

Abstract
Aims
Lymphatic vessel dysfunction is an emerging component of metabolic diseases and can lead to tissue lipid accumulation, dyslipidaemia, and oedema. While lymph leakage has been implicated in obesity and hypercholesterolaemia, whether similar lymphatic dysfunction exists in diabetes has not been investigated.
Methods and results
To measure the lymphatic integrity of transgenic mice, we developed a new assay that quantifies the solute permeability of murine collecting lymphatic vessels. Compared with age-matched wild-type (WT) controls, the permeability of collecting lymphatics from diabetic, leptin receptor-deficient (db/db) mice was elevated >130-fold. Augmenting nitric oxide (NO) production by suffusion of l-arginine rescued this defect. Using pharmacological tools and eNOS−/− mice, we found that NO increased WT lymphatic permeability, but reduced db/db lymphatic permeability. These conflicting actions of NO were reconciled by the finding that phosphodiesterase 3 (PDE3), normally inhibited by NO signalling, was active in db/db lymphatics and inhibition of this enzyme restored barrier function.
Conclusion
In conclusion, we identified the first lymphatic vascular defect in type 2 diabetes, an enhanced permeability caused by low NO bioavailability. Further, this demonstrates that PDE3 inhibition is required to maintain lymphatic vessel integrity and represents a viable therapeutic target for lymphatic endothelial dysfunction in metabolic disease.
Keywords: Permeability, Barrier function
1. Introduction
Lymphatic vessels are crucial for the absorption of intestinal lipids, antigen and immune cell transport to lymph nodes, fluid homeostasis, and cancer cell dissemination.1 Each of these processes relies on the organized movement of solute, fluid, and cells across the lymphatic vessel wall. Dysregulation of lymphatic vessel integrity causes lymph leakage, which is associated with the development of obesity, atherosclerosis, and oedema, and it is thought to facilitate cancer cell entry into these vessels.1–3 However, an understanding of the mechanisms regulating lymphatic vessel integrity has been elusive due to the lack of approaches to quantify and probe this aspect of lymphatic function, which is likely valuable for diagnosing and treating disorders of the lymphatic vasculature.
Lymphatic vessel leakage is most often observed in mice as a consequence of the targeted mutation of genes required for proper lymphatic vascular development (e.g. Vegfr3, Prox1, Angptl4). In these mutant mice, a milky chylous fluid accumulates in the chest around the lungs and is ultimately lethal. In the adult, however, emerging evidence supports the idea that less severe lymphatic leakage is coupled to metabolic disease. For instance, subtle leakage of lymph promotes obesity in mice lacking one allele of Prox1, the transcription factor that confers lymphatic identity to venous endothelial cells.3–5 Conversely, reduction of vascular and lymphatic leakage through Apln overexpression mitigates subcutaneous adipose deposition in a model of diet-induced obesity.6 In mice deficient for apolipoprotein E (ApoE−/−), hypercholesterolaemia results in leaky and enlarged lymphatics, loss of lymphatic valves, and oedema.7 Most recently, defective lymphatic transport has been linked to impaired reverse cholesterol transport from the tissues8 and from atherosclerotic plaques.9,10 In contrast, lymphatic function in the widespread metabolic disease, diabetes, has not been investigated.
In type 2 diabetes, the vasculature is characterized by impaired endothelial nitric oxide (NO) production that leads to a reduced ability of resistance arteries to vasodilate, promoting hypertension. Whereas the role of NO in arterial vasodilation is well defined, its role in the regulation of vascular permeability has remained controversial for the past two decades, with conflicting reports that NO can either maintain or disrupt vascular barrier function.11–16 Whether, and how, NO regulates lymphatic permeability remains unknown both in health and disease.
To determine whether lymphatic dysfunction is present in diabetes, we developed a new assay to measure the permeability of collecting lymphatic vessels isolated from genetically modified mice. Comparison of collecting lymphatic permeability between wild-type and leptin receptor-deficient mice revealed a dramatic loss of lymphatic vascular integrity in diabetes. This defect could be rescued by increasing the substrate availability for NO synthesis or by inhibiting phosphodiesterase 3, an enzyme normally inhibited by cGMP produced downstream of NO. Unexpectedly, we found that NO elevated the permeability of wild-type lymphatic vessels, whereas it lowered the permeability of diabetic lymphatics. To our knowledge, this is the first demonstration of two opposing roles for NO in the regulation of permeability in one preparation, which in this study depended on the differential activity of a phosphodiesterase enzyme. Thus, the treatments identified here may restore lymphatic function in other models of metabolic diseases where NO production is inhibited.
2. Methods
2.1. Mice
Male wild-type (WT) and endothelial nitric oxide synthase-deficient (eNOS−/−) mice on the C57Bl/6J background were studied at 8–10 weeks of age (Jackson Laboratory, Bar Harbor, ME, USA). Leptin receptor-deficient mice (db/db) and age-matched WT controls on the C57Bl/KsJ background were housed and bred at the Dalton Cardiovascular Research Center (JAX strains: 000642 and 000662, respectively). For the diabetic studies, male and female mice between 20 and 30 weeks of age were studied. Animals were housed under standard conditions with ad libitum access to chow and water. Unfasted blood samples were collected from all db/db mice and their age-matched WT controls by tail snip, and blood glucose was measured using a glucometer and test strips (ReliOn Prime, Bentonville, AR, USA) before surgery. All experimental protocols were reviewed and approved by the University of Missouri Animal Care and Use Committee and conformed to the National Institutes of Health's Guide for the Care and Use of Laboratory Animals.
2.2. Vessel isolation and cannulation
Mice were anaesthetized with pentobarbital sodium (Nembutal, 60 mg/kg, i.p.), and the ventral abdomen was shaved. When mice became non-responsive to toe pinch, the intestines were exteriorized through a 2–3 cm midline incision. Collecting lymphatics in the mesentery were located under a stereomicroscope, excised with microscissors, and placed in Krebs buffer containing 0.5% BSA. After several collecting lymphatics were isolated from the mesentery, the mouse was euthanized with an overdose of pentobarbital (200 mg/kg, i.c.) followed by cervical dislocation. Adipose and connective tissues were carefully microdissected from isolated lymphatics in a Sylgard-coated chamber. Collecting lymphatics (1–2 mm long) were then transferred to an isolated vessel apparatus and tied onto two glass micropipettes (∼80 μm OD each) submerged in Krebs buffer in a 3-mL chamber with a water jacket for temperature control. The perfusion micropipette had two lumens; thus, it was capable of perfusing either unlabeled or fluorescently tagged BSA. The outflow micropipette had a single lumen. After cannulation, the vessel was warmed to 37°C over 45 min on a Zeiss Axiovert 100TV inverted fluorescence microscope.
2.3. Measurement of lymphatic vessel solute permeability
Albumin flux (Js, mmol/s) across the collecting lymphatic wall was measured directly with microscope-based photometry since a fraction of the perfused albumin was labelled with a fluorescent dye, similar to a previous in vivo study.17 Fluorescence intensity was measured within a rectangular region of interest defined by four adjustable leaflets in front of a photometer that sampled light from the vessel lumen and adjacent extravascular space. To control perfusion and pressure, each micropipette was connected to manual water manometers via polyethylene tubing. Two manometers on a switch were connected to each side of the dual-lumen perfusion micropipette, such that when the switches were turned, one side of the pipette or the other selectively perfused the lymphatic vessel without changing intraluminal hydrostatic pressure. This allowed controlled perfusion of either the unlabelled or fluorescent BSA. Based on pilot experiments, all vessels were perfused with 9 cm H2O of pressure on the inlet side and 5 cm H2O on the outflow side to enable rapid changes in the perfusate. To make a measurement, unlabelled BSA was perfused first to obtain the background fluorescence intensity (Figure 1i). Switching to perfusion of the fluorescent BSA caused a rapid, step increase in fluorescence intensity (Io) on the photometer (Figure 1ii). Over time, the fluorescent BSA moved across the lymphatic vessel wall into the bath solution, which caused a gradual but linear increase in photometer voltage (dIf/dt) (Figure 1iii). Changing the perfusate to the unlabelled BSA washed away all fluorescence, returning the photometer voltage to baseline, and allowed repeated measurements to be made and responses to pharmacologic agonists/antagonists to be ascertained. Albumin permeability (Ps, cm/s) was then calculated from a rederived form of Fick's first law relating albumin flux to a constant (i.e. unchanging) surface area (S, cm2) and concentration gradient (ΔC, mmol/mL):
| (1) |
where collecting lymphatic diameter (D, cm) was simultaneously tracked on a transmitted light, near infrared image of the vessel displayed on a computer monitor to ensure that it did not change throughout each recording (Figure 1B). There was no overlap between the near infrared light and the fluorescence emission of Alexa-488 BSA.
Figure 1.
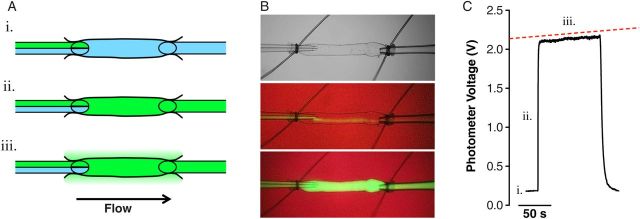
Novel ex vivo approach to measure lymphatic solute permeability. Collecting lymphatic vessels were isolated from the mouse mesentery and cannulated on two micropipettes capable of pressure control. (A) Lymphatic vessels were perfused by a ‘theta’ micropipette that contained two identical solutions, with the exception that one solution contained albumin tagged with a fluorophore. Initially, washout solution (blue) is perfused to obtain a baseline recording on a photometer (i.). Switching to the other side of the micropipette selectively perfuses the fluorescent albumin (green) without a change in intraluminal pressure (ii.). Over time, fluorescent albumin moves across the lymphatic wall (iii.). (B) A brightfield image of a lymphatic vessel is shown (top), followed by a fluorescent image during perfusion of the washout buffer (middle), and perfusion of the fluorescent albumin (bottom). The background is red due to an infrared filter that allows diameter to be measured throughout the recording. (C) A representative digital photometer recording used to calculate vessel permeability. The slope of the steady-state portion of the recording was fit using linear regression (red dashed line).
2.4. Solutions and chemicals
During the experiment, lymphatic vessels were continuously bathed abluminally (0.4 mL/min) with Krebs buffer supplemented with 0.1% BSA that contained (in mmol) 146.9 NaCl, 4.7 KCl, 2 CaCl2.2H2O, 1.2 MgSO4, 1.2 NaH2PO4.H2O, 3 NaHCO3, 1.5 sodium-HEPES, and 5 d-glucose (pH = 7.4 at 37°C). An identical Krebs buffer served as the luminal perfusion solution, but it contained 1% BSA to maintain a concentration gradient across the vessel wall. One side of the dual-lumen micropipette contained this buffer, while the other side contained the same solution except that 5% of the BSA (0.5 mg/mL) was labelled with Alexa-488 fluorescent dye, as in previous publications.17,18
l-NG-nitroarginine methyl ester (l-NAME), l-arginine, and cilostamide (Axxora, Cat no: BML-PD125-0005, Farmingdale, NY, USA) were superfused for 20 min after control measurements and prior to resuming albumin flux measurements. Sodium nitroprusside (SNP) and histamine were added directly to the bath immediately before a single measurement. All drugs were stored in stock concentrations that were diluted in Krebs buffer on the day of use.
2.5. In vivo lymphangiography
To evaluate lymphatic vessel leakage in vivo, lymphangiography was performed on the hindlimbs of both WT and db/db mice. Briefly, the saphenous vein and adjacent popliteal collecting lymphatic vessels were exposed by making an incision in the overlying skin as previously described.19 Evans Blue dye (1% w/v) was then injected into the hind paw subcutaneously. After 5 min, the popliteal collecting lymphatics were imaged under a dissecting microscope, similar to a previously reported fluorescence method.8 Leakage of the dye from the lymphatic vessels into the surrounding tissues was assessed visually for each hind paw.
2.6. Statistical analyses
Data are presented as means ± SEM. For comparison of two groups, a paired or unpaired Student's t-test was used as appropriate. For the rescue experiments, effects were compared within genotypes using two-way ANOVA with Bonferonni's multiple comparisons test, while effects between genotypes were compared using one-way ANOVA with Bonferonni's multiple comparisons test. All tests were performed at a significance level of P < 0.05 using Prism 5 (Graphpad Software Inc., La Jolla, CA, USA). In general, 1–3 collecting lymphatic vessels from each mouse were studied. Fluorescence intensity and vessel diameter data were recorded continuously in a raw text file at 30 Hz, from which single solute flux traces were extracted using Igor Pro (Wavemetrics, Lake Oswego, OR, USA) and imported to Prism to determine the height (Io) and the slope (dIf/dt) of the albumin flux tracings with linear regression. Permeability values were then calculated from these data and lymphatic diameter in MS Excel.
3. Results
3.1. Solute permeability of murine collecting lymphatic vessels
To enable the use of genetically modified mice to study the molecular mechanisms regulating the integrity of the lymphatic vasculature, we developed a new approach for measuring the solute permeability of murine collecting lymphatic vessels. While one of us previously reported the albumin permeability of rat collecting lymphatics in vivo using a microperfusion method,17,18 that approach lacked complete control over pressure and had a low success rate due to large changes in diameter caused by spontaneous contractions. Moreover, collecting lymphatics in mouse mesentery are covered in adipose tissue, hindering their visualization and micropuncture. Overcoming these limitations, collecting lymphatics were microdissected from the mouse mesentery, taking advantage of lymphatics in this tissue bed that lack strong spontaneous contractions. Using this ex vivo approach, permeability could be measured quickly and repeatedly before and after pharmacological intervention using intact lymphatic vessels that had been isolated from either wild-type (WT) or genetically modified mice.
Solute transport across the vascular wall depends on diffusion in addition to the ‘pull’ of water moving through the same pathways, known as ‘convective drag’. Due to this latter effect, the solute permeability (Ps) of blood microvessels appears to be pressure dependent. To define the pressure dependence of murine collecting lymphatic solute transport and choose a criterion pressure for all future measurements of this study, Ps was measured as a function of the net pressure gradient (Figure 2, n = 5). As the pressure gradient favouring fluid filtration was elevated, lymphatic Ps increased gradually. Surprisingly, lymphatic Ps became drastically elevated at the highest pressure gradient of ∼18 cm H2O. In a subset of experiments, after measuring Ps at this pressure gradient, the vessel was returned to the lowest pressure gradient of ∼3 cm H2O after which lymphatic Ps returned to baseline within 10 min, indicating that the endothelial layer was not permanently damaged or disrupted.
Figure 2.

Contribution of diffusion and convection to albumin transport across the lymphatic wall. Lymphatic permeability to albumin (PsBSA) was measured at several different hydrostatic pressures and plotted against the net pressure gradient for filtration, obtained by taking the difference between hydrostatic and oncotic pressure gradients (n = 5). When the standard model describing diffusive to convective transport was fitted to the data (dashed curve), the permeability value at the highest pressure deviated from this model, suggesting that larger pores opened suddenly. Importantly, after vessels were exposed to this high pressure, their permeability returned to normal within minutes at the lowest pressure (gray point). The diffusive permeability (Pd) at the y-intercept was 2.3 × 10−7 cm/s.
The standard model describing the individual contributions of diffusion and convective drag to the solute permeability of blood vessels provides the diffusional permeability (Pd), i.e. the Ps when there is no net filtration of fluid. This model was used previously to estimate Pd for rat lymphatics.17,18 Intriguingly, this model could not fit the data at all pressure gradients; instead, only the measurements made at the three lowest pressure gradients could be fit, suggesting that the highest value is outside the physiological pressure range for mouse collecting lymphatics. When those data are excluded from the analysis, the Pd was calculated as 2.3 × 10−7 cm/s, which compares well to that reported for collecting lymphatic vessels from the rat.17 Collectively, these data demonstrate that the permeability of lymphatic vessels is higher than venular microvessels if measured at the same hydrostatic pressures. To make measurements that approximated the Pd throughout this study, the lowest pressure gradient of 3 cm H2O was subsequently chosen.
3.2. Diabetic mice exhibit a defect in lymphatic permeability regulation
Since leakage of lymph from the lymphatic vasculature appears to be a common feature of metabolic diseases, we hypothesized that lymphatic barrier function would be significantly disrupted in a mouse model of type 2 diabetes. To test this hypothesis, collecting lymphatics from leptin receptor-deficient (db/db) mice and age-matched WT mice on the same genetic background (C57Bl/KsJ) were studied using our ex vivo Ps approach. Notably, db/db homozygous mice begin to develop significant weight gain and hyperglycaemia at ∼8 weeks of age. For this study, vessels were isolated from male and female mice between 20 and 30 weeks of age to ensure that a strong diabetic phenotype was present. As expected, db/db mice were significantly obese and hyperglycaemic compared with control mice (P < 0.0001, Figure 3A and B). After collecting lymphatics from db/db mice were isolated and perfused, leakage of fluorescently labelled albumin from the vessel lumen was visible by eye during the experiment (Figure 3E and G), whereas comparable albumin leakage from WT lymphatics was not observed at any time during experiments (Figure 3D and F). Measurement of db/db collecting lymphatic Ps revealed a dramatic loss of barrier function (299 ± 56 × 10−7 cm/s, n = 23) compared with WT controls (2.2 ± 0.3 × 10−7 cm/s, n = 10) that was statistically significant (P < 0.01, Figure 3C). Data from males and females were pooled, as lymphatic permeability did not differ significantly between the sexes (Figure 4A and B).
Figure 3.

Lymphatic vascular integrity is disrupted in leptin receptor-deficient (db/db) mice. Diabetic db/db mice were obese (A) and hyperglycaemic (B) compared with age-matched wild-type controls on the same genetic background. (C) The albumin permeability (PsBSA) of db/db collecting lymphatics was >130-fold greater than WT controls, indicating a defect in endothelial integrity. Note the logarithmic y-axis scale. *Significantly different from WT control (P < 0.05). Data are from n = 10 WT vessels from 8 mice and n = 23 db/db vessels from 11 mice. Brightfield images of cannulated collecting lymphatics are shown for a WT (D) and db/db (E) vessel. Under epifluorescence, fluorescent albumin leakage from the WT vessel is not visible (F), but it is seen extravasating from the db/db vessel (G). Permeability values for the WT (D and F) and db/db (E and G) collecting lymphatic vessels were 1.1 and 595 × 10−7 cm/s, respectively. Both vessels contained a single valve that was normal in appearance and functional (i.e. remained closed in response to an adverse pressure gradient of 2 cm H2O). Evans Blue dye lymphangiography was performed on WT and db/db mouse hindlimbs (H and I) and demonstrates a lymphatic leakage phenotype is present in vivo in the popliteal collecting lymphatics (n = 3). Fluorescence images were both taken at the same time point. Scale bar is 100 μm in D and F and 200 μm in E and G.
Figure 4.

Lymphatic vessels from db/db mice are significantly enlarged. (A and B) The severe permeability defect of db/db lymphatic vessels was independent of sex (n = 17 males, n = 6 females). (C) When the internal diameter of all vessels was plotted, db/db collecting lymphatics (n = 27) were significantly enlarged compared with WT vessels (n = 11). (D) Diameter was not significantly different between male and female db/db vessels. *Significantly different (P < 0.05). NS, not significantly different.
The hypercholesterolaemia in ApoE−/− mice fed a high-fat diet results in lymphatic leakage along with vessel enlargement and deformed valves.7 To investigate the possibility that collecting lymphatics from db/db mice could display additional lymphatic defects similar to ApoE−/− mice, db/db and WT lymphatic vessel diameters were compared for all vessels studied. Vessels from db/db mice were significantly larger (147 μm, n = 27) compared with controls (119 μm, n = 11, Figure 4C, P < 0.05) and did not differ significantly between male and female db/db mice (Figure 4D). Conversely, lymphatic vessel segments typically contained 1–2 valves, which were always composed of leaflets that were fully mature and closed when exposed to an adverse pressure gradient (Figure 3D and E).
To confirm whether the lymphatic permeability defect that was measured ex vivo was present in vivo, we performed lymphangiography on db/db and WT mice (n = 3) by injecting Evans Blue dye into each hindpaw. As shown in Figure 3H, Evans Blue dye was readily observed in the WT popliteal collecting lymphatics, which did not appear to leak visible quantities of the dye into the surrounding tissues. In contrast, the dye appeared to diffuse from the popliteal lymphatics of db/db mice (Figure 3I) in discrete spots, indicating that these vessels were leaky. Notably, the lymphatic vessels of the popliteal fossa represent a second tissue bed where lymphatic leakage was observed in db/db mice.
3.3. Lymphatic barrier function is rescued by increasing NO bioavailability with l-arginine
Impaired NO formation leads to endothelial dysfunction in the blood vasculature in insulin-resistant diabetes, along with a loss of arterial vasodilation. Several groups have restored arterial vasodilation in diabetes and atherosclerosis by increasing l-arginine levels, the amino acid substrate for NO synthesis.20,21 Therefore, we hypothesized that the collecting lymphatic Ps defect in db/db mice might be rescued by supplementing the bath solution with l-arginine (1 mmol/L). As shown in Figure 5, Ps was assessed first at baseline, again following exposure to l-arginine, and a final time after superfusion of both l-arginine and l-NAME (100 μmol/L). While WT lymphatic vessel Ps was not significantly changed by either treatment, there was a trend for Ps to increase after exposure to l-arginine and decrease upon exposure to both l-arginine and l-NAME (WT: 2.7 ± 0.4 × 10−7 cm/s, l-arginine: 5.3 ± 1.1 × 10−7 cm/s, l-arginine + l-NAME: 1.9 ± 0.7 × 10−7 cm/s, n = 7). In contrast, superfusion of db/db collecting lymphatics with l-arginine significantly reduced their permeability to a level no different from WT, an effect that was reversed upon treatment with l-arginine and l-NAME combined (db/db: 321 ± 84 × 10−7 cm/s, l-arginine: 13.9 ± 5 × 10−7 cm/s, l-arginine + l-NAME: 297 ± 88 × 10−7 cm/s, n = 9, P < 0.05). This reduction in Ps occurred following ∼20 min of l-arginine superfusion and was maintained while l-arginine was present (>1 h). Interestingly, these data suggest that restoration of NO signalling in diabetes rescues lymphatic barrier dysfunction (i.e. lowers solute permeability).
Figure 5.

Impaired nitric oxide bioavailability underlies lymphatic barrier dysfunction in diabetes. Treatment of WT collecting lymphatics with l-arginine (1 mmol/L), a substrate for NO production, did not significantly alter resting permeability to albumin, but tended to increase permeability slightly. Exposure to both l-arginine and l-NAME (100 μmol/L) had no significant effect, but tended to decrease permeability slightly. In contrast, treatment of db/db collecting lymphatics with l-arginine led to a significant rescue in barrier function, indicating that NO production was impaired in db/db lymphatics. Interestingly, replacement of NO reduced permeability in db/db lymphatics, but increased WT permeability. This rescue was reversed by treating db/db lymphatics with l-arginine in combination with l-NAME. *Significantly different (P < 0.05). NS, not significantly different. Data are from n = 7 WT vessels and n = 9 db/db vessels from six mice each.
3.4. NO elevates wild-type collecting lymphatic permeability
NO is a known regulator of microvascular barrier function, but its role in modulating lymphatic vessel permeability has not been investigated previously. Additionally, the actions of NO on blood vascular permeability are the subject of intense debate, because NO has been reported in different laboratories to either increase or decrease blood vessel permeability.11–16 To definitively resolve whether and how NO alters lymphatic vascular permeability, WT collecting lymphatics were tested using a variety of approaches (Figure 6A–D). Pharmacological inhibition of NO synthase (NOS) using the pan-NOS inhibitor l-NG-nitroarginine methyl ester (l-NAME, 100 μmol/L) resulted in a significant five-fold decrease in basal lymphatic Ps (control: 3.3 ± 1.2 × 10−7 cm/s, l-NAME: 0.62 ± 0.2 × 10−7 cm/s, P < 0.05, n = 7, Figure 6A). Demonstrating that this result was not due to off-target effects and depended on the endothelial NOS (eNOS) isoform, collecting lymphatics from eNOS−/− mice displayed a similarly attenuated basal Ps compared with WT controls (WT: 3.2 ± 0.7 × 10−7 cm/s, n = 12; eNOS−/−: 0.66 ± 0.1 × 10−7 cm/s, n = 6, P < 0.05, Figure 6B). Together, these data suggest that eNOS-derived NO elevates lymphatic Ps under basal conditions.
Figure 6.

Nitric oxide increases wild-type (WT) lymphatic vessel permeability. Lymphatic vessel permeability to albumin (PsBSA) was assessed before and after manipulation of nitric oxide signalling. (A) The basal permeability of collecting lymphatics was decreased after application of a pan-inhibitor of nitric oxide synthase (l-NAME, 100 μmol/L). (B) Genetic deletion of eNOS resulted in a similar decrease in collecting lymphatic permeability. (C) Exposure to a nitric oxide donor (SNP, 1 μmol/L) resulted in an increase in lymphatic permeability. (D) Histamine (10 μmol/L) evoked endogenous nitric oxide production that also led to an increase in collecting lymphatic permeability. *Significantly different from control (P < 0.05). Each panel represents n = 6 to 7 vessels from at least six mice.
Because low basal levels of NO might serve a function different from higher NO concentrations, collecting lymphatics were then exposed to SNP (1 μmol/L), a donor of exogenous NO, or to histamine (10 μmol/L), which elicits endogenous NO production. Treatment with SNP (Figure 6C) resulted in a significant six-fold elevation in Ps (control: 3.1 ± 1.3 × 10−7 cm/s, SNP: 19.6 ± 5.4 × 10−7 cm/s, P < 0.05, n = 6). To rule out the possibility that cyanide liberated from SNP was responsible for the increased permeability, we repeated this protocol using diethylamine NONOate (DeaNO, 100 μmol/L), which also increased lymphatic Ps (control: 3.3 ± 0.6 × 10−7 cm/s, DeaNO: 10.3 ± 2.3 × 10−7 cm/s, P < 0.05, n = 6). Histamine similarly produced a transient, but significant, eight-fold increase in Ps (control: 2.5 ± 1.1 × 10−7 cm/s, histamine: 20.7 ± 7.0 × 10−7 cm/s, P < 0.05, n = 7, Figure 6D). Notably, the Ps of isolated coronary venules was reported to increase only two-fold in response to the same concentration of histamine.22 Our finding that lymphatic vessel Ps is more sensitive to histamine than venous Ps is supported by a study showing that cultured lymphatic endothelium has a more robust permeability response to histamine than cultured venous endothelium.23 Surprisingly, while NO decreased the Ps of db/db lymphatics, these experiments firmly establish that WT collecting lymphatics respond to low and high concentrations of endogenous or exogenous NO with an increase in Ps.
3.5. Inhibition of phosphodiesterase 3 restores lymphatic barrier function
Microvascular permeability is determined by the balance of cAMP and cGMP concentrations in the endothelial cell, which are ultimately controlled by the degradation of each second messenger by the relative presence and activity of phosphodiesterase (PDE) enzymes.24,25 To resolve how NO could both increase and decrease permeability in the same preparation, we hypothesized that PDE activation in db/db collecting lymphatics differed from that of WT lymphatics. An elevated basal permeability in the db/db lymphatics could either be a result of increased degradation of cAMP, a molecule that lowers permeability through PKA/Epac1, or due to inhibited degradation of cGMP, which is known to increase permeability through PKG. Since endothelial NO production is impaired in type 2 diabetes, soluble guanylate cyclase activation should be low, with the consequence of low cGMP levels.21 Therefore, we focused on PDEs that degrade cAMP. Intriguingly, PDE3 degrades cAMP, is expressed in endothelium,25 and is normally inhibited by cGMP produced downstream of NO, linking these two opposing pathways. Additionally, PDE3 can be activated by high concentrations of circulating insulin and leptin, as occurs during insulin resistance and diabetes.26,27
To test the hypothesis that PDE3 is activated in db/db collecting lymphatics, thereby degrading cAMP in lymphatic endothelium to raise Ps, the integrity of WT and db/db lymphatics was assessed before and after treatment with the selective, potent PDE3 inhibitor, cilostamide (5 μmol/L, Figure 7). WT collecting lymphatic Ps did not change significantly after exposure to cilostamide (WT: 1.8 ± 0.4 × 10−7 cm/s, cilostamide: 2.6 ± 0.6 × 10−7 cm/s, P = 0.3, n = 6). However, collecting lymphatic Ps was significantly reduced by cilostamide in db/db vessels (db/db: 453 ± 102 × 10−7 cm/s, cilostamide: 9.2 ± 4.3 × 10−7 cm/s, P < 0.05, n = 5) to a value that did not differ from WT controls, an effect similar to l-arginine treatment. Notably, Ps was reduced after ∼20 min of exposure to cilostamide, and, like l-arginine, this effect was maintained for the duration of cilostamide exposure. Interestingly, after db/db lymphatics were rescued with cilostamide, a subset of vessels (n = 2) was exposed to the NO donor, SNP (10 μmol/L), which resulted in an increase in Ps, suggesting that rescued db/db lymphatics respond to NO similarly to WT lymphatics (data not shown). These results indicate that PDE3 is active in db/db, but not WT, lymphatic vessels, and acts as a negative regulator of lymphatic integrity when NO production is impaired. Furthermore, it suggests that restoration of NO signalling reduces the Ps of db/db collecting lymphatics by restoring cGMP to levels sufficient to inhibit PDE3 and raise intracellular cAMP.
Figure 7.

Inhibition of PDE3 rescues lymphatic barrier dysfunction in diabetes. The permeability of collecting lymphatic vessels from WT mice did not change significantly in response to cilostamide (5 μmol/L), a selective PDE3 inhibitor, indicating that PDE3 is normally inactive in WT lymphatics. Notably, PDE3 degrades intracellular cAMP, a second messenger that maintains low endothelial permeability. Conversely, the elevated permeability of collecting lymphatics from db/db mice was significantly reduced to control levels upon exposure to cilostamide. *Significantly different (P < 0.05). Data are from n = 6 WT control vessels and n = 5 db/db vessels from three mice each.
4. Discussion
Here we provide the first measurements of murine collecting lymphatic vessel permeability. Using a new assay that enabled mechanistic studies, we identified a novel lymphatic defect in a mouse model of type 2 diabetes—lymphatic barrier dysfunction. Since this defect was confirmed by in vivo lymphangiography performed in a second tissue bed, lymphatic leakage appeared to be a general feature of a diabetic lymphatic vasculature. Augmenting NO bioavailability restored normal lymphatic barrier function. Interestingly, we showed that NO had the opposite effect on healthy lymphatic vessel permeability. To help explain this dual regulation of permeability, we determined that phosphodiesterase 3 was aberrantly active in diabetic lymphatic vessels due to loss of NO signalling.
Our previous work revealed that healthy collecting lymphatics constitutively leak a portion of the fluid and solute that they transport into the surrounding tissue.17 When integrated with the present findings, it suggests that compromised lymphatic barrier function will lead to severe leakage of lymph into the tissue. Based on an emerging body of literature,3,6–10 we expect that the degree of lymphatic barrier dysfunction in diabetic mice is sufficient to reduce lymph flow, thereby trapping lipids and cholesterol in the tissue. This effect is likely significant, because inhibiting lymphatic transport of cholesterol bound to high-density lipoprotein (HDL-C) from the tissues to the liver (i.e. reverse cholesterol transport) exacerbates atherosclerosis.8–10 Further, these findings link lymphatic endothelial dysfunction to lymph leakage, which leads to tissue adipose deposition, obesity, fibrosis, and inflammation.3,28 Because diabetic patients are at increased risk for dyslipidaemia, atherosclerosis, and oedema, treating lymphatic dysfunction may selectively alleviate these risks. Future studies are required to determine whether rescuing lymphatic dysfunction in diabetes reduces atherosclerotic plaque formation.
Whether NO increases or decreases vascular permeability has been the subject of intense debate.11–16 In the absence of a unifying explanation for the actions of NO on endothelial permeability, these disparate results have been attributed to differences in animal species, tissue beds, techniques to measure permeability, and experimental protocols.12,16 Surprisingly, the idea that NO performs both roles in a context-dependent manner has never been tested. Here, we demonstrate that NO can increase the permeability of healthy lymphatic vessels and, using the same experimental preparation, can reduce lymphatic permeability once it is already elevated by disease. This is in line with literature showing that the barrier-tightening effects of NO only appear after vascular endothelium has been exposed to pro-inflammatory mediators (e.g. platelet activating factor, thrombin),11,14,29 or in culture where vascular endothelial cells assume a pro-inflammatory phenotype and basal permeability is concurrently elevated.24,29
To explain how reduced NO bioavailability leads to elevated lymphatic permeability, we propose that impaired NO production leads to low levels of cGMP, and that this reduction in cGMP relieves the normal inhibition of PDE3 (Figure 8). PDE3 then hydrolyzes cAMP in the cell, a molecule responsible for maintaining a tight endothelial barrier.24,30 Through this mechanism, PDE3 acts as a negative regulator of lymphatic integrity. However, this does not explain why lymphatic vessels in eNOS−/− mice do not also have an elevated permeability. It is very likely that PDE3 not only requires release from inhibition (i.e. low cGMP levels), but also needs phosphorylation to be activated. High insulin or leptin levels, as occur in type 2 diabetes, are likely to be additional signals, and insulin has been reported to phosphorylate and activate PDE3 in multiple tissue types.26,27
Figure 8.

Proposed signalling pathway linking impaired nitric oxide bioavailability to PDE3 function. In healthy lymphatic endothelium (A), basal nitric oxide production leads to cGMP production that then inhibits PDE3 (also known as the cGMP-inhibited PDE). In combination, basal levels of cAMP and cGMP lead to a low basal permeability of ∼2 × 10−7 cm/s. In type II diabetes (B), lymphatic endothelium loses its ability to produce nitric oxide and, as a result, cGMP levels fall. This relieves the inhibition of PDE3 (dashed line). At the same time, high insulin and leptin concentrations are known to phosphorylate and activate PDE3. PDE3 then hydrolyzes intracellular cAMP, which normally maintains a low permeability, leading to loss of endothelial barrier function.
The only study investigating PDE3 inhibition in lymphedema demonstrated that oral administration of cilostazol, a selective PDE3 inhibitor structurally similar to cilostamide, was capable of reversing genetically and surgically induced lymphedema in mice.31 While this recovery was attributed to a lymphangiogenic effect, our data provide an additional mechanism whereby PDE3 inhibition enhances lymphatic vascular integrity during inflammation (i.e. reduces lymph leakage), thereby improving interstitial fluid clearance in those models. Since cilostazol is currently approved by the FDA for treating intermittent claudication in patients, it may represent a viable therapeutic target for treating lymphatic dysfunction in metabolic diseases.
In summary, the present study is the first to quantify lymphatic vessel permeability in mice and demonstrates that lymphatic endothelial dysfunction leads to impaired barrier function. Specifically, we identified a novel lymphatic permeability defect in type 2 diabetes, caused by reduced NO bioavailability and consequent PDE3 activation. Since this approach to assess lymphatic integrity is highly sensitive, it may be useful for identifying new therapeutic targets for treating lymphatic dysfunction in other disease models. As a major symptom of lymphatic dysfunction, enhanced lymphatic leakage may be treated with PDE3 inhibitors in metabolic diseases characterized by impaired endothelial NO synthesis.
Funding
This work was supported by the National Heart, Lung, and Blood Institute at the National Institutes of Health [K99 HL124142 to J.P.S., R01 HL089784 and R01 HL120867 to M.J.D., P01 HL095486 to M.J.D. and M.A.H., and R01 HL085119 to M.A.H.].
Acknowledgements
J.P.S conceived the project, designed, and performed experiments, analyzed data, and wrote and edited the manuscript. M.A.H. provided mice for the diabetic studies, contributed to discussion, edited the manuscript, and approved its final form. M.J.D. contributed to discussion, edited the manuscript, and approved its final form. A portion of this study was presented in talk and poster formats at the joint meeting of the North American Vascular Biology Organization and Microcirculatory Society in Hyannis, Massachusetts, 20–24 October 2013. The authors acknowledge Shan-Yu Ho and Susan Bingaman for their valuable technical assistance, and Professors William Durante and Steve Segal for valuable discussion.
Conflict of interest: none declared.
References
- 1.Alitalo K. The lymphatic vasculature in disease. Nat Med 2011;17:1371–1380. [DOI] [PubMed] [Google Scholar]
- 2.Stacker SA, Williams SP, Karnezis T, Shayan R, Fox SB, Achen MG. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat Rev Cancer 2014;14:159–172. [DOI] [PubMed] [Google Scholar]
- 3.Harvey NL, Srinivasan RS, Dillard ME, Johnson NC, Witte MH, Boyd K, Sleeman MW, Oliver G. Lymphatic vascular defects promoted by Prox1 haploinsufficiency cause adult-onset obesity. Nat Genet 2005;37:1072–1081. [DOI] [PubMed] [Google Scholar]
- 4.Wigle JT, Oliver G. Prox1 function is required for the development of the murine lymphatic system. Cell 1999;98:769–778. [DOI] [PubMed] [Google Scholar]
- 5.Yang Y, García-Verdugo JM, Soriano-Navarro M, Srinivasan RS, Scallan JP, Singh MK, Epstein JA, Oliver G. Lymphatic endothelial progenitors bud from the cardinal vein and intersomitic vessels in mammalian embryos. Blood 2012;120:2340–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sawane M, Kajiya K, Kidoya H, Takagi M, Muramatsu F, Takakura N. Apelin inhibits diet-induced obesity by enhancing lymphatic and blood vessel integrity. Diabetes 2013;62:1970–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim HY, Rutkowski JM, Helft J, Reddy ST, Swartz MA, Randolph GJ, Angeli V. Hypercholesterolemic mice exhibit lymphatic vessel dysfunction and degeneration. Am J Path 2009;175:1328–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim HY, Thiam CH, Yeo KP, Bisoendial R, Hii CS, McGrath KC, Tan KW, Heather A, Alexander JS, Angeli V. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab 2013;17:671–684. [DOI] [PubMed] [Google Scholar]
- 9.Martel C, Li W, Fulp B, Platt AM, Gautier EL, Westerterp M, Bittman R, Tall AR, Chen SH, Thomas MJ, Kreisel D, Swartz MA, Sorci-Thomas MG, Randolph GJ. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J Clin Invest 2013;123:1571–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Randolph GJ, Miller NE. Lymphatic transport of high-density lipoproteins and chylomicrons. J Clin Invest 2014;124:929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hatakeyama T, Pappas PJ, Hobson RW, Boric MP, Sessa WC, Durán WN. Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo. J Physiol 2006;574:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durán WN, Breslin JW, Sánchez FA. The NO cascade, eNOS location, and microvascular permeability. Cardiovasc Res 2010;87:254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kubes P, Granger DN. Nitric oxide modulates microvascular permeability. Am J Physiol 1992;262:H611–H615. [DOI] [PubMed] [Google Scholar]
- 14.Kurose I, Wolf R, Grisham MB, Granger DN. Modulation of ischemia/reperfusion-induced microvascular dysfunction by nitric oxide. Circ Res 1994;74:376–382. [DOI] [PubMed] [Google Scholar]
- 15.Yuan Y, Granger HJ, Zawieja DC, Chilian WM. Flow modulates coronary venular permeability by a nitric oxide-related mechanism. Am J Physiol 1992;263:H641–H646. [DOI] [PubMed] [Google Scholar]
- 16.Yuan SY. New insights into eNOS signaling in microvascular permeability. Am J Physiol Heart Circ Physiol 2006;291:H1029–H1031. [DOI] [PubMed] [Google Scholar]
- 17.Scallan JP, Huxley VH. In vivo determination of collecting lymphatic vessel permeability to albumin: a role for lymphatics in exchange. J Physiol 2010;588:243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scallan JP, Davis MJ, Huxley VH. Permeability and contractile responses of collecting lymphatic vessels elicited by atrial and brain natriuretic peptides. J Physiol 2013;591:5071–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scallan JP, Davis MJ. Genetic removal of basal nitric oxide enhances contractile activity in isolated murine collecting lymphatic vessels. J Physiol 2013;591:2139–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuo L, Davis MJ, Cannon MS, Chilian WM. Pathophysiological consequences of atherosclerosis extend into the coronary microcirculation. Restoration of endothelium-dependent responses by L-arginine. Circ Res 1992;70:465–476. [DOI] [PubMed] [Google Scholar]
- 21.Pieper GM. Review of alterations in endothelial nitric oxide production in diabetes: protective role of arginine on endothelial dysfunction. Hypertension 1998;31:1047–1060. [DOI] [PubMed] [Google Scholar]
- 22.Yuan Y, Granger HJ, Zawieja DC, DeFily DV, Chilian WM. Histamine increases venular permeability via a phospholipase C-NO synthase-guanylate cyclase cascade. Am J Physiol 1993;264:H1734–H1739. [DOI] [PubMed] [Google Scholar]
- 23.Breslin JW. ROCK and cAMP promote lymphatic endothelial cell barrier integrity and modulate histamine and thrombin-induced barrier dysfunction. Lymphat Res Biol 2011;9:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Curry FR, Adamson RH. Vascular permeability modulation at the cell, microvessel, or whole organ level: towards closing gaps in our knowledge. Cardiovasc Res 2010;87:218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Surapisitchat J, Jeon KI, Yan C, Beavo JA. Differential regulation of endothelial cell permeability by cGMP via phosphodiesterases 2 and 3. Circ Res 2007;101:811–818. [DOI] [PubMed] [Google Scholar]
- 26.Hanson MS, Stephenson AH, Bowles EA, Sprague RS. Insulin inhibits human erythrocyte cAMP accumulation and ATP release: role of phosphodiesterase 3 and phosphoinositide 3-kinase. Exp Biol Med 2010;235:256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitamura T, Kitamura Y, Kuroda S, Hino Y, Ando M, Kotani K, Konishi H, Matsuzaki H, Kikkawa U, Ogawa W, Kasuga M. Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine threonine kinase Akt. Mol Cell Biol 1999;19:6286–6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Avraham T, Zampell JC, Yan A, Elhadad S, Weitman ES, Rockson SG, Bromberg J, Mehrara BJ. Th2 differentiation is necessary for soft tissue fibrosis and lymphatic dysfunction resulting from lymphedema. FASEB J 2013;27:1114–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Curry FE, Zeng M, Adamson RH. Thrombin increases permeability only in venules exposed to inflammatory conditions. Am J Physiol Heart Circ Physiol 2003;285:H2446–H2453. [DOI] [PubMed] [Google Scholar]
- 30.Price GM, Chrobak KM, Tien J. Effect of cyclic AMP on barrier function of human lymphatic microvascular tubes. Microvasc Res 2008;76:46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kimura T, Hamazaki TS, Sugaya M, Fukuda S, Chan T, Tamura-Nakano M, Sato S, Okochi H. Cilostazol improves lymphatic function by inducing proliferation and stabilization of lymphatic endothelial cells. J Dermatol Sci 2014;74:150–158. [DOI] [PubMed] [Google Scholar]