membrane binding of the GLA domain.
Ohkubo and Tajkhorshid (2008) explored membrane binding of the fVII GLA using conventional atomistic MD simulations with two alternative methods of triggering membrane binding, and incorporated available experimental data (McCallum et al, 1996; Waters et al, 2006; Colina et al, 2006) into the modeling process. The first method was to remove lipids from the membrane patch below the inserting species, and through successive minimization stages, forcibly insert the protein domain into the bilayer. In the second, the GLA domain was inserted into the bilayer through the use of steered MD (SMD) (Grubmüller et al, 1996; Leech et al, 1996; Izrailev et al, 1997), in which an external applied force drives the insertion over the course of the simulation. The resulting model was unique, in that both the hydrophobic ω-loop and Ca2+ ions contributed substantial interactions with the membrane.
While the devised approach successfully produced the most detailed model of a membrane-bound GLA domain, this approach was not broadly applicable to studying the insertion of other proteins that interact with the membrane. Without a target depth, SMD can easily result in a biased membrane-bound configuration and, due to the slow relaxation time of lipids, equilibration of several hundred nanoseconds or longer may be required to unbias the simulation system. Furthermore, without an applied bias, Ohkubo and Tajkhorshid (2008) did not observe GLA domain insertion into the membrane in over 350 ns of simulation. Finally, the model also depended on a homogeneous lipid composition, a pure PS bilayer, as, again, lateral lipid exchange is a slow process on atomistic MD timescales, and the mixing time for a realistic membrane is rather large, estimated to be on the microsecond timescale by Ingólfsson et al (2014) through coarse-grained simulations.
In order to address these sampling shortcomings, Arcario et al (2011) tested atomistic biphasic solvent systems for their ability to facilitate rapid partitioning of the human protein C (hPrC) GLA domain, an anticoagulant, at the interface between water and a hydrophobic phase. In these biphasic models, hydrophobic compounds whose parameters were available in the CHARMM general force field (Vanommeslaeghe et al, 2010) were constructed into a layer designed to represent the hydrophobic membrane core. The optimal compound had to be largely hydrophobic, liquid under typical simulation conditions, and preferably have a large diffusion constant to enable the membrane constituents to quickly reorganize around an inserting species. To a first approximation, the diffusion constant is inversely proportional to molecular size (Einstein, 1905); however, small hydrophobic compounds are gaseous. This poses a difficult optimization problem, as no natural compound is simultaneously small, hydrophobic, and liquid at ambient conditions, and so a compromise needs to be struck between these three competing goals (Fig. 2). Arcario et al (2011) tested four compounds, including 1,1-dichloroethane (DCLE), dimethyl sulfide (DMS), ethyl propyl ether (EPE), and heptane, which all represent approximate solutions to this problem.
Fig. 2.
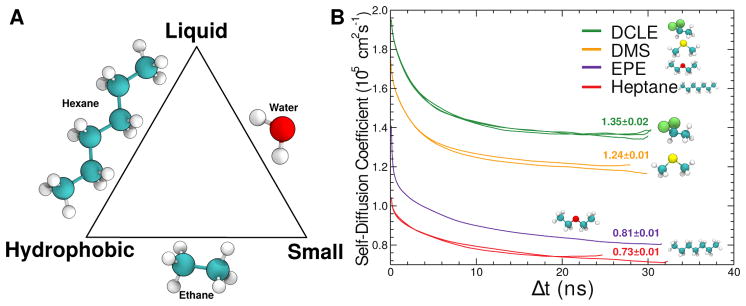
(A) The optimization triangle for desirable membrane mimics, emphasizing that natural compounds can only satisfy 2 of the 3 desired properties. (B) Self-diffusion constants for tested membrane mimics. Smaller compounds diffuse more rapidly, thereby quickly accommodating protein insertion. DCLE is 1,1-dichloroethane, DMS is dimethyl sulfide, and EPE is ethyl propyl ether. Adapted from Arcario et al (2011).
Partitioning of the GLA domain to the water/DCLE interface and partial insertion into the organic phase was fast, occurring spontaneously in under 10 ns, whereas for the other solvents tested, complete and permanent insertion was not observed (Arcario et al, 2011). Additionally, DCLE showed the largest diffusion constant among the compounds tested (Fig. 2), making it the clear choice for use as the membrane core mimic in biphasic simulation system studies. This biphasic insertion model, where a bilayer is modeled as a box of organic solvent, formed the initial step towards the development of the HMMM model.
What was added into the biphasic model formulation by Ohkubo et al (2012) are two layers of surfactants (short-tailed lipids), whose headgroups are identical to those found in physiological phospholipids, but where the acyl tails are truncated beyond the fifth carbon atom. The surfactants spontaneously partition to the interface of the DCLE-water biphasic system, generating a membrane mimetic (Ohkubo et al, 2012). When used to study the insertion of GLA domains, the mimetic enables not only the rapid spontaneous binding of the GLA domain to the membrane, but also recovers the specific Ca2+–headgroup interactions (Fig. 3). This result, combined with the favorable enhanced lipid lateral diffusion and headgroup atomic distributions (Ohkubo et al, 2012), began a push in the field to apply this methodology to proteins which are known or are thought to have substantial membrane interaction (Baylon et al, 2013; Wu and Schulten, 2014; Arcario and Tajkhorshid, 2014; Blanchard et al, 2014; Vermaas and Tajkhorshid, 2014a; Rhéault et al, 2015).
Fig. 3.

Spontaneous binding and insertion of the GLA domain to anionic membranes captured by HMMM. (A) The binding of GLA domains (purple trace) to PS headgroups is mediated by bound Ca2+ ions (purple spheres) and basic side chains (green licorice). Ohkubo and Tajkhorshid (2008) and Ohkubo et al (2012) studied the binding process in both conventional (left) and HMMM (right) bilayers. (B) Penetration depth over time for 10 independent HMMM binding simulations, which all bound to the target membrane within 30 ns and converged to a single insertion depth.
The potential to apply the HMMM model to other membrane-interacting peptides spurred further work, specifically in validating the protein-membrane interaction energetics of the model (Pogorelov et al, 2014). For determining the protein-membrane interaction characteristics, the procedure followed by Pogorelov et al (2014) closely matches that done in prior atomistic (MacCallum et al, 2008) and coarse-grained (Monticelli et al, 2008) computational studies, where the free energy profile (i.e. the potential of mean force, abbreviated PMF) along the membrane normal was computed for amino acid side chain analogues using umbrella sampling (Torrie and Valleau, 1977) and WHAM (Kumar et al, 1992). In general, the accuracy of the HMMM energetics relative to a conventional full membrane representation are on par with or superior to coarse-grained representations (Fig. 4). The accuracy is best on the membrane periphery, where the short-tailed lipids fully represent the atomic structure of a conventional bilayer. In the membrane interior, the fluid nature of the solvent comprising the hydrophobic core allows extra rotational degrees of freedom for bulkier side chains (e.g., Phe and Trp; Fig. 4B) to be populated, which stabilizes them in these regions beyond what is observed in a full lipid bilayer. Additionally, the DCLE solvent has a larger dipole than the lipid acyl tails it is replacing, and as a result stabilizes polar or charged side chains in the membrane interior to a greater extent than is observed in a conventional bilayer. These solvent artifacts can be responsibly managed by geometric constraints on the protein to prevent solvent intercalation where appropriate.
Fig. 4.
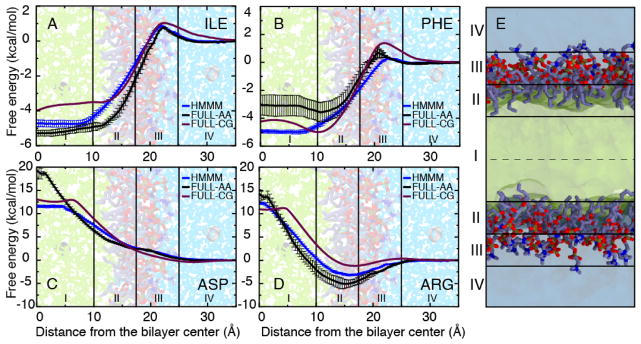
PMFs for representative side chain analogues: aliphatic - Ile (A), hydrophobic - Phe (B), and charged - Asp (C) and Arg (D). Data are presented for the HMMM membrane (blue), FULL-AA (black) (MacCallum et al, 2008), and FULL-CG (purple) (Monticelli et al, 2008). All PMFs are presented by setting the free energy to zero in aqueous solution. Membrane regions I–IV are defined in the panel (E). Error estimates were obtained using bootstrap analysis. Data from Pogorelov et al (2014).
Applications of HMMM to Membrane-Associated Phenomena
What makes these artifacts tolerable is the degree to which the HMMM model accelerates membrane-associated phenomena. Vermaas and Tajkhorshid (2014b) studied a model insertion system, which demonstrated that HMMM bilayers accelerate insertion of a single phospholipid by a factor of 3 by taking advantage of their higher lateral lipid diffusion to “welcome” the inserting species. In membrane-interacting peptide studies where the inserting species is larger, the impact of the accelerated diffusion is enhanced by 1–2 orders of magnitude, depending on the degree of membrane interaction with the inserting species (Ohkubo et al, 2012; Vermaas and Tajkhorshid, 2014a).
The accelerated lipid diffusion and resulting rapid insertion have been used to study membrane binding and membrane activity of various membrane-associated processes (Fig. 5). The applications include developing a model for phospholipid adsorption and desorption mechanics (Vermaas and Tajkhorshid, 2014b), membrane-induced conformational changes that modulate drug and oxygen entry pathways of cytochrome P450 (Baylon et al, 2013) or hemoglobin (Rhéault et al, 2015) and drive domain rearrangement of the F2F3 domains of talin (Arcario and Tajkhorshid, 2014). The HMMM has also been applied to several components of the cellular vesicle fusion machinery, including the Ca2+ sensitive synaptotagmin-1 (Wu and Schulten, 2014), the SNARE component synaptobrevin (Blanchard et al, 2014), and the flexible amphipathic helix α-synuclein (Vermaas and Tajkhorshid, 2014a). These applications will be examined in turn for their key findings in addition to the lessons that can be applied to other systems.
Fig. 5.

Pictorial summary of systems reviewed here studied using the HMMM membrane.
Phospholipid Insertion Mechanics: “One Tail at a Time”
While originally designed as a demonstration of the efficiency of the HMMM on a small test system where direct comparisons to conventional bilayer simulations are possible, the model for phospholipid insertion into biological membranes developed by Vermaas and Tajkhorshid (2014b) also led to novel insight as to the mechanism of phospholipid adsorption from solution, the primary mechanism for phospholipid exchange at low concentrations (McLean and Phillips, 1981; Nichols and Pagano, 1981). This study was complementary to work by Tieleman and Marrink (2006) and Grafmüller et al (2013), which used umbrella sampling simulations to determine the free energy of phospholipid unbinding using the position of the center of mass of the adsorbing or desorbing lipid as the reaction coordinate. Through repeated binding of phospholipids to both conventional and HMMM bilayers, Vermaas and Tajkhorshid (2014b) developed a kinetic model whereby it is demonstrated that realistic binding and unbinding pathways pass through distinct intermediate stages (Fig. 6A), consistent with splayed-tail intermediates observed by Smirnova et al (2010) in vesicle fusion studies.
Fig. 6.

(A) Schematic of the phospholipid insertion process, where the unbound phospholipid (green tails) associates with the membrane reversibly with rate ka, thereafter inserts a single tail with rate k1, followed shortly thereafter by the insertion of the second tail at rate k2 and headgroup reorganization at rate kw. The overall process is governed by the summation of these steps, and occurs at an overall rate kt. (B) Rate comparisons for individual steps from the schematic in (A) between inserting into an HMMM and a conventional full bilayer over different areas per lipid (AL). Each subpanel i–iv compares the rates for a different step, with a consistent color scheme across panels (blue for conventional bilayers, and red for HMMM bilayers). Adapted from Vermaas and Tajkhorshid (2014b). Copyright 2014 American Chemical Society.
The stages enumerated by Vermaas and Tajkhorshid (2014b) are roughly analogous to the process of putting on pants in the morning, in that it occurs one acyl tail at a time. The phospholipid first associates to the bilayer, and may bind and unbind reversibly looking for a suitable insertion location, much like when the reader may sort through their pants to select their favorite pair. After association, the phospholipid inserts its acyl tails in sequence irreversibly in the same manner as one would step into a pair of pants. Finally, the phospholipid puts on its belt when the headgroup reorients and relaxes within the bilayer.
This model is consistent between both a conventional and a HMMM representation, however by breaking down the process into individual steps, Vermaas and Tajkhorshid (2014b) also determined specifically what part of the overall process was accelerated, and how changing the membrane density impacts kinetics. Both reduced membrane density by increasing the area per lipid (AL) and the switch to a HMMM bilayer accelerated the rate of insertion (Fig. 6B). However digging deeper into the data, what is remarkable is where the acceleration for the HMMM bilayer is taking place. At physiological lipid densities, the HMMM accelerates the association step the most, leaving the rate of the first lipid insertion after association unchanged. The analysis of the second tail insertion rate is more complicated, as there are cases where the first tail inserted while the second acyl tail had not yet associated to the bilayer, and so the insertion rate of the second tail does not match. Vermaas and Tajkhorshid (2014b) concluded that HMMM preferentially accelerates the mechanistically simple part of insertion, namely the searching phase where the inserting species is sampling the bilayer surface for a gap to insert into. When this insight is applied to other systems, it suggests that the mechanism and interactions between the membrane and the inserting species are faithfully represented, with the insertion acceleration solely a result of the increased fluidity of the HMMM bilayer interface.
Increasing Drug Accessibility to Cytochrome P450 Through Membrane Binding
Cytochrome P450 (CYP) 3A4 is the most abundant isoform of the CYP family of enzymes in the human body, and it is involved in the metabolism of both xenobiotics, e.g., drugs, and endogenous compounds such as steroidal hormones. The large range of clinically available drugs metabolized by CYP3A4, more than 50% (Guengerich, 1999), has made this enzyme the target of extensive studies. Importantly, accumulating evidence suggests that interaction of CYP3A4 with the membrane is crucial for the recruitment of its lipophilic substrates to the active site of the enzyme (Williams et al, 2003; Schleinkofer et al, 2005), and therefore membrane binding and protein–lipid interactions might play critical roles in the function of the enzyme. However, details of these interactions were still elusive due to the lack of structural information concerning the membrane–bound form of CYP3A4.
Taking advantage of the enhanced lipid dynamics of the HMMM model, Baylon et al (2013) characterized the membrane-bound form of the CYP3A4 by performing multiple independent simulations in which spontaneous binding of the enzyme to the membrane was consistently captured. Five independent simulations in which the globular domain of CYP3A4 (i.e., without the transmembrane helix) was placed initially away from a PC membrane with different initial orientation were performed. In each case CYP3A4 was observed to bind to the membrane within 10 ns of the simulation. These simulations revealed structural elements located in the globular domain of CYP3A4 that mediate lipid interactions, and contribute to the insertion of the protein in the membrane. The convergence of the membrane-bound model of CYP3A4 was assessed by calculating the orientation of the protein in the membrane, measured as the relative angle of the heme moiety with respect to the membrane normal (Fig. 7), with an average of 72.2 ± 3.2° (Baylon et al, 2013). The calculated average angle supports the convergence of the membrane-bound model, since the final tilt angles deviate only by a few degrees, even after starting from initial orientations in solution that differed by 40°. Additional simulations of the membrane-bound CYP3A4 in which the transmembrane helix was added to the protein revealed that the orientation of the protein is mainly determined by the direct interaction of the globular domain with the membrane and not by the presence of the helix. Crucially, the orientation of HMMM bound CYP3A4 is comparable to linear dichroism measurements performed in parallel to the simulations for nanodisc-bound CYP3A4 (Baylon et al, 2013), where the average tilt angle was found to be 59.7 ± 4.1°, yielding excellent agreement between simulations and experiment.
Fig. 7.

Spontaneous membrane binding of CYP3A4 captured with the HMMM model. (A) Example trajectory of membrane binding. Membrane binding was consistently observed in 5 independent simulations (Mem-1 – Mem-5), permitting the identification of structural elements of CYP3A4 interacting with the membrane (e.g., A-anchor, F′-G′ helices shown in magenta). (B) Insertion of the A-anchor into the membrane (i.e., at or below the PO4 level) was consistently observed in all simulations, and converged to the same range of depths. The average positions of the phosphorus (PO4) and the nitrogen (choline) atoms of the lipid head groups are shown using brown and gray lines, respectively. (C) Each simulation began from a different orientation relative to the membrane, as measured by the heme tilt angle. The orientation for each of the simulations converged to around an average value of 72.2 ± 3.2°. The orientation of the protein was also measured experimentally, with an average tilt angle of 59.7 ± 4.1° (thick and thin dotted lines for average and standard deviation in tilt angle plot, respectively), yielding excellent agreement between simulations and experiment. Adapted from Baylon et al (2013). Copyright 2013 American Chemical Society.
From the simulations, Baylon et al (2013) observed that the interaction of CYP3A4 with the membrane favors transient opening of small access pathways leading to the active site by inducing conformational changes of side chains located at the protein–membrane interface. Interestingly, some of these access tunnels are not observed in the solution simulation of the enzyme, i.e., in the absence the membrane. Based on their location, these tunnels may function as access pathways for compounds in or out of the CYP3A4 active site, and highlight the importance of correctly capturing lipid-protein interactions and atomic detail in understanding the mechanism of this key enzyme in the human body. The resulting membrane-bound structures can be used in subsequent simulations to further probe different functions of CYP3A4 in the membrane with atomic detail, such as the recently characterized mechanism of drug-drug interaction mediated by membrane-bound CYP3A4 (Denisov et al, 2015).
Determination of the Membrane Binding Face of Hemoglobin N
Similar to cytochromes, hemoglobins are soluble proteins that are known to interact with the membrane (Fischer et al, 1975; Pathania et al, 2002). The primary function of hemoglobins are to bind small diatomic molecules, including O2 and NO (Milani et al, 2001; Oliveira et al, 2012; Mairbäurl and Weber, 2012), which, given their largely apolar nature, prefer to partition into the membrane (Subczynski and Hyde, 1983; Shiva et al, 2001). Thus to maximize access to its binding partners, hemoglobins can take advantage of membrane interactions which permit substrate access through one of several pathways previously identified by Daigle et al (2009). However prior to a combined computational and experimental study by Rhéault et al (2015), it was unknown what part of hemoglobin N from Mycobacterium tuberculosis interacted with the bilayer or which substrate access pathway would be preferred to permit efficient NO radical detoxification in a low O2 environment as is found in lesions caused by tuberculosis.
Through the use of the MPEx tool (Snider et al, 2009) and PPM server (Lomize et al, 2012), Rhéault et al (2015) generated initial potential membrane binding orientations for hemoglobin N above the membrane surface. Using the HMMM, Rhéault et al (2015) were able to achieve converged binding depths and orientations, which did not change upon subsequent simulation in a conventional bilayer representation. This understanding meshed with experimental findings that the membrane surface was dehydrated by the approach of the protein as measured by IR spectroscopy, however the simulations provided the atomic details of the interactions between the membrane and protein. This combined computational and experimental approach implicated the acidic residue Asp100 in modulating membrane binding via an electrostatic mechanism (Rhéault et al, 2015).
Membrane-Induced Structural Rearrangement of Talin
Integrins, as cell adhesion proteins, play an important role in many physiological and pathological processes, including cell differentiation, platelet aggregation, and tumor metastasis (Hynes, 2002; Liddington and Ginsberg, 2002; Felding-Habermann et al, 2004; Ratnikov et al, 2005; Ma et al, 2007). These heterodimeric transmembrane proteins are expressed in a default, low-affinity state that must be activated by the cytoskeletal-associated protein, talin (Tadokoro et al, 2003; Campbell and Ginsberg, 2004; Ratnikov et al, 2005; Ma et al, 2007; Moser et al, 2009; Shattil et al, 2010). Talin is composed of two main domains: the rod domain, which interacts with the actin cytoskeleton, and the head domain, which interacts with the cell membrane and activates integrin (Critchley, 2009). It is known that talin must bind to anionic phospholipids prior to activating integrin (Rees et al, 1990; Muguruma et al, 1995; Martel et al, 2001; Anthis et al, 2009; Saltel et al, 2009), demonstrating the importance of the membrane in the integrin activation process. While biochemical data (Martel et al, 2001; Saltel et al, 2009) suggest that the talin F3 subdomain, the subdomain responsible for integrin activation, contacts the membrane surface, based on crystallographic structures it was difficult to explain concomitant binding of both F2 and F3 subdomains to the membrane (Anthis et al, 2009; Elliott et al, 2010). Previous simulations (Kalli et al, 2010, 2013) utilized coarse-grained molecular dynamics to describe the process of membrane binding of talin and might have not been able to fully describe some of the atomic details important to the process. Moreover, while biophysical studies had proposed the presence of a hydrophobic membrane anchor that inserts into the membrane (Dietrich et al, 1993; Tempel et al, 1995; Isenberg and Goldmann, 1998; Seelig et al, 2000), the nature of such an anchor remained elusive.
To understand how the structural and dynamic changes in talin are modulated by membrane binding and to allow for optimal talin interaction with the membrane, Arcario and Tajkhorshid (2014) performed all atom MD simulations with a PS HMMM membrane, resulting in a reproducible model of the membrane-bound talin F2F3 (Fig. 8). The starting talin structure was placed in 5 randomized initial orientations with varying heights above and angles with the PS HMMM bilayer. Across all five simulations, talin bound to the membrane in a consistent manner and in three distinct steps. First, a collection of basic residues in the F2 subdomain, known as the membrane-orientation patch (MOP), is attracted to the anionic membrane surface (Fig. 8B). While this patch had been identified as crucial for talin binding in previous mutagenesis experiments (Wegener et al, 2007; Saltel et al, 2009; Kalli et al, 2010), additional residues belonging to the MOP were identified via these simulations. Following initial association via the MOP, an initially buried, Phe-rich hydrophobic membrane anchor is exposed and inserted into the membrane core (Fig. 8C). Previous experimental studies predicted the existence of just such a hydrophobic anchor (Dietrich et al, 1993; Tempel et al, 1995; Isenberg and Goldmann, 1998; Seelig et al, 2000), however its identification and description had proven elusive both to structural and other computational studies (Anthis et al, 2009; Elliott et al, 2010; Kalli et al, 2010). Finally, a large-scale, interdomain conformational change is induced by the membrane, leading to spontaneous binding of the F3 domain to the anionic membrane (Fig. 8D). This large conformational change produces a membrane-bound state in which the F2 and F3 subdomains interact with the membrane simultaneously, bridging the gap and reconciling previous discrepancies between biochemical and structural studies. Following these local and large-scale conformational changes, talin is optimally positioned to interact with integrin.
Fig. 8.
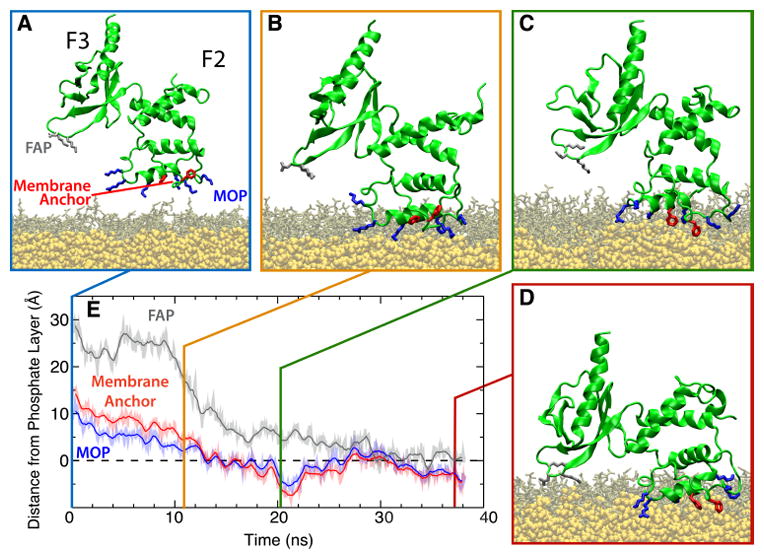
Membrane-binding sequence of the talin F2F3 subdomain. (A) Initial configuration of the talin F2F3 subdomain (green) in the PS HMMM system (DVPS, brown; DCLE, yellow). Residues comprising the MOP (blue), FAP (gray), and Phe-rich membrane anchor (red) are shown. In all images, water and ions have been omitted for clarity. (B–D) The three distinct stages of talin binding to the membrane are demonstrated. Talin is initially attracted to the membrane via MOP residues (B), followed by insertion of the Phe-rich membrane anchor (C), and, finally, a large, interdomain conformational change that brings the F3 subdomain into contact with the membrane surface (D). (E) Timeseries of the height of the MOP, FAP, and Phe-rich membrane anchor with reference to the phosphate layer of the cis leaflet (dashed line) from a representative membrane-binding simulation are shown. See Arcario and Tajkhorshid (2014) for data on all five independent simulations.
In order to confirm the validity of this membrane bound model, Arcario and Tajkhorshid (2014) simulated the membrane-bound talin in a full-tailed, conventional membrane. Starting from a membrane-bound configuration obtained from HMMM simulations, DOPS lipids were matched exactly to existing short-tailed PS HMMM headgroups and the tails oriented towards the center of the membrane, thereby preserving the specific lipid-protein interactions established in HMMM simulations. Over a series of successive steps, the membrane was relaxed around a fixed, membrane-bound talin. Following membrane relaxation, talin was simulated for 100 ns and remained stably bound throughout the simulation, retaining all the molecular interactions observed during the HMMM simulations.
Membrane Curvature Triggered by Synaptotagmin I
Neurotransmitter release is mediated by a Ca2+-triggered membrane fusion event that fuses synaptic vesicles to the presynaptic membrane. During vesicle release, synaptotagmin I (Syt1), acts as a Ca2+ sensor protein, binding to and bending the presynaptic membrane with its C2B domain, thereby regulating membrane fusion (Hui et al, 2009; Martens et al, 2007). With the help of MD simulations employing the HMMM model, Wu and Schulten (2014) proposed a molecular mechanism for how the C2B domain of Syt1 bends the presynaptic membrane and facilitates membrane fusion. Upon membrane binding, C2B moves its C-terminal helix towards the membrane, and have both its Ca2+ loop and the C-terminal helix shallowly insert into an anionic membrane. This shallow insertion causes a pressure imbalance between the two membrane leaflets and thus causes membrane bending, which might facilitate the fusion event.
Wu and Schulten (2014) applied the HMMM model (Ohkubo et al, 2012) to the first stage of the simulations to accelerate C2B membrane binding and lipid redistribution around the domain, leading to spontaneous C2B binding within 30 ns. Importantly, this study is an example of a mixed bilayer HMMM application, where PS and PC headgroups were mixed in a 1:1 ratio to observe lipid redistribution of PS around the inserting C2B domain, which was found to concentrate around the Ca2+-binding loops and lysine residues in the C-terminal helix. In simulations without anionic lipids, the Ca2+ loop does not bind to the membrane. Both the hydrophobic lipid tails and DCLE interact with the hydrophobic residues in the C-terminal helix, resulting a ~ 40 × 15 Å2 region devoid of lipid head groups around the helix in the cis membrane leaflet (Fig. 9). Such lipid behavior has also been reported in many membrane-active peptides, such as melittin (Bernèche et al, 1998), influenza hemagglutinin (Lagüe et al, 2005; Larsson and Kasson, 2013), and has been proposed to facilitate membrane fusion (Smirnova et al, 2010; Risselada et al, 2011; Larsson and Kasson, 2013).
Fig. 9.
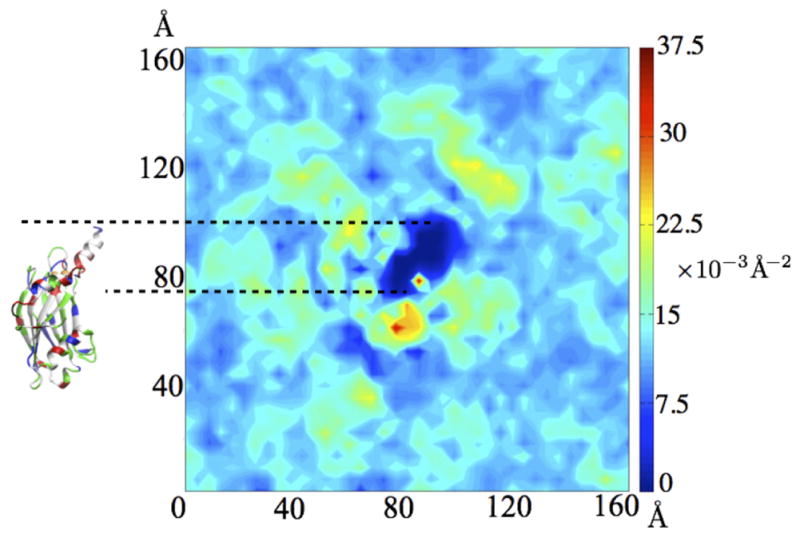
Two-dimensional lipid head group density of the cis leaflet around synaptotagmin C2B domain (top view) resulting from simulations employing the HMMM model. The C2B domain with its protruding C-terminal helix is shown to scale on the left. The color of the density plot reflects the lipid occupancy per unit area, measured using the phosphorous atom of the lipid headgroup.
In the second phase, the HMMM lipids were converted back to full lipids, in order to recover membrane mechanical properties necessary for reliable description of protein induced membrane bending. Membrane bending within the HMMM is unusual in HMMM bilayers, as the organic solvents decouples the top and bottom leaflets from one another, and generally localizes bending to the cis-leaflet. With the C2B domain already inserted in the first step, the C2B domain bends the presynaptic membrane spontaneously within 100 ns with a ~102 nm diameter membrane curvature. Due to the shallow insertion of the C-terminal helix into the membrane, only lipid head groups are displaced from the insertion site, while lipid tails in the cis membrane leaflet approach the helix hydrophobic residues and become disordered. Coupling with the resulting void in the cis leaflet, lipids in the trans leaflet adopt a more stretched and ordered conformation. The difference in lipid-tail ordering between the proximal and distal membrane leaflets generates a pressure imbalance across the bilayers, which serves as the driving force for C2B-driven membrane bending.
Membrane Origins of Structural Heterogeneity in α-Synuclein
α-Synuclein (αS) is a 14 kDa protein that is unstructured in solution, but forms helices when it binds to anionic membranes (Burré et al, 2013; Lokappa et al, 2014). Membrane-induced conformational changes of αS are of particular interest, given the connection between αS misfolding and aggregation and Parkinson’s disease (Singleton et al, 2003; Charras et al, 2004). Experiments have identified two predominant states for monomeric αS bound to anionic membranes, a horseshoe-like broken helix identified with NMR and EPR (Ulmer et al, 2005; Drescher et al, 2008; Rao et al, 2010) where a kink brings together the two helical segments, and an extended kink-free helix conformation without a kink postulated by ESR and DEER experiments (Jao et al, 2004; Georgieva et al, 2008; Jao et al, 2008; Trexler and Rhoades, 2009). It has been suggested that the extended and broken-helix conformations could interconvert (Georgieva et al, 2010; Robotta et al, 2011; Lokappa and Ulmer, 2011), potentially in a lipid-dependent fashion (Borbat et al, 2006; Middleton and Rhoades, 2010). Outside of coarse-grained simulations (Braun et al, 2012; Braun and Sachs, 2015), this conformational diversity had not been sampled, and presented a prime candidate for using the HMMM model to investigate the specific lipid interactions that determine the membrane-bound structure of αS.
Unlike in prior atomistic simulations where αS needed to be placed within the bilayer (Perlmutter et al, 2009), the HMMM model allowed Vermaas and Tajkhorshid (2014a) to observe spontaneous binding and insertion of αS to the bilayer and arrive at an unbiased pool of membrane bound αS conformers. The pool had a mean insertion depth consistent with prior simulation and experimental findings (Jao et al, 2004; Perlmutter et al, 2009), although the variability observed in the HMMM systems was larger. Vermaas and Tajkhorshid (2014a) also noted that interaction between αS and PS broke apart interhelical hydrogen bonds between the N- and C-terminal helices in a subset of the simulations. Intercalation of PS between the helices provides alternative binding partners for basic residues, driving the helices apart (Fig. 10). In a conventional bilayer, mixing occurs too slowly to effectively sample the formation of PS microclusters underlying this process. In addition to using the HMMM to enhance lipid sampling, it was critical that the simulations were repeated several times, given the stochastic nature of the process. Only 20 % of the trials demonstrated a semi-extended conformation where the inter-helix angle (θh) are greater than 47° (Fig. 10).
Fig. 10.
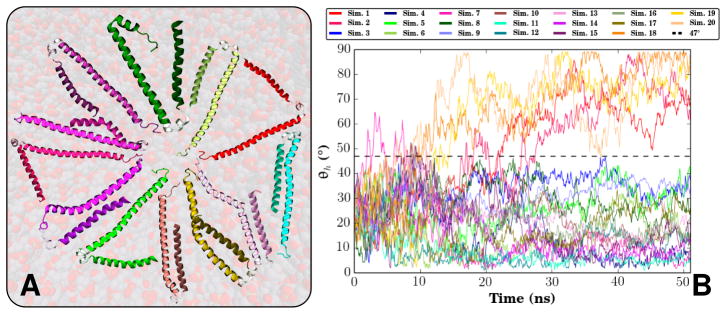
(A) Selected end states of the αS monomers across the 20 HMMM membrane-binding simulations conducted, highlighting the variability of αS when bound to the membrane. (B) Inter-helix angle (θh) for all 20 membrane-binding simulations. θh is defined by the angle between the two vectors formed by a linear interpolation of the α carbons of the N- and C-helices. θh distinguishes between two states, a broken helix state where θh never exceeds 47°, and a semi-extended intermediate state where the inter-helix angle is approximately 90°. Originally published in Vermaas and Tajkhorshid (2014a), used with permission from Elsevier B.V.
To distinguish these weak signals that occur infrequently, at least 10, and preferably 20 independent simulations need to be run to ensure that the observed behavior is a significant result. With the rapid insertion enabled by the HMMM, this level of statistical significance can be obtained with modest computational investment, estimated by Vermaas and Tajkhorshid (2014a) to be an order of magnitude less than what is required in a conventional lipid bilayer for the same degree of natural variability. Even with the advent of special purpose machines such as Anton (Shaw et al, 2008, 2014), using the HMMM allows researchers to see rare events at the membrane interface more frequently and gather superior statistics on the process of interest.
Acrobatics of the Synaptobrevin Transmembrane Domain
SNARE complexes, the machinery of neuronal exocytosis, are composed of fusion proteins located at both the plasma (syntaxin, SNAP-25) and vesicular (synaptobrevin) membrane surfaces (Fasshauer et al, 1997; Südhof, 2004; Südhof and Rothman, 2009). These proteins initially embedded in different (vesicular and plasma) membranes must intimately interact, forming a 120 Å-long triple helix, in order to prime the neurotransmitter-filled vesicle for fusion with the neuronal plasma membrane (Sutton et al, 1998). Synaptobrevin comprises the vesicular component of the SNARE complex and is responsible for targeting a particular vesicle to the plasma membrane and priming it for membrane fusion. While the post-fusion structure of the SNARE complex is well studied (Brunger, 2005; Jahn and Scheller, 2006; Südhof and Rothman, 2009; Stein et al, 2009) and known to stand parallel with the membrane normal (Sutton et al, 1998) with multiple interhelical interactions apparent (Brunger et al, 2009), the details of the pre-fusion state of the individual SNARE proteins is not as well understood.
Studies on the transmembrane (TM; residues 95–116) and linker (residues 85–94) regions have suggested a large tilt (~30–50°) of the linker region of synaptobrevin with respect to the membrane normal (Kweon et al, 2003; Bowen and Brunger, 2006). Conflicting theories on how to explain this large tilt have emerged, however, with some expressing that the linker region is a continuous helix (Bowen and Brunger, 2006) and some arguing for a disordered structure (Durrieu et al, 2009) allowing flexibility between the TM and cytosolic (residues 1–84) domains. The structure of the TM and linker regions is extremely important to the function of synaptobrevin as minor alterations drastically affect the fusion potential of the SNARE complex (McNew et al, 1999; Kweon et al, 2003; Ngatchou et al, 2010). The challenge in finding the spontaneous native tilt of synaptobrevin using MD simulations, however, is the extended time scale associated with lipid diffusion. Tilting of a transmembrane helix requires significant rearrangement of lipids in both membrane leaflets.
To overcome these limitations, Blanchard et al (2014) carried out simulations using the HMMM model with short-tailed PC lipids to attempt to distinguish between these two hypotheses. This study represents the first example of a trans-membrane application of the HMMM model. Five independent simulations, with the initial tilt angle of synaptobrevin ranging from −20° to +40°, were performed, each simulation converging to a tilt angle of 40.1±3.4° within 50 ns (Fig. 11B). The tilt angle resulting from these simulations is within the range provided by previous structural studies (Kweon et al, 2003; Bowen and Brunger, 2006), and the fact that the independent simulations converge from a wide range of starting configurations removes the possibility that the final tilt angle was biased by its initial orientation. Blanchard et al (2014) also identified specific motifs of charged and aromatic residues that dictate the native tilt of synaptobrevin, offering mechanistic insight into why previous mutants had diminished fusion ability (Kweon et al, 2003).
Fig. 11.
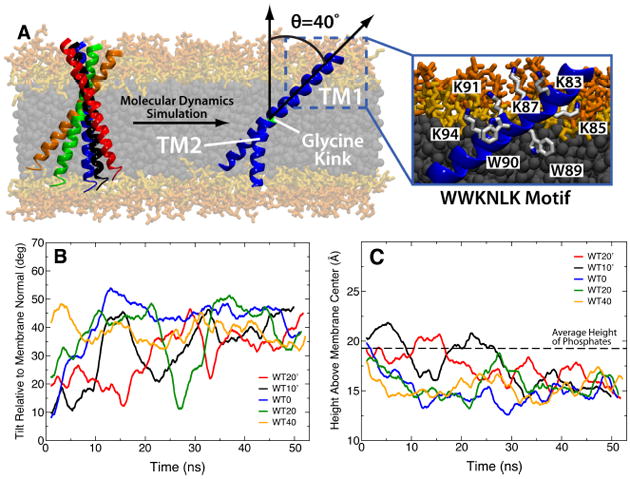
Native tilt angle of the synaptobrevin TM domain. (A) The five initial conditions of synaptobrevin tilt simulated are shown together with the final structure. All simulations converged to an average tilt of 40.1±3.4° with the two segments of the helix (TM1 and TM2) mechanically decoupled by the G100 “glycine kink”. A detailed image of the specific interactions leading to the well-defined orientation of TM1 are shown in the inset. Plots of the angle relative to membrane normal (B) and height of tryptophan residues above membrane center (C) are shown for each of the five simulations, with the color of the trace in each plot matching the color of the helix in (A). In (C), the black dashed line represents the average height of phosphates in the leaflet closest to the tryptophans (upper leaflet in (A)) across all simulations.
Additionally, because of the fluidity of the DCLE layer, Blanchard et al (2014) were able to observe significant dynamics of the TM segment of synaptobrevin on the MD timescales that contain structural implications for previous experimental findings. In particular, while the “linker” region remains completely helical, consistent with circular dichroism (CD) studies (Bowen and Brunger, 2006), a glycine residue at the 100 position disrupts the helical structure and allows the lower portion of the TM domain (TM2) to move independently of the upper portion (TM1) (Fig. 11A). Thus, while TM1 adopts a clearly defined angle relative to the membrane normal, TM2 does not and explores much more space. This exact behavior had been proposed by Bowen and Brunger (2006) as a possibility to explain large discrepancies in CD signals between the “linker” and TM regions of synaptobrevin. The structural findings made possible by the HMMM remained unchanged when Blanchard et al (2014) built a conventional bilayer model starting from the HMMM endpoint, highlighting the general applicability of the HMMM to study particular elements of transmembrane systems.
Best Practices
If these example applications have inspired you to use the HMMM in your own research, there are several key points where the setup of HMMM simulations is different from a conventional membrane simulation. The most consequential difference is that use of the HMMM requires a membrane with fixed surface area, either by running at constant volume or a constant area barostat. In a conventional constant-ratio barostat, the surfactant-like headgroups may cause membrane expansion (Fig. 12A). For membrane-binding studies, this expansion is beneficial to a degree, as it both increases lateral lipid diffusion modestly (Fig. 12B) and creates space for an incoming protein. However if left unchecked the expansion may result in a sparse membrane, and so prior HMMM simulations used a fixed area ensemble, with the pressure controlled only by varying the dimension parallel to the membrane normal (Ohkubo et al, 2012; Baylon et al, 2013; Vermaas and Tajkhorshid, 2014b,a; Blanchard et al, 2014; Arcario and Tajkhorshid, 2014). A good starting area per lipid considers the estimated volume of lipids displaced by the incoming protein. Even though it is not possible to know a priori how deep the penetration of the protein into the membrane will be, a reasonable rule of thumb is that the area per lipid initially should be approximately 5–10 % higher than what is observed in a conventional bilayer, which results in a final effective area per lipid that is nearly physiological (Baylon et al, 2013; Vermaas and Tajkhorshid, 2014a).
Fig. 12.
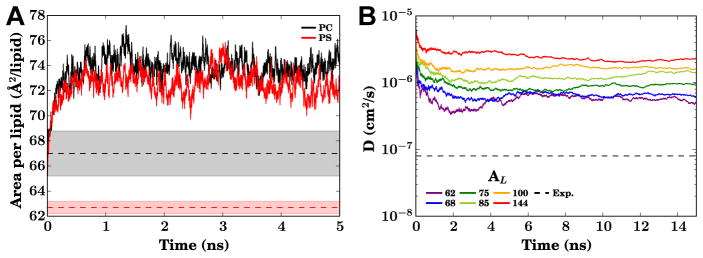
(A) The evolution of the area per lipid of HMMM bilayers in a constant ratio barostat for both PC (black) and PS (red) headgroups, highlighting the bilayer expansion possible due to the shorter acyl tails. For reference, the range of experimental areas per lipid (AL) determined through a combination of X-ray and neutron scattering methods by Fogarty et al (2015) are reported as shaded regions, with the mean as a dashed line. (B) Lateral lipid diffusion constant (DL) time evolution for PC HMMM bilayers over a range of AL maintained by a fixed-area barostat. The dashed line in this panel represents the DL of ~ 8 × 10−8 cm2s−1 for DPPC lipids(Klauda et al, 2006; Wohlert and Edholm, 2006), consistent with most conventional bilayers. The simulations behind this data were conducted using NAMD 2.10 (Phillips et al, 2005) on bilayers constructed using CHARMM-GUI (Jo et al, 2008, 2009; Wu et al, 2014) at 310 K.
In dense membranes, the short acyl tails of the headgroup surfactants may also cause the headgroups to go into solution. While partitioning into the aqueous phase is a natural surfactant-like behavior expected for short-tailed lipids, this is generally undesirable for a bilayer mimic, as it reduces the number of lipids in the membrane. In order to address this behavior, a mild 0.1 kcal/mol/Å2 restraint along the membrane normal can be applied to the carbonyl carbon of the acyl tails (C21 and C31 in CHARMM parlance). With this restraint, the z-positions of the membrane components still readily fluctuates by approximately 3.5 Å in each direction at room temperature, resulting in excellent overlap between the membrane profiles of corresponding atoms in HMMM and conventional bilayers. As for the requirement for fixed area ensembles, these recommendations can be adjusted to suit the system, such as using a physiological lipid density for transmembrane peptides which are already inserted (Blanchard et al, 2014).
For larger transmembrane systems than those discussed in this review, special care needs to be taken to avoid intercalation of organic solvent into the transmembrane domain. Since DCLE is slightly polar, it can and will disrupt the interactions that hold together transmembrane proteins if left unchecked. This can be addressed by specialized in silico solvent models currently in development. However for many applications it is sufficient to restrain the transmembrane domain to retain its shape through the use of harmonic restraints on the atomic positions to keep the protein packed correctly. Such restraints have minimal bearing on the lateral diffusion rate of the lipids around the protein, and do not prevent specific lipid binding sites around the protein from being explored.
In terms of constructing HMMM systems, the step by step procedure is virtually identical to how one would construct a conventional bilayer. For typical situations, where the interest is on sampling an unbiased range of conformations, it is important to generate a diverse starting pool of initial bilayers so that the final state is not unduly biased by the starting geometry of the bilayer. The simplest means of manufacturing HMMM bilayers with diverse lipid compositions is to use the HMMM Builder as recently implemented in CHARMM-GUI (Jo et al, 2008, 2009; Wu et al, 2014). An HMMM bilayer can also be constructed from a conventional bilayer by replacing the entire acyl tail beyond carbon 5 with an organic solvent. Special care, however, should be taken in packing in the right amount of organic solvent, as overfilling or underfilling the bilayer interior can lead to unstable bilayers. In order to match membrane volumes between HMMM and conventional bilayers, 3 DCLE molecules should be added for every 14 acyl tail carbons that have been removed, plus any additional adjustments that need to be made to create a higher AL if desired:
Here, M is the number of DCLE molecules to be added to the bilayer center, C is the number of acyl tail carbons removed from the conventional bilayer, and is the ratio of the AL (area per lipid) between the target HMMM bilayer and the conventional bilayer initially generated.
The final point is that the HMMM excels in the initial sampling phases, but should be converted back into a conventional bilayer at the end prior to extensive equilibrium simulations. In general, it is preferred that the existing short acyl tails from the HMMM lipids are preserved during this conversion process, since the existing tails will not already clash with the other membrane components. Vermaas and Tajkhorshid (2014a) describes an additional procedural enhancement through the use of the organic solvent heavy atom coordinates to seed the positions of the acyl tails around the inserted species, however in many instances using internal coordinate tables to extend the tails results in acceptable starting geometries for continued simulation (Baylon et al, 2013; Wu and Schulten, 2014).
Future Directions
Along with experimental characterization of additional peripheral proteins whose binding to membranes is controlled by the lipid composition and with functional assays further establishing the regulatory roles of specific lipid molecules in membrane protein function in general, the HMMM has a bright future as a method to complement these findings with a mechanistic description of the specific lipid-protein interactions at work. The model itself is also expected to evolve, both through the addition of designer in silico solvents that ameliorate deficiencies in the representation of the hydrophobic cores of the membrane, and new problems in membrane biology to which the HMMM could be applied.
Many of these applications move beyond the realm of peripheral proteins, e.g., into transmembrane systems. One such application is to use the HMMM to identify lipid-specific binding sites on integral membrane proteins with affordable computational effort, as the accelerated lateral lipid diffusion would allow for more rapid sampling on easily accessible computational timescales. Other pressing challenges in membrane biophysics, such as ion-induced lipid clustering and protein-induced enrichment of specific lipids in its surrounding micro-environment, would continue to significantly benefit from the rapid sampling of lipid-protein interactions the HMMM provides. In particular, when used in conjunction with the recent advent of routine μs simulation, the HMMM can begin to tackle challenges long beyond the reach of conventional MD, such as the assembly of membrane-embedded super-complexes at an atomic level of detail. Thus, the number of applications of the HMMM will only continue to grow into the future.
Acknowledgments
This work was supported in part by the National Institutes of Health (Grants R01-GM101048, R01-GM086749, U54-GM087519, and P41-GM104601 to E.T.) and XSEDE compute resources (grant TG-MCA06N060 to E.T. and grant TG-MCB130112 to T.V.P.). J.V.V. acknowledges support from the Sandia National Laboratories Campus Executive Program, which is funded by the Laboratory Directed Research and Development (LDRD) Program. Sandia is a multiprogram laboratory managed and operated by Sandia Corporation, a wholly owned subsidiary of Lockheed Martin Corporation, for the US Department of Energy’s National Nuclear Security Administration under Contract No. DE-AC04-94AL85000, and previous support from the DOE CSGF Fellowship (DE-FG02-97ER25308). M.J.A. acknowledges past support from the NSF GRF Program. Z.W. acknowledges support from the NSF-funded Center of Physics in Living Cell (NSF PHY1430124). T.V.P. is grateful for the support from the Illinois Campus Research Board. T.V.P. was a Faculty Fellow of the National Center for Supercomputing Applications when this work was compleleted.
Footnotes
Compliance with Ethical Standards
The authors declare no conflicts of interest. This original review has not been submitted to any other journal for consideration. This is a review of prior work funded publicly, and appropriate figures have been reused or adapted with permission from the original authors and publishers. The nature of the presented work is purely computational and does not include human or animal subjects.
References
- Andersen OS, Keoppe RE., II Bilayer thickness and membrane protein function: An energetic perspective. Annu Rev Biophys Biomol Struct. 2007;36:107–130. doi: 10.1146/annurev.biophys.36.040306.132643. [DOI] [PubMed] [Google Scholar]
- Anthis NJ, Wegener KL, Ye F, Kim C, Goult BT, Lowe ED, Vakonakis I, Bate N, Critchley DR, Ginsberg MH, Campbell ID. The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J. 2009;28:3623–3632. doi: 10.1038/emboj.2009.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcario MJ, Tajkhorshid E. Membrane-induced structural rearrangement and identification of a novel membrane anchor in Talin F2F3. Biophys J. 2014;107(9):2059–2069. doi: 10.1016/j.bpj.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcario MJ, Ohkubo YZ, Tajkhorshid E. Capturing spontaneous partitioning of peripheral proteins using a biphasic membrane-mimetic model. J Phys Chem B. 2011;115:7029–7037. doi: 10.1021/jp109631y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton GS, Voth GA. Systematic multiscale simulation of membrane protein systems. Curr Opin Struct Biol. 2009;19:138–144. doi: 10.1016/j.sbi.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton GS, Lyman E, Voth GA. Hierarchical coarse-graining strategy for protein-membrane systems to access mesoscopic scales. Faraday Discuss. 2010;144:347–357. doi: 10.1039/b901996k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylon JL, Lenov IL, Sligar SG, Tajkhorshid E. Characterizing the membrane-bound state of cytochrome P450 3A4: structure, depth of insertion, and orientation. J Am Chem Soc. 2013;135(23):8542–8551. doi: 10.1021/ja4003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernèche S, Nina M, Roux B. Molecular dynamics simulation of melittin in a dimyristoylphosphatidylcholine bilayer membrane. Biophys J. 1998;75:1603–1618. doi: 10.1016/S0006-3495(98)77604-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard AE, Arcario MJ, Schulten K, Tajkhorshid E. A highly tilted membrane configuration for the pre-fusion state of synaptobrevin. Biophys J. 2014;107:2112–2121. doi: 10.1016/j.bpj.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbat P, Ramlall TF, Freed JH, Eliezer D. Inter-helix distances in lysophos-pholipid micelle-bound α-Synuclein from pulsed ESR measurements. J Am Chem Soc. 2006;128:10,004–10,005. doi: 10.1021/ja063122l. [DOI] [PubMed] [Google Scholar]
- Bowen M, Brunger AT. Conformation of the synaptobrevin transmembrane domain. Proc Natl Acad Sci USA. 2006;103:8378–8383. doi: 10.1073/pnas.0602644103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun AR, Sachs JN. α-synuclein reduces tension and increases undulations in simulations of small unilamella vesicles. 2015 doi: 10.1016/j.bpj.2015.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun AR, Sevcsik E, Chin P, Rhoades E, Tristram-Nagle S, Sachs JN. α-synuclein induces both positive mean curvature and negative gaussian curvature in membranes. J Am Chem Soc. 2012;134:2613–2620. doi: 10.1021/ja208316h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun AR, Lacy MM, Ducas VC, Rhoades E, Sachs JN. α-Synuclein-induced membrane remodeling is driven by binding affinity, partition depth, and interlea et order asymmetry. J Am Chem Soc. 2014;136(28):9962–9972. doi: 10.1021/ja5016958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger A, Weninger K, Bowen M, Chu S. Single-molecule studies of the neuronal SNARE fusion machinery. Annu Rev Biochem. 2009;78:903–28. doi: 10.1146/annurev.biochem.77.070306.103621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT. Structure and function of SNARE and SNARE-interacting proteins. Quart Rev Biophys. 2005;38:1–47. doi: 10.1017/S0033583505004051. [DOI] [PubMed] [Google Scholar]
- Bu L, Im W, Brooks CL., III Membrane assembly of simple helix homo-oligomers studied via molecular dynamics simulations. Biophys J. 2007;92:854–863. doi: 10.1529/biophysj.106.095216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher D, Hsu YH, Mouchlis VD, Dennis EA, McCammon JA. Insertion of the ca2+-independent phospholipase a2 into a phospholipid bilayer via coarse-grained and atomistic molecular dynamics simulations. PLoS Comput Biol. 2013;9(7):e1003, 156. doi: 10.1371/journal.pcbi.1003156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J, Vivona S, Diao J, Sharma M, Brunger AT, Südhof TC. Properties of native brain α-synuclein. Nature. 2013;498:E4–E6. doi: 10.1038/nature12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell ID, Ginsberg MH. The talin-tail interaction places integrin activation on FERM ground. Trends Biochem Sci. 2004;29:429–435. doi: 10.1016/j.tibs.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Charras GT, Williams BA, Sims SM, Horton MA. Estimating the sensitivity of mechanosensitive ion channels to membrane strain and tension. Biophys J. 2004;87:2870–2884. doi: 10.1529/biophysj.104.040436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Im W, Brooks CL., III Balancing solvation and intramolecular interactions: Toward a consistent generalized Born force field. J Am Chem Soc. 2006;128:3728–3736. doi: 10.1021/ja057216r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colina CM, Venkateswarlu D, Duke R, Perera L, Pedersen LG. What causes the enhancement of activity of factor VIIa by tissue factor? J Thromb Haem. 2006;4:2726–2729. doi: 10.1111/j.1538-7836.2006.02222.x. [DOI] [PubMed] [Google Scholar]
- Critchley DR. Biochemical and structural properties of the integrin-associated cytoskeletal protein talin. Annu Rev Biophys. 2009;28:235–254. doi: 10.1146/annurev.biophys.050708.133744. [DOI] [PubMed] [Google Scholar]
- Daigle R, Rousseau JA, Guertin M, Lagüe P. Theoretical investigations of nitric oxide channeling in Mycobacterium tuberculosis truncated hemoglobin n. Biophys J. 2009;97(11):2967–2977. doi: 10.1016/j.bpj.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J Am Chem Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- Denisov IG, Grinkova YV, Baylon JL, Tajkhorshid E, Sligar SG. Mechanism of drug-drug interactions mediated by human cytochrome P450 CYP3A4 monomer. Biochemistry. 2015;54(13):2227–2239. doi: 10.1021/acs.biochem.5b00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich C, Goldmann W, Sackmann E, Isenberg G. Interaction of NBD-talin with lipid monolayers. FEBS Lett. 1993;324:37–40. doi: 10.1016/0014-5793(93)81527-7. [DOI] [PubMed] [Google Scholar]
- Drescher M, Veldhuis G, van Rooijen BD, Milikisyants S, Subramaniam V, Huber M. Antiparallel arrangement of the helicies of vesicle-bound α-Synuclein. J Am Chem Soc. 2008;130:7796–7797. doi: 10.1021/ja801594s. [DOI] [PubMed] [Google Scholar]
- Durrieu MP, Bond PJ, Sansom MSP, Lavery R, Baaden M. Coarse-grain simulations of the R-SNARE fusion protein in its membrane environment detect long-lived conformational sub-states. ChemPhysChem. 2009;10:1548–1552. doi: 10.1002/cphc.200900216. [DOI] [PubMed] [Google Scholar]
- Einstein A. Über die von der molekularkinetischen Theorie der Wärme geforderte Bewegung von in ruhenden Flüssigkeiten suspendierten Teilchen. Ann d Phys. 1905;17:549–560. doi: 10.1002/andp.19053220806. [DOI] [Google Scholar]
- Elliott PR, Goult BT, Kopp PM, Bate N, Grossmann JG, Roberts GC, Critchley DR, Barsukov IL. The structure of the talin head reveals a novel extended conformation of the FERM domain. Structure. 2010;18:1289–1299. doi: 10.1016/j.str.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman DM. Membranes are more mosaic than fluid. Nature. 2005;438:578–580. doi: 10.1038/nature04394. [DOI] [PubMed] [Google Scholar]
- Fagerberg L, Jonasson K, von Heijne G, Uhlén M, Berglund L. Prediction of the human membrane proteome. Proteomics. 2010;10(6):1141–1149. doi: 10.1002/pmic.200900258. [DOI] [PubMed] [Google Scholar]
- Falls LA, Furie BC, Jacobs M, Furie B, Rigby AC. The ω-loop region of the human prothrombin γ-carboxyglutamic acid domain penetrates anionic phospholipid membranes. J Biol Chem. 2001;276:23,895–23,902. doi: 10.1074/jbc.M008332200. [DOI] [PubMed] [Google Scholar]
- Fasshauer D, Otto H, Eliason WK, Jahn R, Brunger AT. Structural changes are associated with soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor complex formation. J Biol Chem. 1997;272:28,036–28,041. doi: 10.1074/jbc.272.44.28036. [DOI] [PubMed] [Google Scholar]
- Feig M, Brooks CL., III Recent advances in the development and application of implicit solvent models in biomolecule simulations. Curr Opin Struct Biol. 2004;14:217–224. doi: 10.1016/j.sbi.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Felding-Habermann B, Lerner RA, Lillo A, Zhuang S, Weber MR, Arrues S, Gao C, Mao S, Saven A, Janda KD. Combinatorial antibody libraries from cancer patients yield ligand-mimetic Arg-Gly-Asp-containing immunoglobulins that inhibit breast cancer metastasis. Proc Natl Acad Sci USA. 2004;101:17,210–17,215. doi: 10.1073/pnas.0407869101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer S, Nagel RL, Bookchin RM, Jr, EFR, Tellez-Nagel I. The binding of hemoglobin to membranes of normal and sickle erythrocytes. Biochim Biophys Acta Biomembr. 1975;375(3):422–433. doi: 10.1016/0005-2736(75)90357-0. [DOI] [PubMed] [Google Scholar]
- Fogarty JC, Arjunwadkar M, Pandit SA, Pan J. Atomically detailed lipid bilayer models for the interpretation of small angle neutron and x-ray scattering data. Biochim Biophys Acta Biomembr. 2015;1848(2):662–672. doi: 10.1016/j.bbamem.2014.10.041. [DOI] [PubMed] [Google Scholar]
- Gennis RB. Biomembranes: Molecular Structure and Function. Springer-Verlag; New York: 1989. [Google Scholar]
- Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D. Membrane-bound alpha-synuclein forms an extended helix: Long-distance pulsed ESR measurements using vesicles, bicelles and rod-like micelles. J Am Chem Soc. 2008;130:12,856–12,857. doi: 10.1021/ja804517m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D. The lipid-binding domain of wild type and mutant α-synuclein: Compactness and interconversion between the broken and extended helix forms. J Biol Chem. 2010;285:28,261–28,274. doi: 10.1074/jbc.M110.157214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goult BT, Bate N, Anthis NJ, Wegener KL, Gingras AR, Patel B, Barsukov IL, Campbell ID, Roberts GC, Critchley DR. The structure of an interdomain complex that regulates talin activity. J Biol Chem. 2009;284:15,097–15,106. doi: 10.1074/jbc.M900078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafmüller A, Lipowsky R, Knecht V. Effect of tension and curvature on the chemical potential of lipids in lipid aggregates. Phys Chem Chem Phys. 2013;15(3):876–881. doi: 10.1039/c2cp43018e. [DOI] [PubMed] [Google Scholar]
- Grecco HE, Schmick M, Bastiaens PIH. Signaling from the living plasma membrane. Cell. 2011;144(6):897–909. doi: 10.1016/j.cell.2011.01.029. [DOI] [PubMed] [Google Scholar]
- Grubmüller H, Heymann B, Tavan P. Ligand binding and molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 1996;271:997–999. doi: 10.1126/science.271.5251.997. [DOI] [PubMed] [Google Scholar]
- Guengerich PF. Cytochrome P450 3A4: regulation and role in drug metabolism. Annual Review of Pharmacology and Toxicology. 1999;39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- Hui E, Johnson CP, Yao J, Dunning FM, Chapman ER. Synaptotagmin-mediated bending of target membrane is a critical step in Ca2+-regulated fusion. Cell. 2009;138:709–721. doi: 10.1016/j.cell.2009.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. Integrins: Bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Im W, Brooks CL., III Interfacial folding and membrane insertion of designed peptides studied by molecular dynamics simulations. Proc Natl Acad Sci USA. 2005;102:6771–6776. doi: 10.1073/pnas.0408135102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingólfsson HI, Melo MN, van Eerden FJ, Arnarez C, López CA, Wassenaar TA, Periole X, De Vries AH, Tieleman DP, Marrink SJ. Lipid organization of the plasma membrane. J Am Chem Soc. 2014;136(14):14,554–14,559. doi: 10.1021/ja507832e. [DOI] [PubMed] [Google Scholar]
- Isenberg G, Goldmann WH. Peptide-specific antibodies localize the major lipid binding sites of talin dimers to oppositely arranged N-terminal 47 kDa subdomains. FEBS Lett. 1998;426:165–170. doi: 10.1016/s0014-5793(98)00336-6. [DOI] [PubMed] [Google Scholar]
- Izrailev S, Stepaniants S, Balsera M, Oono Y, Schulten K. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys J. 1997;72:1568–1581. doi: 10.1016/S0006-3495(97)78804-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izvekov S, Voth GA. Multiscale coarse graining of liquid-state systems. J Chem Phys. 2005;123:134, 105. doi: 10.1063/1.2038787. [DOI] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs - engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- Jao CC, Der-Sarkissian A, Chen J, Langen R. Structure of membrane-bound α-synuclein studied by site-directed spin-labeling. Proc Natl Acad Sci USA. 2004;101:8331–8336. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc Natl Acad Sci USA. 2008;105(50):19,666–19,671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J Comp Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Jo S, Lim JB, Klauda JB, Im W. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys J. 2009;97:50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli AC, Wegener KL, Goult BT, Anthis NJ, Campbell ID, Sansom MS. The structure of the talin/integrin complex at a lipid bilayer: An NMR and MD simulation study. Structure. 2010;18:1280–1288. doi: 10.1016/j.str.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli AC, Campbell ID, Sansom MSP. Conformational changes in talin on binding to anionic phospholipid membranes facilitate signaling by integrin transmembrane helices. PLoS Comput Biol. 2013;9(10):e1003, 316. doi: 10.1371/journal.pcbi.1003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalid S, Bond PJ. Multiscale molecular dynamics simulations of membrane proteins. Methods in Molecular Biology. 2013;924:635–657. doi: 10.1007/978-1-62703-017-5_25. [DOI] [PubMed] [Google Scholar]
- Klauda JB, Brooks BR, Pastor RW. Dynamical motions of lipids and a finite size effect in simulations of bilayers. J Chem Phys. 2006;125:144, 710. doi: 10.1063/1.2354486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose C, Surma MA, Simons K. Organellar lipidomics – background and perspectives. Curr Opin Cell Biol. 2013;25:406–413. doi: 10.1016/j.ceb.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Kohout SC, Corbalán-García S, Gómez-Fernández JC, Falke JJ. C2 domain of protein kinase Cα: Elucidation of the membrane docking surface by site-directed fluorescence and spin labeling. Biochemistry. 2003;42:1254–1265. doi: 10.1021/bi026596f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comput Chem. 1992;13:1011–1021. [Google Scholar]
- Kweon DH, Kim CS, Shin YK. Regulation of neuronal SNARE assembly by the membrane. Nat Struct Biol. 2003;10:440–447. doi: 10.1038/nsb928. [DOI] [PubMed] [Google Scholar]
- Lagüe P, Roux B, Pastor RW. Molecular dynamics simulations of the influenza hemagglutinin fusion peptide in micelles and bilayers: Conformational analysis of peptide and lipids. J Mol Biol. 2005;354:1129–1141. doi: 10.1016/j.jmb.2005.10.038. [DOI] [PubMed] [Google Scholar]
- Lai CL, Landgraf KE, Voth GA, Falke JJ. Membrane docking geometry and target lipid stoichiometry of membrane-bound PKC C2 domain: A combined molecular dynamics and experimental study. J Mol Biol. 2010;402:301–310. doi: 10.1016/j.jmb.2010.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson P, Kasson PM. Lipid tail protrusion in simulations predicts fusogenic activity of influenza fusion peptide mutants and conformational models. PLoS Comput Biol. 2013;9:e1002, 950. doi: 10.1371/journal.pcbi.1002950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leech J, Prins J, Hermans J. SMD: Visual steering of molecular dynamics for protein design. IEEE Comp Sci Eng. 1996;3(4):38–45. [Google Scholar]
- Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- Liddington RC, Ginsberg MH. Integrin activation takes shape. J Cell Biol. 2002;158:833–839. doi: 10.1083/jcb.200206011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SW, Kochendoerfer GG, Carrol KS, Wang D, Mathies RA, Sakmar TP. Mechanisms of spectral tuning in blue cone visual pigments. Visible and Raman spectroscopy of blue-shifted rhodopsin mutants. J Biol Chem. 1998;273:24,583–24,591. doi: 10.1074/jbc.273.38.24583. [DOI] [PubMed] [Google Scholar]
- Lindahl E, Sansom MSP. Membrane proteins: molecular dynamics simulations. Curr Opin Struct Biol. 2008;18:425–431. doi: 10.1016/j.sbi.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Lokappa SB, Ulmer TS. Alpha-synuclein populates both elongated and broken helix states on small unilamellar vesicles. J Biol Chem. 2011;286(24):21,450–21,457. doi: 10.1074/jbc.M111.224055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokappa SB, Suk JE, Balasubramanian A, Samanta S, Situ AJ, Ulmer TS. Sequence and membrane determinants of the random coil-helix transition of α-synuclein. J Mol Biol. 2014;426(10):2130– 2144. doi: 10.1016/j.jmb.2014.02.024. [DOI] [PubMed] [Google Scholar]
- Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. OPM database and ppm web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:D370–376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Qin J, Plow E. Platelet integrin αIIbβ3: Activation mechanisms. J Thromb Haem. 2007;5:1345–1352. doi: 10.1111/j.1538-7836.2007.02537.x. [DOI] [PubMed] [Google Scholar]
- MacCallum JL, Bennett WFD, Tieleman DP. Distribution of amino acids in a lipid bilayer from computer simulations. Biophys J. 2008;94:3393–3404. doi: 10.1529/biophysj.107.112805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mairbäurl H, Weber RE. Oxygen transport by hemoglobin. Compr Physio. 2012;2(2):1463–1489. doi: 10.1002/cphy.c080113. [DOI] [PubMed] [Google Scholar]
- Marrink SJ, Tieleman DP. Perspective on the MARTINI model. Chemical Society Reviews. 2013;42:6801–6822. doi: 10.1039/c3cs60093a. [DOI] [PubMed] [Google Scholar]
- Marrink SJ, de Vries AH, Mark AE. Coarse grained model for semiquantitative lipid simulations. J Phys Chem B. 2004;108:750–760. [Google Scholar]
- Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH. The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- Martel V, Racaud-Sultan C, Dupe S, Marie C, Paulhe F, Galmiche A, Block MR, Albiges-Rizo C. Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J Biol Chem. 2001;276:21,217–21,227. doi: 10.1074/jbc.M102373200. [DOI] [PubMed] [Google Scholar]
- Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316:1205–1208. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- McCallum CD, Hapak RC, Neuenschwander PF, Morrissey JH, Johnson AE. The location of the active site of blood coagulation factor VIIa above the membrane surface and its reorientation upon association with tissue factor. J Biol Chem. 1996;271:28,168–28,175. doi: 10.1074/jbc.271.45.28168. [DOI] [PubMed] [Google Scholar]
- McCallum CD, Su B, Neuenschwander PF, Morrissey JH, Johnson AE. Tissue factor positions and maintains the factor VIIa active site far above the membrane surface even in the absence of the factor VIIa Gla domain — A fluorescence resonance energy transfer study. J Biol Chem. 1997;272:30,160–30,166. doi: 10.1074/jbc.272.48.30160. [DOI] [PubMed] [Google Scholar]
- McLean LR, Phillips MC. Mechanism of cholesterol and phosphatidylcholine exchange or transfer between unilamellar vesicles. Biochemistry. 1981;20:2893–2900. doi: 10.1021/bi00513a028. [DOI] [PubMed] [Google Scholar]
- McNew JA, Weber T, Engelman DM, Söllner TH, Rothman JE. The length of the flexible SNAREpin juxtamembrane region is a critical determinant of SNARE–dependent fusion. Mol Cell. 1999;4:415–421. doi: 10.1016/s1097-2765(00)80343-3. [DOI] [PubMed] [Google Scholar]
- van Meer G, de Kroon AIPM. Lipid map of the mammalian cell. J Cell Sci. 2011;124(Pt 1):5–8. doi: 10.1242/jcs.071233. [DOI] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton ER, Rhoades E. Effects of curvature and composition on α-Synuclein binding to lipid vesicles. Biophys J. 2010;99:2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani M, Pesce A, Ouellet Y, Ascenzi P, Guertin M, Bolognesi M. Mycobacterium tuberculosis hemoglobin n displays a protein tunnel suited for o2 diffusion to the heme. EMBO J. 2001;20(15):3902–3909. doi: 10.1093/emboj/20.15.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H, Fujimoto Z, Atoda H, Morita T. Crystal structure of an anticoagulant protein in complex with the Gla domain of factor X. Proc Natl Acad Sci USA. 2001;98:7230–7234. doi: 10.1073/pnas.131179698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal J, Zhu X, Cui Q, Yethiraj A. Sequence-dependent interaction of β-peptides with membranes. J Phys Chem B. 2010;114:13,585–13,592. doi: 10.1021/jp1070242. [DOI] [PubMed] [Google Scholar]
- Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink SJ. The MARTINI coarse grained forcefield: Extension to proteins. J Chem Theor Comp. 2008;4:819–834. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- Mosbaek CR, Nolan D, Persson E, Svergun DI, Bukrinsky JT, Vestergaard B. Extensive small-angle X-ray scattering studies of blood coagulation factor VIIa reveal interdomain felxibility. Biochemistry. 2010;49:9739–9745. doi: 10.1021/bi1011207. [DOI] [PubMed] [Google Scholar]
- Moser M, Legate KR, Zent R, Fässler R. The tail of integrins, talin, and kindlins. Science. 2009;324:895–899. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
- Mouritsen OG. Life–as a Matter of Fat. 1. Springer–Verlag; Berlin: 2005. [Google Scholar]
- Muguruma M, Nishimuta S, Tomisaka Y, Ito T, Matsumura S. Organisation of the functional domains in membrane cytoskeletal protein talin. J Biochem. 1995;117:1036–1042. doi: 10.1093/oxfordjournals.jbchem.a124803. [DOI] [PubMed] [Google Scholar]
- Nelsestuen GL. Enhancement of vitamin-K-dependent protein function by modification of the γ-carboxyglutamic acid domain: Studies of protein C and factor VII. Trends Cardiovasc Med. 1999;9:162–167. doi: 10.1016/s1050-1738(99)00024-9. [DOI] [PubMed] [Google Scholar]
- Ngatchou AN, Kisler K, Fang Q, Walter AM, Zhao Y, Bruns D, Sørensen JB, Lindau M. Role of synaptobrevin C terminus in fusion pore formation. Proc Natl Acad Sci USA. 2010;107:18,463–18,468. doi: 10.1073/pnas.1006727107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols JW, Pagano RE. Kinetics of soluble lipid monomer diffusion between vesicles. Biochemistry. 1981;20(10):2783–2789. doi: 10.1021/bi00513a012. [DOI] [PubMed] [Google Scholar]
- Ohkubo YZ, Tajkhorshid E. Distinct structural and adhesive roles of Ca2+ in membrane binding of blood coagulation factors. Structure. 2008;16:72–81. doi: 10.1016/j.str.2007.10.021. [DOI] [PubMed] [Google Scholar]
- Ohkubo YZ, Pogorelov TV, Arcario MJ, Christensen GA, Tajkhorshid E. Accelerating membrane insertion of peripheral proteins with a novel membrane mimetic model. Biophys J. 2012;102:2130–2139. doi: 10.1016/j.bpj.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira A, Singh S, Bidon-Chanal A, Forti F, Martí MA, Boechi L, Estrin DA, Dikshit KL, Luque FJ. Role of PheE15 gate in ligand entry and nitric oxide detoxification function of Mycobacterium tuberculosis truncated hemoglobin n. PLoS One. 2012;7(11) doi: 10.1371/journal.pone.0049291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania R, Navani NK, Rajamohan G, Dikshit KL. Mycobacterium tuberculosis hemoglobin HbO associates with membranes and stimulates cellular respiration of recombinant Escherichia coli. J Biol Chem. 2002;277(18):15,293–15,302. doi: 10.1074/jbc.M111478200. [DOI] [PubMed] [Google Scholar]
- Perlmutter JD, Braun AR, Sachs JN. Curvature Dynamics of α-Synuclein Familial Parkinson Disease Mutants. J Biol Chem. 2009;284:7177–7189. doi: 10.1074/jbc.M808895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comp Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogorelov TV, Vermaas JV, Arcario MJ, Tajkhorshid E. Partitioning of amino acids into a model membrane: Capturing the interface. J Phys Chem B. 2014;118:1481–1492. doi: 10.1021/jp4089113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao JN, Jao CC, Hegde BG, Langen R, Ulmer TS. A combinatorial NMR and EPR apporoach for evaluating the structural ensemble of partially folded proteins. J Am Chem Soc. 2010;132:8657–8668. doi: 10.1021/ja100646t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnikov B, Partridge A, Ginsberg M. Integrin activation by talin. J Thromb Haem. 2005;3:1783–1790. doi: 10.1111/j.1538-7836.2005.01362.x. [DOI] [PubMed] [Google Scholar]
- Rees D, Ades SE, Singer S, Hynes RO. Sequence and domain structure of talin. Nature. 1990;347:685–689. doi: 10.1038/347685a0. [DOI] [PubMed] [Google Scholar]
- Rhéault JF, Gagné E, Guertin M, Lamoureux G, Auger M, Lagüe P. Molecular model of hemoglobin N from Mycobacterium tuberculosis bound to lipid bilayers: A combined spectroscopic and computational study. Biochemistry. 2015;54(11):2073–2084. doi: 10.1021/bi5010624. [DOI] [PubMed] [Google Scholar]
- Risselada H, Kutzner C, Grubmuller H. Caught in the act: Visualization of SNARE-mediated fusion events in molecular detail. ChemBioChem. 2011;12:1049–1055. doi: 10.1002/cbic.201100020. [DOI] [PubMed] [Google Scholar]
- Robotta M, Braun P, van Rooijen B, Subramaniam V, Huber M, Drescher M. Direct evidence of coexisting horseshoe and extended helix conformations of membrane-bound alpha-synuclein. ChemPhysChem. 2011;12:267–269. doi: 10.1002/cphc.201000815. [DOI] [PubMed] [Google Scholar]
- Saltel F, Mortier E, Hytönen VP, Jacquier MC, Zimmermann P, Vogel V, Liu W, Wehrle-Haller B. New PI(4,5)P2- and membrane proximal integrin-binding motifs in the talin head control β3-integrin clustering. J Cell Biol. 2009;187:715–731. doi: 10.1083/jcb.200908134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio JL, Gerla MJ, Klosea C, Ejsingb CS, Beugc H, Simonsa K, Shevchenko A. Membrane lipidome of an epithelial cell line. Proc Natl Acad Sci USA. 2011;108:1903–1907. doi: 10.1073/pnas.1019267108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleinkofer K, Sudarko, Winn PJ, Lüdemann SK, Wade RC. Do mammalian cytochrome P450s show multiple ligand access pathways and ligand channelling? EMBO Reports. 2005;6(6):584–9. doi: 10.1038/sj.embor.7400420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig A, Blatter XL, Frentzel A, Isenberg G. Phospholipid binding of synthetic talin peptides provides evidence for an intrinsic membrane anchor of talin. J Biol Chem. 2000;275:17,954–17,961. doi: 10.1074/jbc.M002264200. [DOI] [PubMed] [Google Scholar]
- Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: The end game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw DE, Deneroff MM, Dror RO, Kuskin JS, Larson RH, Salmon JK, Young C, Batson B, Bowers KJ, Chao JC, Eastwood MP, Gagliardo J, Grossman JP, Ho CR, Ierardi DJ, Kolossváry I, Klepeis JL, Layman T, McLeavey C, Moraes MA, Mueller R, Priest EC, Shan Y, Spengler J, Theobald M, Towles B, Wang SC. Anton, a special-purpose machine for molecular dynamics simulation. Comm ACM. 2008;51:91–97. [Google Scholar]
- Shaw DE, Grossman J, Bank JA, Batson B, Butts JA, Chao JC, Deneroff MM, Dror RO, Even A, Fenton CH, et al. Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis. IEEE Press; 2014. Anton 2: raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer; pp. 41–53. [Google Scholar]
- Shelley JC, Shelley MY, Reeder RC, Bandyopadhyay S, Moore PB, Klein ML. Simulations of phospholipids using a coarse grain model. J Phys Chem B. 2001;105:9785–9792. [Google Scholar]
- Shiva S, Brookes PS, Patel RP, Anderson PG, Darley-Usmar VM. Nitric oxide partitioning into mitochondrial membranes and the control of respiration at cytochrome c oxidase. Proc Natl Acad Sci USA. 2001;98(13):7212–7217. doi: 10.1073/pnas.131128898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer S, Nicolson G. The fluid mosaic model of the structure of cell membranes. Science. 1972;173:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. α-synuclein locus triplication causes parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Smirnova YG, Marrink SJ, Lipowsky R, Knecht V. Solvent-exposed tails as prestalk transition states for membrane fusion at low hydration. J Am Chem Soc. 2010;132(19):6710–6718. doi: 10.1021/ja910050x. [DOI] [PubMed] [Google Scholar]
- Snider C, Jayasinghe S, Hristova K, White SH. MPEx: A tool for exploring membrane proteins. Prot Sci. 2009;18:2624–2628. doi: 10.1002/pro.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, Weber G, Wahl MC, Jahn R. Helical extension of the neuronal SNARE complex into the membrane. Nature. 2009;460:525–528. doi: 10.1038/nature08156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subczynski WK, Hyde JS. Concentration of oxygen in lipid bilayers using a spin-label method. Biophys J. 1983;41(3):283–286. doi: 10.1016/S0006-3495(83)84439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Südhof TC, Rothman J. Membrane fusion: Grappling with SNARE and SM proteins. Science. 2009;323:474–7. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, dePereda JM, Ginsberg MH, Calderwood DA. Talin binding to integrin β tails: A final common step in integrin activation. Science. 2003;302:103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- Tempel M, Goldmann WH, Isenberg G, Sackmann E. Interaction of the 47-kDa talin fragment and the 32-kDa vinculin fragment with acidic phospholipids: A computer analysis. Biophys J. 1995;69:228–241. doi: 10.1016/S0006-3495(95)79894-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieleman DP, Marrink SJ. Lipids out of equilibrium: Energetics of desorption and pore mediated flip-flop. J Am Chem Soc. 2006;128:12,462–12,467. doi: 10.1021/ja0624321. [DOI] [PubMed] [Google Scholar]
- Torrie GM, Valleau JP. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J Comp Phys. 1977;23:187–199. [Google Scholar]
- Trexler AJ, Rhoades E. α-Synuclein binds large unilamellar vesicles as an extended helix. Biochemistry. 2009;48:2304–2306. doi: 10.1021/bi900114z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, MacKerell AD., Jr CHARMM General Force Field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comp Chem. 2010;31(4):671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermaas JV, Tajkhorshid E. Conformational heterogeneity of α-synuclein in membrane. Biochim Biophys Acta Biomembr. 2014a;1838(12):3107–3117. doi: 10.1016/j.bbamem.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermaas JV, Tajkhorshid E. A microscopic view of phospholipid insertion into biological membranes. J Phys Chem B. 2014b;118:1754–1764. doi: 10.1021/jp409854w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters EK, Yegneswaran S, Morrissey JH. Raising the active site of factor VIIa above the membrane surface reduces its procoagulant activity but not factor VII autoactivation. J Biol Chem. 2006;281:26,062–26,068. doi: 10.1074/jbc.M604915200. [DOI] [PubMed] [Google Scholar]
- Wegener KL, Partridge AW, Han J, Pickford AR, Liddington RC, Ginsberg MH, Campbell ID. Structural basis of integrin activation by talin. Cell. 2007;128:171–182. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- Williams PA, Cosme J, Ward A, Angove HC, Matak Vinković D, Jhoti H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature. 2003;424(6947):464–8. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- Wohlert J, Edholm O. Dynamics in atomistic simulations of phospholipid membranes: Nuclear magnetic resonance relaxation rates and lateral diffusion. J Chem Phys. 2006;125:204–703. doi: 10.1063/1.2393240. [DOI] [PubMed] [Google Scholar]
- Wu E, Cheng X, Jo S, Rui H, Song K, Dávila-Contreras E, Qi Y, Lee J, Monje-Galvan V, Venable R, Klauda J, Im W. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J Comp Chem. 2014;35(27):1997–2004. doi: 10.1002/jcc.23702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Schulten K. Synaptotagmin’s role in neurotransmitter release likely involves Ca2+-induced conformational transition. Biophys J. 2014;107:1156–1166. doi: 10.1016/j.bpj.2014.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim MA, Goh KI, Cusick ME, Barabasi AL, Vidal M. Drug–target network. Nat Biotechnol. 2007;25:1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]