

Abstract
Background:
46,XY disorders of sex development (DSD) comprise a heterogeneous group of congenital conditions. Mutations in a variety of genes can affect gonadal development or androgen biosynthesis/action and thereby influence the development of the internal and external genital organs.
Objective:
The objective of the study was to identify the genetic cause in two 46,XY sisters of a consanguineous family with DSD and gonadal tumor formation.
Methods:
We used a next-generation sequencing approach by exome sequencing. Electrophysiological and high-resolution ultrasound examination of peripheral nerves as well as histopathological examination of the gonads were performed.
Results:
We identified a novel homozygous R124Q mutation in the desert hedgehog gene (DHH), which alters a conserved residue among the three mammalian Hedgehog ligands sonic hedgehog, Indian hedgehog, and desert hedgehog. No other relevant mutations in DSD-related genes were encountered. The gonads of one patient showed partial gonadal dysgenesis with loss of Leydig cells in tubular areas with seminoma in situ and a hyperplasia of Leydig cell-like cells expressing CYP17A1 in more dysgenetic parts of the gonad. In addition, both patients suffer from a polyneuropathy. High-resolution ultrasound revealed a structural change of peripheral nerve structure that fits well to a minifascicle formation of peripheral nerves.
Conclusion:
Mutations in DHH play a role in 46,XY gonadal dysgenesis and are associated with seminoma formation and a neuropathy with minifascicle formation. Gonadal dysgenesis in these cases may be due to impairment of Sertoli cell-Leydig cell interaction during gonadal development.
Disorders of sex development (DSD) comprise a heterogeneous group of congenital conditions affecting the development of the internal and external genital organs. The current classification is based on karyotype and phenotype. It describes three different categories: the disorders due to atypical sex chromosome numbers, including mosaicism, and 46,XX and 46,XY DSD (1). A distinction is made between those conditions affecting gonadal development (gonadal dysgenesis) and those affecting directly hormone synthesis and action. This is based on the understanding of whether a global gonadal failure is present or whether a specific condition affecting steroidogenesis is the underlying cause of DSD.
46,XY DSD with female phenotype are clinically classified as gonadal dysgenesis if a uterus can be detected, and as a likely disorder of androgen biosynthesis or action in case of absence of a uterus, despite the fact that in essence the diagnosis of gonadal dysgenesis can only be made on the basis of histological evaluation of the gonads. Endocrine assessment can give further guidance. The genetic causes of 46,XY gonadal dysgenesis provide new insight into the cascade of gonadal development.
Groundbreaking was the discovery of the sex determining region Y (SRY) gene as an essential factor to initiate testicular development from a bipotent gonad (2). However, less than 15% of all 46,XY complete gonadal dysgenesis cases are found to carry relevant SRY mutations (3). The role of further genes, especially of defined mutations in Wilms tumor 1 (WT1) and SRY box 9 (SOX9), has been described with partial and complete gonadal dysgenesis, in association with renal or bone abnormalities (4–8). An important gene involved in gonad formation as well as the initiation of steroidogenesis is nuclear receptor subfamily 5 group A member 1 (NR5A1), encoding the steroidogenic-factor 1 (SF-1). Homozygous or heterozygous mutations are associated with a highly variable phenotype, ranging from simple hypospadias or even infertility in 46,XY males to unequivocally females with the 46,XY karyotype, with and without a variable degree of adrenal insufficiency (9). SF-1 is a pivotal factor in both the determination of the gonad and sex differentiation by initiation of steroidogenesis and insulin-like 3 (INSL3) expression within the Leydig cells or anti-Mullerian hormone expression in Sertoli cells (10).
In the physiology of the developmental cascade, once SRY has played its role as a genetic switch toward testis development, the differentiation of Sertoli cells is initiated by the up-regulation of SOX9 and its maintenance by the subsequent initiation of feed-forward loops via fibroblast growth factor 9 and prostaglandin D2 (11–14). Sertoli cells trigger the differentiation of peritubular myoid cells and Leydig cells through the secretion of desert hedgehog (DHh) (15, 16). DHh is the least studied member of the three mammalian Hedgehog proteins [sonic hedgehog (SHh); Indian hedgehog (IHh), and DHh] known to act as morphogenetic ligands in developmental aspects. In mice, Dhh is expressed just after the onset of sex determination in Sertoli cells [at embryonic day 11.5] acting as a morphogenetic regulator upon peritubular myoid cells and fetal Leydig cell differentiation (17, 18). Once secreted from the Sertoli cells, it acts on its target cells via a receptor called Patched1, expressed in interstitial cells (17, 18). Knockout of Dhh in mice affects male sex development, but interestingly the phenotype of Dhh null mice depends on their genetic background that ranges from defects in spermatogenesis to feminized males lacking adult type Leydig cells, displaying defects in peritubular myoid cells, malformed testis cords, abundant fibrotic tissue in the interstitium, and defects in the development of peripheral nerve sheaths (15–19).
In humans, mutations in DHH have been described in a small number of patients with gonadal dysgenesis. The first patient described presented with a homozygous missense mutation associated with partial gonadal dysgenesis and minifascicular polyneuropathy (20). Later a few patients with homozygous DHH mutations and complete gonadal dysgenesis also have been reported (21, 22). A recently described mutation in the Hedgehog acetyl-transferase gene (HHAT), which is required for N-terminal palmitoylation of Hedgehog impedes Hedgehog protein signaling and leads to a syndromic form of 46,XY DSD with complete gonadal dysgenesis and skeletal malformations (23). The male patient had dysplastic immature seminiferous tubules delimited with a dense interstitial compartment containing a greatly reduced portion of steroidogenic cells, indicated by CYP11A1 expression.
Here we describe two sisters with 46,XY DSD with a homozygous mutation in DHH, who both developed gonadal dysgenesis and seminoma and demonstrated a polyneuropathy.
Patients and Methods
Patients
The two patients were born in Syria to parents who were first cousins. Patient 1 reported that she received surgery in early childhood because of an inguinal hernia. The family moved to Germany during her childhood of the siblings; however, her parents and further siblings were not available for visits or analysis. Further early history of the siblings was uneventful.
Patient 1 was first seen at an outside institution because of primary amenorrhea at the age of 17 years. At this time, apparently very little breast development and pubic hair was seen and hypergonadotropic hypogonadism was noted (FSH 79 IU/L and LH 27 IU/L, estradiol not measurable). The external genitalia were of female appearance without any signs of virilization. A 46,XY karyotype was analyzed. On vaginoscopy, a vagina of 3–4 cm in length and normal width without an upper vagina and cervix (portio) was noted. Gonads were not visualized on laparoscopy. This was initially thought to be due to the suspected removal of the gonads in infancy. On the basis of lacking Mullerian remnants and little pubic hair growth, androgen insensitivity was assumed, but analysis of the androgen receptor gene was unremarkable. Estrogen replacement therapy was initiated, and the patient developed normal female secondary sex characteristics as well as normal pubic hair development. Sexual intercourse was possible without any further procedures. At the age of 30 years, a right inguinal hernia occurred and a gonad was seen on herniotomia, which was initially placed back into the abdomen. Apparently only the left gonad had been removed in infancy. Due to recurrent inguinal pain, the right gonad was removed and histology demonstrated a seminoma (T1, N0, M0, R0) (see Figure 2) and chemotherapy with carboplatin was administered.
Figure 2.
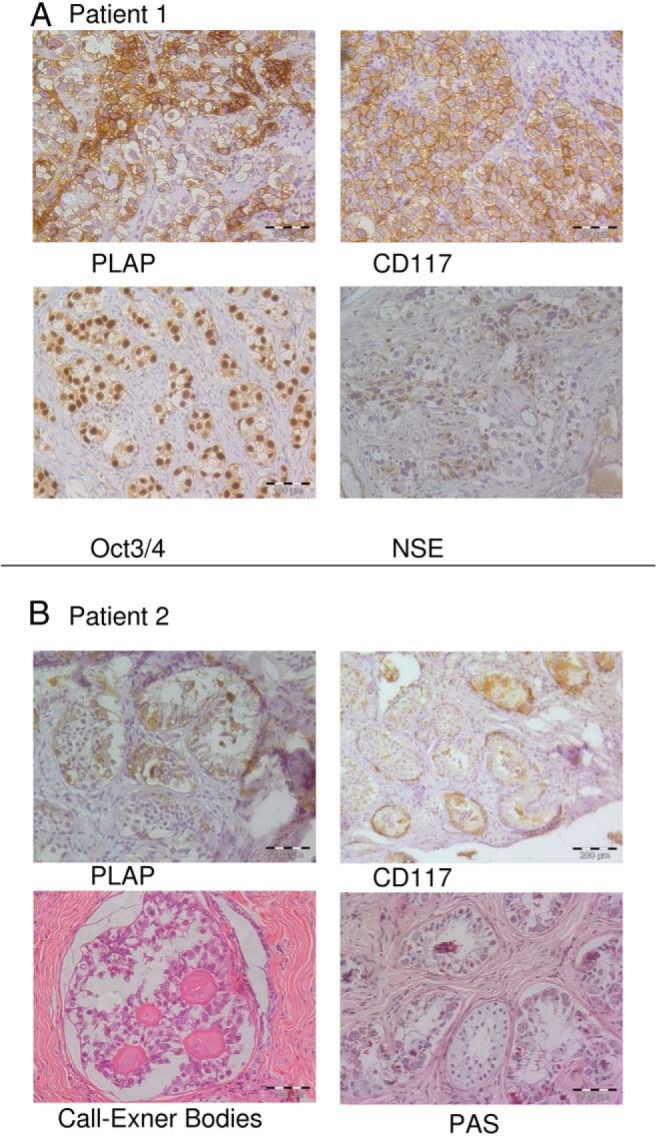
Immunohistological evaluation of the testicular tissue. A, Patient 1, Invasive seminoma/dysgerminoma. No seminiferous tubules could be seen. Testicular tissue was replaced by seminoma cells expressing PLAP (alkaline phosphatase, placental) and CD117 (KIT, v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog). A strong expression of Oct3/4 (POU class 5 homeobox 1) and a granular expression of NSE [enolase 2 (gamma, neuronal)] of seminoma cells were seen. B, Patient 2, Seminoma in situ. Seminiferous tubules including Sertoli cells and some germ cells are surrounded by irregular masses of fibrotic tissue. Within the seminiferous tubules, atypical proliferating cells positive for PLAP and CD117 were detected. Periodic acid-Schiff (PAS) staining showed germ cells with granular glycogen-rich cytoplasm. At another site a gonadoblastoma with Call Exner bodies was seen.
The main reason for consultation in our DSD clinic at the age of 34 years was lack of a diagnosis leading to uncertainty in respect of prognosis. Clinical examination of the breast and pubic hair development according to stage Tanner 5 was recorded. No signs of virilization were observed. Gynecological examination again did not show a portio and no uterus-like structures were visualized on ultrasound. The external genitalia were of normal female appearance. She was under replacement therapy with estradiol valerate and cyproterone acetate, and all measured hormone values were within the normal female reference ranges (LH 5.9 U/L, FSH 8.8 U/L, dehydroepiandrosterone sulfate 2.26 mg/L, T 0.073 μg/L, ACTH 20.3 ng/L, cortisol 471 mmol/L). The patient reported numbness of the feet and hand, which had begun in her early 20s, which increased and ascended the extremities over time.
Patient 2, a younger sister of patient 1 also revealed a 46,XY karyotype when presenting with primary amenorrhea at the age of 17 years at an outside institution. The phenotype was similar to patient 1 with little breast development, scarce pubic hair, female external genitalia, and a blind-ending vagina without Mullerian remnants. Under the assumption of a defect in androgen biosynthesis, gonadectomy was performed to prevent unwanted virilization as well as possible malignant transformation of the gonad. Hormone analysis prior to surgery showed high LH (18.5 IU/L) and FSH (36.8 IU/L) values but a low T value (1.3 nmol/L). The patient received vaginoplasty using the laparoscopic Vecchietti procedure in combination with gonadectomy. Histological evaluation of the gonads showed a seminoma in situ with a beginning invasion of the stroma in the right testis and a circumscribed gonadoblastoma. The patient was first seen at the age of 30 years in our institution due to lack of a definitive diagnosis. At that time she received a substitution with estradiol valerate and cyproterone acetate for suspected hirsutism, which we did not confirm. Clinical investigation revealed a normal body hair distribution, no apparent signs of virilization, Tanner stages B5, and pH 4–5. Gynecological examination demonstrated a vagina of 4–5 cm in length, normal width, and no portio. Laboratory values showed LH 2.6 U/L, FSH 7.76 U/L, estradiol 49.8 pg/mL, T 0.057 μg/L, dehydroepiandrosterone sulfate 1.82 ng/mL, and cortisol 373 mmol/L.
In both sisters, clinical neurological examination revealed signs of a glove and stocking like polyneuropathy. Muscle tendon reflexes at the arm were very weak and absent at the legs. Reduced surface sensitivity of feet and hands and a reduced vibration sensitivity of 7/8 at the finger end joints and of 5/8 at the ankles were found. Atrophy of the extensor digitorum brevis muscle at both feet was noted. The remaining neurological examination was normal in both.
Both sisters gave written consent as recommended by the Ethical Committee of the University of Luebeck to further extensive genetic studies on a scientific basis.
Exome capture and sequencing
Exomes were captured in solution using SureSelect XT Human All Exon V4 enrichment kit (Agilent) and sequenced as 100 bp paired ends on a HiSeq2000 System (Illumina), generating 11.02 and 9.08 Gb of sequence data with an average read depth of 127 x and 105 x on target regions. Sequences were aligned to the human reference genome (GRCh37/hg19) using a Burrows-Wheeler aligner (24). Single-nucleotide variants and small insertions and deletions were called with SAMtools and Pindel (and filtered to leave only those variants with a minor allele frequency of less than 1%). Annotation was performed with custom scripts.
Immunohistochemistry
After gonadectomy, the testes had been fixed in 4% paraformaldehyde and embedded in paraffin. Antibodies and pretreatments used to stain sections in our study are summarized in Supplemental Table 1. To reduce nonspecific background due to endogenous peroxidase, the slides were treated with hydrogen peroxide. Bound antibodies were visualized using an UltraVision detection system (Thermo Scientific) with diaminobenzidine and Fast Red staining.
Electrophysiology and high-resolution ultrasound of peripheral nerves
Routine diagnostic for detection of a polyneuropathy was performed. At least one motor and sensory nerve conduction velocity and a sensory nerve action potential and compound muscle action potential and one F-Wave latency from the arms and legs was captured.
High-resolution ultrasound (3–13 mHz matrix transducer; MyLab Alpha) was performed of several peripheral nerves of the extremities and of the brachial plexus of the right side.
Structural analysis
The computer program Pymol was used for both the structural analysis and generation of Figure 5 (25).
Figure 5.
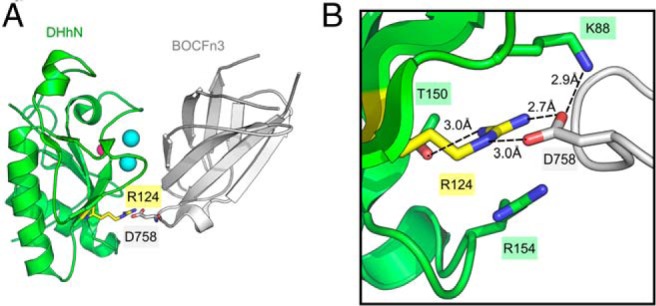
Binding of DHhN to BOC. A, Ribbon diagram of the complex between the DHhN (green) with the third FNIII repeat of BOC (BOCFn3) (gray) (PDB code 3N1G). Represented as spheres are calcium ions (cyan) and zinc ion (pink). R124 of DHhN (yellow) and D758 of BOCFn3 are shown as sticks. B, Close-up view of the binding interface between DHhN (green) and BOCFn3 (white). Residues involved in a hydrogen-bonding network around R124 (yellow) are shown in sticks, with nitrogen atoms colored blue and oxygen atoms red. Potential hydrogen bonds are indicated by dashed lines and the distances between atoms are measured in Ångstroms.
Results
Next generation sequencing
Exome sequencing revealed a homozygous c.371G>A mutation in exon 2 of the desert hedgehog gene (DHH; NM_021044.2; NC_000012.11:g.49485105C>T) responsible for a p.R124Q (reference sequence NP_066382.1) mutation in the autocatalytically spliced and secreted N-terminal part of the protein (DHhN). Next generation sequencing results were verified by Sanger sequencing of amplified genomic DNA of the patients using the primer sequences published previously by Umehara et al (20) (Figure 1). The mutation is novel and not listed in SNPdb, 1000 Genomes Project, or Exome Sequencing Project ESP6500, or the Exome Aggregation Consortium, which covers more than 60 000 exomes (26). All pathogenicity prediction algorithms, ie, SIFT, PolyPhen2, MutationTaster, and CADD, consider this mutation as damaging, probably damaging, or disease causing. No other relevant mutation was detected in currently known DSD-related genes.
Figure 1.
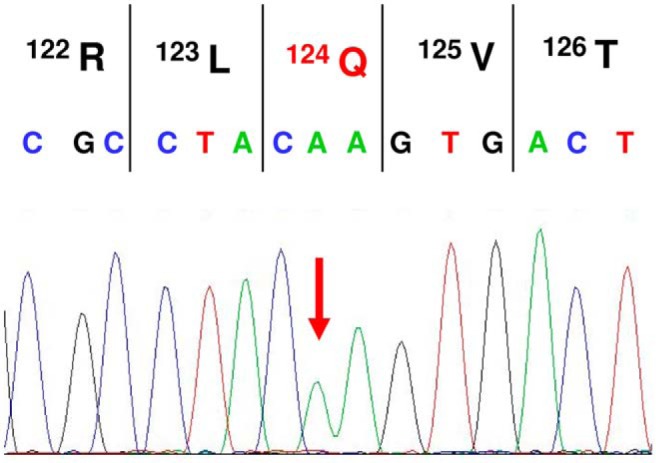
Verification of the homozygous R124Q mutation by Sanger sequencing in patient 1.
Histological evaluation
Patient 1
Histopathological analysis of the right gonad revealed a classical invasive seminoma. Neither seminiferous tubules nor Leydig cells were detectable. Seminoma cells showed strong octamer-binding transcription factor (Oct)-3/4 (POU class 5 homeobox 1) and placental/germ alkaline phosphatase (PLAP; alkaline phosphatase, placental) expression (Figure 2A). Only dispersed single cells were calretinin (calbindin 2) positive. Germ cells expressed CD117 (KIT, v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog) (Figure 2A) and weak PGP9.5 (ubiquitin carboxyl-terminal esterase L1). Seminoma cells displayed granular expression of neuron-specific enolase (NSE) [enolase 2 (gamma, neuronal)] (Figure 2A) and a weak E-cadherin [cadherin 1, type 1, E-cadherin (epithelial)] expression.
Patient 2
Parts of the gonads show testicular tissue with seminiferous tubules including Sertoli cells and some germ cells. The tubules are surrounded by irregular masses of fibrotic tissue. Epididymal tissue was also developed. Other parts of the gonad resemble a streak gonad. At the right gonad, a fallopian tube was found side by side with epididymal tissue (Supplemental Figure 1).
Within the seminiferous tubules, atypical proliferating cells positive for CD117 and PLAP were detected indicating a seminoma/dysgerminoma in situ (Figure 2B). These atypical germ cells were also positive for OCT3/4 and showed a weak staining of NSE. Within some tubules a hyperplasia of Sertoli cells was visible. At the right testis, a beginning invasion of seminoma cells was visible (data not shown). A nodular hyperplasia of Leydig cell-like cells positive for NSE and melan A could be detected. These cells strongly express CYP17A1 (steroid 17α-hydroxylase and 17,20-lyase) (Figure 3A) but not INSL3 (Figure 3B) and are located in highly fibrous tissue with rare and atrophic seminiferous tubules. No CYP17A1- or INSL3-positive cells were observed in the interstitial tissue between seminiferous tubules of areas with seminoma in situ (Figure 3C). At another site an area with a gonadoblastoma with Call Exner bodies was seen (Figure 2B).
Figure 3.
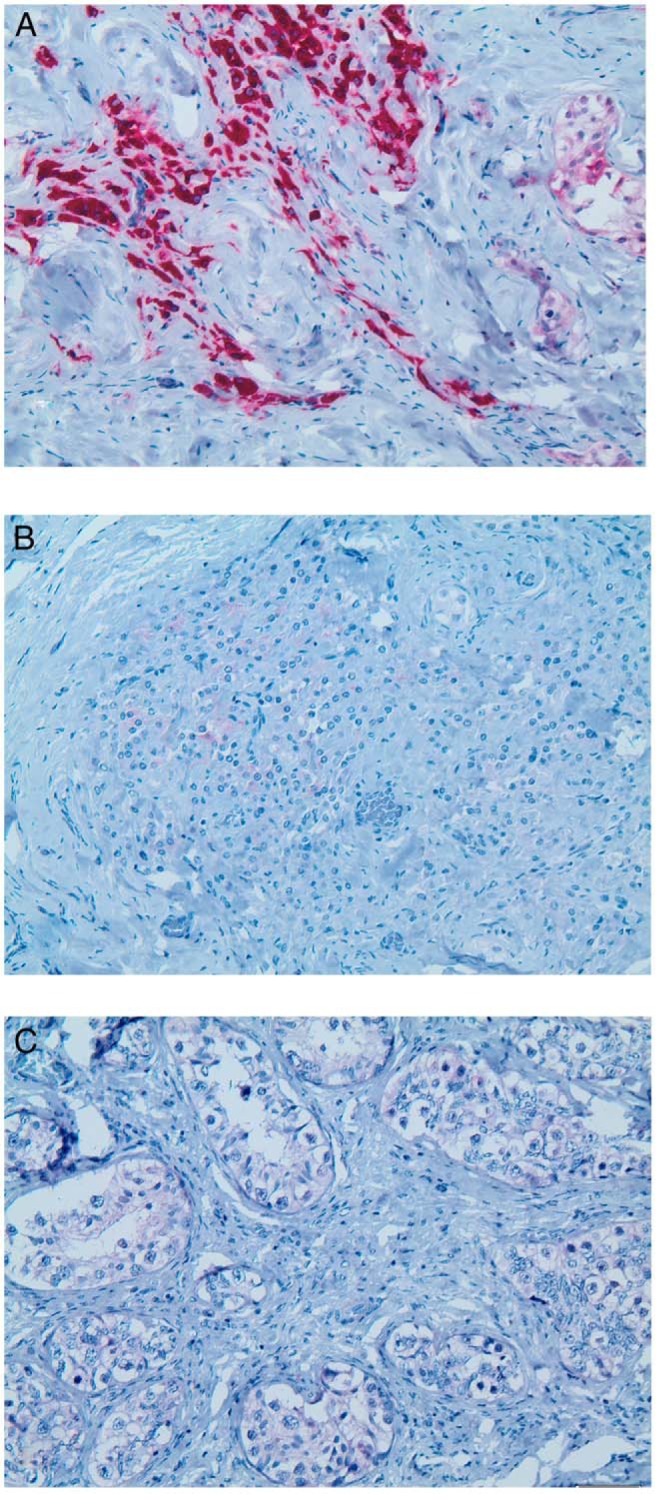
Expression of CYP17A1 and INSL3 in the gonad of patient 2. A, Nodular Leydig cell-like cells within the unstructured dysgenetic part of the gonad with rare and atrophic tubuli show a strong CYP17A1 expression. B, Only single cells show faint expression of INSL3. C, In contrast, no CYP17A1-expressing Leydig cells could be detected in the interstitium between the seminiferous tubules of areas with seminoma in situ.
Electrophysiology
In both patients electrophysiology revealed signs of a mixed axonal and demyelinating sensory motor polyneuropathy with reduced compound muscle action potentials and reduced or absent sensory nerve action potential but also prolonged F-wave latencies and reduced motor nerve conduction velocity. The values are given in Supplemental Table 2.
High-resolution ultrasound
In both subjects the findings were similar. The median and ulnar nerve at the right arm and the peronael and tibial nerve at the left as well as the popliteal and the sural nerve at the right distal lower leg showed an increased echogenicity. The typical structure of a peripheral nerve at the extremities, with a honeycomb-like pattern of hypoechogenic fasciles with a surrounding endoneural and perineural epineurium, was absent. Even very small hypoechogenic dots in the nerves may indicate that there are minifascicles. A comparison of a normal median nerve and the typical finding of the peripheral nerves are shown in Figure 4. The echotexture of the brachial plexus was inconspicuously hypoechogenic-like in controls.
Figure 4.
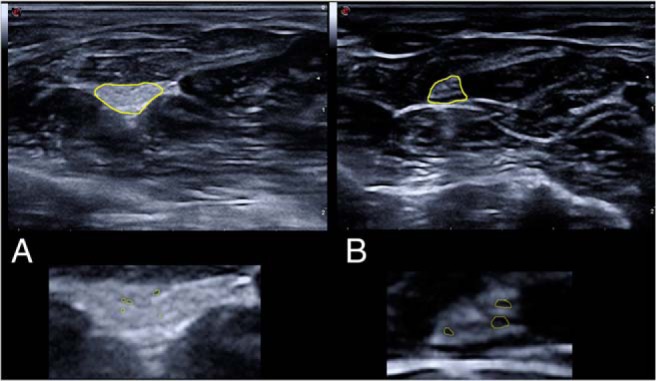
Cross-sectional ultrasound pictures at the distal third of the lower arm of patient 1 (A) and an age-matched control (B) are shown. The median nerve is marked in yellow in the upper part of the figure. In the lower half of the figure, the median nerves are shown enlarged. The median nerve of patient 1 (and all other investigated nerves at the extremities) showed a loss of the typical fascicular honeycomb-like structure and only very little hypoechogenic dots that may correspond with the formation of minifasciles. Fascicles are marked in yellow.
Discussion
Both sisters had been diagnosed with a 46,XY DSD with a presumed autosomal-recessive underlying cause due to their consanguinity. The history of a previous surgical intervention in Syria had obscured the diagnosis of a global gonadal failure and had favored suspicion for Leydig cell failure, also on the basis of a lack of uterine development. A candidate gene for the 46,XY DSD was found only on exome sequencing and revised the diagnosis to gonadal dysgenesis due to a homozygous mutation in DHH. This demonstrates the need for multiple gene analysis in conditions like DSD because the clinical presentation is highly variable and not specific, even for the main categories according to the currently used classification.
In retrospect, this diagnosis of gonadal dysgenesis was of elemental importance to the patients because both developed a germ cell tumor and suffer from polyneuropathy. In patient 1 seminoma cells replaced the gonadal tissue and no testicular structures were visible. Patient 2 developed seminoma in situ indicated by seminiferous tubules with proliferating germ cells positive for OCT3/4 and PLAP and an invasion of these cells into the stroma of the right testis. So far, only six patients with homozygous mutations in the DHH gene have been reported (20–22). One of them developed bilateral streak gonads with bilateral dysgerminoma and one developed bilateral streak gonads with bilateral gonadoblastoma. Together with the two patients reported here, a great proportion (four of eight) of the patients developed seminoma/dysgerminoma/gonadoblastoma, indicating a high incidence of germ cell cancer in patients with homozygous DHH mutations as also seen in other cases of gonadal dysgenesis with different causes.
Both of our patients have a glove and stocking-like sensory impairment as described in the first reported case and electropysiological measurements verified a neuropathy in both. These two additional cases strengthen the evidence that DHh signaling is important for peripheral nerve conduction. Recent studies suggest that DHh regulates myelination in the peripheral nervous system through primary cilia (27). The gonadal phenotype of patients with homozygous DHH mutations varied between partial gonadal dysgenesis with an immature uterus, testis on one side and a streak gonad on the other side (20) and complete gonadal dysgenesis with streak gonads and Mullerian structures, ie, a hypoplastic uterus or fallopian tubes (21, 22). This is a common finding in the variability of partial gonadal dysgenesis. Our patients did not develop a uterus, but patient 2 presented a fallopian tube side by side with an epididymis at the right gonad (Supplemental Figure 1), indicating an incomplete regression of Mullerian derivates, despite an observed Sertoli cell hyperplasia. This indicates that DHh may also have a small intracrine impact on anti-Mullerian hormone expression from Sertoli cells. Within the dysgenetic part of the gonad, a nodular Leydig cell hyperplasia was seen, strongly expressing CYP17A1 (Figure 3), a key enzyme in the androgen biosynthesis pathway (28).
In contrast, no CYP17A1-expressing cells could be detected in the interstitial tissue (Figure 3). Staining of INSL3, a second Leydig cell marker crucial for testicular descent (29), was negative and indicates that the CYP17A1-expressing cells are not fully functional Leydig cells. The decrease instead of the absence of fetal Leydig cells in Dhh−/− mice was thought to be a possible compensation by other hedgehog ligands (17, 30). Shh expression was observed in the epididymis of fetal Dhh−/− mice (18), and possibly SHh had led to the hyperplasia of the CYP17A1-expressing cells in the dysgenetic part of the gonad. Nevertheless, the very low T values (1.3 ng/L) measured just before gonadectomy supports a disruption of the androgen biosynthesis pathway. Recently a homozygous missense mutation in the Dhh gene was mapped in a testicular feminized rat strain. These rats showed a reduced number of fetal Leydig cells and a lack of adult-type Leydig cells as well as loss of testicular descent (31). In contrast to the mouse strains, all homozygous male rats showed a feminized phenotype without apparent differences, even in different genetic backgrounds. This demonstrates the importance of delicate genetic and histological investigations to confirm a diagnosis of gonadal dysgenesis due to the insecurity of proof of Mullerian structures with clinical investigations or imaging.
Testicular descent is a continuous process that is often divided into two main phases: the transabdominal phase, depending on INSL3 for induction of the male gubernaculums, and an androgen-dependent inguinoscrotal phase. The impact of DHh on Leydig cell development has been shown not only by gene deletions but also by ectopic activation of the Hedgehog pathway in SF-1-positive somatic precursor cells of fetal ovaries. These cells differentiate into fetal Leydig cells that produce androgens and express Insl3, leading to virilization and descent of the ovaries (30). Our patients presented with inguinal gonads, and both INSL3 expression (evidenced by immunostaining, Figure 3) and T production (evidenced by low serum T levels measured just before gonadectomy) have been strongly affected by inappropriate Leydig cell differentiation.
Hedgehog signaling is mediated by the hedgehog coreceptors: the cell adhesion molecule, down-regulated by oncogenes (CDO), and Brother of CDO (BOC). CDO and BOC bind all mammalian Hedgehog proteins in a conserved manner (32). The mutation found in the two sisters described here, R124Q, sits at the interface between DHhN and BOC (32). R124 is conserved in the N-terminal domains of each of the mammalian hedgehogs (DHh, SHh, and the N-terminal domain of IHh) and forms hydrogen bonds with a conserved aspartate in both CDO (D872) or BOC (D758) (32). R124 also forms an internal bond to T150 of DHhN, which may stabilize the protein (Figure 5). Hedgehog-interacting protein (HHIP) is a negative regulator of hedgehog signaling and uses a binding surface on the N-terminal hedgehog that overlaps with the binding surface used by CDO and BOC (33–35). Crystal structures of SHhN bound to HHIP show that R123 of SHhN (corresponding to R124 of DHhN) interacts with the L2 loop of HHIP and provides a complementary charge for E380 in HHIP (36). R124Q might affect interactions with multiple partners that either positively or negatively regulate hedgehog signaling, and the phenotypic effect might be a complex mixture of gain and loss of function (35). Heterozygous mutations in this area of IHh including R128Q (corresponding to R124 in DHhN) have been linked to brachydactyly type A1 (37) and illustrate the importance of this conserved residue in hedgehog signaling.
Four of the described six patients with homozygous mutations in DHH leading to gonadal dysgenesis harbor either frame shift mutations (p.Q21fsX71 and p.L363CfsX4) or an alteration of the start codon (p.M1?) and are predicted to abolish DHhN protein (Table 1). The missense mutation P162L and small deletion p.E91del detected in the two further patients have been analyzed by protein modeling and are predicted to alter the binding interface to CDO and BOC (22, 38). With respect to the gonadal phenotype of Dhh−/− mice and unaffected fathers carrying heterozygous mutations in DHH, we conclude that loss of function in DHh signaling is responsible for the phenotype of 46,XY patients with gonadal dysgenesis. This is also supported by experiments in mice, showing that ectopic activation of the hedgehog pathway in ovaries leads to the appearance of Leydig cells and descent of the ovaries but leaves the female reproductive system intact (30).
Table 1.
Homozygous Mutations in DHH
| DHh Mutation | Protein | Phenotype | Age, y | Karyotype | Gonads | Reference |
|---|---|---|---|---|---|---|
| c.2T>C | p.M1? | PGD and minifascicular neuropathy | 27 | 46,XY | Testis on one side and streak gonad on other side | (20) |
| c.57_60dupAGCC | p.Q21fsX71 | CGD | 17 | 46,XY | Hypoplastic uterus with streak ovaries by ultrasound | (22) |
| c.271_273delGAG | p.E91del | CGD | 26 | 46,XY | Bilateral streak gonads | (22) |
| c.371G>A | p.R124Q | PGD and neuropathy | 23 | 46,XY | Testis on both sides, seminoma in situ, gonadoblastoma | This study |
| c.371G>A | p.R124Q | PGD and neuropathy | 30 | 46,XY | Testis on one side, seminoma | This study |
| c.485T>C | p.L162P | CGD | 16 | 46,XY | Bilateral streaks | (21) |
| c.1086delG | p.L363CfsX4 | CGD | 19 | 46,XY | Bilateral streaks with bilateral gonadoblastoma | (21) |
| c.1086delG | p.L363CfsX4 | CGD | 26 | 46,XY | Bilateral streaks with bilateral dysgerminoma | (21) |
The finding of two further patients with gonadal dysgenesis carrying a homozygous mutation in the DHH gene and the immunohistological findings that show a loss of adult Leydig cells in the interstitium support the role of DHh in Leydig cell differentiation and implicate an important role of DHH mutations in DSD with suspected gonadal dysgenesis, particularly in consanguineous families.
Acknowledgments
We thank Tim M. Strom (Institute of Human Genetics, Helmholtz Zentrum München, München, Germany) for performing the exome sequencing. We are grateful to the family for participating in the study, to D. Struve and M. Erdmann-Jensko for excellent technical assistance, and Daniel J. Leahy for critical reading of the manuscript.
This work was supported by the intramural funding of the University of Luebeck SPP “Medizinische Genetik.” J.M.K. was supported by National Institutes of Health Grant R01 HD055545 (to Daniel J. Leahy).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BOC
- Brother of CDO
- CDO
- cell adhesion molecule, down-regulated by oncogenes
- DHh
- desert hedgehog
- DHhN
- N-terminal part of the DHh
- DSD
- disorders of sex development
- HHIP
- Hedgehog-interacting protein
- IHh
- Indian hedgehog
- INSL3
- insulin-like 3
- NSE
- neuron-specific enolase
- Oct
- octamer-binding transcription factor
- PLAP
- placental/germ alkaline phosphatase
- SF-1
- steroidogenic-factor 1
- SHh
- sonic hedgehog
- SHhN
- N-terminal part of the SHh
- SOX9
- SRY box 9.
References
- 1. Hughes IA, Houk C, Ahmed SF, Lee PA. Consensus statement on management of intersex disorders. Arch Dis Child. 2006;91:554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sinclair AH, Berta P, Palmer MS, et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature. 1990;346:240–244. [DOI] [PubMed] [Google Scholar]
- 3. Bashamboo A, McElreavey K. Gene mutations associated with anomalies of human gonad formation. Sex Dev. 2013;7:126–146. [DOI] [PubMed] [Google Scholar]
- 4. Foster JW, Dominguez-Steglich MA, Guioli S, et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature. 1994;372:525–530. [DOI] [PubMed] [Google Scholar]
- 5. Huang B, Wang S, Ning Y, Lamb AN, Bartley J. Autosomal XX sex reversal caused by duplication of SOX9. Am J Med Genet. 1999;87:349–353. [DOI] [PubMed] [Google Scholar]
- 6. Wagner T, Wirth J, Meyer J, et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell. 1994;79:1111–1120. [DOI] [PubMed] [Google Scholar]
- 7. Thong MK, Scherer G, Kozlowski K, Haan E, Morris L. Acampomelic campomelic dysplasia with SOX9 mutation. Am J Med Genet. 2000;93:421–425. [PubMed] [Google Scholar]
- 8. Kohler B, Biebermann H, Friedsam V, et al. Analysis of the Wilms' tumor suppressor gene (WT1) in patients 46,XY disorders of sex development. J Clin Endocrinol Metab. 2011;96:E1131–E1136. [DOI] [PubMed] [Google Scholar]
- 9. Ferraz-de-Souza B, Lin L, Achermann JC. Steroidogenic factor-1 (SF-1, NR5A1) and human disease. Mol Cell Endocrinol. 2011;336:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mello MP, Franca ES, Fabbri HC, Maciel-Guerra AT, Guerra-Junior G. Multifunctional role of steroidogenic factor 1 and disorders of sex development. Arq Bras Endocrinol Metab. 2012;55:607–612. [DOI] [PubMed] [Google Scholar]
- 11. Kim Y, Bingham N, Sekido R, Parker KL, Lovell-Badge R, Capel B. Fibroblast growth factor receptor 2 regulates proliferation and Sertoli differentiation during male sex determination. Proc Natl Acad Sci USA. 2007;104:16558–16563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim Y, Kobayashi A, Sekido R, et al. Fgf9 and Wnt4 act as antagonistic signals to regulate mammalian sex determination. PLoS Biol. 2006;4:e187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wilhelm D, Hiramatsu R, Mizusaki H, et al. SOX9 regulates prostaglandin D synthase gene transcription in vivo to ensure testis development. J Biol Chem. 2007;282:10553–10560. [DOI] [PubMed] [Google Scholar]
- 14. Wilhelm D, Martinson F, Bradford S, et al. Sertoli cell differentiation is induced both cell autonomously and through prostaglandin signaling during mammalian sex determination. Dev Biol. 2005;287:111–124. [DOI] [PubMed] [Google Scholar]
- 15. Clark AM, Garland KK, Russell LD. Desert hedgehog (Dhh) gene is required in the mouse testis for formation of adult-type Leydig cells and normal development of peritubular cells and seminiferous tubules. Biol Reprod. 2000;63:1825–1838. [DOI] [PubMed] [Google Scholar]
- 16. Pierucci-Alves F, Clark AM, Russell LD. A developmental study of the Desert hedgehog-null mouse testis. Biol Reprod. 2001;65:1392–1402. [DOI] [PubMed] [Google Scholar]
- 17. Yao HH, Whoriskey W, Capel B. Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev. 2002;16:1433–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bitgood MJ, Shen L, McMahon AP. Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr Biol. 1996;6:298–304. [DOI] [PubMed] [Google Scholar]
- 19. Parmantier E, Lynn B, Lawson D, et al. Schwann cell-derived Desert hedgehog controls the development of peripheral nerve sheaths. Neuron. 1999;23:713–724. [DOI] [PubMed] [Google Scholar]
- 20. Umehara F, Tate G, Itoh K, et al. A novel mutation of desert hedgehog in a patient with 46,XY partial gonadal dysgenesis accompanied by minifascicular neuropathy. Am J Hum Genet. 2000;67:1302–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Canto P, Soderlund D, Reyes E, Mendez JP. Mutations in the desert hedgehog (DHH) gene in patients with 46,XY complete pure gonadal dysgenesis. J Clin Endocrinol Metab. 2004;89:4480–4483. [DOI] [PubMed] [Google Scholar]
- 22. Das DK, Sanghavi D, Gawde H, Idicula-Thomas S, Vasudevan L. Novel homozygous mutations in Desert hedgehog gene in patients with 46,XY complete gonadal dysgenesis and prediction of its structural and functional implications by computational methods. Eur J Med Genet. 2011;54:e529–e534. [DOI] [PubMed] [Google Scholar]
- 23. Callier P, Calvel P, Matevossian A, et al. Loss of function mutation in the palmitoyl-transferase HHAT leads to syndromic 46,XY disorder of sex development by impeding hedgehog protein palmitoylation and signaling. PLoS Genet. 2014;10:e1004340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schrödinger LLC. The PyMOL molecular graphics system, version 1.7.2. 2014. https://www.pymol.org/
- 26. Exome Aggregation Consortium. ExAC Browser (Beta). 2015. http://exac.broadinstitute.org/ Accessed January 15, 2015.
- 27. Yoshimura K, Takeda S. Hedgehog signaling regulates myelination in the peripheral nervous system through primary cilia. Differentiation. 2012;83:S78–S85. [DOI] [PubMed] [Google Scholar]
- 28. Miller WL. The syndrome of 17,20 lyase deficiency. J Clin Endocrinol Metab. 2012;97:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nef S, Parada LF. Cryptorchidism in mice mutant for Insl3. Nat Genet. 1999;22:295–299. [DOI] [PubMed] [Google Scholar]
- 30. Barsoum IB, Bingham NC, Parker KL, Jorgensen JS, Yao HH. Activation of the Hedgehog pathway in the mouse fetal ovary leads to ectopic appearance of fetal Leydig cells and female pseudohermaphroditism. Dev Biol. 2009;329:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kawai Y, Noguchi J, Akiyama K, et al. A missense mutation of the Dhh gene is associated with male pseudohermaphroditic rats showing impaired Leydig cell development. Reproduction. 2011;141:217–225. [DOI] [PubMed] [Google Scholar]
- 32. Kavran JM, Ward MD, Oladosu OO, Mulepati S, Leahy DJ. All mammalian Hedgehog proteins interact with cell adhesion molecule, down-regulated by oncogenes (CDO) and brother of CDO (BOC) in a conserved manner. J Biol Chem. 2010;285:24584–24590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bishop B, Aricescu AR, Harlos K, O'Callaghan CA, Jones EY, Siebold C. Structural insights into hedgehog ligand sequestration by the human hedgehog-interacting protein HHIP. Nat Struct Mol Biol. 2009;16:698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chuang PT, McMahon AP. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature. 1999;397:617–621. [DOI] [PubMed] [Google Scholar]
- 35. Beachy PA, Hymowitz SG, Lazarus RA, Leahy DJ, Siebold C. Interactions between Hedgehog proteins and their binding partners come into view. Genes Dev. 2010;24:2001–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bosanac I, Maun HR, Scales SJ, et al. The structure of SHH in complex with HHIP reveals a recognition role for the Shh pseudo active site in signaling. Nat Struct Mol Biol. 2009;16:691–697. [DOI] [PubMed] [Google Scholar]
- 37. Byrnes AM, Racacho L, Grimsey A, et al. Brachydactyly A-1 mutations restricted to the central region of the N-terminal active fragment of Indian Hedgehog. Eur J Hum Genet. 2009;17:1112–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Castro JJ, Mendez JP, Coral-Vazquez RM, et al. In vitro and molecular modeling analysis of two mutant desert hedgehog proteins associated with 46,XY gonadal dysgenesis. DNA Cell Biol. 2013;32:524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]