

Abstract
The availability of reference genome sequences, especially the human reference, has revolutionized the study of biology. However, whilst the genomes of some species have been fully sequenced, a wide range of biological problems still cannot be effectively studied for lack of genome sequence information. Here, I identify neglected areas of biology and describe how both targeted species sequencing and more broad taxonomic surveys of the tree of life can address important biological questions. I enumerate the significant benefits that would accrue from sequencing a broader range of taxa, as well as discuss the technical advances in sequencing and assembly methods that would allow for wide-ranging application of whole-genome analysis. Finally, I suggest that in addition to “Big Science” survey initiatives to sequence the tree of life, a modified infrastructure-funding paradigm would better support reference genome sequence generation for research communities most in need.
Biology fundamentals from the genome reference
Freely available whole-genome reference sequences – the genome sequences in the public domain (Table 1) with annotated gene models and viewable in browsers – have been so immensely successful, valuable, and accessible that they are now taken for granted in many research communities. Despite what is clearly a paradigm shift, the number of available sequences is actually quite low, and access to well-annotated genomes is limited. For example, some relatively common model organisms have only incomplete or poorly annotated genomes, such as maize, and others have no publicly available genome, including Xenopus laevis, the sequence of which is still awaiting publication. Here, I propose that additional references surveying the tree of life are a necessary foundation for the study of biology in the 21st century and will enable biology to transcend its observational roots and become more of an engineering discipline. I begin by illustrating the extent of the transformation genome references enable in biology by noting the successes and techniques brought about by the sequencing of the human genome. I then discuss how reference genome sequences could bring about a similar revolution for the remainder of the tree of life.
Table 1.
Where are the reference genomes?
| Focus | Database | URL | Notes |
|---|---|---|---|
| All Sequences | NCBI genbank | www.ncbi.nlm.nih.gov/genbank/ | The International Nucleotide Sequence Database Collection (INSDC) collects all sequences |
| EMBL-ENA | www.ebi.ac.uk/ena | ||
| DNA Databank of Japan | www.ddbj.nig.ac.jp | ||
| Genome Annotation Portals | Ensemble Genomes | http://ensemblgenomes.org | |
| NCBI-Refseq/entrez | www.ncbi.nlm.nih.gov/refseq/ | ||
| Example Large Community based Databases | UCSC Genome Browsers | http://genome.ucsc.edu | Focused on Mammals These model organism based databases link genome and gene sequences to other reagents and mutant lines, publications and, for E. coli, systems biology |
| Mouse Genome Informatics | www.informatics.jax.org | ||
| Flybase | http://flybase.org | ||
| Wormbase | www.wormbase.org | ||
| Saccharomyces Genome db | www.yeastgenome.org | ||
| EcoCyc E. coli database | http://ecocyc.org | ||
| Plant genome database | www.plantgdb.org | ||
| Ortholog databases | OrthoDB | http://orthodb.org | Rapid lookup of orthologous genes across many species |
| PhylomeDB | http://phylomedb.org |
In assessing the impact of the human reference sequence, it is instructive to remember a time when the number of human protein coding genes was thought to be as high as 120,000 (although sensible approaches placed the number lower [1]). A GeneSweep pool [2] was held at the 2000 Cold Spring Harbor Laboratory Biology of Genomes meeting, and all estimates of human gene number – by the world’s assembled genomics experts – were significantly higher than the actual number revealed in 2003, which has since been refined down further [3]. The genome sequencing revolution is still in its infancy; however we must acknowledge it as the major driver of biology since the start of the 21st century. Much of the credit for these successes is due to the US National Human Genome Research Institute (NHGRI) and its surrounding community, whose leadership has driven sequencing technology, investigation of genome biology, and general human and model organism biology for the past two decades.
Reference genomes also enable analysis of RNAseq data. In the human genome, we now contrast protein coding sequence comprising ~1% of the genome with extensive transcription of large amounts of the genome and the assignment of function to as much as possibly 80% of the genome [4]. Combining RNAseq and a reference to align that data to enabled the discovery of new classes of non-coding RNA such as ~8,000 human long non-coding RNAs (lncRNAs) [5]. The genome sequence is also the structural framework for the transcriptional machinery and the source of information to be transcribed. The ENCODE project extended our functional understanding of the human reference genome by annotating transcription-factor binding sites, enhancers, chromatin accessibility and modification patterns, and the identification of eQTLs. These have facilitated deeper understanding of epigenetic regulation of RNA processing, non-coding RNA, and regulatory networks, and sparked the growing appreciation for the importance of the three dimensional structure of the functioning cellular genome [4]. Overall, observational descriptions of the human genome have resolved previous misunderstandings (such as gene number) and unknowns (such as transcriptional capability), but most importantly they provide the necessary foundation for current and future progress in fundamental biology and clinical medicine.
Technology Designed Around the Human Reference Genome Leads the Way
Humans, like much of the tree of life, do not share the traits of classic genetic models such as Drosophila, mice, and yeast, which have short life spans and whose gene expression can be experimentally controlled. Thus, human genetic analyses based on short-read alignment to reference genomes are directly applicable to the majority of species. For example, re-sequencing a single patient can identify natural Mendelian causative alleles or de novo mutations. Sequencing 2,000 exomes from patients referred to a medical genetics clinic led to a diagnosis for 25% of patients [6]. Genome sequencing of individuals is routine in model organisms [7] [8, 9], but has also been used for other species, such as dogs [10] where it was used to identify mutations underlying the neurodegenerative disorder neuronal ceroid lipofuscinosis and shed light on the same disease in humans. Genome-wide association studies (GWAS) based on SNP, exome, and genome sequencing of cohorts have contributed to our understanding of complex disease genetics identifying over 15,000 regions associated with the majority of common human diseases [11]. GWAS is also applicable to quantitative traits in non-model species including crops [12] and farm animals for agricultural traits such as fertility and milk production [13, 14]. Single-cell sequencing and alignment of resultant short reads to the human reference has primarily been used to understand how mutation variation and mutant cell lineage within human tumors affects cancer treatment [15]. The same technique also enables molecular study of individual microbial species that cannot be grown outside of microbial communities [16]. Population sequencing can identify (and sometimes date) recent selection on genomes such as altitude adaptation (see [17] for review) and convergent adaptation of human lactase persistence in both Africa and Europe ~7,000 years ago [18]. In birds, population sequencing associated selection of the ALX1 crainofacial transcription factor to beak shape, clarifying species delineations in Galapagos island finches [19]. Sequencing domesticated dog populations identified selection on nervous system development genes for behavior and genes enabling adaptation to a starch-rich diet, both crucial for domestication [20]. Genome sequencing of ancient Neanderthal DNA [21] identified remnants of historical gene flow from Neanderthal, Denisovan populations, and possibly Homo erectus, into H. sapiens. Similarly, investigation of small genomic regions containing yellow skin chicken domestication genes in DNA from 280 BC dated fixation of domestication alleles to the last 500 years [22]. Sequence from a 600,000-year-old horse bone preserved in permafrost [23] changed divergence time estimates for the horse lineage, and identified putative domestication loci.
A small sampling of life
A measure of the incredible success of genome references is that for many researchers their availability is taken for granted: it’s assumed that the sequence of gene X, its paralogs, alternative splice forms, and its chromosomal location are all known. It is important to remember, however, that THE VAST MAJORITY OF SPECIES CANNOT BE STUDIED EFFECTIVELY DUE TO LACK OF A GENOME REFERENCE. The extent of reference sequence coverage of the eukaryotes is shown in Fig. 1. Within the relatively well-studied vertebrates, fifty percent of primate families have a reference, comprehensive sampling of bird species has recently started [24], and the mammals are well covered, but reptiles and amphibians have extremely few genome references. Outside of the vertebrates, there is a dearth of genomes throughout the tree of life. Approximately half of the insect orders have no representative genome. The water flea Daphnia [25] has the only high quality crustacean genome available. The myrapods are represented by a single centipede genome [26], Chicilierates (spiders, mites and ticks) are currently represented by only three published genomes, the agricultural pest spider mite [27], a social spider, and tarantula [28]. Outside of the arthropods, invertebrate genome representation drops again. Whilst there is at least one or two of each invertebrate phylum, that it is the equivalent of having a chicken and a fish sequence be the closest representative to the human sequence. For example, the mollusks, among the most diverse animal phyla, are currently represented by a limpet, a polycheat, and a leech [29]. Whilst this is a start (and an excellent scientific paper), it is not useful for those studying cephlapods such as octopi and cuttlefish for their alien intelligence, LCD skins, and camouflage ability – the closest related genome sequence is 400My diverged. There are roughly as many plant as mammalian genomes, despite plants being a taxonomic kingdom, and mammals being only a class. Less charismatic micro-fauna are also poorly represented with the exception of prokaryotes where small genome size makes cost-effective genome sequencing routinely the first analysis performed.
Figure 1. Current Genome Sequences Across the Eukaryotes.
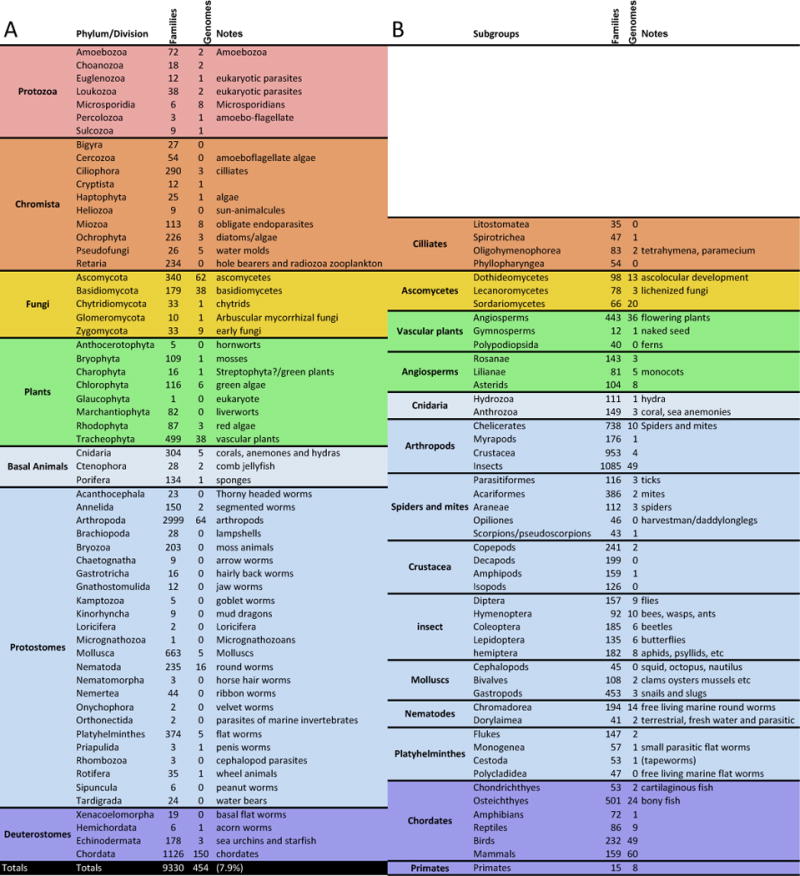
Numbers of eukaryotic taxonomic families represented with a reference genome assembly in NCBI. A: listed by phylum. B: Breakouts for phyla with especially large numbers of taxa. The vast majority of these reference genomes are in draft status, as very few large eukaryotic genomes have been finished. Some are of low quality with particularly short contigs. NCBI was searched using the web-link: http://www.ncbi.nlm.nih.gov/taxonomy/?term=family%5Brank%5D+taxonomy_assembly_exp%5B filter%5D+Araneae%5Borgn%5D in March 2015, where the term Araneae can be replaced with other taxonomic terms in the database. This returns a list and number of families with a genome assembly. Note that the described prokaryotes and archaea have representative genome sequences for essentially all described families. Although the numbers are smaller due to the need to culture a prokaryote before assigning an official species, this fact speaks to the immense utility of genome sequences when studying the microbial world as well as the ease and low cost of generating effectively finished references. By contrast only 7.9% of eukaryotic families have a representative genome sequence, and, of course, a far smaller percentage of genera and species.
Gaps in genome reference sampling cause gaps in biological understanding
The absence of these genome references is not just slowing research into specific questions; it is precluding a complete description of the molecular underpinnings of biology necessary for a true understanding of life on our planet. At a basic level, there is a need for continued improvement of taxonomic description. Although it is over 250 years of since Linnaeus’ Species Plantarum, the taxonomic tree is not fully nailed down and contains many controversial nodes [30]. For example, the initial sequence from the Honeybee genome project quickly showed that the hymenoptera (ants, bees, and wasps) rather than the coleopteran (beetles) are basal in the holometabola, in contrast to the previous view [31]. The information from more reference genomes will go beyond taxonomy, though. The planetary gene list is required for improved understanding of our ecosystems, as it underlies the metabolic capacity of trophic levels within food chains and biomes and determines the rates of material transfer between them [32]. The “pan genome” reference sequence of the dominant ocean bloom forming phytoplankton Emiliania huxleyi [33] shows varying strain gene content around a common genomic core. These genic differences underlie different metabolic capacities for processes such as carbon fixation, release of CO2 during the calcification of exoskeletons, affects on atmospheric sulfur, and adaptation to different oceanic environments such as low phosphorous.
The core set of common orthologous genes in different groups is the basis of a true understanding of the mechanistic requirements of life. Understanding the interactions between, and functions of, these genes often comes from the study of lethal mutations in model organisms, but is also the basis of engineering artificial life such as Mycoplasma mycoides JCVI-syn1.0 with it’s 1Mb artificially generated genome [34], which is helping to define the minimal essential gene set for a free living bacteria. Outside of the core set of orthologous genes, rapidly evolving genes with little orthologous sequence are often under intense selection for interaction with other organisms, including molecular warfare between attacking species and defending immune systems, such as bacterial antigens and immune recognition molecules in plants [35], genes expressed due to environmental interactions such as in the crustacean water flea [25], chemical warfare with complex venom mixtures from many species [28, 36, 37], and more intimate symbioses between species such as the arthropod formation of plant galls [38] and metabolic connections between aphids and their microbial symbionts [39]. Bio-prospecting these specialized molecules is key to unlocking the pharmacology of the planet.
Beyond the innate utility of the gene set, additional high quality comparative reference genomes are required to further understand the connection between genotype and phenotype. How have alterations around the core animal developmental program produced the many different morphologies and phenotypes of life on earth? Comparative genomics can help answer these questions as genome analysis of marine mammals from three orders identified convergently evolving genes for adaptation to the marine habitat [40]. Comparative genomics has also identified signals of convergent evolution in echo location [41] and stickleback adaptation to fresh water [42]. Reconstructed ancestral genomes and gene sets showing the evolutionary accumulation order of novel developmental components, for example showing how the two duplications and successive gene loss in the vertebrate lineage enabled increased specialization [43], and more recently initial genomic analysis of the living fossil horseshoe crab shows evidence of a whole-genome duplication in the chelicerate lineage [44]. Ultimately, understanding details of the cumulative nature of gene sets and their internal connections will improve understanding of epistasis, pleiotropy, and developmental robustness. This leads naturally to questions about the evolutionary history of life on earth. Ancestral gene sets, both coding and non-coding, provide one of the longest telescopes into the earliest stages of life. New sub disciplines – Evolutionary Cell Biology (ECB) [45] and Evolutionary Systems Biology (ESB) [46, 47] – are trying to understand the evolution and workings of the cellular machinery. An early success of ECB identified a fifth adaptin complex for protein transport between intracellular compartments that was previously suggested and dismissed in human, before sequence conservation across the eukaryotes eventually connected the protein to hereditary spastic paraplegia [48].
Gene orthologies and comparative genomics are unifying forces of biology
The unifying theme of biology is evolutionary conservation of the gene set and the resultant proteins that make up the biochemical and structural networks of cells and organisms throughout the tree of life. Whole-genome sequences and their derived protein coding sequences make this fact more abundantly clear with each passing year, with conserved signals in both RNA and protein coding genes observable from the earliest glimmerings of life. Multiple groups have tried to define the gene set of the last universal common ancestor (LUCA) found in extant species: 80 clusters of orthologous genes (COGs) were found to be present in every genome available in 2003 [49]. Since then, new estimates have ranged between 66 and 571 COGs depending on the methods used [50].
Similar analyses at other points in the tree of life include the Last Eukaryotic Common Ancestor (LECA) and the ancestral gene set of the ur-bilaterian (Figure 2). Ogura et al. investigated ancestral gene sets “at the split of plant-animal-fungi and the divergence of bilaterian animals”, estimating an increase of ~4,108 COGs from 2,469 at the plant-animal-fungi split to 6,577 in the ancestral gene set of the bilateria [51]. Gene orthology is the rule, not the exception: Waterhouse et al. looked at 95 eukaryotic species and found that 86% of over 1.3 million protein coding genes could be placed in orthologous groups [52]. Thus the large majority of genes and their protein products can be productively studied across wide swaths of taxonomic space.
Figure 2. A Taxonomic approach to sampling the tree of life.
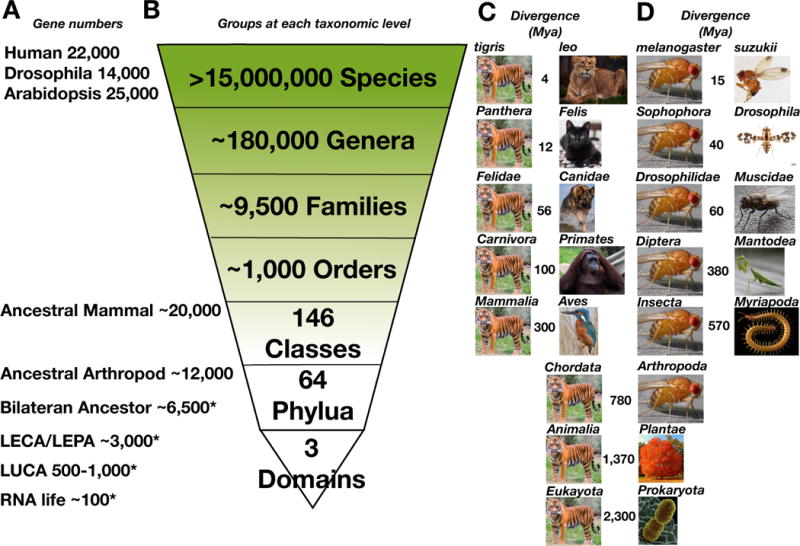
This diagram indicates the number of taxonomic groups at different Linnaean levels throughout the tree of life. A. Gene number (extant species) or * estimates of Clusters of Orthologous Genes (COGs) at ancestral nodes (LUCA: Last Universal Common ancestor; LECA: Last Eukaryotic Common Ancestor, LPCA: Last Prokaryotic Common Ancestor). Notably, a large proportion of genes are orthologously conserved from early in evolution enabling common study of gene products and processes in many different organisms. B. The number of taxanomic groups at different Linnaean levels throughout the tree of life. Note the rapid expansion of representative group numbers above the family level in genera and species, suggesting that taxonomy is a cost effective survey approach. C. Vertebrate and D. Invertebrate example divergence times at the taxonomic levels identified in B. Note that the mammals suffer somewhat from taxonomic inflation, but also that this reflects human interests and is not necessarily problematic. Photo credits: “Tiger – National Zoo 2011” by Ron Cogswell, “Female Indian Lion” by Steve Wilson, “Cat – black cat at the London Cat Café” by Tom Godber, Dog “In memoriam “Moja Vom Dorrequelle” by Harold Meerveld, Organatang: “Evolution of Expression” by Kabilan Subramanian, Kingfisher “Common Kingfisher (Alcedo atthis)” by Ron Knight, Fly “Drosophila immigrans” by John Tann, Tree “Flaming Orange Red Autumn Tree” by Joel יוֹאֵל, Prokaryote “Klebsiella pneumoniae Bacterium – Colorized scanning electron micrograph showing carbapenem-resistant Klebsiella pneumoniae interacting with a human neutrophil.” by NIAID, Drosophila suzukii “Spotted-wing Drosophila (Drosophila suzukii) male” by Martin Cooper, “praying mantis” by ShivaShankar, “Musca domestica” by Joan Quintana, all obtained via Flicker, CC 2.0 License. “Centipede: adult female Strigamia maritima with 94 legs” by Carlo Brena with permission and “Drosophila Grimshawi” from flybase.
Whilst these gene sequence orthologies join researchers across the whole of biology, cross-species substitution of genes is the strongest argument for their unifying force in biology. Because of their shared origin MANY GENES ARE FUNCTIONALLY INTERCHANGEABLE BETWEEN SPECIES. A famous example is the Drosophila Pax6/eyeless gene, which will work when expressed in mice and Xenopus, and vice-versa. (See Walter Gehring’s excellent review of the evolution of vision published shortly before his death [53]). Thus this gene, and its many conserved downstream cis-regulatory target sequences, can be studied in any species since the ~780Mya divergence of the protostomes and deuterostomes. Downstream genes are also well conserved with 69% of eye-expressed genes in the octopus having eye expression in human eyes [53]. Another classic example is the original identification of the human CDC2 gene by complementation of a cdc2 mutant strain of fission yeast [54] – at an evolutionary separation of ~1,200 million years [55]. Disease-relevant examples include Drosophila γ-secretase, which correctly processes human amyloid precursor protein thus enabling relevant protostome models of Alzheimer’s disease (see [56] for review). Finally, note that even mis-folded proteins can work across species: the prion causing bovine spongiform encephalopathy causes Creutzfeldt-Jakob disease in humans [57]. Orthologous genes with orthologous function give additional value to large mutation collections aiming for comprehensive gene coverage in model species such as zebrafish, [58], Drosophila [59], mouse [60], C.elegans [61], yeast [62], and beetle [63], because fundamentally we are all studying a single conserved gene set of life.
Comparing references at different evolutionary distances links phenotype and genotype, and the identification of selected genes and elements requires comparative genomics [64, 65]. For example, comparison of closely related primate sequences identified an 81bp human-specific gain-of-function developmental enhancer conserved in primates but with 13 substitutions in humans that is likely involved in the evolution of the opposable thumb [66]. Within primate comparisons also identified accelerated evolution of FOXP2 in the human lineage, which likely played a role in the development of human speech and language [67]. Comparing more diverged mammalian sequences identified genes critical for the marine mammal lifestyle as mentioned above [40], but also identified 4.2% of the human genome under evolutionarily constraint at a resolution of 12 bases [68]. Deep analysis of protein sequence in evolutionary time is used by tools such as Pfam, [69], PolyPhen [70], SIFT [71], Evolutionary Trace [72], and evolutionary action equations [73] to detect functionally significant changes and understand the medical significance of human polymorphisms.
Surveying life on planet earth is practical today
Although there are many sequencing projects underway (Box 1) that may be reaching their own goals, it seems that more could be achieved through greater coordination. The total surface of the earth is only 510.1×106 km2, which can be circumnavigated by commercial aircraft in just two days. In the same way that Google and others have mapped the surface of our planet, it is now technically and financially possible to survey the genomic tree of life before extensive taxa are lost due to further habitat destruction. The taxonomist and museum communities have a much broader working view of life on earth than those of us working on specific problems in medicine or model organisms. This comprehensive and global view inspired the Global Genome Initiative (GGI) [80], which aims to “preserve and understand the genomic diversity of life on earth”. The critical insight is that the number of taxonomic groups decreases rapidly as you ascend the Linnaean taxonomic categories (Figure 2). Thus, despite multiple millions of species, there are approximately 180,000 described genera, only 9,500 described families, and only 1,400 orders (a pre-publication family list is available at the GGI knowledge portal: http://ggi.eol.org/downloads). The GGI is underway and aims to collect genomic material for at least one representative of half of the described genera. Due to the immense biodiversity available in many locations, it is likely that collection in less than 100 carefully selected locations around the world will achieve this goal. Storage will be for both ongoing research, and for the longer-term role of museums as curators for future research opportunities. A database has already been created to coordinate this international effort by collaborators in the associated Global Genome Biodiversity Network [81]. The BGI has a similar initiative in the new China National Genebank. “Barcode” sequencing of GGI samples is planned to provide non-experts with a cost effective tool to identify species down to the genus level. It is also important to sample DNA from endangered species prior to extinction. Whilst the re-animation of lost species is likely not practical, their genomic histories and innovations can be captured in vitro even if the viable wild populations are lost. Finally, note that these projects do not aim to sequence the genomes or transcriptomes of these species at this time. However, moving up a taxonomic level from genera to family comprehensively surveys life on earth with only 10,000 representative taxa. This number is less than that already proposed for vertebrates, invertebrates, plants and others, and should be compared to proposals to clinically sequence large cohorts such as 100,000 UK citizens [82].
Box 1. Initiatives to date fall into the trap of balkanizing biology.
In contrast to the emerging idea of gene orthology uniting many areas of biology, ongoing survey initiatives to produce reference genomes for portions of the tree of life demonstrate the balkanized nature of research communities and their funding agencies. The Genome 10K project “to embark for the first time on a truly comprehensive study of vertebrate evolution” [74] whilst exciting and extremely worthy, is of limited use to research communities not studying vertebrates. “Me too” initiatives include the i5K Project to sequence “5000 arthropods of medical, agricultural, industrial, ecological and scientific importance” [75]. The US funded National Plant Genome Initiative is making progress, especially with the iPlant database [76], but the objectives of its latest five-year plan no longer include sequencing plant genomes [77]. Perhaps the greatest ambition for sampling the tree of life is the BGI’s 1000 plant and animal genomes project and related support of the Genome 10K project. Despite these efforts, there are still many holes in the initiatives. GIGA, the global invertebrate genomics alliance [78], was formed to fill the invertebrate non-arthropod hole in these efforts. Overall, communication between these initiatives is poor, funding is insufficient and even databases are balkanized. Despite a great number of them, only the NCBI, DDBJ, and EMBL serve all species [79]. This balkanization is also due to the different missions of funding agencies. Above I noted the success of the NHGRI in driving genome sequencing to date, but whilst the NHGRI is a bastion of support for basic biology, its mission to improve human health necessarily focuses its research funding. The NSF has provided some funding for plant genomes, but less for animals. The USDA has an obvious mandate for agricultural organisms, but not beyond those. By contrast BGI has taken a species of interest approach, with its 1,000 animal and plant genomes project. Both international and funding agency boundaries have tended to reinforce the isolation among research communities such that it will take collaboration and investment around a common goal to systematically sample the genomes of life on earth.
What will we learn from a genomic survey of life on earth?
Whilst the author is fond of “stamp collecting”, there are many good reasons to expand the reference sequences that underlie biological research (Table 2). We have already learned groups of reference sequences are more powerful than single references. For example, reference sequencing of 48 birds surveying avian biology provided insights and a research foundation for all aspects of avian biology including the evolution of feathers, flight, pneumatic bones, beaks, vocal learning, genome compactness, and more [24]. Figure 3. Illustrates a selection of biological insights from recent reference genomes showing the explosive impact they can have across biology. Given the rapid and far-reaching success of researching species with references, we can identify some low-hanging fruit:
Additional reference sequences will “enable single nucleotide resolution of conserved regulatory sequences in human and other sequenced model genomes enhancing our understanding of non-coding GWAS hits” [68].
Reference sequences add value to model systems (including those used for developmental biology studies, genetics, neuroscience and behavioral science research, population genetics, and understanding disease) and expand the number of model species that can be productively studied.
The wide availability of reference sequences accelerates the identification of therapeutic molecules and targets for intervention against pathogens and vectors.
A genomic survey of life on earth would discover and help elucidate true genomic innovation such as the origin of proteins, biochemical pathways, and the core metazoan developmental program.
Identification of the genomic basis of phenotypic innovations at different scales from major taxonomic innovations (such as multi-cellularity or adaptive immune system or lifespan) to those occurring on a shorter time scale (such as differences between closely related species).
The reconstruction of ancestral genomes will enable better identification of orthologs, and in parallel, surveying genomes will also identify the boundaries of the universal gene/protein sequence space.
Delineating the temporal order of the ancestral presence and absence of genes and their interactions – physical and genetic – sets bounds on models of epistasis and developmental robustness in our efforts to understand the evolutionary underpinnings of quantitative genetics and common disease.
At the most basic level, reference genomes allow species identification and delineation. Genome references thus underlie studies on speciation, gene flow, and hybridization, accelerate the identification of gene products of practical use (medical and industrial), and provide a new set of universal identification tools for conservation biology.
Table 2.
What can I do with my reference genome?
| Input data | Enabled methods |
|---|---|
| A Single Reference Genome -> The Annotated Geneset and The Molecular Biology Toolkit | Global Gene annotation – ortholog and paralog (and pseudo gene) identification, enabling protein expression Protein sequence identification enabling databases for proteome MS-MS analysis Gene family delineation Gene content and life style correlation DNA Methylation epigenetic analysis Comprehensive quantitative transcriptional analysis Transgenic manipulation of organisms and/or cell lines – CRISPR, RNAi, knockdown Possible RNAi bio-pesticide control measures ncRNA gene model identification Metabolic network analysis |
| Add Sequences of Individuals -> Population and Quantitative Genomics | GWAS (genome scan) for quantitative traits/complex disease Quantitative trait loci mapping using crosses Extreme phenotype sequencing for quantitative trait mapping Rapid Mutation Mapping – ems in model species, Mendelian variation in non-model species Identification of genes and regions under evolutionary selection Estimation of historical population sizes using the PSMC model [106] Expression QTL identification Marker informed rapid breeding for desirable traits |
| Additional Nearby References -> Short Range Comparative Genomics | FST analysis determining regions of differentiation between populations. Identification of convergently evolving genes associated with specific phenotypes Identification of cis-regulatory elements by evolutionary constraint Identification of genes underlying taxon specific traits |
| Genome References Survey of Life on Earth -> Unified study of Biology | Comprehensive survey of evolutionary innovation Comprehensive temporal mapping of evolutionary innovation Large scale correlation of gene content and life style Orthodb and Phylomdb identification and delineation of orthologous genes Ancestral genome reconstruction Acceleration of total biological research output |
Figure 3. Biological insights from sixteen recent reference eukaryotic genomes.

Although reference genome sequences alone do not generate high profile publications, the biological insights enabled by new reference genomes continue to excite. A The dominant bloom forming phytoplankton coccolithophore Emiliania huxleyi’s “pan genome” shows gene content plasticity between morphologically and ecologically different strains affecting the carbon cycle and enabling formation of large-scale episodic blooms under a wide variety of environmental conditions around the globe [33]. B The 20Gb Norway Spruce genome has grown by gradual accumulation of LTR TEs not whole-genome duplication and contains > 22,000 lncRNAs [93] C The genome of Amborella tricopoda – the remaining sister species to all extant flowering plants – identifies a whole-genome duplication prior to angiosperm formation likely responsible for 1179 gene lineages associated with the origin of the angiosperms including genes for flowering, wood formation, and response to stress [94]. D The coffee genome shows independent duplication of caffeine production genes in comparison to tea and cocca showing convergent evolution of caffeine synthesis [95]. E The genome of ciliate Oxytricha trifallax, a complex eukaryote, is split into approximately 16,000 nanochromosomes amplified to high copy number (~2,000) with commensurate expansion of telomere end binding proteins likely involved in processing tens of millions of telomeres per cell [96]. F A rosette shaped colony of the choanoflagellate Salpingoeca rosetta genome. Its genome encodes conserved cytokinesis genes including septins that are shared with the metazoans helping to illuminate metazoan multicellular evolution and mechanisms [97]. G Two recent ctenophore (comb jelly) genomes Mnemiopsis leidyi (shown) and Pleurobrachia bachei have illuminated the early evolution of cell type and neural systems. Ctenophores are the earliest lineage of the metazoan and are missing hox genes, and classical neurotransmitter pathways suggesting that ctenopore nervous systems evolved independently and enabling the observation that sponges likely lost neural cells [98, 99]. H The genomes of three spiralian species, the owl limpet (Lottia gigantean -shown), a marine polychaete (Capitella teleta) and a freshwater leech (Helobdella robusta) have provided an initial entry into the previously un-sampled mollusks and annelids. These genomes show more similarity to the invertebrate deuterostomes such as Amphioxus than flies and nematodes and expand the catalog of genes present in the last bilaterian ancestor [29]. I The myriapods invaded the land independently from chelicerates (spiders and mites) and insects. This is shown in the genome of the centipede Strigamia maritima with features such as independently evolved olfaction receptors and the use of paralogs rather than alternate splicing to generate gene diversity seen most prominently in the immunity gene dscam [26] J The genome of the Hessian fly Mayetiola destructor illuminated the genic adaptations to a gall forming lifestyle identifying >1,000 putative gall effector genes and the role of a 426 ubiquitin E3 ligase mimicking gene family in plant gall formation [38]. K Pregnant tsetse fly. The genome of the infamous obligate blood feeder and vector of tranpansomiasis, Glossina morsitans, in addition to containing genes related to its blood feeding lifestyle, also revealed genes underlying milk glands and lactation for its intrauterine larval development and nourishment by glandular secretions [100]. L The essentially finished genome of the zebra fish vertebrate model Danio rerio holds at least one ortholog of 71.4% of human genes dramatically underscoring the value of comparative model systems [101]. M The African coelacanth (Latimeria chalumnae) genome showed changes in genes and regulatory elements involved in immunity, nitrogen excretion, and the development of fins, tail, ear, eye, brain, and olfaction during the vertebrate adaptation to land. It also identified the ling fish and not the coelacanth as the closest living relative to the tetrapods [102]. N The genome of the Western painted turtle, Chrysemys picta bellii, shows an extraordinary slow rate of sequence evolution and allowed identification of genes involved in tolerance to freezing and oxygen deprivation [103]. O Male peregrine falcon, representing just one of 48 bird species sequenced by Zhang et Al. [24]. This comparative genomics tour de force resolved long standing questions in avian phylogeny, identified genes underlying vocal learning, skeletal pneumatization, volume constant lung, feathers, toothlessness, a diversity of dietary specializations, opsins for tetra chromatic vision, and evolutionary loss of the right ovary in highly constrained genomes. P Recent non-human primate genomes include the gibbon [104] and the Marmoset (pictured) that provided insights into diminutive size and frequent twinning [105]. Photo attributions: A: Alison R. Taylor (University of North Carolina Wilmington Microscopy Facility) Emiliania huxleyi – single-celled marine phytoplankton that produce calcium carbonate scales (coccoliths). A scanning electron micrograph of a single coccolithophore cell. CC 2.5 licence. B: F.D. Richards (via flickr) Norway Spruce CC 2.0 license. C: Scott Zona (via flickr) Male flowers of Amborella trichopoda, CC 2.0 licence. D: Jeevan Jose, Kerala, India Coffea canephora Pierre ex A. Froehner CC Attribution-ShareAlike 4.0 International License. E: User:Gustavocarra wikimedia commons, Scanning electron microscope view of Oxytricha trifallax, Public Domain. F: Mark J. Dayel, S. rosetta colony scanning electron micrograph, CC 3.0 attribution share a like license. G: Vidar A from Gozo, Malta, via wikimedia commons, Mnemiopsis leidyi – Oslofjord, Norway.jpg, CC 2.0 license. H: Jerry Kirkhart -originally posted to Flickr as Owl Limpet, Lottia gigantea, CC 2.0 license. I: Carlo Brena Centipede S. martima gift. J: PD-USGOV-USDA-ARS, Hessian fly, GNU Free Documentation public domain. K: Geoffrey M. Attardo, Female pregnant tsetse, Glossina morsitans morsitans CC 2.5 license. L: kamujp (via flickr) Danio Rerio, CC 2.0 license, M: Mordecai, 1998 File:El-celacanto.jpg, CC Attribution-Share Alike 4.0 International license. N: Oregon Department of Fish & Wildlife, Western painted turtle hatchlings (cropped to focus on a single hatchling) CC 2.0 license. O: Author U.S. Fish and Wildlife Service Headquarters, Male peregrine falcon, CC 2.0 license. P: Leszek Leszczynski, Marmoset, CC 2.0 license.
Finally, genome references massively accelerate non-model-organism research. Non-model-organism research, despite making progress, is losing ground relative to research on species with available genomes. The expansion of experiments made possible by a reference sequence highly disadvantages grants studying species without references. Worse, students trained on these species cannot use the latest technologies requiring references, and are thus a decade behind the state of the art. The Insect Genetic Technologies Research Co-ordination Network [83] is one effort to address the non-model-organisms genomic tools training gap with workshops, protocols, and grants for peer-to-peer training in new techniques. However, the fact remains that without genome reference sequences most genetic technologies have significantly less utility.
Towards robust de-novo genome sequencing
At the core of our ability to generate a broad survey of the taxa on earth is cost, both in dollars and time (Box 2). Multiple new competing technologies have dramatically improved the quality and robustness of genome assembly, enabling genome reference sampling of the tree of life. To date, both size constraints and technical difficulties in robust assembly of polymorphic and repetitive genomes from cost-effective short reads have slowed the production of de-novo genome sequences. The “draft” genome references produced (excepting bacterial and other small genomes) have many gaps and are not appropriate for long-term archiving in databases and museums. Although genome size estimates are not comprehensive, extrapolating known genome sizes to families with unknown genome sizes by taxonomic position allows a 2Gb estimation of the average genome size (data not shown) with a range from ~5Mb for prokaryotes, to ~100Gb [84]. Sequencing costs have focused de-novo references on the smallest genomes with slow progression to the largest, however perhaps a financial target of $10,000 for the robust assembly of the average 2Gb genome would focus the community in the same way the $1,000 human genome target has. Current short read costs for a 2Gb draft genome are below $10,000, but higher quality “archival” references are required. Although analysis costs will become a larger proportion of total reference cost, the marginal cost per software analysis is potentially very low, demonstrating the importance of shared high quality annotation and analysis software.
Box 2. Funding for new genome references.
The large-scale survey and equipment grant paradigms
Although there is clearly a need for funding and coordinating current large-scale genome initiatives to fully survey the tree of life (Box 1), there is also a need for smaller funding opportunities for individual groups to accelerate research and improve the scientific return on research expenditures from funding agencies. Funding for the production of genome references is currently extremely difficult, as these tasks do not fit the hypothesis-driven research paradigm that drives much of science, and are disadvantaged compared to grants where genome resources and techniques are utilized. There is however, a natural alignment with a different type of proposal – the infrastructure/equipment grant. Infrastructure grants provide materials and tools to enhance research productivity, and the equipment or resource is expected to provide utility for some time beyond the initial investment. High quality genome references, annotations, and resources can be generated for less cost than say, a good microscope. Like other infrastructure investments, it is more cost effective to give researchers the tools they need, rather than pay students and postdocs for multiple additional years working around the lack of a resource. Resources such as iPlant [76] can provide toolsets and computational power to small communities ensuring high quality datasets. As it is standard practice to deposit data at the NCBI and its partners, these infrastructure reference genomes are a lasting electronic resource for researchers worldwide, not just in the grant-receiving institution. The competitive grant mechanism is also the best way to identify the most underserved communities, where genome sequences will enable the most cost-effective enhancement of scientific return.
The most promising avenue to robust turnkey high-quality assembly is long sequence reads. Pacific Biosciences long reads routinely enable finished bacterial and other small de-novo genome references [85] and can be used for de-novo genome assembly of larger genome references including human [86]. Oxford Nanopore is currently producing extremely long reads (up to 100kb) in beta testing, although read quality from single molecules remains a challenging problem. Illumina synthetic long reads now enable the generation and assembly of long reads from only 500ng of DNA, and have proven effective with highly polymorphic genomes [87] and working with repeats [88], and new companies such as 10× Genomics (Pleasanton, CA) and Dovetail Genomics (Santa Cruz, CA) are innovating in this area. Aside from long reads, assembly of cost-effective Illumina reads is continuing to improve. Discovar [89] uses 250bp Illumina reads and produces more contiguous assemblies than Allpaths-LG, which it replaced, while requiring only 60× genome coverage sequence as input. Platanus [90] is a new assembler designed specifically for polymorphic genome datasets – a major source of gaps in current genome assemblies. Finally, validation of genome assembly has often been neglected for cost reasons. Optical mapping has been available for over a decade, but BioNano genomics has made such genome assembly validation and chromosome arm length scaffolding cost effective [91]. Chromatin sequencing also enables chromosome arm length scaffolding and validation of genome assembly [92]. Together, these techniques promise a robust cost-effective turnkey de-novo genome references in the near term.
Concluding remarks
De-novo reference genome sequencing is not an end, but rather the foundational necessity for productive biological and medical research. It enhances, rather than replaces, other areas of biological enquiry. The potential for de-novo reference genome sequences combined with high-throughput biology technologies to cost effectively accelerate all biological research has not been utilized significantly beyond model organisms, and is currently restraining progress in many areas of biological research. Genome references and the resultant orthologous gene sets will illuminate the single tree of life on our planet, the study of which can potentially unify researchers studying different species around the common core of all biology.
Highlights.
We propose to comprehensively survey genome sequences of life on earth
Sequencing taxa at the family level greatly reduces the required number of species
New sequencing technologies enable cost effective global genome surveying
Both big science initiatives and small genome infrastructure funding is needed
Acknowledgments
I would like to thank Jeffrey Rogers, Shelley Sazer and Rhiannon Macrae for discussion and help with editing. This work was supported by NHGRI grant U54 HG003273. The author would also like to thank the many people who made images used in the figures available for sharing in the public domain under Creative Commons licenses.
Biography
Stephen (fringy) Richards studies genomics at the Baylor College of Medicine, Human Genomics Sequencing Center (BCM-HGSC). He also indulges in the old fashioned hobby of collecting postage stamps. If, within 3 years of the publication date, you wish to send him a postcard with an interesting current stamp on it, he will return the favor with an interesting current US stamp. Author address: Stephen Richards N1501.01 Alkek building, Human Genome Sequencing Center, Baylor College of Medicine, 1 Baylor Plaza, Houston, TX 77030, USA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ewing B, Green P. Analysis of expressed sequence tags indicates 35,000 human genes. Nature genetics. 2000;25:232–234. doi: 10.1038/76115. [DOI] [PubMed] [Google Scholar]
- 2.Pennisi E. Human genome. A low number wins the GeneSweep Pool. Science. 2003;300:1484. doi: 10.1126/science.300.5625.1484b. [DOI] [PubMed] [Google Scholar]
- 3.Clamp M, et al. Distinguishing protein-coding and noncoding genes in the human genome. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19428–19433. doi: 10.1073/pnas.0709013104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cabili MN, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes & development. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Y, et al. Molecular Findings Among Patients Referred for Clinical Whole-Exome Sequencing. Jama. 2014 doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leshchiner I, et al. Mutation mapping and identification by whole-genome sequencing. Genome research. 2012;22:1541–1548. doi: 10.1101/gr.135541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blumenstiel JP, et al. Identification of EMS-induced mutations in Drosophila melanogaster by whole-genome sequencing. Genetics. 2009;182:25–32. doi: 10.1534/genetics.109.101998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haelterman NA, et al. Large-scale identification of chemically induced mutations in Drosophila melanogaster. Genome research. 2014;24:1707–1718. doi: 10.1101/gr.174615.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo J, et al. A CLN8 nonsense mutation in the whole genome sequence of a mixed breed dog with neuronal ceroid lipofuscinosis and Australian Shepherd ancestry. Molecular genetics and metabolism. 2014;112:302–309. doi: 10.1016/j.ymgme.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 11.Welter D, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic acids research. 2014;42:D1001–1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang X, Han B. Natural variations and genome-wide association studies in crop plants. Annual review of plant biology. 2014;65:531–551. doi: 10.1146/annurev-arplant-050213-035715. [DOI] [PubMed] [Google Scholar]
- 13.Luo W, et al. Genome-wide association study of porcine hematological parameters in a Large White × Minzhu F2 resource population. International journal of biological sciences. 2012;8:870–881. doi: 10.7150/ijbs.4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olsen HG, et al. Genome-wide association mapping in Norwegian Red cattle identifies quantitative trait loci for fertility and milk production on BTA12. Animal genetics. 2011;42:466–474. doi: 10.1111/j.1365-2052.2011.02179.x. [DOI] [PubMed] [Google Scholar]
- 15.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blainey PC. The future is now: single-cell genomics of bacteria and archaea. FEMS microbiology reviews. 2013;37:407–427. doi: 10.1111/1574-6976.12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheviron ZA, Brumfield RT. Genomic insights into adaptation to high-altitude environments. Heredity. 2012;108:354–361. doi: 10.1038/hdy.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tishkoff SA, et al. Convergent adaptation of human lactase persistence in Africa and Europe. Nature genetics. 2007;39:31–40. doi: 10.1038/ng1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamichhaney S, et al. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature. 2015 doi: 10.1038/nature14181. [DOI] [PubMed] [Google Scholar]
- 20.Axelsson E, et al. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature. 2013;495:360–364. doi: 10.1038/nature11837. [DOI] [PubMed] [Google Scholar]
- 21.Prufer K, et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature. 2014;505:43–49. doi: 10.1038/nature12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Girdland Flink L, et al. Establishing the validity of domestication genes using DNA from ancient chickens. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:6184–6189. doi: 10.1073/pnas.1308939110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orlando L, et al. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature. 2013;499:74–78. doi: 10.1038/nature12323. [DOI] [PubMed] [Google Scholar]
- 24.Zhang G, et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science. 2014;346:1311–1320. doi: 10.1126/science.1251385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colbourne JK, et al. The ecoresponsive genome of Daphnia pulex. Science. 2011;331:555–561. doi: 10.1126/science.1197761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chipman AD, et al. The First Myriapod Genome Sequence Reveals Conservative Arthropod Gene Content and Genome Organisation in the Centipede Strigamia maritima. PLoS biology. 2014;12:e1002005. doi: 10.1371/journal.pbio.1002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grbic M, et al. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature. 2011;479:487–492. doi: 10.1038/nature10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanggaard KW, et al. Spider genomes provide insight into composition and evolution of venom and silk. Nature communications. 2014;5:3765. doi: 10.1038/ncomms4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simakov O, et al. Insights into bilaterian evolution from three spiralian genomes. Nature. 2013;493:526–531. doi: 10.1038/nature11696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pace NR. Mapping the tree of life: progress and prospects. Microbiology and molecular biology reviews: MMBR. 2009;73:565–576. doi: 10.1128/MMBR.00033-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savard J, et al. Phylogenomic analysis reveals bees and wasps (Hymenoptera) at the base of the radiation of Holometabolous insects. Genome research. 2006;16:1334–1338. doi: 10.1101/gr.5204306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Falkowski PG, et al. The microbial engines that drive Earth’s biogeochemical cycles. Science. 2008;320:1034–1039. doi: 10.1126/science.1153213. [DOI] [PubMed] [Google Scholar]
- 33.Read BA, et al. Pan genome of the phytoplankton Emiliania underpins its global distribution. Nature. 2013;499:209–213. doi: 10.1038/nature12221. [DOI] [PubMed] [Google Scholar]
- 34.Gibson DG, et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 2010;329:52–56. doi: 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- 35.Jones JD, Dangl JL. The plant immune system. Nature. 2006;444:323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- 36.Vonk FJ, et al. The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:20651–20656. doi: 10.1073/pnas.1314702110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warren WC, et al. Genome analysis of the platypus reveals unique signatures of evolution. Nature. 2008;453:175–183. doi: 10.1038/nature06936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao C, et al. A Massive Expansion of Effector Genes Underlies Gall-Formation in the Wheat Pest Mayetiola destructor. Current Biology. 2015 doi: 10.1016/j.cub.2014.12.057. in Press. [DOI] [PubMed] [Google Scholar]
- 39.Wilson AC, et al. Genomic insight into the amino acid relations of the pea aphid, Acyrthosiphon pisum, with its symbiotic bacterium Buchnera aphidicola. Insect molecular biology. 2010;19(Suppl 2):249–258. doi: 10.1111/j.1365-2583.2009.00942.x. [DOI] [PubMed] [Google Scholar]
- 40.Foote AD, et al. Convergent evolution of the genomes of marine mammals. Nature genetics. 2015 doi: 10.1038/ng.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parker J, et al. Genome-wide signatures of convergent evolution in echolocating mammals. Nature. 2013;502:228–231. doi: 10.1038/nature12511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones FC, et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature. 2012;484:55–61. doi: 10.1038/nature10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dehal P, Boore JL. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS biology. 2005;3:e314. doi: 10.1371/journal.pbio.0030314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nossa CW, et al. Joint assembly and genetic mapping of the Atlantic horseshoe crab genome reveals ancient whole genome duplication. GigaScience. 2014;3:9. doi: 10.1186/2047-217X-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lynch M, et al. Evolutionary cell biology: two origins, one objective. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:16990–16994. doi: 10.1073/pnas.1415861111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Medina M. Genomes, phylogeny, and evolutionary systems biology. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(Suppl 1):6630–6635. doi: 10.1073/pnas.0501984102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soyer OS, O’Malley MA. Evolutionary systems biology: what it is and why it matters. BioEssays: news and reviews in molecular, cellular and developmental biology. 2013;35:696–705. doi: 10.1002/bies.201300029. [DOI] [PubMed] [Google Scholar]
- 48.Hirst J, et al. The fifth adaptor protein complex. PLoS biology. 2011;9:e1001170. doi: 10.1371/journal.pbio.1001170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harris JK, et al. The genetic core of the universal ancestor. Genome research. 2003;13:407–412. doi: 10.1101/gr.652803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goldman AD, et al. LUCApedia: a database for the study of ancient life. Nucleic acids research. 2013;41:D1079–1082. doi: 10.1093/nar/gks1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ogura A, et al. Estimation of ancestral gene set of bilaterian animals and its implication to dynamic change of gene content in bilaterian evolution. Gene. 2005;345:65–71. doi: 10.1016/j.gene.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 52.Waterhouse RM, et al. Correlating traits of gene retention, sequence divergence, duplicability and essentiality in vertebrates, arthropods, and fungi. Genome biology and evolution. 2011;3:75–86. doi: 10.1093/gbe/evq083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gehring WJ. The evolution of vision. Wiley interdisciplinary reviews. Developmental biology. 2014;3:1–40. doi: 10.1002/wdev.96. [DOI] [PubMed] [Google Scholar]
- 54.Lee MG, Nurse P. Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature. 1987;327:31–35. doi: 10.1038/327031a0. [DOI] [PubMed] [Google Scholar]
- 55.Kumar S, Hedges SB. TimeTree2: species divergence times on the iPhone. Bioinformatics. 2011;27:2023–2024. doi: 10.1093/bioinformatics/btr315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prussing K, et al. Drosophila melanogaster as a model organism for Alzheimer’s disease. Molecular neurodegeneration. 2013;8:35. doi: 10.1186/1750-1326-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barria MA, et al. Exploring the zoonotic potential of animal prion diseases: in vivo and in vitro approaches. Prion. 2014;8:85–91. doi: 10.4161/pri.28124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kettleborough RN, et al. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature. 2013;496:494–497. doi: 10.1038/nature11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bellen HJ, et al. The Drosophila gene disruption project: progress using transposons with distinctive site specificities. Genetics. 2011;188:731–743. doi: 10.1534/genetics.111.126995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bradley A, et al. The mammalian gene function resource: the International Knockout Mouse Consortium. Mammalian genome: official journal of the International Mammalian Genome Society. 2012;23:580–586. doi: 10.1007/s00335-012-9422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thompson O, et al. The million mutation project: a new approach to genetics in Caenorhabditis elegans. Genome research. 2013;23:1749–1762. doi: 10.1101/gr.157651.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giaever G, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 63.iBeetle-Consortium. IBeetle-Base 2014 [Google Scholar]
- 64.Barsh GS, Andersson L. Evolutionary genomics: Detecting selection. Nature. 2013;495:325–326. doi: 10.1038/495325a. [DOI] [PubMed] [Google Scholar]
- 65.Nielsen R, Hubisz MJ. Evolutionary genomics: detecting selection needs comparative data. Nature. 2005;433:E6. doi: 10.1038/nature03222. discussion E7–8. [DOI] [PubMed] [Google Scholar]
- 66.Prabhakar S, et al. Human-specific gain of function in a developmental enhancer. Science. 2008;321:1346–1350. doi: 10.1126/science.1159974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Preuss TM. Human brain evolution: from gene discovery to phenotype discovery. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(Suppl 1):10709–10716. doi: 10.1073/pnas.1201894109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lindblad-Toh K, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Finn RD, et al. The Pfam protein families database. Nucleic acids research. 2010;38:D211–222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nature methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kumar P, et al. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 72.Mihalek I, et al. A family of evolution-entropy hybrid methods for ranking protein residues by importance. Journal of molecular biology. 2004;336:1265–1282. doi: 10.1016/j.jmb.2003.12.078. [DOI] [PubMed] [Google Scholar]
- 73.Katsonis P, Lichtarge O. A formal perturbation equation between genotype and phenotype determines the Evolutionary Action of protein-coding variations on fitness. Genome research. 2014 doi: 10.1101/gr.176214.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Genome, K.C.o.S. Genome 10K: a proposal to obtain whole-genome sequence for 10,000 vertebrate species. The Journal of heredity. 2009;100:659–674. doi: 10.1093/jhered/esp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Robinson GE, et al. Creating a Buzz About Insect Genomes. Science. 2011;331:1386. doi: 10.1126/science.331.6023.1386. [DOI] [PubMed] [Google Scholar]
- 76.Goff SA, et al. The iPlant Collaborative: Cyberinfrastructure for Plant Biology. Frontiers in plant science. 2011;2:34. doi: 10.3389/fpls.2011.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Council, N.S.a.T, editor. The Interagency Working Group on Plant Genomics. NATIONAL PLANT GENOME INITIATIVE FIVE-YEAR PLAN: 2014–2018. 2014. [Google Scholar]
- 78.GIGA. GIGA. 2012 http://giga.nova.edu.
- 79.Brunak S, et al. Nucleotide sequence database policies. Science. 2002;298:1333. doi: 10.1126/science.298.5597.1333b. [DOI] [PubMed] [Google Scholar]
- 80.Global Genomes Initiative Golbal Genomes Initiative. http://www.mnh.si.edu/ggi/index.html.
- 81.Droege G, et al. The Global Genome Biodiversity Network (GGBN) Data Portal. Nucleic acids research. 2014;42:D607–612. doi: 10.1093/nar/gkt928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Genomics England. The 100,000 genomes project. 2014 http://www.genomicsengland.co.uk/the-100000-genomes-project/
- 83.O’Brochta D. 2014 http://igtrcn.org.
- 84.Gregory TR. Animal Genome Size Database 2015 [Google Scholar]
- 85.Chin CS, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature methods. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 86.Biosciences, P. Data Release: ~54× Long-Read Coverage for PacBio-only De Novo Human Genome Assembly. 2014 http://blog.pacificbiosciences.com/2014/02/data-release-54x-long-read-coverage-for.html.
- 87.Voskoboynik A, et al. The genome sequence of the colonial chordate. Botryllus schlosseri. eLife. 2013;2:e00569. doi: 10.7554/eLife.00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McCoy RC, et al. Illumina TruSeq Synthetic Long-Reads Empower De Novo Assembly and Resolve Complex, Highly-Repetitive Transposable Elements. PloS one. 2014;9:e106689. doi: 10.1371/journal.pone.0106689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weisenfeld NI, et al. Comprehensive variation discovery in single human genomes. Nature genetics. 2014 doi: 10.1038/ng.3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kajitani R, et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome research. 2014;24:1384–1395. doi: 10.1101/gr.170720.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lam ET, et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nature biotechnology. 2012;30:771–776. doi: 10.1038/nbt.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Burton JN, et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature biotechnology. 2013;31:1119–1125. doi: 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nystedt B, et al. The Norway spruce genome sequence and conifer genome evolution. Nature. 2013;497:579–584. doi: 10.1038/nature12211. [DOI] [PubMed] [Google Scholar]
- 94.Amborella Genome P. The Amborella genome and the evolution of flowering plants. Science. 2013;342:1241089. doi: 10.1126/science.1241089. [DOI] [PubMed] [Google Scholar]
- 95.Denoeud F, et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science. 2014;345:1181–1184. doi: 10.1126/science.1255274. [DOI] [PubMed] [Google Scholar]
- 96.Swart EC, et al. The Oxytricha trifallax macronuclear genome: a complex eukaryotic genome with 16,000 tiny chromosomes. PLoS biology. 2013;11:e1001473. doi: 10.1371/journal.pbio.1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fairclough SR, et al. Premetazoan genome evolution and the regulation of cell differentiation in the choanoflagellate Salpingoeca rosetta. Genome biology. 2013;14:R15. doi: 10.1186/gb-2013-14-2-r15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Moroz LL, et al. The ctenophore genome and the evolutionary origins of neural systems. Nature. 2014;510:109–114. doi: 10.1038/nature13400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ryan JF, et al. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science. 2013;342:1242592. doi: 10.1126/science.1242592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.International Glossina Genome, I. Genome sequence of the tsetse fly (Glossina morsitans): vector of African trypanosomiasis. Science. 2014;344:380–386. doi: 10.1126/science.1249656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Howe K, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496:498–503. doi: 10.1038/nature12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Amemiya CT, et al. The African coelacanth genome provides insights into tetrapod evolution. Nature. 2013;496:311–316. doi: 10.1038/nature12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shaffer HB, et al. The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome biology. 2013;14:R28. doi: 10.1186/gb-2013-14-3-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Carbone L, et al. Gibbon genome and the fast karyotype evolution of small apes. Nature. 2014;513:195–201. doi: 10.1038/nature13679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Marmoset Genome S, Analysis C. The common marmoset genome provides insight into primate biology and evolution. Nature genetics. 2014;46:850–857. doi: 10.1038/ng.3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]