

Abstract
In multicellular organisms, individual cells have evolved to sense external and internal cues in order to maintain cellular homeostasis and survive under different environmental conditions. Cells efficiently adjust their metabolism to reflect the abundance of nutrients, energy and growth factors. The ability to rewire cellular metabolism between anabolic to catabolic processes is critical for cells to thrive. Thus, cells have developed, through evolution, metabolic networks that are highly plastic and tightly regulated to meet the requirements necessary to maintain cellular homeostasis. The plasticity of these cellular systems is tightly regulated by complex signaling networks that integrate the intracellular and extracellular information. The coordination of signal transduction and metabolic pathways is essential in maintaining a healthy and rapidly responsive cellular state.
Introduction
Living organisms require a constant supply of energy to maintain cell and organ function. Thus, an adequate balance between energy production and energy expenditure is essential to maintain cellular homeostasis. This is achieved by the regulation of the dynamics between the combustion of fuel sources to produce energy (catabolism), and their ability to utilize energy to synthesize macromolecules (anabolism). The importance of the balance between these two processes becomes apparent when the metabolic differences between growing cells and differentiated/quiescent cells are examined. To support growth and proliferation, cells rewire their metabolism to promote anabolic processes that synthesize the macromolecules (proteins, carbohydrates, lipids and nucleic acids) required for generating a daughter cell. On the other hand, most tissues are comprise of differentiated and non-dividing cells, thus their metabolism is normally wired towards catabolic processes that provide energy to sustain cellular integrity and function. Maintaining this delicate balance is one of the most important requirements of life. Thus, it comes as no surprise that eukaryotic cells have evolved to constantly and carefully modulate these processes in response to the ever-changing conditions.
In multicellular organisms, cells must be responsive to systemic cues of the physiological state to maintain energetic and cellular stability in addition to sensing the immediate environment. This is achieved through the ability of the cells to sense secreted factors (e.g. cytokines, growth factors, hormones) that, upon binding to a cell surface receptor, initiate signaling cascades that transduce information and regulate metabolism. Moreover, to ensure that balance between both the availability of nutrients and the cellular capacity to use them effectively is maintained, cells can also sense intracellular metabolite concentrations to fine-tune the signaling networks independently of the environment. Many recent findings have highlighted the fact that metabolites serve as indicators of the metabolic state of the cell, that transduce this information through regulation of pro-translational modifications, such as acetylation, methylation and glycosylation, that regulate the activities of several signaling molecules and transcriptional regulators (not discussed further here, for review on this topic see [1,2]).
Understanding this intricate bidirectional relationship is a challenge due to its complexity, but one that is vital for understanding the principles of cellular homeostasis. Such knowledge will be of enormous benefit to determining how diseases develop as well as how to treat them.
Anabolic rewiring induced by PI3K/Akt and Ras/ERK signaling
Growth factors, hormones and nutrient signals provide the information required to rewire intermediate metabolism towards anabolism, thereby supporting cell growth and proliferation. The signaling framework downstream of these stimuli is primarily defined by two highly conserved and critical pathways, the phosphatidylinositol-3-kinase (PI3K)/Akt and the extracellular signal-regulated kinase - mitogen-activated protein kinase (ERK-MAPK) signaling cascades (Fig.1).
Fig. 1. Anabolic rewiring induced by PI3K/Akt, Ras/ERK and mTORC1 signaling.
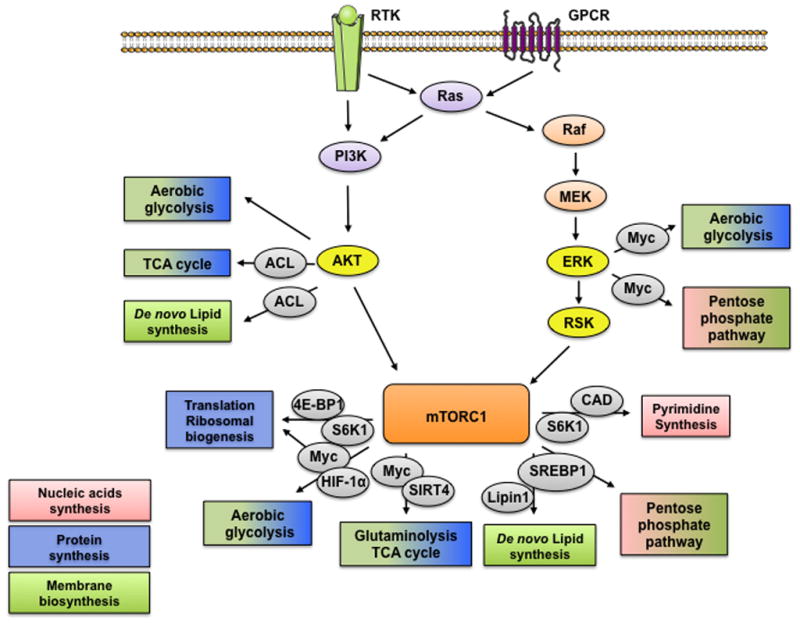
Extracellular signals activate two major signaling cascades controlled by the activation of PI3K and Ras. PI3K and Ras regulate Akt and ERK, which in turn induce changes in intermediate metabolism to promote anabolic processes. In addition, they also induce the activation of mTORC1, thus further supporting the rewiring of cellular metabolism towards anabolic processes. Through various mechanisms Akt, ERK and mTORC1 stimulate mRNA translation, aerobic glycolysis, glutamine anaplerosis, lipid synthesis, the pentose phosphate and pyrimidine synthesis, thus producing the major components necessary for cell growth and proliferation.
PI3K/Akt signaling-induced Anabolic Reprogramming
Growth factors and other ligands activate PI3K signaling upon binding and consequent activation of their cell surface receptors, such as receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs). This leads to the phosphorylation of membrane phosphatidylinositiol lipids and the recruitment and activation of several protein kinases, which perpetuate the extracellular signals to modulate intracellular processes [3,4]. One of the most critical signal propagators regulated by PI3K signaling is protein kinase B/Akt [3,4]. Indeed, Akt rewires metabolism in response to environmental cues by three distinct means; i) by the direct phosphorylation and regulation of metabolic enzymes, ii) by activating/inactivating metabolism altering transcriptional factors, and iii) by modulating other kinases that themselves regulate metabolism [5].
Akt regulates glucose metabolism, inducing both glucose uptake and glycolytic flux by increasing the expression of the glucose transporter genes and regulating the activity of glycolytic enzymes, respectively [6–8]. Morever, the ability of Akt to induce glycolysis is also mediated by the regulation of Hexokinase (HK). HK performs the first step of glycolysis. Akt has been shown to regulate the ability of HK-II to interact with the mitochondria, and thus promotes glucose carbon to be oxidized through glycolysis [9]. By regulating glycolysis, Akt might be involved in regulating the tricarboxylic acid (TCA) cycle activity via the malate/aspartate and glycerol-phosphate shuttles. In addition to glucose metabolism, Akt also directly phosphorylates and activates ATP-citrate lyase (ACL) [10]. ACL promotes the production of acetyl-coA in the cytosol from citrate generated in the TCA cycle [11]. Cytosolic acetyl-coA is vital for de novo lipid synthesis, as it can initiate and/or elongate fatty acids chains [11], thus linking Akt signaling to lipid synthesis.
Moreover, Akt also regulates the transcription factor c-Myc, a key transcriptional factor that promotes anabolic processes, through phosphorylation and inactivation of a negative regulator of c-Myc, glycogen synthase kinase-3 (GSK3) [12]. Together these findings demonstrate that upon stimulation, the PI3K/AKT pathway rewires cells from catabolic to anabolic metabolism (Fig.1).
Ras/ERK signaling cascades and its consequences for anabolism
Extracellular cues also lead to the activation of the small GTPase, Ras. Like PI3K, the Ras family (H-, K- and N-Ras) is activated downstream of cell surface receptors. Ras activation involves its transition to a GTP-bound state, which initiates signal transduction through several pathways, of which the ERK-MAPK signaling cascade is the best characterized [13].
Taking into consideration the key role of Ras in orchestrating biological responses to stimuli that induce cell growth and proliferation, Ras stands out as a possible key driver of anabolic reprogramming. In support of this, Ras has been shown to decouple glucose and glutamine metabolism, thus diverting these carbon sources to anabolic pathways to support cell growth and proliferation [14]. Ras signaling enhances glucose uptake and glycolytic flux, but decreases glucose entry into the TCA cycle [14,15]. This increased flux through glycolysis has been shown to fuel anabolic processes by diverting glucose-derived carbon to the non-oxidative arm of the PPP, thus supporting nucleotide biosynthesis [16]. Interestingly, the mechanisms behind these effects of Ras were found to be through ERK stabilization of c-Myc, which increases the expression of enzymes involved in these pathways [16]. In addition, ERK also induces the flux of glucose-derived carbon towards biosynthetic pathways by phosphorylating and inducing nuclear translocation of the anabolism-related version of pyruvate kinase, pyruvate kinase M2 (PKM2) [17]. While Ras signaling diverts the glucose-derived carbon flux away from the TCA cycle, it also promotes the utilization of glutamine for anaplerosis and the maintenance of redox potential [14,18]. Thus, activation of Ras makes the cells more dependent on glutamine as a source of carbon and nitrogen for anabolic processes [14,18].
Together, these reports have shown that activation of Ras/ERK signaling cascade rewires cells towards anabolism, to promote synthesis of building blocks and energy necessary for cell growth and proliferation (Fig.1).
Mechanistic target of rapamycin (mTOR) as the master regulator of anabolic reprogramming
Despite the direct effects of PI3K/AKT and Ras/ERK on metabolism, activation of mTOR by these pathways seems to account for a large proportion of their metabolic contributions. mTOR exists in two functionally and structurally distinct complexes mTOR complex 1 (mTORC1) and 2 (mTORC2). Of the two complexes, mTORC1 seems to have the most direct influence in the maintenance of energetic balance [19]. The PI3K-Akt and Ras/ERK pathways are potent activators of mTORC1 activity, through the negative regulation of tuberous sclerosis complex 2 (TSC2), a major inhibitor of mTORC1 activation. Akt directly phosphorylates TSC2 at multiple sites [20]. ERK1/2 induce the phosphorylation of TSC2 through its downstream target p90 ribosomal S6 Kinase (RSK) at some Akt as well as at novel sites [21]. These phosphorylation events release TSC2-mediated inhibition of the GTPase Ras homolog enriched in brain (RHEB), thus allowing RHEB to activate mTORC1 [22]. Moreover, both ERK and RSK promote mTORC1 activity by phosphorylating raptor, a key substrate-binding element of the mTORC1 complex [23,24]. Importantly, mTORC1 is also considered a major nutrient sensor as its activity is regulated by the availability of amino acids and glucose [25,26]. Thus, the ability of mTORC1 to integrate mitogenic signals with the nutritional status of the cell makes it a critical rheostat for the maintenance of metabolic balance and cellular homeostasis [26].
In the presence of nutrients and growth factors, mTORC1 drives ATP-consuming cellular processes necessary for cells to grow and proliferate (Fig. 2). mTORC1 also regulates protein synthesis by inducing mRNA translation and ribosome biogenesis [27,28] through its canonical substrates S6 kinases (S6Ks) and the inhibitory eIF4E-binding proteins (4EBPs) [29]. Interestingly, mTORC1 has been shown to also increase the efficiency of proteasome-mediated protein degradation to maintain proteostasis and sustain the increase in protein synthesis [30]. In addition to protein synthesis, mTORC1 has been recently implicated in the regulation of other major metabolic pathways of the cell, including lipid and nucleic acid synthesis, glycolysis, glutaminolysis, TCA cycle and oxidative phosphorylation, further supporting the idea of mTORC1 as a master regulator of metabolism [26,31].
Fig. 2. Regulation of intermediate metabolism by nutrient and energy sensors.
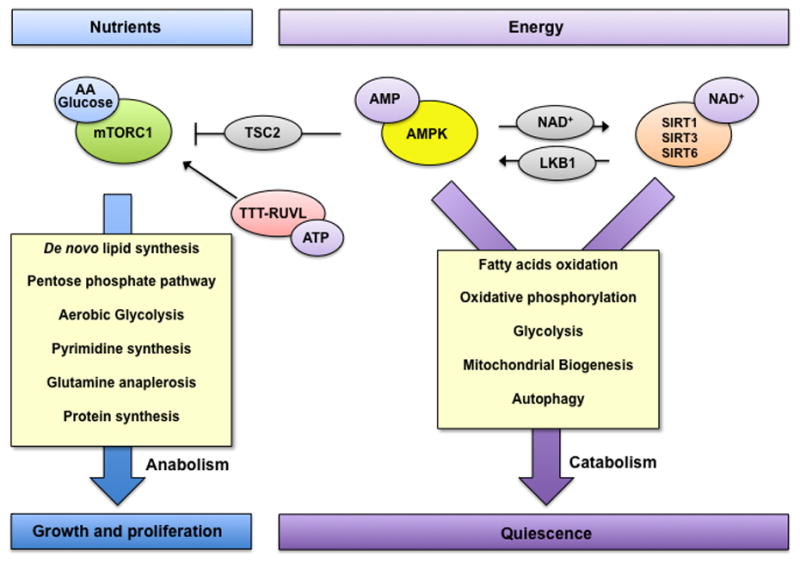
Nutrient and energy-responsive pathways fine-tune the output of signaling cascades, allowing for the correct balance between the availability of nutrients and the cellular capacity to use them effectively. AMPK and SIRT1 respond to the energy status of the cells through sensing of AMP and NAD+ levels respectively. When energy is scarce these sensors are activated inducing a rewiring of intermediate metabolism to catabolic processes in order to produce energy and restore homeostasis. When nutrients (such as glucose and amino acids) and energy are available, AMPK, SIRT1, SIRT3 and SIRT6 are repressed and mTORC1 is active, thus promoting a shift towards anabolic processes and energy production. These networks of signaling cascades, their interconnection and regulation allow the cells to maintain energetic balance and allow for the physiological adaptation to the ever-changing environment.
The ability of mTORC1 to regulate these pathways has been largely attributed to the regulation of key metabolic-related transcription factors. However, recent reports have also identified post-translational mechanisms [32,33]. Indeed, through regulation of 4EBP1 and S6K1, mTORC1 can promote the translation of hypoxia-inducible factor 1α (HIF1α) and c-Myc, thereby inducing the expression of glycolytic enzymes, glucose transporters and inhibiting the glucose-derived carbon flux through the TCA cycle [34,35]. This diverts the glucose-derived carbon from the TCA cycle to biosynthetic pathways, which promote cell growth. Consistent with this notion, mTORC1 signaling induces the oxidative arm of the pentose phosphate pathway (PPP) through increasing the expression of the rate-limiting enzyme, glucose-6-phosphate dehydrogenase (G6PD) thus increasing the generation of ribose (essential for nucleotide synthesis) and NADPH (essential for lipid synthesis) [35]. Moreover, mTORC1 through S6K1, regulates de novo pyrimidine synthesis by phosphorylating and activating the carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (CAD) [33,36]. CAD catalyzes the first three steps in de novo pyrimidine synthesis, thus providing a direct link between mTORC1 and an increase in the production of nucleotides [33,36].
c-Myc and mTORC1 are potent regulators of glutamine-mediated anabolic processes. c-Myc induces the expression of several proteins essential for glutamine anaplerosis, such as glutaminase (GLS) and the glutamine transporters [37]. Cells with mTORC1 active have been reported to be addicted to glutamine [38], indicating that glutamine is a key carbon source for mTORC1-related metabolic rewiring. Interestingly, mTORC1 has been recently shown to enhance c-Myc translation efficiency through S6K1, and consequently increase GLS activity [39] and many other c-Myc targets. mTORC1 also induces the activity of the mitochondrial glutamate dehydrogenase (GDH) through inhibition of SIRT4 transcription, a known regulator of GDH activity [40], supporting the role of mTORC1 in inducing glutamine anaplerosis to replenish the TCA cycle and anabolic processes.
In addition to increasing the activity of HIF1α and c-Myc, mTORC1 activation also leads to increased sterol regulatory element binding proteins (SREBP) activity [35]. SREBPs orchestrate the ability of the cells to synthesize lipids, as they induce the global expression of enzymes involved in de novo fatty acid synthesis [41]. In addition, mTORC1 also regulates SREBPs activity by inducing the phosphorylation and inhibition of LIPIN1, a phosphatase that inhibits SREBPs activity [42], thus directly linking mTORC1 to lipid synthesis.
mTORC1 signaling, therefore, is a critical regulator of anabolic processes that fuel cell growth and proliferation (Fig.1).
Fine-tuning signaling networks and catabolic rewiring through energetic sensors
Energetic homeostasis is regulated by both nutrient availability and energy demand, which are constantly changing. Therefore, cells have evolved multiple nutrient- and energy-sensing pathways to recognize the level of intracellular nutrients (such as mTORC1, described above) and energetic status (AMP/ATP ratio, NAD+/NADH). In times of nutrient deprivation or energetic deficit, nonessential ATP consumption is inhibited and energy stores are mobilized for catabolic processes. The best known regulators of these processes are AMP-activated protein kinase (AMPK), and Silent information regulator 1 (SIRT1) [43–45]. Under low-energy conditions, AMPK and SIRT1 are activated by increases in intracellular AMP and NAD+ levels, respectively. Once activated AMPK and SIRT1 switch on catabolic pathways that generate ATP while switching off anabolic pathways and other ATP-consuming processes, thus restoring the energy balance [43–45]. The complementary effects of AMPK and SIRT1 suggest that cells evolved both enzymes to work in a coordinated fashion. Thus, AMPK and SIRT1 are able to regulate each other [46] and are both frequently required to stimulate major pathways [47,48].
AMPK and SIRT1 coordinate the increase in the ability of the cells to oxidize fatty acids, thus fueling mitochondrial oxidative phosphorylation and the generation of ATP in an efficient manner [49]. AMPK promotes fatty acids oxidation (FAO) by regulation and activation of the peroxisome proliferator-activated receptor alpha (PPARα), a key transcriptional regulator of FAO [50]. AMPK also phosphorylates and inactivates acetyl- coenzyme A (CoA) carboxylase (ACC)-2, thus releasing the inhibitory pressure of malonyl-CoA from the uptake of fatty acids by the mitochondria for β-oxidation [51]. Importantly, in addition to increasing FAO, both AMPK and SIRT1 repress the ability of cells to synthesize fatty acids, by inhibiting the actions of SREBP1c [52,53].
SIRT1 and AMPK also have an important role in the regulation of glucose metabolism. AMPK induces glucose uptake and its oxidation through the glycolytic pathway, through regulation of glucose transporters and 6-phosphofructo-2-kinase/fructose-2,6-biphosphate [54,55]. Moreover, AMPK blocks glucose uptake through inducing a thioredoxin-interacting protein-dependent regulation of GLUT1 [56]. SIRT1 promotes the carbon flux from glucose to enter the TCA cycle by repressing HIF-1α, thus feeding oxidative phosphorylation (OXPHOS) in the mitochondria [57,58]. This suggests that SIRT1 and AMPKs actions complement each other, ensuring that the glucose that enters cells is used to produce ATP through oxidative phosphorylation and preventing it from entering biosynthetic pathways, such as the PPP. In addition, AMPK and SIRT1 regulate the CREB-regulated transcription co-activator2 (CRTC2), thus repressing gluconeogenesis [59,60].
SIRT1 and AMPK are also both necessary for the activation of the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) [49]. Following phosphorylation by AMPK, SIRT1 deacetylates PGC-1α and leads to its full activation, thus inducing the expression of genes related with FAO and mitochondrial biogenesis [48,61]. Importantly, this ability to activate mitochondrial biogenesis is central to the metabolic rewiring induced by AMPK and SIRT1 as it generates increased capacity for the oxidative catabolism of both glucose and fatty acids.
In addition to SIRT1, other sirtuin family members also play a role in regulation of metabolism. Particularly SIRT3 and SIRT6 have been shown to regulate glycolysis and TCA cycle through HIF-1α and c-Myc [62–64]. SIRT3 also contributes for catabolic processes by promoting oxidative phosphorylation through direct deacetylation of OXPHOS components [65,66]. On the other hand, SIRT4 has been shown to negatively regulate FAO [67,68], as well as to promote glutamine anaplerosis [40], suggesting a potential role for this sirtuin in promoting anabolic processes.
As a major regulator of anabolic processes, mTORC1’s activity is also indirectly regulated by the energetic state of the cells. AMPK phosphorylates and stimulates the activity of TSC2, thus repressing mTORC1 signaling [69]. AMPK also directly phosphorylates raptor, a critical component of mTORC1, to suppress mTORC1 signaling [70]. Moreover, energy depletion also inhibits mTORC1 function in an AMPK-independent manner. The AAA+ ATPase-containing complex Tel2-Tti-Tti2-RUVBL1/2 (TTT-RUVBL1/2) responds to cellular energy state and directly regulates the functional assembly of mTORC1 [71], however the mechanism of energy sensing for this process remains to be elucidated. Thus, AMPK, SIRT1 and the TTT-RUVBL complex fine-tune signaling transduction in accordance to the energetic state of the cell, regulating the balance between anabolic and catabolic processes, thereby maintaining cellular homeostasis (Fig.2).
Conclusions
Cellular homeostasis is maintained in coordination with extracellular cues (such as growth factors and nutrients) and intracellular metabolite concentrations. The interplay among all these factors coordinate complex signal transduction networks that perpetuate the information and rewire the metabolism of the cells. Taking into consideration the fact that cells are highly plastic and constantly exposed to a multitude of signals, an important question emerges. How are these pathways coordinated by the small number of upstream signaling regulators in response to the diverse intra and extra-cellular signals? The response is still largely unknown, but surely part of the answer must be based on how these conserved pathways integrate their actions, their crosstalk and how they are regulated. Importantly, the notion that intracellular metabolite levels are potent regulators of signaling pathways should also be taken into account. This is an important area of research that has emerged recently, with numerous metabolites being described to regulate signaling cascades, thus contributing to the maintenance of energetic balance. Therefore, the understanding of these signaling cascades and their ability to fine-tune the balance between catabolism and anabolism is extremely important for understanding the development of metabolic-related diseases. An in depth study of these signal integration mechanisms is therefore an attractive area for further investigation. Furthermore, the knowledge gained may yield important therapeutic targets for drug development for use in a multitude of metabolic diseases.
Highlights.
Signaling networks intracellular and extracellular cues to maintain homeostasis
PI3K/AKT and Ras/ERK signaling induces anabolic reprogramming
mTORC1 is a master node of signaling integration that promotes anabolism
AMPK and SIRT1 fine tune signaling networks in response to energetic status
Acknowledgments
We apologize to those whose work was not discussed and cited in this review due to limitations in space and scope. We thank Dr. Michal Nagiec, Dr. Eric Bell, Dr. Sang Gyun Kim and Gwen Buel for kindly providing helpful discussions and comments on this manuscript. J.B. is an Established Investigator of the LAM Foundation. NIH Grants GM51405, CA46595 and HL121266 provide research support for the Blenis laboratory.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Wellen KE, Thompson CB. A two-way street: Reciprocal regulation of metabolism and signalling. Nature reviews Molecular cell biology. 2012;13 (4):270–276. doi: 10.1038/nrm3305. [DOI] [PubMed] [Google Scholar]
- 2.Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell metabolism. 2012;16(1):9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vanhaesebroeck B, Stephens L, Hawkins P. Pi3k signalling: The path to discovery and understanding. Nature reviews Molecular cell biology. 2012;13 (3):195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 4.Hemmings BA, Restuccia DF. Pi3k-pkb/akt pathway. Cold Spring Harbor perspectives in biology. 2012;4(9):a011189. doi: 10.1101/cshperspect.a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whiteman EL, Cho H, Birnbaum MJ. Role of akt/protein kinase b in metabolism. Trends in endocrinology and metabolism: TEM. 2002;13(10):444–451. doi: 10.1016/s1043-2760(02)00662-8. [DOI] [PubMed] [Google Scholar]
- 6.Kim DI, Lim SK, Park MJ, Han HJ, Kim GY, Park SH. The involvement of phosphatidylinositol 3-kinase /akt signaling in high glucose-induced downregulation of glut-1 expression in arpe cells. Life sciences. 2007;80 (7):626–632. doi: 10.1016/j.lfs.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 7.Roberts DJ, Tan-Sah VP, Smith JM, Miyamoto S. Akt phosphorylates hk-ii at thr-473 and increases mitochondrial hk-ii association to protect cardiomyocytes. The Journal of biological chemistry. 2013;288(33):23798–23806. doi: 10.1074/jbc.M113.482026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase b and other protein kinases of the insulin signaling cascades. The Journal of biological chemistry. 1997;272(28):17269–17275. doi: 10.1074/jbc.272.28.17269. [DOI] [PubMed] [Google Scholar]
- 9.Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB, Hay N. Hexokinase-mitochondria interaction mediated by akt is required to inhibit apoptosis in the presence or absence of bax and bak. Molecular cell. 2004;16(5):819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 10.Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of atp-citrate lyase as a protein kinase b (akt) substrate in primary adipocytes. The Journal of biological chemistry. 2002;277(37):33895–33900. doi: 10.1074/jbc.M204681200. [DOI] [PubMed] [Google Scholar]
- 11.Zaidi N, Swinnen JV, Smans K. Atp-citrate lyase: A key player in cancer metabolism. Cancer research. 2012;72(15):3709–3714. doi: 10.1158/0008-5472.CAN-11-4112. [DOI] [PubMed] [Google Scholar]
- 12.Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. The Journal of biological chemistry. 2003;278(51):51606–51612. doi: 10.1074/jbc.M310722200. [DOI] [PubMed] [Google Scholar]
- 13.Morrison DK. Map kinase pathways. Cold Spring Harbor perspectives in biology. 2012;4(11) doi: 10.1101/cshperspect.a011254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G, Chiaradonna F. Oncogenic k-ras decouples glucose and glutamine metabolism to support cancer cell growth. Molecular systems biology. 2011;7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiaradonna F, Sacco E, Manzoni R, Giorgio M, Vanoni M, Alberghina L. Ras-dependent carbon metabolism and transformation in mouse fibroblasts. Oncogene. 2006;25(39):5391–5404. doi: 10.1038/sj.onc.1209528. [DOI] [PubMed] [Google Scholar]
- 16*.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149(3):656–670. doi: 10.1016/j.cell.2012.01.058. In this report authors show for the first time that activation of oncogenic K-Ras induces a glucose-associated metabolic rewiring towards anabolic processes, fueling pancreatic tumor cell growth and proliferation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, Lu Z. Erk1/2-dependent phosphorylation and nuclear translocation of pkm2 promotes the warburg effect. Nature cell biology. 2012;14(12):1295–1304. doi: 10.1038/ncb2629. Here it is reported for the first time a direct link between ERK signaling and metabolic regulation. Authors show that ERK2 directly phosphorylates PKM2, thus inducing anabolic glucose metabolism and sustaining biosyntetic pathways. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18**.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, et al. Glutamine supports pancreatic cancer growth through a kras-regulated metabolic pathway. Nature. 2013;496(7443):101–105. doi: 10.1038/nature12040. This work shows a novel pathway by which glutamine fuels anabolic processes in response to K-Ras activation. The authors demonstrate that glutamine-derived aspartate is metabolized to pyruvate through the malate-aspartate shuttle providing redox potential and sustaining the TCA cycle, thus supporting K-Ras mediated-anabolic reprogramming and cell growth and proliferation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laplante M, Sabatini DM. Mtor signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai SL, Tee AR, Short JD, Bergeron JM, Kim J, Shen J, Guo R, Johnson CL, Kiguchi K, Walker CL. Activity of tsc2 is inhibited by akt-mediated phosphorylation and membrane partitioning. The Journal of cell biology. 2006;173(2):279–289. doi: 10.1083/jcb.200507119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal s6 kinase. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(37):13489–13494. doi: 10.1073/pnas.0405659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang J, Manning BD. The tsc1-tsc2 complex: A molecular switchboard controlling cell growth. The Biochemical journal. 2008;412(2):179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carriere A, Romeo Y, Acosta-Jaquez HA, Moreau J, Bonneil E, Thibault P, Fingar DC, Roux PP. Erk1/2 phosphorylate raptor to promote ras-dependent activation of mtor complex 1 (mtorc1) The Journal of biological chemistry. 2011;286(1):567–577. doi: 10.1074/jbc.M110.159046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carriere A, Cargnello M, Julien LA, Gao H, Bonneil E, Thibault P, Roux PP. Oncogenic mapk signaling stimulates mtorc1 activity by promoting rsk-mediated raptor phosphorylation. Current biology : CB. 2008;18(17):1269–1277. doi: 10.1016/j.cub.2008.07.078. [DOI] [PubMed] [Google Scholar]
- 25.Howell JJ, Manning BD. Mtor couples cellular nutrient sensing to organismal metabolic homeostasis. Trends in endocrinology and metabolism: TEM. 2011;22 (3):94–102. doi: 10.1016/j.tem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dibble CC, Manning BD. Signal integration by mtorc1 coordinates nutrient input with biosynthetic output. Nature cell biology. 2013;15(6):555–564. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holz MK, Ballif BA, Gygi SP, Blenis J. Mtor and s6k1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123(4):569–580. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 28.Tcherkezian J, Cargnello M, Romeo Y, Huttlin EL, Lavoie G, Gygi SP, Roux PP. Proteomic analysis of cap-dependent translation identifies larp1 as a key regulator of 5'top mrna translation. Genes & development. 2014;28(4):357–371. doi: 10.1101/gad.231407.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma XM, Blenis J. Molecular mechanisms of mtor-mediated translational control. Nature reviews Molecular cell biology. 2009;10(5):307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 30**.Zhang Y, Nicholatos J, Dreier JR, Ricoult SJ, Widenmaier SB, Hotamisligil GS, Kwiatkowski DJ, Manning BD. Coordinated regulation of protein synthesis and degradation by mtorc1. Nature. 2014 doi: 10.1038/nature13492. The authors show that in addition to the well established role of mTORC1 in protein synthesis, mTORC1 signaling also regulates proteosomal-mediated protein degradation, thus promoting proteostasis and therefore sustaining protein homeostasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Howell JJ, Ricoult SJH, Ben-Sahra I, Manning BD. A growing role for mtor in promoting anabolic metabolism. Biochemical Society transactions. 2013;41:906–912. doi: 10.1042/BST20130041. [DOI] [PubMed] [Google Scholar]
- 32.Yecies JL, Manning BD. Transcriptional control of cellular metabolism by mtor signaling. Cancer research. 2011;71(8):2815–2820. doi: 10.1158/0008-5472.CAN-10-4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33*.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mtor and s6k1. Science. 2013;339(6125):1323–1328. doi: 10.1126/science.1228792. This work, together with Robitaille et al (Ref. 33), show for the first time a posttranslational mechanisms of regulation of anabolic metabolism by mTORC1 signaling. These reports show that mTORC1, through S6K1, phosphorylates CAD inducing pyrimidine byosinthesis, thus fueling nucleic acid biosynthesis and promoting anabolic growth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.West MJ, Stoneley M, Willis AE. Translational induction of the c-myc oncogene via activation of the frap/tor signalling pathway. Oncogene. 1998;17 (6):769–780. doi: 10.1038/sj.onc.1201990. [DOI] [PubMed] [Google Scholar]
- 35.Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma QC, Gorski R, Cleaver S, Heiden MGV, et al. Activation of a metabolic gene regulatory network downstream of mtor complex 1. Molecular cell. 2010;39(2):171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, Prescianotto-Baschong C, Sauer U, Jenoe P, Hall MN. Quantitative phosphoproteomics reveal mtorc1 activates de novo pyrimidine synthesis. Science. 2013;339(6125):1320–1323. doi: 10.1126/science.1228771. [DOI] [PubMed] [Google Scholar]
- 37.Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. C-myc and cancer metabolism. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(20):5546–5553. doi: 10.1158/1078-0432.CCR-12-0977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choo AY, Kim SG, Vander Heiden MG, Mahoney SJ, Vu H, Yoon SO, Cantley LC, Blenis J. Glucose addiction of tsc null cells is caused by failed mtorc1-dependent balancing of metabolic demand with supply. Molecular cell. 2010;38 (4):487–499. doi: 10.1016/j.molcel.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Csibi A, Lee G, Yoon S-O, Tong H, Ilter D, Elia I, Fendt S-M, Roberts TM, Blenis J. The mtorc1/s6k1 pathway regulates glutamine metabolism through the eif4b-dependent control of c-myc translation. Current Bioloby. 2014 doi: 10.1016/j.cub.2014.08.007. In press. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40**.Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A, Morrison T, Henske EP, et al. The mtorc1 pathway stimulates glutamine metabolism and cell proliferation by repressing sirt4. Cell. 2013;153(4):840–854. doi: 10.1016/j.cell.2013.04.023. This report shows for the first time that mTORC1 depends on glutamine anaplerosis to replenish the TCA cycle intermediates and provide redox potential to fuel anabolic processes, thus promoting cell growth and proliferation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eberle D, Hegarty B, Bossard P, Ferre P, Foufelle F. Srebp transcription factors: Master regulators of lipid homeostasis. Biochimie. 2004;86(11):839–848. doi: 10.1016/j.biochi.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 42.Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, Sabatini DM. Mtor complex 1 regulates lipin 1 localization to control the srebp pathway. Cell. 2011;146(3):408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hardie DG, Ross FA, Hawley SA. Ampk: A nutrient and energy sensor that maintains energy homeostasis. Nature reviews Molecular cell biology. 2012;13 (4):251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang HC, Guarente L. Sirt1 and other sirtuins in metabolism. Trends in endocrinology and metabolism: TEM. 2014;25(3):138–145. doi: 10.1016/j.tem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haigis MC, Sinclair DA. Mammalian sirtuins: Biological insights and disease relevance. Annual review of pathology. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, Ido Y. Ampk and sirt1: A long-standing partnership? American journal of physiology Endocrinology and metabolism. 2010;298(4):E751–760. doi: 10.1152/ajpendo.00745.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J. Interdependence of ampk and sirt1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell metabolism. 2010;11(3):213–219. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. Ampk regulates energy expenditure by modulating nad+ metabolism and sirt1 activity. Nature. 2009;458(7241):1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Canto C, Auwerx J. Pgc-1alpha, sirt1 and ampk, an energy sensing network that controls energy expenditure. Current opinion in lipidology. 2009;20(2):98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee WJ, Kim M, Park HS, Kim HS, Jeon MJ, Oh KS, Koh EH, Won JC, Kim MS, Oh GT, Yoon M, et al. Ampk activation increases fatty acid oxidation in skeletal muscle by activating pparalpha and pgc-1. Biochemical and biophysical research communications. 2006;340(1):291–295. doi: 10.1016/j.bbrc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 51.Hardie DG, Pan DA. Regulation of fatty acid synthesis and oxidation by the amp-activated protein kinase. Biochemical Society transactions. 2002;30(Pt 6):1064–1070. doi: 10.1042/bst0301064. [DOI] [PubMed] [Google Scholar]
- 52.Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, Gao B, et al. Ampk phosphorylates and inhibits srebp activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell metabolism. 2011;13(4):376–388. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ponugoti B, Kim DH, Xiao Z, Smith Z, Miao J, Zang M, Wu SY, Chiang CM, Veenstra TD, Kemper JK. Sirt1 deacetylates and inhibits srebp-1c activity in regulation of hepatic lipid metabolism. The Journal of biological chemistry. 2010;285(44):33959–33970. doi: 10.1074/jbc.M110.122978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L. Phosphorylation and activation of heart pfk-2 by ampk has a role in the stimulation of glycolysis during ischaemia. Current biology : CB. 2000;10(20):1247–1255. doi: 10.1016/s0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- 55.Xi X, Han J, Zhang JZ. Stimulation of glucose transport by amp-activated protein kinase via activation of p38 mitogen-activated protein kinase. The Journal of biological chemistry. 2001;276(44):41029–41034. doi: 10.1074/jbc.M102824200. [DOI] [PubMed] [Google Scholar]
- 56.Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, Shen CH, Wen J, Asara J, McGraw TE, Kahn BB, et al. Ampk-dependent degradation of txnip upon energy stress leads to enhanced glucose uptake via glut1. Molecular cell. 2013;49(6):1167–1175. doi: 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Molecular cell. 2010;38(6):864–878. doi: 10.1016/j.molcel.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 58**.Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, et al. Declining nad(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155(7):1624–1638. doi: 10.1016/j.cell.2013.11.037. Here, the authors demonstrate that the energy sensor SIRT1 regulates HIF-1alpha to fine-tune the balance between anabolic and catabolic processes. This work shows for the first time the importance of maintaining this balance for the aging process. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, Schenk S, Milne J, Meyers DJ, Cole P, Yates J, 3rd, Olefsky J, et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456(7219):269–273. doi: 10.1038/nature07349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee JM, Seo WY, Song KH, Chanda D, Kim YD, Kim DK, Lee MW, Ryu D, Kim YH, Noh JR, Lee CH, et al. Ampk-dependent repression of hepatic gluconeogenesis via disruption of creb. Crtc2 complex by orphan nuclear receptor small heterodimer partner. The Journal of biological chemistry. 2010;285(42):32182–32191. doi: 10.1074/jbc.M110.134890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61*.Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS, Hubbard BP, et al. Sirt1 is required for ampk activation and the beneficial effects of resveratrol on mitochondrial function. Cell metabolism. 2012;15(5):675–690. doi: 10.1016/j.cmet.2012.04.003. This report shows the importance of the crosstalk between the energy sensors SIRT1 and AMPK to the regulation of catabolic processes and maintenance of cellular homeostasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bell EL, Emerling BM, Ricoult SJ, Guarente L. Sirt3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ros production. Oncogene. 2011;30(26):2986–2996. doi: 10.1038/onc.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB, Pandolfi PP, et al. Sirt3 opposes reprogramming of cancer cell metabolism through hif1alpha destabilization. Cancer cell. 2011;19(3):416–428. doi: 10.1016/j.ccr.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber D, Cosentino C, et al. The histone deacetylase sirt6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151(6):1185–1199. doi: 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vassilopoulos A, Pennington JD, Andresson T, Rees DM, Bosley AD, Fearnley IM, Ham A, Flynn CR, Hill S, Rose KL, Kim HS, et al. Sirt3 deacetylates atp synthase f1 complex proteins in response to nutrient- and exercise-induced stress. Antioxidants & redox signaling. 2014;21(4):551–564. doi: 10.1089/ars.2013.5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Finley LW, Haas W, Desquiret-Dumas V, Wallace DC, Procaccio V, Gygi SP, Haigis MC. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PloS one. 2011;6(8):e23295. doi: 10.1371/journal.pone.0023295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Laurent G, de Boer VC, Finley LW, Sweeney M, Lu H, Schug TT, Cen Y, Jeong SM, Li X, Sauve AA, Haigis MC. Sirt4 represses peroxisome proliferator-activated receptor alpha activity to suppress hepatic fat oxidation. Molecular and cellular biology. 2013;33(22):4552–4561. doi: 10.1128/MCB.00087-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Laurent G, German NJ, Saha AK, de Boer VC, Davies M, Koves TR, Dephoure N, Fischer F, Boanca G, Vaitheesvaran B, Lovitch SB, et al. Sirt4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl coa decarboxylase. Molecular cell. 2013;50(5):686–698. doi: 10.1016/j.molcel.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Inoki K, Zhu T, Guan KL. Tsc2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 70.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. Ampk phosphorylation of raptor mediates a metabolic checkpoint. Molecular cell. 2008;30(2):214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71**.Kim SG, Hoffman GR, Poulogiannis G, Buel GR, Jang YJ, Lee KW, Kim BY, Erikson RL, Cantley LC, Choo AY, Blenis J. Metabolic stress controls mtorc1 lysosomal localization and dimerization by regulating the ttt-ruvbl1/2 complex. Molecular cell. 2013;49(1):172–185. doi: 10.1016/j.molcel.2012.10.003. This work shows that the TTT-RUVBL1/2 complex regulates the functional assembly of mTORC1. The authors show that the TTT-RUVLB1/2 is an ATP-dependent complex that responds to the metabolic state of the cell, thus further supporting the idea of mTORC1 as a cellular rheostat that dictates cellular fate. [DOI] [PMC free article] [PubMed] [Google Scholar]