

Abstract
Physical inactivity in older adults is a risk factor for developing glucose intolerance and impaired skeletal muscle function. Elevated inflammation and ceramide biosynthesis have been implicated in metabolic disruption and are linked to Toll-like receptor (TLR)/myeloid differentiation primary response 88 (MyD88) signaling. We hypothesize that a physical inactivity stimulus, capable of inducing glucose intolerance, would increase skeletal muscle inflammation and ceramide biosynthesis signaling and that this response would be regulated by the TLR/MyD88 pathway. Therefore, we subjected wild-type (WT) and MyD88−/− mice to hindlimb unloading (HU) for 14 days or an ambulatory control period. We observed impaired glucose uptake, muscle insulin signaling (p-Akt), and increased markers of NF-κB signaling (p-IκBα), inflammation (p-JNK, IL-6), TLR4, and the rate-limiting enzyme of ceramide biosynthesis, SPT2, with HU WT (P < 0.05), but not in HU MyD88−/− mice. Concurrently, we found that 5 days of bed rest in older adults resulted in whole body glucose dysregulation, impaired skeletal muscle insulin signaling, and upregulation of muscle IL-6 and SPT2 (P < 0.05). Post-bed rest TLR4 abundance was tightly correlated with impaired postprandial insulin and glucose levels. In conclusion, MyD88 signaling is necessary for the increased inflammation, ceramide biosynthesis signaling, and compromised metabolic function that accompanies physical inactivity.
Keywords: metabolic disruption, insulin resistance, TLR4, bed rest
reduced levels of ambulatory activity contribute considerably to the worldwide development of metabolic disease, including glucose intolerance, insulin resistance, and type 2 diabetes (1, 4). Older adults are at risk for adopting physically inactive lifestyles (32), often precipitated by functional deficits following repeated bouts of bed rest/hospitalization (8, 23) coupled with slow/incomplete muscle recovery from injury or illness-related disuse (47). A repeated observation in older adults following a bout of short-term physical inactivity is the rapid deterioration of whole body insulin sensitivity, glucose uptake, and lean mass (8, 13). Because skeletal muscle plays a significant role in postprandial glucose disposal and whole body insulin sensitivity (19, 46), skeletal muscle dysfunction can have a profound impact on the development of metabolic disorders. Therefore, a further understanding of the cellular events responsible for altered postprandial glucose homeostasis is needed within this vulnerable population.
A novel candidate mechanism that may contribute to physical inactivity-induced insulin resistance is activation of the Toll-like receptor/myeloid differentiation primary response 88 (TLR/MyD88) pathway and subsequent increase in inflammatory pathways and ceramide accrual within skeletal muscle tissue. Mammalian TLRs, such as TLR4, are transmembrane proteins predominantly found at the surface of immune cells and nonimmune tissues (e.g., skeletal muscle) and play a pivotal role as a first line of defense in pathogen recognition and innate immunity (49). The most recognized cellular response to TLR agonists (e.g., LPS/endotoxin, saturated fatty acids) is an upregulation of proinflammatory cytokines mediated by NF-κB and MAP kinase signaling (e.g., JNK) (37, 38, 45). Importantly, a dysregulated inflammatory response can lead to impaired insulin signaling (28) and is noted to be chronically elevated with insulin resistance, obesity, and diabetes. Of the proteins associated with TLR signaling, MyD88 is a common, yet indispensable, cytoplasmic adaptor protein of most of the TLR subfamily members (31, 39). This was well demonstrated nearly 15 years ago when Kawai et al. (31) demonstrated that MyD88−/− mice have a blunted response to endotoxin compared with WT mice. Therefore, investigating MyD88 is a logical first-step approach linking skeletal muscle TLR/MyD88 signaling to physical inactivity-induced glucose intolerance.
Another recently recognized action of TLR/MyD88 signaling is de novo synthesis of the bioactive sphingolipid ceramide (11, 26). In 2011, Holland et al. (26) reported that TLR4-deficient mice are resistant to a lipid-induced skeletal muscle ceramide accumulation. Moreover, LPS increased myotube mRNA levels of critical enzymes involved in ceramide biosynthesis (e.g., SPT2), whereas knockdown of NF-κB signaling reversed this transcriptional response (26). Ceramides regulate many important cellular functions such as apoptosis and cellular differentiation, proliferation, and growth (5). However, ceramide overabundance in muscle cells can antagonize insulin signaling (11, 27, 42) and is linked with many metabolic abnormalities (2, 3, 12, 25, 48).
The role of TLR/MyD88 signaling in mediating impaired insulin responsiveness and glucose handling during physical inactivity is unknown. Therefore, in light of important work implicating inflammation and ceramides with metabolic disruption, the purpose of this study was to determine whether the TLR/MyD88 pathway is important in regulating inflammation, ceramide biosynthesis signaling, and impaired insulin signaling in muscle following physical inactivity in both rodent and human models.
MATERIALS AND METHODS
Animals.
Female C57BL/6J wild-type (WT) and MyD88−/− mice (C57BL/6J genetic background; Jackson Laboratories) 8–12 wk old were housed in a conventional animal house and maintained on a 12:12-h light-dark cycle and temperature controlled environment (22–23°C). The WT and MyD88−/− mice were divided into two groups: ambulatory controls [(CON): WT, n = 10; MyD88−/−, n = 6] and hindlimb unloading [(HU): WT, n = 10; MyD88−/−, n = 6]. Animal weights (and postintervention) can be found in Table 1. All animals were handled in accordance with the National Institutes of Health Guide to the Care and Use of Laboratory Animals. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Utah (no. 13-11001).
Table 1.
Body and muscle weight and fasting physiological measurements from control (CON) and hindlimb unloaded (HU) WT and MyD88−/− mice
| WT CON (n = 10) | WT HU (n = 10) | MyD88−/− CON (n = 6) | MyD88−/− HU (n = 6) | |
|---|---|---|---|---|
| Body weight, g | 19.95 ± 0.03 | 20.43 ± 0.04 | 19.13 ± 0.08 | 18.98 ± 0.06 |
| Soleus muscle weight, g | 0.0075 ± 0.0006 | 0.0034 ± 0.0005*† | 0.0079 ± 0.0008 | 0.0059 ± 0.0005* |
| Plantaris muscle weight, g | 0.0145 ± 0.0027 | 0.0064 ± 0.0009*† | 0.0111 ± 0.0009 | 0.0089 ± 0.0006* |
| Fasting glucose, mg/dl | 92 ± 6 | 102 ± 6 | 92 ± 3 | 97 ± 6 |
| Fasting insulin, μU/ml | 7.2 ± 0.3 | 18.2 ± 0.7*† | 7.4 ± 0.4 | 8.9 ± 0.5 |
| HOMA-IR | 1.2 ± 0.1 | 2.9 ± 0.2*† | 1.1 ± 0.1 | 1.4 ± 0.1* |
Data are means ± SE.
Different from respective CON (P < 0.05);
different from MyD88 HU (P < 0.05). Note: fasting insulin and HOMA-IR were only n = 5 for WT and n = 4 for MyD88.
Hindlimb unloading protocol.
Animals assigned to CON were able to freely ambulate in their cage (2–3 animals/cage) and have ad libitum access to food (standard chow) and water during the 14-day experimental period. For the HU group, animals underwent hindlimb suspension (2 animals/cage) using a modified unloading method based on the traditional NASA Morey-Holton design for studying disuse-atrophy in rodents (20). Briefly, a sterile surgical steel suture was inserted into the animal's tail while the animal was anesthetized. The steel suture was then shaped into a ring for later suspension onto a steel bar with a swivel. After a 5-day recovery period, the animals were suspended off the ground by their hindlimbs (30°) for 14 days. Animals had access to a 360° perimeter within the cage and were able to reach food and water. All mice were monitored at least once daily for behavior and for food and water intake. Body weights were monitored daily to ensure that mice were not experiencing excessive weight loss due to malnutrition or dehydration, and there was no weight loss evident in the HU animals (Table 1). On day 13, CON and HU mice were fasted for 6 h and received an intraperitoneal glucose injection (1 g/kg body wt ip). Blood glucose levels (Bayer Contour) via a tail vein were determined immediately before (0) and 5, 15, 30, 60, and 120 min following the glucose injection. On day 14, animals were fasted for 6 h and euthanized with CO2, and paired soleus and plantaris muscles were rapidly dissected, frozen in liquid nitrogen, and stored at −80°C for later analysis. Additionally, a sample of blood was collected before euthanizing the animals for measurement of insulin (Insulin ELISA Kit; Crystal Chem, Chicago, IL).
Participants.
Nine healthy, recreationally active older adults (7 F, 2 M; 66 ± 1 yr; BMI 25 ± 1 kg/m2) were recruited from the Salt Lake City area by posted flyers and radio advertisement (Table 2). Inclusion criteria included age 60–75 yr, BMI <30 kg/m2, and normal insulin sensitivity and glucose tolerance as determined from a medical history and blood screening. Subjects were excluded if they had diabetes, untreated hypo/hyperthyroidism, kidney, liver, or respiratory disease, uncontrolled hypertension, vascular disease, cancer, or history of stroke. All subjects read and signed the informed consent. This study was approved by the University of Utah Institutional Review Board (no. 00050933) and conformed to the Declaration of Helsinki and Title 45, US Code of Federal Regulations, Part 46, “Protection of Human Subjects”.
Table 2.
Subject characteristics and physiological parameters before and after bed rest
| Pre Bed Rest | Post Bed Rest | P Value | |
|---|---|---|---|
| Body weight, kg | 71.6 ± 3.9 | 70.0 ± 4.0 | <0.01 |
| Total lean mass, % | 60.3 ± 2.0 | 59.1 ± 2.0 | <0.01 |
| Total fat mass, % | 36.6 ± 2.0 | 37.7 ± 2.0 | <0.01 |
| Fasting glucose, mg/dl | 92.5 ± 1.8 | 93.2 ± 1.5 | 0.64 |
| Fasting insulin, μU/ml | 5.7 ± 0.4 | 6.2 ± 0.6 | 0.54 |
| HOMA-IR | 1.3 ± 0.2 | 1.4 ± 0.3 | 0.50 |
| Glucose AUC (2 h OGTT), mg/dl | 486.0 ± 2.6 | 544.2 ± 2.4 | 0.04 |
| Insulin AUC (2 h OGTT), μU/ml | 99.3 ± 2.3 | 157.5 ± 3.6 | 0.06 |
| Matsuda Index | 9.4 ± 0.6 | 7.7 ± 0.6 | 0.02 |
| NEFA, mmol/l | 0.42 ± 0.04 | 0.40 ± 0.09 | 0.84 |
| NEFA, %change | — | 1.10 ± 0.17 | 0.71 |
| Endotoxin, EU/ml | 0.58 ± 0.37 | 0.72 ± 0.14 | 0.46 |
| Endotoxin, %change | — | 91.2 ± 45.5 | 0.09 |
| High-density lipoprotein (HDL), mg/dl | 55.8 ± 3.1 | 49.8 ± 2.5 | 0.02 |
| Low-density lipoprotein (LDL), mg/dl | 92.7 ± 7.9 | 97.9 ± 10.1 | 0.38 |
| Very low-density lipoprotein, mg/dl | 18.7 ± 1.4 | 21.8 ± 4.7 | 0.43 |
| LDL/HDL ratio | 1.7 ± 0.2 | 2.0 ± 0.2 | 0.04 |
| Total cholesterol, mg/dl | 167.1 ± 8.8 | 169.2 ± 10.3 | 0.71 |
| Triglycerides, mg/dl | 93.4 ± 7.1 | 109.8 ± 23.7 | 0.40 |
Data are means ± SE from n = 9 older adults.
Bed rest protocol.
Prior to experimental bed rest and after an overnight fast, subjects arrived at the University of Utah Center for Clinical and Translational Sciences (CCTS) Clinical Research Unit and underwent a dual-energy X-ray absorptiometry (DEXA) scan for assessment of body composition and an oral glucose tolerance test (OGTT). Approximately 1–2 wk later, subjects participated in a 5-day/4-night bed rest protocol at the CCTS. Subjects remained in their hospital bed during the entire duration of the 5-day study except, when needed, subjects were assisted by nursing staff to access the toilet or sink (using a wheelchair). Three meals a day were prepared by a research dietician and were composed of a macronutrient composition of 15% protein, 55% carbohydrate, and 30% fat. Daily caloric intake was determined by body weight and corrected for physically inactive individuals. Standard of care for bedridden patients was followed during this 5-day protocol as we have done previously (17). This included 24-h nursing supervision, daily safety blood draws, intermittent lower leg compression devices, and daily visits by a physical therapist to administer passive range of motion in the arms and legs. On day 4, the DEXA scan and OGTT test were repeated under the same conditions as before. On day 1 and day 5 of bed rest and after an overnight fast, a muscle sample was obtained from the vastus lateralis by percutaneous needle biopsy, as we have done previously (15). The tissue sample was cleaned with saline, and visible fat and connective tissue were quickly removed. Tissue was frozen in liquid nitrogen and stored at −80°C for later analysis.
Human serum analysis.
Serum collected during the OGTT and was assessed for glucose using a standard glucose analyzer (YSI, Yellow Springs, OH). Insulin (EMD Millipore, Billerica, MA), lipopolysacharride (LPS, LAL assay; Lonza, Walkersville, MD) and nonesterified fatty acids [NEFA-HR (2); Wako Chemicals, Richmond, VA] were determined according to manufacturers' instructions. Insulin and glucose values during fasting were used to calculate HOMA-IR during fasting, and postprandial glucose and insulin values were used to calculate the Matsuda Index, a measure of peripheral (i.e., muscle) insulin sensitivity calculated from fasting glucose and insulin responses and following the OGTT. Serum or plasma was delivered to ARUP (Associated Regional and University Pathologists) Laboratories for specific lab analysis for HDL, LDL, total cholesterol, and triglycerides.
Immunoblotting.
The relative abundance of target proteins was determined in muscle samples via immunoblotting, as we have done previously (17). Briefly, tissue samples were homogenized 1:10 (wt/vol) using a glass tube and mechanically-driven pestle grinder in an ice-cold buffer containing 50 mM Tris (pH 7.5), 250 mM mannitol, 40 mM NaF, 5 mM pyrophosphate, 1 mM EDTA, 1 mM EGTA, and 1% Triton X-100 with a protease inhibitor cocktail. Homogenates were centrifuged at 1,500 g for 10 min at 4°C. After centrifugation, the supernatant was collected and the protein concentration determined using the Bradford technique. Proteins from the supernatant fraction were separated via polyacrylamide gel electrophoresis, transferred onto a polyvinylidene difluoride membrane, and incubated with primary and secondary antibodies directed against the proteins of interest. Membranes were exposed on a ChemiDoc XRS (Bio-Rad) and quantified with Image Lab software (Bio-Rad). The specific antibodies used to detect target proteins were TLR4, phospho-nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IκBα Ser32/36), total IkBα, phospho-c Jun NH2-terminal kinase (JNK; Thr183/Tyr185), total JNK, interleukin-6 (IL-6), serine palmitoyltransferase-2 (SPT2), phospho-Akt (Ser473/Thr308), total Akt, protein tyrosine phosphatase receptor type C (CD45), and monocyte chemotactic protein-1 (MCP-1). All antibodies were purchased from Cell Signaling Technologies (Danvers, MA) except TLR4, SPT2, CD45, and secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). GAPDH (Cell Signaling Technologies) was used to verify equal protein loading in each lane. We also included an internal control on each gel to normalize each protein and to correct for gel-to-gel variability.
Gene expression.
RNA was isolated from skeletal muscle samples as reported previously (18). Total RNA was DNase treated, and cDNA was synthesized using commercially available kits (DNA-free; Ambion, Austin, TX) (iScript; Bio-Rad, Hercules, CA). Real-time qPCR was carried out with a CFX Connect real-time PCR cycler (Bio-Rad) combined with SYBR Green fluorescence. Cycle threshold values were normalized to β2-microglobulin (β2M), and then fold change values (vs. Pre-Bed Rest) were calculated using the 2−ΔΔCT method. Primers were custom designed (Beacon Designer) and carefully optimized for myeloid differentiation primary response 88 (MYD88) (NM_001172567) forward AGCCTTATTTCCTAATGGG, reverse GACTTGTCACTGCTGAAG and CD45 (NM_002838) forward AGCAATATCAATTCCTATATGAC, reverse GCACCAAGTGGATTAACA. Primer sequences for IL-6 (NM_000600) and MCP-1 (NM_002982) were generated from PrimerBank (50), while primer sequences for β2M have been published previously (16). All primer sequences were purchased from Life Sciences.
Statistics.
Data are expressed as means ± SE. Statistical analyses were conducted using the Prism program. Statistical analyses for animal experiments were conducted using an analysis of variance (ANOVA). The Student-Newman-Keuls test was used for post hoc analysis. A paired t-test was used to analyze subject characteristics and pre vs. post blood and muscle data from the human experiments. A repeated-measures ANOVA was used to assess differences in serum glucose and insulin following the OGTT. Pearson correlations were used on selected muscle markers and physiological parameters. Area under the curve was calculated using the trapezoid rule. Statistical significance was set a priori at P < 0.05.
RESULTS
Mouse characteristics.
There were no differences in body weight between WT and MyD88−/− CON groups or in response to 14 days of HU (Table 1). In contrast, soleus and plantaris weight in the WT HU group decreased ∼50% compared with WT CON group (P < 0.05) but only ∼25% in the MyD88−/− HU group compared with MyD88−/− CON groups (P < 0.05). Thus, the decrease in muscle weight was significantly greater in WT HU than in the MyD88−/− HU group (P < 0.05). Finally, we found that fasting insulin and HOMA-IR significantly increased in WT HU mice (vs. CON, P < 0.05). Although HOMA-IR was subtly higher in MyD88−/− HU (P < 0.05), WT HU fasting insulin and HOMA-IR responses were significantly greater than those in MyD88−/− HU. Fasting glucose responses were not different between groups.
After 14 days of HU (Fig. 1), blood glucose levels in the WT HU group were significantly elevated above all groups at 15, 30, and 60 min during a GTT (P < 0.05). As a result, glucose AUC for WT HU mice was significantly greater than in all groups (P < 0.05). In contrast, GTT response in MyD88−/− HU was similar to MyD88−/− CON and WT CON. Together, physical inactivity disrupted postprandial glucose uptake and fasting insulin sensitivity in WT but not in MyD88−/− mice.
Fig. 1.
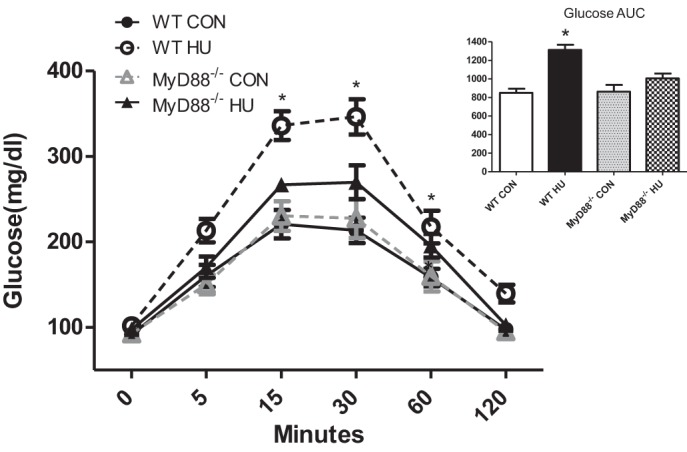
Postprandial glucose tolerance in response to physical inactivity in wild-type (WT) and myeloid differentiation primary response 88 (MyD88)−/− mice. Data (means ± SE) represent blood glucose before (0 min) and during a 2-h glucose tolerance test (GTT) from WT ambulatory control mice (WT CON; ●, solid line, n = 10), WT hindlimb unloaded mice (WT HU; ○, dashed line, n = 10), MyD88 knockout ambulatory control mice (MyD88−/− CON; △, dashed line, n = 6), and MyD88 knockout hindlimb unloaded mice (MyD88−/− HU; ▲, solid line, n = 6). Right: glucose AUC calculated from each of the mouse groups (WT CON, open bar; WT HU, filled bar; MyD88−/− CON gray bar; MyD88−/− HU, checkered bar). All groups had increased glucose levels during the GTT that returned to baseline by 120 min. *Difference at 15, 30, and 60 min and AUC between WT HU vs. all groups (P < 0.05).
Mouse skeletal muscle immunoblotting.
Expression of several proteins related to inflammatory and ceramide biosynthesis pathways was significantly increased after 14 days of HU in WT soleus muscle compared with WT CON), including TLR4 (Fig. 2A; ∼70%), IL-6 (∼500%; Fig. 2D), and SPT2 (∼90%; Fig. 2E) (all P < 0.05). Immunoblotting also revealed activation of NF-κB and JNK signaling pathways after HU, including phosphorylation of IκBα at Ser32/36 (Fig. 2B; ∼60%), and JNK at Thr183/Tyr185 (Fig. 2C; ∼90%). Also, phosphorylation of Akt at Ser473 (Fig. 2F) decreased by ∼50% after HU in WT mice (P < 0.05). Importantly, soleus protein expression and phosphorylation of TLR4, IκBα (Ser32/36), JNK (Thr183/Tyr185), IL-6, SPT2, and Akt (Ser473) were unchanged in MyD88−/− mice (vs. all groups) after 14 days of HU. There were no changes in total protein levels for any of the phosphospecific proteins (P > 0.05; data not shown). Results in plantaris were similar to soleus data except that there were no changes in JNK phosphorylation after HU for both WT and MyD88−/− mice (Fig. 3). We also evaluated protein expression for CD45, a cell surface marker of immune cells, and a chemokine (MCP-1) in soleus skeletal muscle homogenate samples. We found that CD45 protein expression increased in WT HU mice (P < 0.05) vs. all groups (WT Con: 1.55 ± 0.45; WT HU: 3.02 ± 0.46; MyD88−/− Con: 2.25 ± 0.30; MyD88−/− HU: 1.41 ± 0.26 AU). There were no changes in MCP-1 protein expression between all animal groups (WT Con: 1.61 ± 0.13; WT HU: 1.70 ± 0.11; MyD88−/− Con: 1.49 ± 0.15; MyD88−/− HU: 1.18 ± 0.18 AU).
Fig. 2.

Soleus muscle Toll-like receptor (TLR)/MyD88 signaling in response to physical inactivity in WT and MyD88−/− mice. Data (means ± SE) represent immunoblot protein expression in soleus muscle for WT CON (open bar, n = 10), WT HU (filled bar, n = 10), MyD88−/− CON (gray bar, n = 6), MyD88−/− HU (checkered bar, n = 6). A: TLR4. B: nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IκBα Ser32/36). C: c-Jun NH2-terminal kinase (JNK Thr183/Tyr185). D: IL-6. E: serine palmitoyltransferase-2 (SPT2). F: Akt Ser473. Protein expression data were normalized to internal control and are presented in arbitrary units (AU). *Difference between WT HU and all experimental groups (P < 0.05). Representative immunoblot images are presented in replicate. GAPDH was used to verify equal sample loading. Any adjustments to digital images do not alter the information contained therein.
Fig. 3.

Plantaris muscle TLR/MyD88 signaling in response to physical inactivity in WT and MyD88−/− mice. Data (means ± SE) represent immunoblot protein expression in plantaris muscle for WT CON (open bar, n = 10), WT HU (filled bar, n = 10), MyD88−/− CON (gray bar, n = 6), MyD88−/− HU (checkered bar, n = 6). A: TLR4. B: IκBα Ser32/36. C: JNK Thr183/Tyr185. D: IL-6. E: SPT2. F: Akt Ser473. Protein expression data were normalized to internal control and are presented in AU. *Difference between WT HU and all experimental groups (P < 0.05). #Difference between WT HU and WT CON (P < 0.05). Representative immunoblot images are presented in replicate. GAPDH images were used to verify equal sample loading. Any adjustments to digital images do not alter the information contained therein.
Human tissue composition and blood data.
Five days of bed rest in the older adult human research subjects resulted in a decrease in percent lean body mass (∼0.8%), an increase in percent fat mass (∼1.1%), and an overall decrease in body weight (∼1.6kg) (P < 0.05) (Table 2). There were no differences in fasting levels of serum glucose or insulin or the calculated HOMA-IR after bed rest. In contrast, the Matsuda Index was lower after bed rest (P < 0.05). Serum levels of NEFA, reported as either absolute or percent change from pre-bed rest, did not change as a result of 5 days of bed rest. Similarly, absolute serum endotoxin levels did not change as a result of bed rest; however, there was a tendency for the percentage of endotoxin levels to increase (P = 0.09). Finally, HDL-cholesterol levels and the HDL/LDL ratio decreased after bed rest (P < 0.05), but bed rest did not alter LDL-cholesterol, total cholesterol, or triglycerides.
After bed rest, glucose levels were higher at 60 and 90 min during the OGTT compared with before bed rest (Fig. 4A; P < 0.05). As a result, glucose AUC was significantly increased after bed rest (P < 0.05). Serum insulin levels after glucose challenge were elevated after bed rest at all time points (Fig. 4B; P < 0.05), and total insulin AUC tended to be higher after bed rest (P = 0.06).
Fig. 4.

Postprandial glucose and insulin responses before and after bed rest. Serum glucose (mg/dl; A) and insulin (μU/ml; B) levels in response to an oral GTT before (○, solid line) and after (●, dotted line) bed rest in older adults (means ± SE; n = 9). Blood was sampled before (0) and 30, 60, 90, and 120 min after ingestion of 75 g of Glucola. Right: AUC calculated for serum glucose and insulin before and after bed rest. *Different from 0 min (P < 0.05). #Different from Pre (P < 0.05). $P = 0.06 vs. Pre.
Human skeletal muscle immunoblotting and gene expression.
Following bed rest in older adults, protein expression for TLR4 (Fig. 5A) tended to increase by ∼35% (P = 0.08), SPT2 protein (Fig. 5B) increased by ∼40% (P < 0.05), MYD88 mRNA expression (Fig. 5C) decreased ∼25%, IL-6 mRNA expression (Fig. 5D) increased by ∼200%, and phosphorylation of Akt at Ser473 (Fig. 5E) decreased by ∼20% (P < 0.05). There were no differences in the phosphorylation of Akt at Thr308 (Fig. 5F; P = 0.14). Neither CD45 (1.35 ± 0.18 fold change from Pre; P = 0.19) or MCP-1 (1.36 ± 0.23 fold change from Pre; P = 0.27) mRNA expression was altered after 5 days of bed rest (data not shown). Total protein levels for Akt did not change as a result of 5 days of bed rest (P = 0.40).
Fig. 5.

Human skeletal muscle TLR4/MyD88 signaling responses before and after bed rest. Vastus lateralis skeletal muscle was sampled from older adult participants (means ± SE; n = 9) before and after 5 days of bed rest. Data represent skeletal muscle: TLR4 protein expression (A), SPT2 protein expression (B), MYD88 mRNA expression (C), IL-6 mRNA expression (D), phospho-Akt Ser473 (E), and phospho-Akt Thr308 (F) expression. Data are expressed as fold change from Pre Bed Rest (set at 1). Images above panels are representative immunoblots for the respective proteins (in duplicate). *Different from Pre Bed Rest (P < 0.05). GAPDH images were used to verify equal sample loading. Any adjustments to digital images do not alter the information contained therein.
Correlations.
Finally, we determined the relationship of two of our notable skeletal muscle findings with glucose uptake impairment in older adults after bed rest. We found that, after 5 days of bed rest, TLR4 protein expression was inversely correlated with the Matsuda Index (Fig. 6A) and positively correlated with insulin AUC (Fig. 6C) (P < 0.05) but not glucose AUC (Fig. 6B). In contrast, SPT2 protein expression was not related to either of these physiological parameters (Fig. 6, D–F). Additionally, HOMA-IR was not correlated with TLR4 (R = 0.23) or SPT2 (R = 0.40) protein content (not shown).
Fig. 6.

Relationship between human skeletal muscle TLR4 and SPT2 protein expression and changes in the Matsuda Index and glucose and insulin levels in response to a 2-h OGTT. Data are Pearson correlations (R value) for the following dependent variables from healthy older adults (n = 9) and are plotted as fold change from Pre Bed Rest. A: TLR4 vs. Matsuda Index. B: TLR4 vs. glucose AUC. C: TLR4 vs. insulin AUC. D: SPT2 vs. Matsuda Index. E: SPT2 vs. glucose AUC. F: SPT2 vs. insulin AUC. R and P values are inset within panels.
DISCUSSION
In this study, we identified that the MyD88 pathway is a major regulator of downstream skeletal muscle cellular signaling and peripheral metabolic disruption (e.g., impaired glucose uptake, insulin resistance) caused by physical inactivity. Specifically, we noted that MyD88 signaling in mice in response to 14 days of HU was responsible for increased inflammation (i.e., JNK, IL-6), ceramide biosynthesis signaling (i.e., SPT2), and TLR4 protein expression within skeletal muscle homogenates. In an attempt to ascertain whether these findings in mice could be translated to humans, we evaluated similar end points in older adult skeletal muscle samples before and after 5 days of bed rest. Interestingly, skeletal muscle SPT2 protein and IL-6 mRNA expression increased in older adults after bed rest, while increased TLR4 was strongly correlated with worsened postprandial insulin responses and the Matsuda Index. Together, these novel data support the hypothesis that MyD88 signaling is an important mechanism for altered glucose handling, inflammation, and ceramide biosynthesis signaling following short-term physical inactivity.
This is the first study to identify altered MyD88 signaling as a mediator of disrupted glucose uptake initiated by physical inactivity. Namely, a whole body knockout of MyD88 completely nullified the peripheral metabolic disruption (glucose intolerance, insulin resistance) caused by 14 days of hindlimb unloading in mice. Decreased muscle contractile activity is one of two major metabolic factors (along with nutritional lipid overload) that play undisputed roles in metabolic disruption (1). It has been known for some time that short-term controlled bed rest (6) or reduced physical activity (40) significantly disrupts whole body insulin sensitivity and glucose uptake in young adults. Few studies have examined the short-term consequences of physical inactivity on insulin sensitivity and glucose tolerance in older adults (8, 13). In this study, we show that older adults developed glucose intolerance (and other indexes of peripheral metabolic disruption; decreased HDL-C) in less than 5 days of bed rest. This is in agreement with Coker et al. (13), who observed insulin-mediated suppression of glucose uptake after 10 days of bed rest in older adults. Our data are important and clinically relevant because 1) they clearly demonstrate the metabolic potency of very short-term bouts of inactivity in older adults, 2) a majority of older adults hospitalized for acute illness typically spend ∼5 days with low amounts of ambulatory activity during hospitalization (22), and 3) according to our animal data, which is strongly supported by our bed rest model, TLR4/MyD88 signaling may partly mediate glucose intolerance with physical inactivity in older adults, and therefore this pathway could represent a future target of pharmacological intervention.
Another major finding in this study was that MyD88 signaling was responsible for the physical inactivity-induced upregulation of inflammation and ceramide biosynthesis signaling in whole skeletal muscle tissue homogenates. Proinflammatory cytokines and lipid intermediates, such as ceramides, are well known to interfere with insulin signaling (9, 28, 42) and therefore may have been responsible for reduced Akt phosphorylation (∼50% in mouse and ∼20% in human) observed in our physical inactivity experiments. Previous studies of physical inactivity in rodents (34) and humans (7, 33) have observed decreased skeletal muscle insulin/Akt signaling and GLUT4 content, but surprisingly, upstream mechanisms have not been addressed. In this study, we uniquely show that MyD88 signaling increased the expression of SPT2, a rate-limiting enzyme that catalyzes the conversion of palmitoyl CoA and serine into ceramide. Because SPT2 plays a pivotal role in de novo ceramide accumulation, this enzyme has served as a reputable target (via myriocin) to prevent increases in ceramide and restore insulin sensitivity in various tissues (26, 51). Even though our human bed rest model was substantially shorter in duration compared with the animal experiment (for ethical considerations), 5 days of bed rest was sufficient to increase skeletal muscle SPT2 in older adults, suggesting that ceramide levels may be heightened with bed rest. The shorter duration of bed rest may have also contributed to the unexpected decrease in MYD88 mRNA expression. It is difficult to interpret the MYD88 mRNA data observed after bed rest, since activation of TLR/MyD88 signaling is primarily regulated by protein interactions/dimerization steps rather than changes in MYD88 abundance levels. It is possible that a decrease in MYD88 mRNA may be a compensatory mechanism to slow overactivation of TLR/MyD88 signaling and metabolic perturbations during the early stages of inactivity (5 days bed rest). Clearly, a more thorough investigation of the various downstream signaling molecules combined with more appropriate methods of evaluation (coimmunoprecipitation) will be necessary to conduct in future human inactivity studies. Together, our data imply that physical inactivity increased a critical enzyme (i.e., SPT2) associated with de novo ceramide synthesis and is mediated by MyD88 signaling.
Since there are multiple TLRs that lie upstream of MyD88, we cannot conclude from these experiments which specific receptor is responsible for downstream MyD88 signaling. However, given our observations of increased TLR4 expression in muscle after HU in mice, we suspect TLR4 is at least partially responsible, although other TLRs may be cocontributors. In fact, a previous study in humans showed a transcriptional increase in not only skeletal muscle TLR4 but also TLR5 and -6 in response to a 2-day lipid infusion (29). Nonetheless, the importance of TLR4 in metabolic disruption is well documented, especially under conditions of endotoxin or palmitate stimulation or following a high-fat diet (26, 29, 36, 43). In fact, in a cross-sectional study, Reyna et al. noted an increase in skeletal muscle TLR4 protein content in obese and diabetic individuals (vs. lean), a response that was correlated with the HOMA-IR, a measure of hepatic fasting insulin sensitivity (44). In older adults, we noted a tendency (P = 0.08) for skeletal muscle TLR4 protein abundance to increase after 5 days of bed rest (∼35%). This was similar, although not as robust, to the increased TLR4 expression we reported previously after a 7-day bed rest study in older adults (18). Even though the increase in TLR4 after 5 days of bed rest was not correlated with HOMA-IR, our data were strongly correlated with the Matsuda Index, a measure of peripheral (i.e., muscle) insulin sensitivity calculated from the OGTT, emphasizing the importance of skeletal muscle and the TLR4/MyD88 pathway in regulating postprandial glucose uptake following short-term physical inactivity. Interestingly, SPT2 was not correlated to these physiological parameters in older adults after bed rest. Since the turnover of ceramide is regulated by multiple de novo synthesis enzymes (SPT, CERS, DES) and breakdown enzymes (10), it would be premature to discredit SPT2 as an important precursor to glucose intolerance in older adults with bed rest. Instead, future studies are needed to determine if changes in SPT2 activity, or a composite of several ceramide-related enzymes, in mice and humans translate into muscle ceramide accumulation with physical inactivity.
An unexpected finding was that the physical inactivity-induced increase in TLR4 protein content was completely blocked in MyD88−/− mice after 14 days of HU even though MyD88 is downstream of TLR4. Previous work has shown that JNK is capable of regulating the transcription of TLR1 in macrophages (30), suggesting that MyD88 signaling may induce a positive feedback loop to further enhance sensitivity to certain TLR ligands. Therefore, knockout of MyD88 (and therefore reduced JNK phosphorylation) may help explain the reduction in muscle TLR4 protein content in MyD88−/− mice with unloading, but this remains to be determined.
Finally, our animal and human physical inactivity experimental models induced robust atrophy similar to previous reports (13, 18, 20). However, an interesting observation was that MyD88−/− mice were partially protected from muscle loss during 14 days of hindlimb unloading. It is not entirely surprising that MyD88−/− mice were only partially resistant to disuse atrophy, since TLR4 signaling can occur through a MyD88-independent pathway (e.g., TRIF) (52). Currently, few mechanistic data exist linking regulation of muscle cell size with MyD88 signaling. Previous studies have demonstrated that endotoxin/LPS, a specific TLR4 ligand, caused a reduction in skeletal muscle protein synthesis in rodents possibly by increasing proinflammatory cytokine levels (35). We speculate that blockade of NF-κB signaling (via IκBα) in MyD88−/− mice and attenuation of an increase in proinflammatory cytokines (IL-6) perhaps resulted in inhibition of protein synthesis (14, 24). In support, we previously noted increased IL-6 expression in older adults after 7 days of bed rest (18) and a reduced amino acid-stimulated protein synthesis response (15). Another possible explanation for partial atrophy protection in MyD88 KO mice may be through TNF receptor adaptor protein 6 (TRAF6). TRAF6 serves as an adaptor molecule and signal transducer downstream of MyD88. TRAF6 may mediate muscle atrophy by augmenting muscle proteolysis via autophagy and the ubiquitin proteasome system. In support, TRAF6 knockout mice have considerably less atrophy and muscle expression of MuRF1, MAFbx, and autophagy-related proteins (vs. littermate TRAF6 floxed mice) in response to 14 days of denervation (41). Identifying the precise mechanisms that mediate physical inactivity-induced muscle atrophy through the TLR/MyD88 signaling pathway is warranted.
In conclusion, we show for the first time that MyD88 is an important mechanistic link between physical inactivity and glucose intolerance, and this may be a result of a skeletal muscle environment characterized by MyD88-dependent activation of inflammation and ceramide biosynthesis signaling (Fig. 7). Our findings in older adult skeletal muscle after bed rest support these findings. Future studies are warranted to examine various ceramide species and lipid intermediates following physical inactivity in mouse and human skeletal muscle as well as the contribution of immune cells to metabolic dysruption. For example, we observed an increase in immune cell infiltration following 14 days of hindlimb unloading in WT mice (but not in MyD88 mice), suggesting that perhaps altered MyD88 signaling of immune cells within skeletal muscle (in lieu of or in addition to altered MyD88 signaling in skeletal muscle cells) may contribute to glucose intolerance and altered skeletal muscle signaling. Evidence supports that infiltration of macrophages in skeletal muscle following a high-fat diet may contribute to insulin resistance (21). Taken together, these data will be important in the development of future pharmacological approaches aimed at attenuating TLR/MyD88 signaling and maintenance of postprandial glucose tolerance in physically inactive older adults.
Fig. 7.
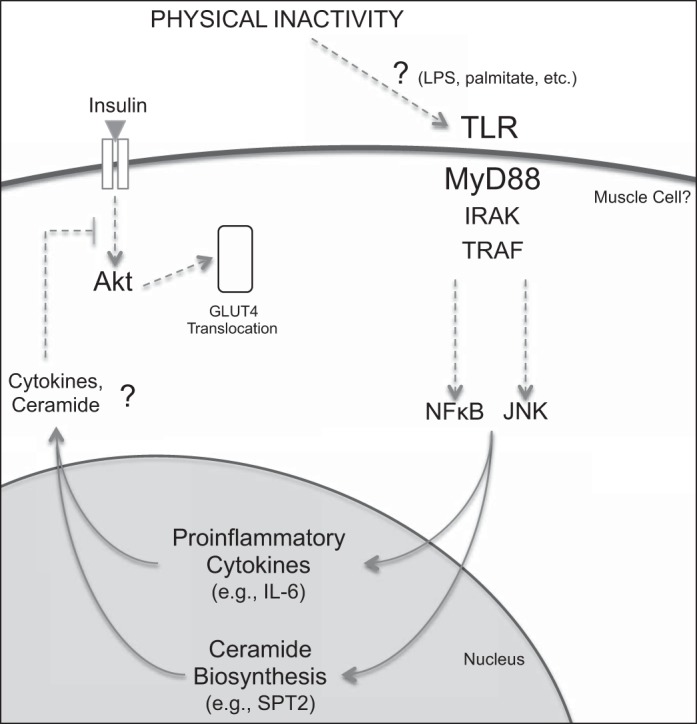
Proposed mechanistic role of MyD88 signaling-mediated metabolic disruption caused by physical inactivity. Global knockdown of MyD88 protected skeletal muscle from physical inactivity-induced increase in NF-κB and JNK signaling, increased IL-6 and SPT2 protein expression, decreased Akt phosphorylation, and glucose intolerance. Remaining to be determined are the upstream ligand(s) and the specific TLR(s) that bridge physical inactivity and activation of MyD88 signaling and whether an increase in SPT2 translates into ceramide species accumulation. Moreover, further research is warranted to investigate the cell source of disrupted MyD88 signaling (e.g., skeletal muscle, immune cells). Dotted line represents indirect regulation and question mark represents unknown mechanism.
GRANTS
This work was supported by grants from the National Institute on Aging (K01-AG-038556), the National Center for Advancing Translational Sciences (UL1-TR000105), and University of Utah's Diabetes and Metabolism Center.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: O.S.K., R.E.T., K.M.B., M.R., B.T.B., and M.J.D. performed experiments; O.S.K. and K.M.B. analyzed data; O.S.K., J.D.S., T.J., B.T.B., D.A.M., R.M.O., and M.J.D. interpreted results of experiments; O.S.K. prepared figures; O.S.K. and M.J.D. drafted manuscript; O.S.K., K.M.B., J.D.S., T.J., B.T.B., D.A.M., R.M.O., and M.J.D. edited and revised manuscript; O.S.K., R.E.T., K.M.B., M.R., J.D.S., T.J., B.T.B., D.A.M., R.M.O., and M.J.D. approved final version of manuscript; R.M.O. and M.J.D. conception and design of research.
ACKNOWLEDGMENTS
We thank the CCTS nursing, dietary, and medical staff for their assistance with the muscle biopsies, blood sampling, and patient care during the inpatient and outpatient visits. We also thank the animal facility staff of the University of Utah for the animal support during these experiments. Finally, we are grateful for the time and effort put forth by each participant in this study.
REFERENCES
- 1.Anonymous. Global Strategy on Diet, Physical Activity and Health. World Health Organization, 2004. [Google Scholar]
- 2.Adams JM 2nd, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, Mandarino LJ. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 53: 25–31, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Amati F, Dube JJ, Alvarez-Carnero E, Edreira MM, Chomentowski P, Coen PM, Switzer GE, Bickel PE, Stefanovic-Racic M, Toledo FG, Goodpaster BH. Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: another paradox in endurance-trained athletes? Diabetes 60: 2588–2597, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bankoski A, Harris TB, McClain JJ, Brychta RJ, Caserotti P, Chen KY, Berrigan D, Troiano RP, Koster A. Sedentary activity associated with metabolic syndrome independent of physical activity. Diabetes Care 34: 497–503, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartke N, Hannun YA. Bioactive sphingolipids: metabolism and function. J Lipid Res 50 Suppl: S91–S96, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergouignan A, Rudwill F, Simon C, Blanc S. Physical inactivity as the culprit of metabolic inflexibility: evidence from bed-rest studies. J Appl Physiol (1985) 111: 1201–1210, 2011. [DOI] [PubMed] [Google Scholar]
- 7.Bienso RS, Ringholm S, Kiilerich K, Aachmann-Andersen NJ, Krogh-Madsen R, Guerra B, Plomgaard P, van Hall G, Treebak JT, Saltin B, Lundby C, Calbet JA, Pilegaard H, Wojtaszewski JF. GLUT4 and glycogen synthase are key players in bed rest-induced insulin resistance. Diabetes 61: 1090–1099, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breen L, Stokes KA, Churchward-Venne TA, Moore DR, Baker SK, Smith K, Atherton PJ, Phillips SM. Two weeks of reduced activity decreases leg lean mass and induces “anabolic resistance” of myofibrillar protein synthesis in healthy elderly. J Clin Endocrinol Metab 98: 2604–2612, 2013. [DOI] [PubMed] [Google Scholar]
- 9.Chavez JA, Holland WL, Bar J, Sandhoff K, Summers SA. Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J Biol Chem 280: 20148–20153, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab 15: 585–594, 2012. [DOI] [PubMed] [Google Scholar]
- 11.Chavez JA, Summers SA. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch Biochem Biophys 419: 101–109, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Coen PM, Hames KC, Leachman EM, DeLany JP, Ritov VB, Menshikova EV, Dube JJ, Stefanovic-Racic M, Toledo FG, Goodpaster BH. Reduced skeletal muscle oxidative capacity and elevated ceramide but not diacylglycerol content in severe obesity. Obesity (Silver Spring) 21: 2362–2371, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coker RH, Hays NP, Williams RH, Xu L, Wolfe RR, Evans WJ. Bed rest worsens impairments in fat and glucose metabolism in older, overweight adults. J Gerontol Series A Biol Sci Med Sci 69: 363–370, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooney R, Kimball SR, Eckman R, Maish G, Shumate M 3rd, Vary TC. TNF-binding protein ameliorates inhibition of skeletal muscle protein synthesis during sepsis. Am J Physiol Endocrinol Metab 276: E611–E619, 1999. [DOI] [PubMed] [Google Scholar]
- 15.Drummond MJ, Dickinson JM, Fry CS, Walker DK, Gundermann DM, Reidy PT, Timmerman KL, Markofski MM, Paddon-Jones D, Rasmussen BB, Volpi E. Bed rest impairs skeletal muscle amino acid transporter expression, mTORC1 signaling, and protein synthesis in response to essential amino acids in older adults. Am J Physiol Endocrinol Metab 302: E1113–E1122, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drummond MJ, Miyazaki M, Dreyer HC, Pennings B, Dhanani S, Volpi E, Esser KA, Rasmussen BB. Expression of growth-related genes in young and older human skeletal muscle following an acute stimulation of protein synthesis. J Appl Physiol 106: 1403–1411, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drummond MJ, Timmerman KL, Markofski MM, Walker DK, Dickinson JM, Jamaluddin M, Brasier AR, Rasmussen BB, Volpi E. Short-term bed rest increases TLR4 and IL-6 expression in skeletal muscle of older adults. Am J Physiol Regul Integr Comp Physiol 305: R216–R223, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drummond MJ, Timmerman KL, Markofski MM, Walker DK, Dickinson JM, Jamaluddin M, Brasier AR, Rasmussen BB, Volpi E. Short-term bed rest increases TLR4 and IL-6 expression in skeletal muscle of older adults. Am J Physiol Regul Integr Comp Physiol 305: R216–R223, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrannini E, Simonson DC, Katz LD, Reichard G Jr, Bevilacqua S, Barrett EJ, Olsson M, DeFronzo RA. The disposal of an oral glucose load in patients with non-insulin-dependent diabetes. Metab Clin Exper 37: 79–85, 1988. [DOI] [PubMed] [Google Scholar]
- 20.Ferreira JA, Crissey JM, Brown M. An alternant method to the traditional NASA hindlimb unloading model in mice. J Vis Exp 2011 Mar 10;(49). pii: 2467. doi: 10.3791/2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fink LN, Costford SR, Lee YS, Jensen TE, Bilan PJ, Oberbach A, Bluher M, Olefsky JM, Sams A, Klip A. Pro-inflammatory macrophages increase in skeletal muscle of high fat-Fed mice and correlate with metabolic risk markers in humans. Obesity (Silver Spring) 22: 747–757, 2014. [DOI] [PubMed] [Google Scholar]
- 22.Fisher SR, Kuo YF, Graham JE, Ottenbacher KJ, Ostir GV. Early ambulation and length of stay in older adults hospitalized for acute illness. Arch Intern Med 170: 1942–1943, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher SR, Kuo YF, Sharma G, Raji MA, Kumar A, Goodwin JS, Ostir GV, Ottenbacher KJ. Mobility after hospital discharge as a marker for 30-day readmission. J Gerontol Series A Biol Sci Med Sci 68: 805–810, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haddad F, Zaldivar F, Cooper DM, Adams GR. IL-6-induced skeletal muscle atrophy. J Appl Physiol 98: 911–917, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Haus JM, Kashyap SR, Kasumov T, Zhang R, Kelly KR, Defronzo RA, Kirwan JP. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 58: 337–343, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, Pagliassotti MJ, Scherer PE, Summers SA. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest 121: 1858–1870, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5: 167–179, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 271: 665–668, 1996. [DOI] [PubMed] [Google Scholar]
- 29.Hussey SE, Lum H, Alvarez A, Cipriani Y, Garduno-Garcia J, Anaya L, Dube J, Musi N. A sustained increase in plasma NEFA upregulates the Toll-like receptor network in human muscle. Diabetologia 57: 582–591, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Izadi H, Motameni AT, Bates TC, Olivera ER, Villar-Suarez V, Joshi I, Garg R, Osborne BA, Davis RJ, Rincon M, Anguita J. c-Jun N-terminal kinase 1 is required for Toll-like receptor 1 gene expression in macrophages. Infect Immun 75: 5027–5034, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11: 115–122, 1999. [DOI] [PubMed] [Google Scholar]
- 32.Krentz AJ, Viljoen A, Sinclair A. Insulin resistance: a risk marker for disease and disability in the older person. Diabet Med 30: 535–548, 2013. [DOI] [PubMed] [Google Scholar]
- 33.Krogh-Madsen R, Thyfault JP, Broholm C, Mortensen OH, Olsen RH, Mounier R, Plomgaard P, van Hall G, Booth FW, Pedersen BK. A 2-wk reduction of ambulatory activity attenuates peripheral insulin sensitivity. J Appl Physiol (1985) 108: 1034–1040, 2010. [DOI] [PubMed] [Google Scholar]
- 34.Kump DS, Booth FW. Alterations in insulin receptor signalling in the rat epitrochlearis muscle upon cessation of voluntary exercise. J Physiol 562: 829–838, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lang CH, Frost RA, Jefferson LS, Kimball SR, Vary TC. Endotoxin-induced decrease in muscle protein synthesis is associated with changes in eIF2B, eIF4E, and IGF-I. Am J Physiol Endocrinol Metab 278: E1133–E1143, 2000. [DOI] [PubMed] [Google Scholar]
- 36.Liang H, Hussey SE, Sanchez-Avila A, Tantiwong P, Musi N. Effect of lipopolysaccharide on inflammation and insulin action in human muscle. PLos One 8: e63983, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol 10: 2327–2334, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masharani UB, Maddux BA, Li X, Sakkas GK, Mulligan K, Schambelan M, Goldfine ID, Youngren JF. Insulin resistance in non-obese subjects is associated with activation of the JNK pathway and impaired insulin signaling in skeletal muscle. PLos One 6: e19878, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 278: 1612–1615, 1997. [DOI] [PubMed] [Google Scholar]
- 40.Olsen RH, Krogh-Madsen R, Thomsen C, Booth FW, Pedersen BK. Metabolic responses to reduced daily steps in healthy nonexercising men. JAMA 299: 1261–1263, 2008. [DOI] [PubMed] [Google Scholar]
- 41.Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y, Kumar A. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol 191: 1395–1411, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Powell DJ, Turban S, Gray A, Hajduch E, Hundal HS. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem J 382: 619–629, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Radin MS, Sinha S, Bhatt BA, Dedousis N, O'Doherty RM. Inhibition or deletion of the lipopolysaccharide receptor Toll-like receptor-4 confers partial protection against lipid-induced insulin resistance in rodent skeletal muscle. Diabetologia 51: 336–346, 2008. [DOI] [PubMed] [Google Scholar]
- 44.Reyna SM, Ghosh S, Tantiwong P, Meka CS, Eagan P, Jenkinson CP, Cersosimo E, Defronzo RA, Coletta DK, Sriwijitkamol A, Musi N. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes 57: 2595–2602, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116: 3015–3025, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stuart CA, Shangraw RE, Prince MJ, Peters EJ, Wolfe RR. Bed-rest-induced insulin resistance occurs primarily in muscle. Metab Clin Exper 37: 802–806, 1988. [DOI] [PubMed] [Google Scholar]
- 47.Suetta C, Frandsen U, Mackey AL, Jensen L, Hvid LG, Beyer ML, Petersson SJ, Schroder HD, Andersen JL, Aagaard P, Schjerling P, Kjaer M. Aging is associated with diminished muscle re-growth and myogenic precursor cell expansion in the early recovery phase after immobility-induced atrophy in human skeletal muscle. J Physiol 591: 3789–37804, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Summers SA, Nelson DH. A role for sphingolipids in producing the common features of type 2 diabetes, metabolic syndrome X, and Cushing's syndrome. Diabetes 54: 591–602, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Takeda K, Akira S. TLR signaling pathways. Sem Immunol 16: 3–9, 2004. [DOI] [PubMed] [Google Scholar]
- 50.Wang X, Spandidos A, Wang H, Seed B. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res 40: D1144–D1149, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Watson ML, Coghlan M, Hundal HS. Modulating serine palmitoyl transferase (SPT) expression and activity unveils a crucial role in lipid-induced insulin resistance in rat skeletal muscle cells. Biochem J 417: 791–801, 2009. [DOI] [PubMed] [Google Scholar]
- 52.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301: 640–643, 2003. [DOI] [PubMed] [Google Scholar]