

Abstract
Aims
Depressed sarcoplasmic reticulum (SR) Ca2+ cycling, a universal characteristic of human and experimental heart failure, may be associated with genetic alterations in key Ca2+-handling proteins. In this study, we identified a novel PLN mutation (R25C) in dilated cardiomyopathy (DCM) and investigated its functional significance in cardiomyocyte Ca2+-handling and contractility.
Methods and results
Exome sequencing identified a C73T substitution in the coding region of PLN in a family with DCM. The four heterozygous family members had implantable cardiac defibrillators, and three developed prominent ventricular arrhythmias. Overexpression of R25C-PLN in adult rat cardiomyocytes significantly suppressed the Ca2+ affinity of SR Ca2+-ATPase (SERCA2a), resulting in decreased SR Ca2+ content, Ca2+ transients, and impaired contractile function, compared with WT-PLN. These inhibitory effects were associated with enhanced interaction of R25C-PLN with SERCA2, which was prevented by PKA phosphorylation. Accordingly, isoproterenol stimulation relieved the depressive effects of R25C-PLN in cardiomyocytes. However, R25C-PLN also elicited increases in the frequency of Ca2+ sparks and waves as well as stress-induced aftercontractions. This was accompanied by increased Ca2+/calmodulin-dependent protein kinase II activity and hyper-phosphorylation of RyR2 at serine 2814.
Conclusion
The findings demonstrate that human R25C-PLN is associated with super-inhibition of SERCA2a and Ca2+ transport as well as increased SR Ca2+ leak, promoting arrhythmogenesis under stress conditions. This is the first mechanistic evidence that increased PLN inhibition may impact both SR Ca2+ uptake and Ca2+ release activities and suggests that the human R25C-PLN may be a prognostic factor for increased ventricular arrhythmia risk in DCM carriers.
Keywords: Dilated cardiomyopathy, Calcium cycling, Mutation
1. Introduction
Dilated cardiomyopathy (DCM), a common cause of heart failure, is characterized by dilation and impaired contraction of the left ventricle. Multiple aetiologies may underlie DCM, including rare variants in more than 30 genes, which initiate diverse pathophysiological mechanisms leading to left-ventricular dysfunction or arrhythmia and increased morbidity and mortality.1
A common clinical hallmark and characteristic of failing cardiomyocytes is the defective Ca2+ homeostasis, exhibited by a prolonged decay time of intracellular Ca2+ transient and changes in systolic and diastolic Ca2+ levels.2–4 The prolonged Ca2+-transient decay time and increased diastolic Ca2+ levels may result from impaired ryanodine receptor function, decreased sarcoplasmic reticulum Ca2+-ATPase (SERCA2a) levels, and augmented SERCA2a inhibition by phospholamban (PLN).5 PLN is a prominent regulator of Ca2+ cycling and a primary mediator of the β-adrenergic effects resulting in enhanced cardiac output.6,7 In the dephosphorylated state, PLN inhibits SERCA2a and shifts its Ca2+ activation towards lower apparent Ca2+ affinity. However, upon PKA-mediated phosphorylation, the inhibition on SERCA2a by PLN is relieved and its Ca2+ affinity is increased.7 Thus, PLN plays a key role in the regulation of Ca2+ reuptake by SERCA2a to induce relaxation and decrease diastolic Ca2+ levels. It is also a prominent mediator of the β-adrenergic stimulatory responses in the heart.7
The important role of PLN in cardiac function has prompted searches for human PLN genetic variants, which may be associated with DCM. Indeed, four mutations have been identified in the coding region of PLN. The first mutation was V49G, which resulted in potent inhibition of the Ca2+ affinity of SERCA2a. Cardiac overexpression of the V49G mutant PLN in mouse led to super-inhibition of cardiac contractility and remodelling, which progressed to DCM and early mortality.8 The second mutation identified had a stop codon for Leucine 39 (L39X-PLN) with onset of DCM and heart failure during the teenage years.9 The third human mutation, R9C, had no effects on SERCA2a activity under basal conditions but appeared to trap PKA, which blocked PKA-mediated phosphorylation of even wild-type PLN. The detrimental effects of such chronic inhibition of SERCA2a activity resulted in DCM.10 The fourth PLN mutation was a deletion of Arg-14 (Arg14Del).11 It was found that heterozygous carriers exhibited inherited DCM with left-ventricular dilation, contractile dysfunction, variable conduction system disease with episodes of malignant ventricular arrhythmias in some carriers and death by middle age.12,13 The mechanism underlying the detrimental effects of this human mutation involves super-inhibition of SERCA2a activity, likely mediated through a disturbance in the structure of PLN.11 These findings add to accumulating evidence that myocellular calcium dysregulation caused by mutations in human PLN is sufficiently deleterious to cause DCM and initiate heart failure.
In this study, we report a new mutation (R25C) in the coding region of the human PLN gene (PLN), identified in a pedigree with DCM that also showed prominent ventricular arrhythmia and need for implantable cardiac defibrillators (ICDs).14,15 Exome sequencing of affected family revealed that they had a R25C-PLN mutation, which was associated with super-inhibition of SERCA2a, depressed myocyte contractile and Ca2+-kinetic parameters and increased arrhythmias in cardiomyocytes. The mechanisms underlying the detrimental effects of this mutation include enhanced interaction of R25C-PLN mutant with SERCA2a, leading to super-inhibition of SERCA2a activity and increased Cam kinase II (CaMKII) activity associated with hyper-phosphorylation of Serine 2814 in RyR.
2. Methods
For details regarding methods, refer to the Supplementary material online, Methods.
2.1. Identification of a PLN mutation and generation of adenoviruses
For the human study, written informed consent was obtained and methods were conducted in accordance with the Declaration of Helsinki, and the study was approved by the Institutional Review Boards of Oregon Health and Science University and the University of Miami in FL, USA. Genomic DNA was extracted using standard procedures and PLN exomes were sequenced.15,16 Adenoviruses expressing green fluorescent protein (Ad.GFP), WT-PLN (Ad.WT), and R25C-PLN (Ad.R25C) were generated.9
2.2. Myocyte culture and quantitative immunoblotting
Animals were handled according to the Institutional Animal Care and Use Committee at the University of Cincinnati. Myocytes from adult male rats were isolated,17 cultured for 2 h and infected with adenoviruses. At 24 h post-infection, we determined PLN levels by western blots and initial rates of SR Ca2+ uptake.18 Data were analysed by non-linear regression using the OriginLab 5.1 program.
2.3. Cardiomyocyte contractility and Ca2+ transients
Contractions were obtained at 0.5 Hz and Ca2+ kinetics were determined in cells loaded with Fura-2 /AM and excited at 340 and 380 nm.
2.4. HEK 293 cells, immunofluorescence, and immunoprecipitation
HEK 293 cells were transfected with GFP-PLN and SERCA2 constructs,19 fixed, blocked, and incubated with SERCA2 antibody. Samples were counterstained with Alexa Fluor anti-mouse 568 secondary antibody, mounted with Vectashield medium containing DAPI and analysed by confocal microscopy. In parallel, transfected HEK 293 cells were lysed and processed for immunoprecipitation.
2.5. Measurement of Ca2+ sparks, waves, and diastolic SR Ca2+ leak
Spontaneous Ca2+ sparks and waves were obtained in quiescent cells loaded with Rhod-2 AM. Diastolic SR Ca2+ leak was measured using the tetracaine method.20
2.6. Aftercontractions in rat cardiomyocytes
Rat ventricular myocytes were paced at 2 Hz in the presence of 1 µmol/L ISO. After 2 or 3 trains of stimulation, pacing was stopped and spontaneous aftercontractions within 2–5 s were recorded.
2.7. CaMKII activity assay
Cell lysates from infected myocytes were processed for CaMKII activity (Cyclex Kit, MBL International, Woburn, MA, USA).
2.8. Statistics
Results are expressed as mean ± SEM. Comparisons were evaluated by one-way ANOVA and a post-hoc Tukey test. Fisher's exact test was used for calcium sparks, waves, and after contractions. Values of P < 0.05 were considered significant.
3. Results
3.1. Identification of a novel PLN mutation in a familial DCM pedigree
Forty-eight variants, meeting our established criteria for exome sequencing analysis,15 were identified in two sisters (III.1 and III.3) in our family pedigree (Figure 1). Of these 48, three were in previously identified DCM genes, including a novel heterozygous PLN mutation that resulted in change of arginine into cysteine at position 25, and two missense mutations in Titin (TTN) (A3656S and P7178S). Because it is known that even rare (<0.5%) TTN missense mutations are prevalent in control samples,16,21 these missense variants were considered unlikely to be the cause of DCM. The PLN mutation had high conservation scores (Phastcons 1; GERP 4.31), and based on other heterozygous PLN missense mutations causing DCM,8–11 it was hypothesized that this mutation also may alter PLN activity. None of the genes harbouring the remaining 45 variants were associated with known cardiovascular disorders. No additional PLN variants were identified in 16 other families.15
Figure 1.
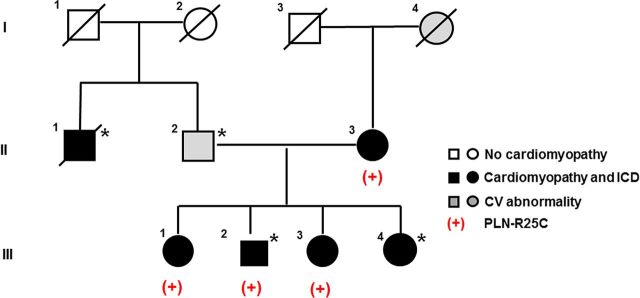
Familial DCM Pedigree with a PLN mutation. Squares represent males and circles represent females. Slash denotes deceased. Darkened symbols indicate idiopathic DCM with implantable cardiac defibrillator (ICD) and grey symbols represent a significant cardiovascular abnormality. Open symbols represent negative cardiovascular history. The presence of the PLN mutation is denoted with (+) and the presence of the LMNA mutation14 with asterisk.
A pedigree with the PLN mutation status is shown in Figure 1. Sanger sequencing confirmed that the PLN mutation was present in the sisters (III.1 and III.3), the proband (III.2) who also carried a previously identified LMNA mutation,15 as well as in his mother (II.3), who had DCM but was not found to carry the LMNA mutation.15
3.2. Clinical characteristics
Clinical characteristics of family members with the PLN mutation are provided (Table 1). The proband (III.2) presented at age 47 with sudden cardiac arrest and was diagnosed with non-ischaemic DCM upon resuscitation. An ICD was placed from which he has received multiple appropriate shocks. Approximately 10 years later, he required heart transplantation due to worsening heart failure. Two of his sisters were also found to harbour the R25C-PLN mutation. His older sister (III.1) was first screened clinically at age 51 and was found to have left-ventricular enlargement but preserved systolic function. Despite already receiving an angiotensin receptor blocker (ARB), 2 years later her ventricular function had deteriorated. Non-sustained ventricular tachycardia (NSVT) was found on Holter monitoring, and an ICD was placed prior to the onset of any heart failure. She was treated with full-dose beta blockade and an ARB. Over the course of the following 10 years, two cardioversions were required due to persistent atrial fibrillation. The proband's younger sister (III.3), who also had NSVT and required an ICD, was diagnosed with DCM at age 45 after presenting with signs of heart failure. Their mother (II.3), who had complained of palpitations since age 47, had premature ventricular contractions at age 57. She was diagnosed with DCM at 71 and had multiple PVCs, couplets, and NSVT for years, for which an ICD was placed at age 84 because of progressive pump dysfunction. She progressed to chronic atrial fibrillation and received multiple cardioversions, and died from heart failure at age 90.
Table 1.
Clinical characteristics of family members with the R25C-PLN mutation
| Pedigree position | DCM | Age of diagnosis, years | LVEDD, mm (Z-score) | LVEF, % | ECG/Arrhythmia | Comment |
|---|---|---|---|---|---|---|
| II.3 | Yes | 71 | 60 (4.1) | 35 | AF, PVCs, LBBB, ICD | HF |
| III.1 | Yes | 53 | 58 (3.4) | 40 | AF, 1AVB, VT, ICD | HF |
| III.2 | Yes | 47 | 72 (4.85) | 28 | AF, RBBB, PM/ICD VT, SCD |
Heart transplant at 58 years |
| III.3 | Yes | 45 | 59.6 (3.99) | 17 | NSVT, PM/ICD | HF |
LVEDD, left-ventricular end-diastolic dimension by echocardiography; Z-score, number of standard deviations of the LVEDD above the population mean; EF, left-ventricular ejection fraction; AF, atrial fibrillation; PVCs, premature ventricular contractions; LBBB, left bundle branch block; ICD, implantable cardiac defibrillator; HF, heart failure; 1AVB, first-degree atrioventricular block; VT, ventricular tachycardia; RBBB, right bundle branch block; PM, pacemaker; SCD, sudden cardiac death; NSVT, non-sustained ventricular tachycardia.
3.3. Expression of mutant PLN in adult cardiomyocytes and Ca2+ uptake assay
To determine whether the inhibitory effects of PLN on cardiac contractility are modified by the R25C mutation, recombinant adenoviruses encoding GFP, WT-PLN, or R25C-PLN were transduced into adult rat ventricular myocytes. The protein levels of PLN in infected cardiomyocytes were assessed by SDS–PAGE and western blots. Quantitative immunoblotting of cell lysates revealed a similar ratio of PLN pentamers to monomers in mutant PLN (85.8%) cardiomyocytes, compared with WT-PLN (86.6%) and GFP (84.9%) groups (Figure 2A). Furthermore, upon boiling the cardiac homogenates prior to SDS–PAGE, the mutant could be dissociated into monomers, similar to WT-PLN. There were no significant changes in the SERCA2a protein levels in the infected mutant cardiomyocytes (Figure 2A). When the apparent PLN/SERCA2a ratio was calculated, it was found to be a 1.9-fold in R25C myocytes, compared with the GFP group, and this ratio was similar to that for WT-PLN (1.93) (Figure 2B).
Figure 2.
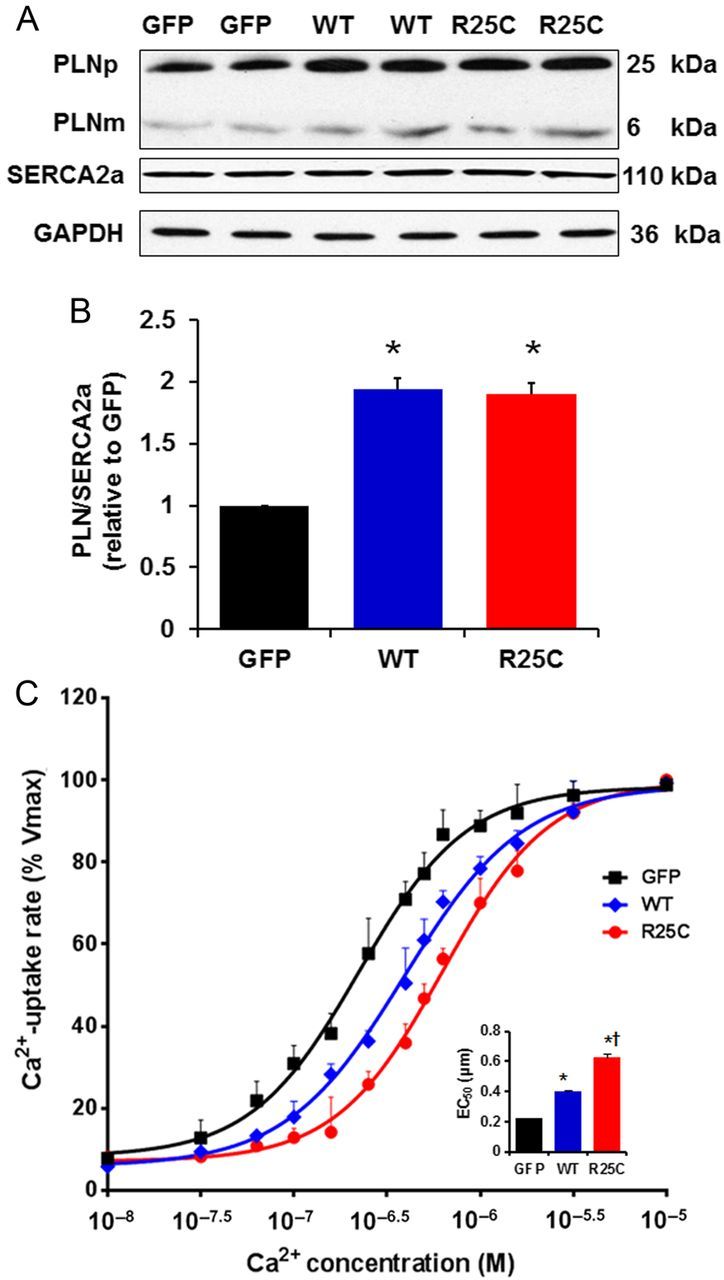
Quantitative immunoblots from infected cardiomyocytes and Ca2+ uptake assays. (A) Representative blots of PLN and SERCA. PLNp, pentameric PLN; PLNm, monomeric PLN; (B) PLN protein levels in GFP, WT and R25C cardiomyocytes expressed as relative ratio of PLN/SERCA2a (n = 4 hearts); (C) Effects of wild-type and mutant R25C-PLN on the apparent Ca2+ affinity of SERCA2a. After 24-h infection with adenoviruses, cardiomyocytes were homogenized and the initial rates of oxalate-supported SR Ca2+ uptake were measured. Data are expressed as per cent of maximal uptake rates in each group (Vmax: 99 ± 4 in GFP, 101 ± 3 in WT, and 96 ± 5 in R25C, nmol/mg/min). Inset: The average EC50 values for the three groups (n = 6 hearts). *P < 0.05, vs. GFP; †P < 0.05, vs. WT. Values are mean ± SE.
To examine the functional significance of the R25C substitution in PLN and its impact on SERCA2a regulation, the initial rates of ATP-dependent, oxalate-facilitated SR Ca2+ uptake were measured in cell lysates from infected cardiomyocytes. There were no significant differences in the maximal rates of SR Ca2+ uptake between GFP, WT, or R25C groups. However, the EC50 value for Ca2+ dependence of Ca2+ uptake was significantly higher in WT-PLN cells (0.40 ± 0.03 µM), compared with the GFP group (0.22 ± 0.02 µM), consistent with our previous findings.22 Interestingly, R25C-PLN further increased the SERCA2a EC50 value to 0.63 ± 0.07 µM (Figure 2C), Thus, the mutant PLN is a super-inhibitor of the affinity of SERCA2a for Ca2+ compared with WT-PLN.
3.4. Functional analysis of the R25C-PLN mutant
Overexpression of WT-PLN has been shown to lead to significant depression of cardiomyocyte function.22 To determine whether the super-inhibitory effects of the R25C mutation on SERCA2a activity translated into alterations at the cellular level, the contractile parameters of infected cardiomyocytes were assessed. The resting cell length in mutant PLN infected myocytes was not altered compared with the WT-PLN or GFP groups. However, the amplitude of basal cell contraction (fractional shortening, FS%) was more depressed in myocytes overexpressing R25C mutant PLN (60%) than in myocytes overexpressing WT-PLN (76%), compared with GFP control (100%) (Figure 3A and B). The maximal velocities of cardiomyocyte shortening and re-lengthening were also more depressed in myocytes with mutant PLN (+dL/dt: 52%; −dL/dt: 46%) than in cells with WT-PLN (+dL/dt: 67%; −dL/dt: 66%), compared with GFP controls (100%) (Figure 3C and D).
Figure 3.

Contractile parameters in Ad.GFP, Ad.WT-PLN, and Ad.R25C-PLN infected cardiomyocytes. (A) Representative cell-shortening traces of Ad.GFP, Ad.WT-PLN, and Ad.R25C-PLN cardiomyocytes (cell-length trace represents the percentage of resting cell length); (B) Fractional shortening (FS%); (C) Maximum rates of contraction (+dL/dt); (D) Maximum rates of relengthening (−dL/dt) (20–25 cells were measured per experiment or each heart; n = 4 hearts for GFP, WT-PLN, and R25C-PLN groups). *P < 0.05, vs. GFP; †P < 0.05, vs. WT. Values are mean ± SE.
3.5. Overexpression of R25C-PLN significantly suppresses Ca2+ transients, delays the rate of Ca2+ removal, and elevates intracellular diastolic Ca2+
To determine whether the observed alterations in myocyte mechanics reflected similar alterations in Ca2+ handling, intracellular Ca2+ transients in infected cardiomyocytes were measured by use of the Fura-2/AM fluorescence indicator (2 µM). Our results demonstrate that the amplitude of Ca2+ transients was reduced by 55% in mutant-PLN and by 37% in WT-PLN, compared with GFP cardiomyocytes (Figure 4A and B). The time to 50% decay of the Ca2+ signal was prolonged by 65% in cardiomyocytes overexpressing mutant and by 32% in cardiomyocytes overexpressing WT-PLN, relative to GFP group (Figure 4C). In addition, intracellular diastolic Ca2+ was also analysed and it was found to be increased by 18% in R25C-PLN compared with WT-PLN or GFP cardiomyocytes (Figure 4D). Thus, the R25C-PLN mutant depressed mechanical and Ca2+ kinetic parameters and increased diastolic Ca2+ levels, consistent with the enhanced inhibition of SERCA2a.
Figure 4.
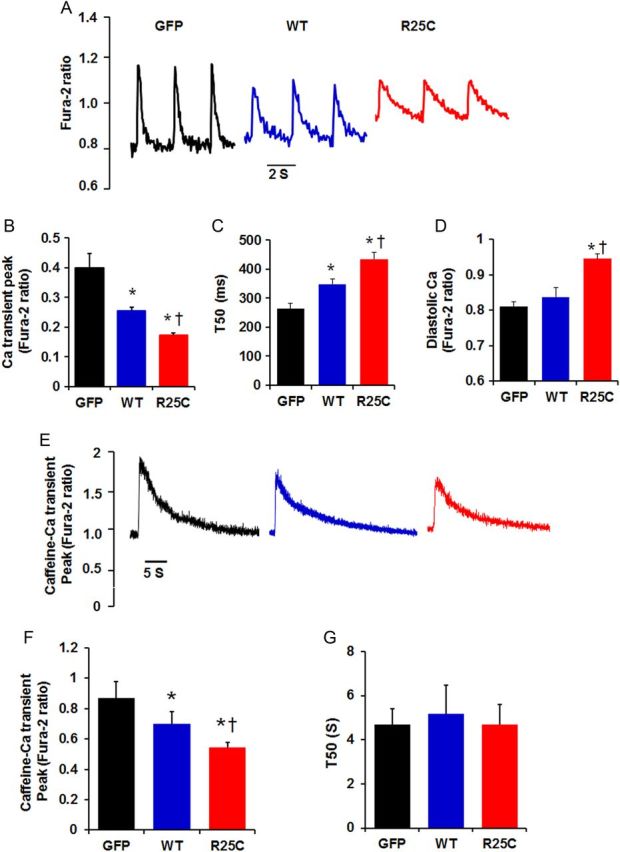
Ca2+ kinetics in Ad.GFP, Ad. WT-PLN, and Ad.R25C-PLN cardiomyocytes. Infected myocytes were incubated with Fura-2/AM for half an hour and Ca2+ transients were measured. (A) Representative tracings of Ca2+ transients; (B) Ca2+ transient amplitude in infected cardiomyocytes; (C) Time to 50% decay (T50) of Ca2+ signal; (D) Intracellular diastolic Ca2+; (E) Representative tracings of caffeine (10 mM)-induced Ca2+ transient; (F) Caffeine-induced Ca2+ transient amplitude; (G) Time to 50% decay (T50) of caffeine-induced Ca2+ transient peak (20–25 cells were measured per experiment or each heart; n = 4 hearts for GFP, WT-PLN, and R25C-PLN groups). *P < 0.05, vs. GFP; †P < 0.05, vs. WT. Values are mean ± SE.
The effects of mutant-PLN on intracellular Ca2+ transients prompted further studies on the influence of R25C-PLN on SR Ca2+ load. It was observed that the caffeine-induced Ca2+ transient peak was reduced by 37% in R25C-PLN cardiomyocytes and by 20% in WT-PLN cardiomyocytes, compared with the GFP group (Figure 4E and F), indicating a super-inhibitory effect of mutant-PLN on SR Ca2+ content. However, the time constant for 50% decay of the caffeine-induced Ca2+ transient (T50), which mainly reflects Ca2+ extrusion by the sodium/calcium exchanger (NCX), was similar between the three groups (Figure 4G).
3.6. Isoproterenol stimulation relieves the inhibitory effects of R25C-PLN
Phospholamban has been postulated to be phosphorylated during β-adrenergic stimulation, resulting in relief of its inhibitory effects on SERCA2a and cardiac function.7 To determine whether the super-inhibitory effects of mutant PLN could be also relieved by β-agonists, cardiac myocytes infected with Ad.GFP, Ad.WT-PLN, or Ad.R25C-PLN were stimulated with isoproterenol and their mechanical and Ca2+ kinetic parameters were assessed. It is interesting to note that maximal stimulation, obtained at 100 nmol isoproterenol, resulted in complete reversal of the inhibitory effects of wild-type or mutant PLN. Similarly, under maximal isoproterenol stimulation, the inhibition on the amplitude of systolic Ca2+ transient and the rate of decay of this signal were fully reversed in cardiomyocytes overexpressing mutant PLN. The maximally stimulated values were similar among the three groups (see Supplementary material online, Figure S1).
3.7. The mutant R25C-PLN exhibits enhanced association to SERCA2 and PKA abrogates this effect
To gain insights into the mechanisms associated with increased inhibition of SERCA2a by R25C-PLN, we co-expressed SERCA2 with either WT or mutant PLN in HEK cells to evaluate their interactions in the absence of endogenous proteins. For this, we generated a GFP-WT-PLN or GFP-R25C-PLN constructs and transiently co-transfected these along with SERCA2 in HEK 293 cells. Initial immunofluorescence studies indicated that, similar to GFP-WT-PLN, the GFP-R25C-PLN mutant exhibited co-localization with SERCA2 in ER (Figure 5A). Subsequent immunoprecipitation studies from co-transfected cells revealed a significant increase in the levels of SERCA2 in the R25C-PLN sample, compared with WT-PLN (Figure 5B and C), indicating enhanced formation of the SERCA2/R25C-PLN protein complex. Interestingly, parallel experiments in cell lysates, that had been previously phosphorylated by PKA, revealed that phosphorylation of R25C-PLN abolishes the increased interaction of this mutant with SERCA2, as similar levels of SERCA2 were observed in the phosphorylated R25C-PLN and WT-PLN protein complexes (Figure 5D and E). These findings are consistent with relief of the mutant PLN super-inhibitory effects on contractility and Ca2+-cycling upon Iso-stimulation of cardiomyocytes (see Supplementary material online, Figure S1).
Figure 5.
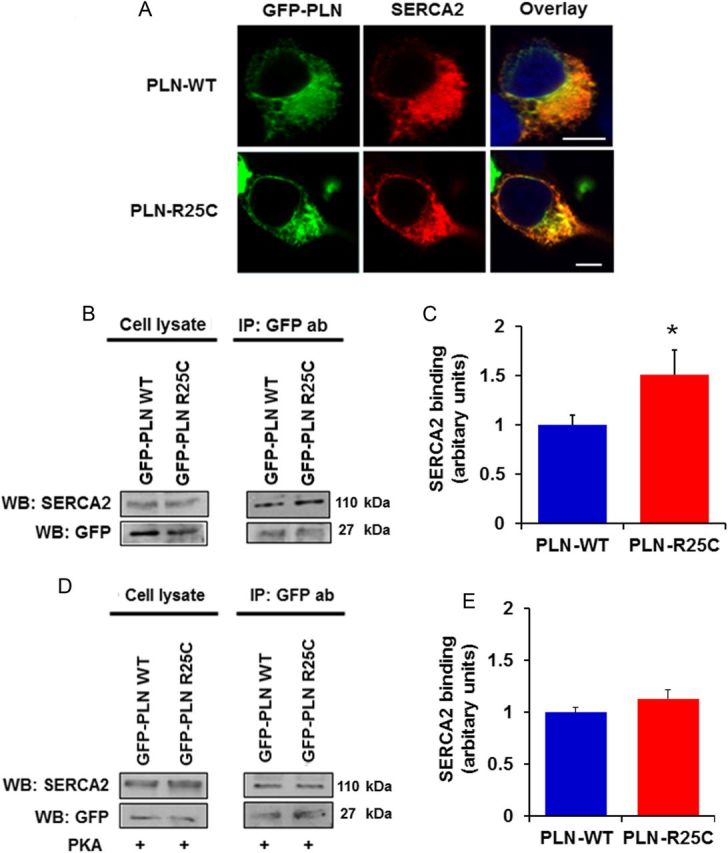
(A) The R25C-PLN mutant co-localizes with SERCA2 in transfected HEK 293 cells, similar to WT-PLN. Nuclei are stained with DAPI. Scale bar, 5 μm; (B and C) R25C-PLN mutant exhibits enhanced association to SERCA2. Immunoprecipitation assays in HEK 293 cells that co-express GFP-PLN and SERCA2 were performed using GFP antibody. Quantification of SERCA2 levels revealed a significant increase in the SERCA2/R25C-PLN protein complex compared with GFP-WT-PLN (n = 4) values are means ± SE; *P < 0.05, compared with WT-PLN); (D and E) enhanced binding of R25C-PLN with SERCA2a was abolished upon PKA phosphorylation. Immunoprecipitations were performed in lysates from HEK 293 transfected cells that had been previously phosphorylated with PKA. Western blot analysis (D) determined similar levels of SERCA2 in both WT-PLN and R25C-PLN samples and quantitative analysis (E) showed no difference in SERCA2 binding between WT-PLN and R25C-PLN (n = 4).
3.8. Increased Ca2+ sparks, waves, diastolic SR Ca2+ leak, and stress-induced aftercontractions in R25C-PLN cardiomyocytes
As described earlier, all R25C affected members in the family pedigree developed cardiac arrhythmias (Figure 1 and Table 1). The molecular trigger for arrhythmia is enhanced SR Ca2+ leak, evidenced by increases in Ca2+ sparks or waves at the cardiomyocyte level.23 To determine the effect of R25C on SR Ca2+ release, we examined Ca2+ spark properties in intact quiescent cells. The line-scan and three-dimensional images for Ca2+ sparks are presented in Figure 6A. It was observed that spark frequency was two-fold in the mutant PLN cardiomyocytes, compared with WT-PLN or GFP cardiomyocytes (Figure 6B). Next, the frequency of spontaneous Ca2+ waves was examined in GFP, WT-PLN, and R25C-PLN myocytes. It was found that Ca2+ waves were developed in 40% of R25C cardiomyocytes, compared with 4% of WT-PLN cells and 6% of GFP cells (Figure 6C and D). Thus, R25C-PLN increases the frequency of Ca2+ sparks and Ca2+ waves in cardiomyocytes.
Figure 6.

Ca2+ sparks, waves, diastolic SR Ca2+ leak, and stress-induced aftercontractions (Acs) in GFP, WT-PLN, and R25C-PLN cardiomyocytes. (A) Representative line-scan and three-dimensional (3D) images of Ca2+ sparks acquired in infected cardiomyocytes; (B) Cumulative data on Ca2+ spark frequency; (C) Representative line-scan and 3D images of Ca2+ waves acquired in R25C-PLN cardiomyocytes; (D) Percentage of cells showing Ca2+ waves (10–15 cells were measured per experiment or each heart; n = 6 hearts for GFP, WT-PLN, and R25C-PLN groups); (E) Representative traces of SR Ca2+ leak were obtained from the three groups. Ca2+ leak was determined as the tertacaine sensitive drop in diastolic −2 ratio; (F) Comparison of average diastolic SR Ca2+ leak; (G) Quantification of leak/SR load relationships in GFP, WT, and R25C myocytes (ratio of twitch Ca2+ transient/caffeine-induced Ca2+ transient (10–12 cells were measured per experiment or each heart; n = 4 hearts for GFP, WT-PLN and R25C-PLN groups); *P < 0.05, vs. WT and GFP. Values are mean ± SE. (H) Representative traces of Acs; (I) Percentage of the infected cardiomyocytes that developed Acs (10–12 cells were measured per experiment or each heart; n = 6 hearts for GFP, WT-PLN, and R25C-PLN groups). *P < 0.05, vs. GFP and WT. Values are mean ± SE.
Recent studies have suggested that RyR2-mediated Ca2+ leak occurs in part as Ca²+ sparks, although there is RyR-mediated and Ca2+ spark-independent leak.24 Therefore, total diastolic SR Ca²+ leak was also measured using the tertacaine protocol25 (Figure 6E and F). The ratio of SR Ca2+ leak to SR Ca2+ load was significantly larger in R25C cardiomyocytes, compared with GFP and WT cardiomyocytes (Figure 6G), suggesting that R25C-PLN increases the SR Ca2+ leak.
Next, we determined the role of R25C-PLN under stress conditions by measuring the frequency of aftercontractions in GFP, WT-PLN, and R25C cardiomyocytes at 2-Hz field stimulation in the presence of 1 µM Isoproterenol. Spontaneous aftercontractions occurred in 74% of R25C cells within 5 s after pacing was stopped, compared with 17% of WT-PLN and 16% of GFP cells (Figure 6H and I). Taken together, these findings suggest that overexpression of R25C-PLN enhances the propensity for spontaneous Ca2+ release from the SR, resulting in increased susceptibility to arrhythmia.
3.9. Increased SR Ca2+ leak and arrhythmias in R25C myocytes are associated with augmented phosphorylation of RyR2 at serine-2814
We then investigated whether the increased SR Ca2+ leak in R25C cardiomyocytes was associated with increase in the phosphorylation of RyR2. Previous studies have shown that enhanced phosphorylation of Ser2808 (PKA site) and Ser2814 (CaMKII site) in RyR2 can regulate RyR2 function.26,27 Thus, we examined the phosphorylation state of RyR2 in R25C cardiomyocytes, by performing western blots with phospho-specific antibodies against the RyR2 Ser2808 and Ser2814 sites. It was observed that R25C cells had significantly increased phosphorylation levels of RyR at Ser2814, but not at Ser2808 (Figure 7A–C). This prompted us to determine the activity of CaMKII in cell lysates from GFP, WT-PLN, and R25C cardiomyocytes, using a non-radiographic CaMKII ELISA. We found that the activity of CaMKII in R25C myocytes was increased to two-fold compared with WT-PLN or GFP groups (Figure 7D), consistent with the increase in diastolic Ca2+ in these cells (Figure 4D). To further confirm this finding, phosphorylation of CaMKII at Thr286 residue, which represents permanent activation of the CaMKII enzyme, was determined in cell lysates from GFP, WT-PLN, and R25C cardiomyocytes using western blots. It was observed that the level of phosphorylation of CaMKII at Thr286 was considerably higher in R25C cells, compared with the WT-PLN or GFP groups (Figure 7E and F). Interestingly, the increased CaM kinase activity did not reflect altered phosphorylation of PLN (see Supplementary material online, Figure S2) in the mutant cells, suggesting that mutant PLN either altered CaMKII binding or increased PLN-phosphatase activity. To further examine the contribution of CaMKII mediated phosphorylation of RyR2 in the R25C-induced increases of Ca2+ sparks and waves, KN93, the selective inhibitor of CaMKII was utilized in parallel studies with KN92 as a control. We found that KN93 completely abrogated the increased Ca2+ spark frequency (Figure 7G), indicating that the aberrant SR Ca2+ release was contributed by the increased CaMKII phosphorylation of RyR. Furthermore, KN93 fully abolished the Ca2+ waves, elicited by R25C-PLN (Figure 7H). These data suggest that the aberrant SR Ca2+ leak and resultant arrhythmia were associated with increased CaMKII activity and hyper-phosphorylation of RyR at Ser2814.
Figure 7.
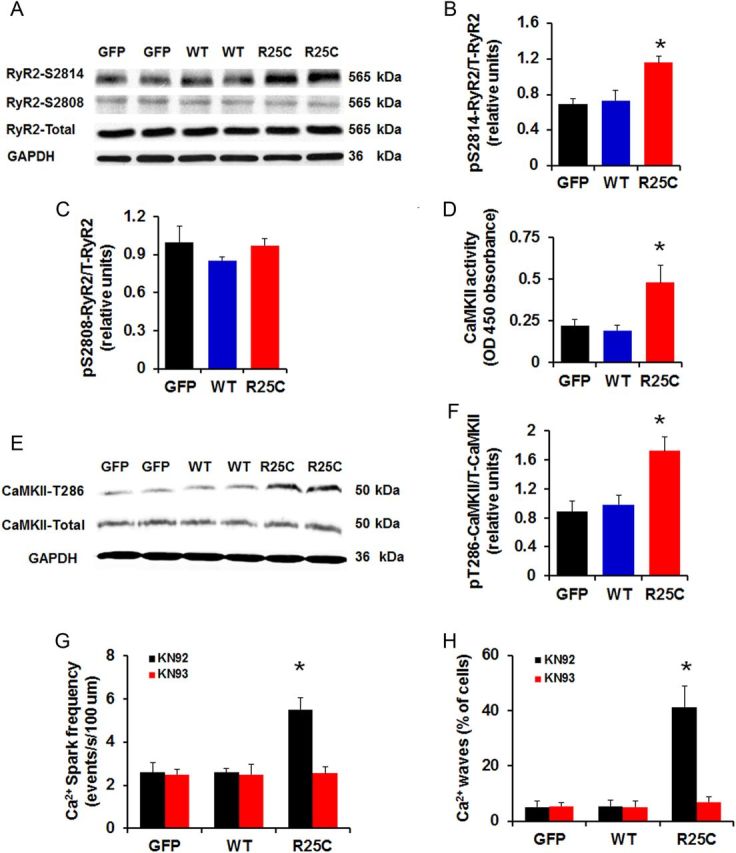
Phosphorylation of RyR2 and CaMKII activity. (A) Representative blots of phosphorylation and total levels of RyR2; (B and C) Percentage of phosphorylated Ser2808 (pSer2808) and Ser2814 (pSer2814) RyR2 in infected cardiomyocytes (n = 6 hearts); (D) CaMKII activity in GFP, WT, and R25C cardiomyocytes (n = 7 hearts); (E) Representative blots of phosphorylation and total levels of CaMKII; (F) Percentage of phosphorylated Thr286 (pT286) CaMKII in infected cardiomyocytes (n = 4 hearts); (G and H) Ca2+ spark frequency and percentage of Ca2+ waves recorded in GFP, WT, and R25C cardiomyocytes in the absence or presence of CaMKII inhibitor KN93 (1 µmol/L), with KN92 (1 µmol/L) used as a control (15–20 cells were measured per experiment or each heart; n = 4 hearts for GFP, WT-PLN, and R25C-PLN groups). *P < 0.05, vs. WT-Basal, †P < 0.05, vs. R25C-Basal. Values are mean ± SE.
4. Discussion
In the current study, we identified a novel R25C-PLN mutation in familial DCM, which acts as a super-inhibitor of SERCA2a and results in decreased SR Ca2+ content, Ca2+ transients, and impaired contractile function. The depressed SR Ca2+ resequestration is associated with increased CaMKII activity and hyper-phosphorylation of RyR2 at Ser2814, leading to aberrant Ca2+-leak and after-contractions under stress conditions. This is the first evidence that an experiment by nature to alter PLN function results in increased SR Ca2+-leak and propensity to arrhythmia, a significant component of the cardiovascular phenotype observed in the carriers of the PLN mutation.
Our findings provide valuable new insights into the established paradigm of DCM gene/phenotype relationships. We1,28 and others have suggested that the vast majority of DCM cases and families, shown to have genetic cause, appear monophenotypic-a classic ‘pure’ DCM with no atypical cardiovascular or syndromic features. For these families, ventricular or supraventricular arrhythmias have been attributed to a generic progression towards advanced heart failure with its manifold cellular abnormalities, including increased susceptibility to arrhythmia rather than a gene-specific abnormality. Dissecting between the DCM-elicited arrhythmias and those resulting from a specific molecular defect remains challenging, although the exception has been the category of ‘DCM with prominent arrhythmia’,28 assigned predominantly to variants in LMNA, but also observed with variants in SCN5A29 or DES.30 Our current work augments this established gene/phenotype paradigm, as previously reported rare variants in PLN have been associated with predominantly monophenotypic DCM, while the R25C PLN variant leads to promiment arrhythmias, which we suggest may be due in part to novel, specific mechanisms. These PLN-mutation carriers presented with arrhythmia in middle-age and in the setting of DCM, characterized by an ‘adult-onset’, as commonly observed in most of genetic cardiomyopathy. Another recent report with the R14del-PLN variant, a founder mutation in a large number of Dutch individuals, also suggested a propensity to arrhythmia, although a key predictor of arrhythmia was reduced systolic function (<45%).13
Previous studies regarding the role of super-inhibitor PLN mutants in mouse models suggested that the depressed SERCA2 activity and contractility were associated with hypertrophic remodelling and ventricular failure over the long term.31,32 Similar to those studies, our functional data further support the role of PLN mutations in DCM. The mechanisms underlying the super-inhibitory activity of R25C-PLN appear to include increased association or interaction of this mutant with SERCA2a, resulting in increased inhibition of SR Ca2+-transport. Indeed, depressed SR Ca2+ cycling is a major characteristic of human and experimental heart failure. Accordingly, cardiac overexpression33 or gene transfer34 of SERCA2a in rat models of heart failure not only significantly improved cardiac contractile function, but increased survival and cardiac metabolism. Similarly, inhibition of PLN activity resulted in enhanced SR Ca2+ cycling and cardiac contractility34,35 as well as attenuated heart-failure progression.36 In addition, RyR inhibition by calstabin improved cardiac function in failing hearts.23 These studies suggest that disturbed SR Ca2+ cycling may serve as a root cause of heart failure and this hypothesis has been well supported by recent studies in human patients.37,38
Interestingly, the R25C-PLN was also associated with increased Ca2+ sparks, waves, and stress-induced after-contractions, which may have contributed to the clinical features of cardiac arrhythmia in R25C carriers at the cellular level. The underlying pathways appear to involve increased inhibition of SERCA2a by mutant PLN, resulting in elevated diastolic Ca2+ and activation of CaMKII39 (Figure 4D). Actually, several studies in neuron cells and cardiomyocytes have shown that sustained intracellular Ca2+ elevation enhances CaMKII auto-phosphorylation and increases Ca2+/calmodulin-independent kinase activity.40–42 The activation of CaMKII reflected enhanced phosphorylation of RyR2 at Ser2814, leading to increases in SR Ca2+ leak and arrhythmia, consistent with previous studies.42–44 Indeed, inhibition of CaMKII activity by KN93 abrogated the increases in Ca2+-sparks and Ca2+-waves, elicited by R25C-PLN. Notably, overexpression of WT-PLN in cardiomyocytes, which resulted in a smaller degree of SERCA2a inhibition than R25C-PLN, did not significantly alter diastolic Ca2+ levels, CaMKII activity or phosphorylation of RyR2 at Ser2814. These findings indicate that inhibition of SERCA2a beyond a threshold point has detrimental effects through increased diastolic Ca2+ levels and CaMKII activation.
The mechanisms associated with the super-inhibitory effects of R25C-PLN are likely mediated through a disturbed conformational structure of PLN. Arg25 is highly conserved across species and this residue supplies a positive charge, which is important for maintaining the hinge region of PLN in the hydrophilic cytosolic environment. Thus, the Arg25 replacement with cysteine, a non-polar amino acid, could destabilize the hinge angle, leading to conformational changes that enhance the association between PLN and SERCA2a.45 Interestingly, the super-inhibitory effects of mutant PLN were completely relieved upon isoproterenol stimulation, which also reduced the binding of R25C-PLN with SERCA2 to similar levels as WT-PLN, indicating that this mutation did not alter the ability of PLN to get phosphorylated. Future studies on the three-dimensional structure of mutant PLB and SERCA2a will provide insights into the mechanisms by which R25C-PLN exerts its inhibitory effects and may contribute to design of appropriate molecules to relieve PLB inhibition on SERCA2a and improve function in DCM carriers.
In summary, the current study demonstrates that a newly identified human R25C-PLN mutant is associated with DCM and increased propensity to arrhythmias. Our findings support the view that suppression of Ca2+-resequestration by the SR may result in depressed contractility as well as activation of other detrimental pathways, which further exacerbate impaired Ca2+-handling, resulting in arrhythmias and deteriorative remodelling. Thus, the mutation reported in this study, together with the previously reported human PLN mutations, point to the paramount importance of genetic and cardiac screening for these rare exome variants that disrupt normal homeostatic mechanisms for calcium cycling in the human heart.
5. Limitations
Other factors, including environmental or other genetic perturbations, may have also contributed to the R25C-PLN human phenotype. However, the clinical, pedigree, and functional data are compelling and congruent with prior reports of PLN mutations causing DCM.
Supplementary material
Supplementary Material is available at Cardiovascular Research online.
Funding
This work was supported by the National Institutes of Health (HL26057 and HL64018 to E.G.K., HL58626 to R.E.H.); and the European Community's Seventh Framework Program (FP7/2007-2013 under grant agreement no. HEALTH-F2-2009-241526, EUTrigTreat to E.G.K.)
Acknowledgements
We thank the families and referring physicians for their participation in the Familial Dilated Cardiomyopathy Research Program.
Conflict of interest: none declared.
References
- 1.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 2013;10:531–547. [DOI] [PubMed] [Google Scholar]
- 2.Beuckelmann DJ, Nabauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 1992;85:1046–1055. [DOI] [PubMed] [Google Scholar]
- 3.Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, Morgan JP. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res 1987;61:70–76. [DOI] [PubMed] [Google Scholar]
- 4.Hasenfuss G. Alterations of calcium-regulatory proteins in heart failure. Cardiovasc Res 1998;37:279–289. [DOI] [PubMed] [Google Scholar]
- 5.Meyer M, Schillinger W, Pieske B, Holubarsch C, Heilmann C, Posival H, Kuwajima G, Mikoshiba K, Just H, Hasenfuss G. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation 1995;92:778–784. [DOI] [PubMed] [Google Scholar]
- 6.MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol 2003;4:566–577. [DOI] [PubMed] [Google Scholar]
- 7.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res 2012;110:1646–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haghighi K, Schmidt AG, Hoit BD, Brittsan AG, Yatani A, Lester JW, Zhai J, Kimura Y, Dorn GW, II, MacLennan DH, Kranias EG. Superinhibition of sarcoplasmic reticulum function by phospholamban induces cardiac contractile failure. J Biol Chem 2001;276:24145–24152. [DOI] [PubMed] [Google Scholar]
- 9.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW, II, MacLennan DH, Kremastinos DT, Kranias EG. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest 2003;111:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 2003;299:1410–1413. [DOI] [PubMed] [Google Scholar]
- 11.Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA, Fan GC, Tsiapras D, Parekh RR, Dorn GW, II, MacLennan DH, Kremastinos DT, Kranias EG. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci USA 2006;103:1388–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Posch MG, Perrot A, Geier C, Boldt LH, Schmidt G, Lehmkuhl HB, Hetzer R, Dietz R, Gutberlet M, Haverkamp W, Ozcelik C. Genetic deletion of arginine 14 in phospholamban causes dilated cardiomyopathy with attenuated electrocardiographic R amplitudes. Heart Rhythm 2009;6:480–486. [DOI] [PubMed] [Google Scholar]
- 13.van Rijsingen IA, van der Zwaag PA, Groeneweg JA, Nannenberg EA, Jongbloed JD, Zwinderman AH, Pinto YM, Lekanne Dit Deprez RH, Post JG, Tan HL, de Boer RA, Hauer RN, Christiaans I, van den Berg MP, van Tintelen JP, Wilde AA. Outcome in Phospholamban R14del Carriers: Results of a Large Multicentre Cohort Study. Circ Cardiovasc Genet 2014;7:455–465. [DOI] [PubMed] [Google Scholar]
- 14.Cowan J, Li D, Gonzalez-Quintana J, Morales A, Hershberger RE. Morphological analysis of 13 LMNA variants identified in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Circ Cardiovasc Genet 2010;3:6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, Rahko PS, Hershberger RE. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J 2008;156:161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Norton N, Li D, Rampersaud E, Morales A, Martin ER, Zuchner S, Guo S, Gonzalez M, Hedges DJ, Robertson PD, Krumm N, Nickerson DA, Hershberger RE. Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ Cardiovasc Genet 2013;6:144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu ST, Shen YF, Liu GS, Lei CH, Tang Y, Wang JF, Yang YJ. Altered intracellular Ca2+ regulation in chronic rat heart failure. J Physiol Sci 2010;60:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo W, Grupp IL, Harrer J, Ponniah S, Grupp G, Duffy JJ, Doetschman T, Kranias EG. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ Res 1994;75:401–409. [DOI] [PubMed] [Google Scholar]
- 19.Arvanitis DA, Vafiadaki E, Fan GC, Mitton BA, Gregory KN, Del Monte F, Kontrogianni-Konstantopoulos A, Sanoudou D, Kranias EG. Histidine-rich Ca-binding protein interacts with sarcoplasmic reticulum Ca-ATPase. Am J Physiol Heart Circ Physiol 2007;293:H1581–H1589. [DOI] [PubMed] [Google Scholar]
- 20.Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 2006;116:2510–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012;366:619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kadambi VJ, Ponniah S, Harrer JM, Hoit BD, Dorn GW, II, Walsh RA, Kranias EG. Cardiac-specific overexpression of phospholamban alters calcium kinetics and resultant cardiomyocyte mechanics in transgenic mice. J Clin Invest 1996;97:533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wehrens XH, Lehnart SE, Reiken S, van der Nagel R, Morales R, Sun J, Cheng Z, Deng SX, de Windt LJ, Landry DW, Marks AR. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc Natl Acad Sci USA 2005;102:9607–9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zima AV, Bovo E, Bers DM, Blatter LA. Ca(2)+ spark-dependent and -independent sarcoplasmic reticulum Ca(2)+ leak in normal and failing rabbit ventricular myocytes. J Physiol 2010;588:4743–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res 2003;93:592–594. [DOI] [PubMed] [Google Scholar]
- 26.Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen BX, Marks AR. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest 2010;120:4375–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation 2010;122:2669–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hershberger RE, Siegfried JD. Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol 2011;57:1641–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McNair WP, Sinagra G, Taylor MR, Di Lenarda A, Ferguson DA, Salcedo EE, Slavov D, Zhu X, Caldwell JH, Mestroni L. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J Am Coll Cardiol 2011;57:2160–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Spaendonck-Zwarts KY, van Hessem L, Jongbloed JD, de Walle HE, Capetanaki Y, van der Kooi AJ, van Langen IM, van den Berg MP, van Tintelen JP. Desmin-related myopathy. Clin Genet 2011;80:354–366. [DOI] [PubMed] [Google Scholar]
- 31.Zvaritch E, Backx PH, Jirik F, Kimura Y, de Leon S, Schmidt AG, Hoit BD, Lester JW, Kranias EG, MacLennan DH. The transgenic expression of highly inhibitory monomeric forms of phospholamban in mouse heart impairs cardiac contractility. J Biol Chem 2000;275:14985–14991. [DOI] [PubMed] [Google Scholar]
- 32.Zhai J, Schmidt AG, Hoit BD, Kimura Y, MacLennan DH, Kranias EG. Cardiac-specific overexpression of a superinhibitory pentameric phospholamban mutant enhances inhibition of cardiac function in vivo. J Biol Chem 2000;275:10538–10544. [DOI] [PubMed] [Google Scholar]
- 33.Muller OJ, Lange M, Rattunde H, Lorenzen HP, Muller M, Frey N, Bittner C, Simonides W, Katus HA, Franz WM. Transgenic rat hearts overexpressing SERCA2a show improved contractility under baseline conditions and pressure overload. Cardiovasc Res 2003;59:380–389. [DOI] [PubMed] [Google Scholar]
- 34.del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK, Lewandowski ED, Hajjar RJ. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase in a rat model of heart failure. Circulation 2001;104:1424–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.del Monte F, Harding SE, Dec GW, Gwathmey JK, Hajjar RJ. Targeting phospholamban by gene transfer in human heart failure. Circulation 2002;105:904–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoshijima M, Ikeda Y, Iwanaga Y, Minamisawa S, Date MO, Gu Y, Iwatate M, Li M, Wang L, Wilson JM, Wang Y, Ross J, Jr, Chien KR. Chronic suppression of heart-failure progression by a pseudophosphorylated mutant of phospholamban via in vivo cardiac rAAV gene delivery. Nat Med 2002;8:864–871. [DOI] [PubMed] [Google Scholar]
- 37.Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail 2009;15:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M, Hajjar RJ. Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ Res 2014;114:101–108. [DOI] [PubMed] [Google Scholar]
- 39.Zhang T, Brown JH. Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc Res 2004;63:476–486. [DOI] [PubMed] [Google Scholar]
- 40.Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res 2004;94:e61–e70. [DOI] [PubMed] [Google Scholar]
- 41.Eshete F, Fields RD. Spike frequency decoding and autonomous activation of Ca2+-calmodulin-dependent protein kinase II in dorsal root ganglion neurons. J Neurosci 2001;21:6694–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson ME, Braun AP, Wu Y, Lu T, Wu Y, Schulman H, Sung RJ. KN-93, an inhibitor of multifunctional Ca2+/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J Pharmacol Exp Ther 1998;287:996–1006. [PubMed] [Google Scholar]
- 43.Curran J, Tang L, Roof SR, Velmurugan S, Millard A, Shonts S, Wang H, Santiago D, Ahmad U, Perryman M, Bers DM, Mohler PJ, Ziolo MT, Shannon TR. Nitric oxide-dependent activation of CaMKII increases diastolic sarcoplasmic reticulum calcium release in cardiac myocytes in response to adrenergic stimulation. PLoS One 2014;9:e87495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 2013;502:372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kimura Y, Kurzydlowski K, Tada M, MacLennan DH. Phospholamban regulates the Ca2+-ATPase through intramembrane interactions. J Biol Chem 1996;271:21726–21731. [DOI] [PubMed] [Google Scholar]