

Abstract
Nephrotic syndrome (NS) is a clinicopathological entity characterized by proteinuria, hypoalbuminemia, peripheral edema, and hyperlipidemia. It is the most common cause of glomerular disease in children and adults. Although the molecular pathogenesis of NS is not completely understood, data from the study of familial NS suggest that it is a “podocytopathy.” Virtually all of the genes mutated in hereditary NS localize to the podocyte or its secreted products and the slit diaphragm. Since the completion of human genome sequence and the advent of next generation sequencing, at least 29 causes of single-gene NS have been identified. However, these findings have not been matched by therapeutic advances owing to suboptimal in vitro and in vivo models for the study of human glomerular disease and podocyte injury phenotypes. Multidisciplinary collaboration between clinicians, geneticists, cell biologists, and molecular physiologists has the potential to overcome this barrier and thereby speed up the translation of genetic findings into improved patient care.
Keywords: gene mutation, nephrotic syndrome, podocyte
nephrotic syndrome (NS) is a clinicopathological entity characterized by proteinuria, hypoalbuminemia, peripheral edema, and hyperlipidemia. It is the most common cause of glomerular disease in children and adults. The annual incidence of NS in the United States and Europe is estimated at 7/100,000 (10). NS is a manifestation of various pathological changes in the kidney. In clinical practice, NS is classified based on the patient's initial response to corticosteroid therapy (10). Classically, 80% of cases in the pediatric age group are steroid sensitive and are therefore classified as steroid-sensitive nephrotic syndrome (SSNS). The remaining 20% are called steroid-resistant nephrotic syndrome (SRNS). The SRNS variant of the disease is more prevalent in adults and is classically due to the pathological lesions of focal and segmental glomerulosclerosis (FSGS) (14). SRNS is characterized by a rapid progression to end-stage kidney disease (ESKD), and it is the most common glomerular cause of ESKD (14). In 2005, FSGS accounted for 12% of ESKD cases in the United States, costing an estimated $3 billion in Medicare spending (30, 36).
NS as a Podocytopathy
The molecular pathogenesis of NS is not completely understood; however, mounting evidence suggests that NS is due to defects in the glomerular filtration barrier (GFB). The GFB is composed of three layers: the specialized fenestrated endothelial cells and their overlying glycocalyx, the glomerular basement membrane (GBM), and glomerular visceral epithelial cells (i.e., podocytes) whose distal foot processes attach to the GBM (27). The functional integrity of the GFB depends on molecular cross talk between the three layers (27). While all three components are critical for the molecular sieving functions of the GFB, studies of familial NS/FSGS have revealed that the podocyte is the most important component of the GFB in that virtually all the genes mutated in hereditary SRNS localize to the podocyte or its secreted products and the slit diaphragm (SD) (Fig. 1) (1, 2, 8, 9, 25, 26, 31). These observations have given rise to the concept that most NS, especially FSGS, is due to an underlying podocytopathy (6). Podocytes are terminally differentiated cells consisting of a cell body, axon-like primary processes, and laterally radiating secondary foot processes that interdigitate with those of neighboring podocytes to form the highly specialized filamentous structure of the SD.
Fig. 1.

Podocyte foot process architecture and proteins involved in hereditary nephrotic syndromes. The podocyte contains F-actin, myosin (M), and actin-binding proteins synaptopodin (S) and α-actinin-4 (αACTN4). αACTN4 is mutated in focal and segmental glomerulosclerosis (FSGS) type 1. The slit diaphragm contains proteins that include nephrin, podocin, and CD2AP. Nephrin is mutated in congenital nephrotic syndrome of the Finnish type and podocin in steroid-resistant nephrotic syndrome. Neph1 and Neph2 are glomerular slit diaphragm proteins that interact with nephrin. Podocin is a transmembrane, lipid raft-associated protein that associates with nephrin and CD2AP. ANLN interacts with CD2AP, nephrin, and the p85-regulatory subunit of the phosphoinositol-3-kinase. SCARB2 is a lysosomal membrane protein. Zona occludens 1 (ZO-1) is a cell-to-cell junction protein. PLCε1 is an enzyme identified as a cause of diffuse mesangial sclerosis. TTC21β (not shown) is a ciliary protein involved in retrograde transport. Angiotensin receptor 1 is an example of a G protein-coupled receptor (GPCR) and can activate TRPC6. A mutation in TRPC6 results in increased calcium (Ca2+) transients and FSGS. Arhgap24 modulates the activity of Rac1 Rho-GTPase, and ARHGDIA modulates the activity of the Rac1 and Cdc42 Rho-GTPases. CoQ6 is an essential component of the mitochondrial respiratory transport chain that is required for the biosynthesis of CoQ10. ADCK4 is involved in the metabolism of CoQ10. Talin, paxillin, and vinculin (TPV) are connected to laminin-11 via α3-β1-integrin dimers and are involved in anchoring the podocyte to the glomerular basement membrane (GBM). Myo1E is a mediator of F-actin filament organization. The protein phosphatase receptor type O (PTPRO a.k.a. GLEPP1) may regulate podocyte structure and function via its interaction with CD2AP. INF2 is mutated in FSGS and is a regulator of F-actin assembly. The LAMB2 mutation is responsible for Pierson's syndrome and results in diffuse mesangial sclerosis. LCAT deficiency demonstrates lipid deposits in the GBM. LMX1B and WT1 (not shown) are transcription factors mutated in syndromic and non-syndromic proteinuric renal diseases. SMARCAL1 (not shown) is a member of a family of DNA helicases that modulate transcription through the modification of chromatin structure. Focal adhesion kinase (FAK) and p130Cas (CAS) are kinases that regulate actin filament assembly in focal contacts. Image adapted from Comprehensive Clinical Nephrology (Elsevier, 2015) with permission.
Genetics of NS
The completion of the human genome-sequencing project and advances in sequencing technology have accelerated the pace of gene discovery in Mendelian diseases. Insights into NS and other familial kidney diseases have greatly expanded in the era of next-generation sequencing (NGS) technologies. Classically, identification of single-gene causes of NS required the availability of large pedigrees with sufficient power to establish a disease locus, labor-intensive, fine-mapping strategies, and direct sequencing of genes in the refined locus. The first report of a single gene cause of NS appeared in the literature in 1998 when mutations in the nephrin gene (NPHS1) were reported as a cause of early-onset NS (24). Since then, integration of NGS technologies has led to the discovery of at least 29 genetic causes of NS, most of them within the last 10 years (Fig. 2).
Fig. 2.
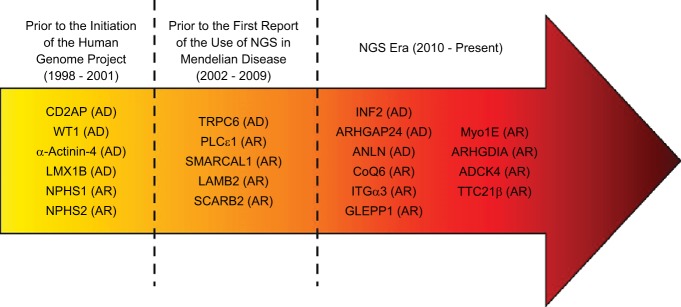
Timeline of gene discovery in hereditary nephrotic syndrome (NS). Gene discovery in NS has accelerated in the era of next generation sequencing (NGS) technology. Nearly 50% of NS genes have been discovered in the past 4 years due to the integration of NGS technologies.
Clinical Translation of Genetic Findings and Barriers to Translation
Identification of novel NS genes have significantly improved our understanding of NS disease mechanisms and led to improved patient outcomes. For example, the discovery of mutations in nephrin has laid the groundwork for understanding the molecular structure of the podocyte SD. Before the identification of nephrin, transmission electron microscopy (TEM) studies revealed that the SD is made up of a “zipper-like” array of regularly spaced cross bridges between interdigitating podocyte foot process (43). Despite the advances in our understanding of glomerular ultrastructure brought about by this study, the molecular composition of the SD and its relevance to podocyte and GFB physiology would remain unknown for nearly three decades. In subsequent years, studies revealed that the cross bridges are formed by homophilic interactions of nephrin (24).
Studies of familial nephrotic syndromes have shown that most of the genes mutated in hereditary NS serve critical functions in the maintenance of the podocyte actin cytoskeleton and signaling in the SD (2, 4, 5, 13, 16, 21, 35, 44). The knowledge gained from these studies and careful phenotyping data have led to the development of a framework for genetic testing of NS that may have relevance in clinical practice (17). Furthermore, it is now known that most cases of genetic NS are resistant to immunosuppression; it is therefore justifiable to modify the intensity and duration of immunosuppression in individuals with monogenic NS (17, 18). This shift in provider practice, informed by basic research discovery, has the potential to spare many patients the untoward side effects associated with prolonged courses of high-dose corticosteroid and other more potent immunosuppressive agents. Also, establishing genetic diagnoses can be helpful in streamlining pre- and post-kidney transplant management and prognostication.
Identification of podocyte dysfunction as central to the pathogenesis of NS has led to the reevaluation of the mechanisms of action of existing therapies for NS. For example, Ransom et al. (32) demonstrated that glucocorticoids prevent or reverse purine aminonucleoside-induced podocyte injury via direct stabilizing effects on the podocyte actin cytoskeleton and upregulation of Rho-GTPase activity. Similarly, Faul et al. and other investigators (3, 11, 39) described the role of cyclosporine A as a direct inhibitor of cathepsin-L-mediated synaptopodin degradation, and Fornoni et al. (12) reported that rituximab ameliorates podocyte actin cytoskeletal dysfunction and apoptosis via inhibition of sphingomyelin phosphodiesterase acid-like 3b depletion and downregulation of acid sphingomyelinase activity. Furthermore, Canaud et al. (7) described a potential mechanism for sirolimus-induced proteinuria via the mTORC2-dependent inhibition of AKT2 phosphorylation in podocytes. Most recently, Hall et al. (22) demonstrated that phosphodiesterase-V inhibitors ameliorated ANG II-induced podocyte dysmotility via the PKG-mediated downregulation of transient receptor potential cation channel 6 (TRPC6) activity. Finally, using a reverse genetics approach, angiopoietin-like 4 (ANGPT4) was identified as a podocyte-secreted inducer of GBM injury and NS, making ANGPT4 a promising novel therapeutic target for minimal-change disease (MCD) (9). Collectively, these findings have broadened our knowledge of viable and pharmacologically modifiable targets within the podocyte and reinforced the value of podocyte-directed therapies in the treatment of NS.
Although outside the scope of this limited perspective piece, genome-wide association studies (GWAS) have also been of value in the study of more common idiopathic forms of NS. The identification of APOL1 variants as risk alleles for FSGS in African Americans (19, 20), HLA-DQA1 and PLA2R variants in association with idiopathic membranous glomerulonephritis (38), and HLA-DQA1 and PLCG2 in association with SSNS (15) have spawned new avenues of investigation which may yield critical insights into disease pathogenesis.
Some of the single-gene defects identified in familial NS studies are potential therapeutic targets. For instance, TRPC6 is a well-recognized cause of familial NS. TRPC6 mutations cause FSGS via a mechanism involving aberrant calcium ion conductance in podocytes (42). Because of its intrinsic and potentially modifiable enzymatic activity, the value of TRPC6 as a potential therapeutic target has attracted considerable interest. However, improvements in our understanding of the mechanisms of TRPC6-mediated podocyte injury in FSGS have not been matched by therapeutic advances. While pharmacological therapies have shown promise in modulating the activity of TRPC6 in vitro (22), these findings have not translated into clinically useful therapeutic tools. Some of the factors responsible for this are 1) the lack of disease-specific cell lines for in vitro modeling and 2) unavailability of suitable animal models for NS. With respect to in vitro modeling, most researchers in this field rely almost exclusively on primary and immortalized podocyte culture for biochemical and molecular genetic analyses (33, 34). Although these mouse- and human-derived immortalized podocyte models have been of benefit, they lack essential podocyte characteristics that may be of importance in achieving a full understanding of the mechanisms of podocyte injury. For instance, conditionally immortalized podocytes do not form SD cross bridges in vitro. Given that a number of the gene mutations relevant to FSGS occur in SD proteins, these models may not allow us to appreciate the global alterations in podocyte structure and signaling caused by such mutations. In addition, molecular alterations such as targeted gene knockdown and exogenous promoter-driven gene overexpression studies in these models may also be problematic as such broad disruptions in the biology of a protein may introduce unintended physiological irregularities due to altered protein localization and stoichiometry, and cytotoxicity. One promising alternative to these strategies may be found in induced pluripotent stem cells (iPS). Directed differentiation of iPS cells into glomerular podocytes has been reported as a potential patient-specific stem cell-based therapy for CKD (37). While there have been no successful clinical applications of this strategy so far in the treatment of kidney disease, this approach may hold promise for the study of hereditary SRNS/FSGS. iPS cells derived from affected patients may provide disease-specific model systems with representative gene and protein expression profiles relevant to the pathogenesis of the disease. This could potentially improve our understanding of disease mechanisms and the evaluation of therapeutic strategies.
For similar reasons, whole animal modeling of NS has been equally challenging. For decades, investigators have relied upon mice for human disease modeling as 99% of mouse genes have a homolog within the human genome (28). Despite this high degree of similarity, mice are often resistant to glomerular injury and fail to recapitulate human phenotypes. In addition, mouse colonies are difficult and costly to establish and maintain. More recently, focus has shifted to the use of zebrafish embryos in the study of human glomerular disease. This is because 71.4% of human genes have at least one ortholog in the zebrafish genome, the zebrafish pronephros is fully developed by 3.5 days postfertilization and has a renal unit that functions similarly to that in humans, and the optical transparency of the zebrafish embryo facilitates high-quality imaging of disease-related changes occurring within pronephric structures (40, 41). Furthermore, zebrafish colonies are less expensive to maintain and different reports have demonstrated that genetic manipulation of genes causing human NS in zebrafish produces phenotypes that are very similar to human disease (40, 41). Therefore, this model holds promise for drug discovery and testing. However, the main limitation of this model system is that it is not amenable to longitudinal study. Development of methods that facilitate gene manipulation in adult fish will go a long way in overcoming these barriers. An emerging technology that holds promise in this regard is the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas gene-editing system. Recently, Irion et al. (23) reported the successful use of CRISPR/Cas to target and repair a truncating mutation of alb (albb4), which produces hypopigmentation in zebrafish. Through coinjection of circularized wild-type alb DNA with the CRISPR/Cas system in zebrafish larvae at the one-cell stage, the authors report roughly 10% germ line transmission of the repaired allele to the next generation (23). Such advances in the use of CRISPR/Cas technology may soon enable us to expand the utility of zebrafish in the modeling of human glomerular diseases and to develop and test novel therapeutic tools for hereditary NS.
Conclusion
In conclusion, genetic studies in the last two decades using forward genetics and NGS has significantly improved our understanding of the pathogenesis of NS. Unfortunately, there is a mismatch between the knowledge and translation to bedside. This is due, in part, to the lack of suitable in vitro and in vivo models for NS. Collaboration between clinicians, geneticists, cell biologists, and molecular physiologists has the potential to overcome this barrier and thereby speed up the translation of genetic findings into improved patient care.
GRANTS
R. Gbadegesin is supported by National Institutes of Health (NIH)/National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grants DK098135-01A1 and DK094987. R. Gbadegesin is the recipient of a Doris Duke Clinical Scientist Development Award, and part of this work was supported by Doris Duke Charitable Foundation Grant 2009033. G. Hall receives support from NIH/NIDDK Duke Training Grant in Nephrology 5T32DK0007731.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: G.H. and R.A.G. provided conception and design of research; G.H. and R.A.G. performed experiments; G.H. and R.A.G. analyzed data; G.H. and R.A.G. interpreted results of experiments; G.H. and R.A.G. prepared Figs.; G.H. and R.A.G. drafted manuscript; G.H. and R.A.G. edited and revised manuscript; G.H. and R.A.G. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the late Michelle P. Winn, MD, for her seminal contribution to the field of nephrology and, most importantly, for being a great teacher and an outstanding mentor.
REFERENCES
- 1.Abrahamson DR, Hudson BG, Stroganova L, Borza DB, St. John PL. Cellular origins of type IV collagen networks in developing glomeruli. J Am Soc Nephrol 20: 1471–1479, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akilesh S, Suleiman H, Yu H, Stander MC, Lavin P, Gbadegesin R, Antignac C, Pollak M, Kopp JB, Winn MP, Shaw AS. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Invest 121: 4127–4137, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bensman A, Niaudet P. Non-immunologic mechanisms of calcineurin inhibitors explain its antiproteinuric effects in genetic glomerulopathies. Pediatr Nephrol 25: 1197–1199, 2010. [DOI] [PubMed] [Google Scholar]
- 4.Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, Dahan K, Gubler MC, Niaudet P, Antignac C. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 24: 349–354, 2000. [DOI] [PubMed] [Google Scholar]
- 5.Boyer O, Benoit G, Gribouval O, Nevo F, Tete MJ, Dantal J, Gilbert-Dussardier B, Touchard G, Karras A, Presne C, Grunfeld JP, Legendre C, Joly D, Rieu P, Mohsin N, Hannedouche T, Moal V, Gubler MC, Broutin I, Mollet G, Antignac C. Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. J Am Soc Nephrol 22: 239–245, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buscher AK, Weber S. Educational paper: the podocytopathies. Eur J Pediatr 171: 1151–1160, 2012. [DOI] [PubMed] [Google Scholar]
- 7.Canaud G, Bienaime F, Viau A, Treins C, Baron W, Nguyen C, Burtin M, Berissi S, Giannakakis K, Muda AO, Zschiedrich S, Huber TB, Friedlander G, Legendre C, Pontoglio M, Pende M, Terzi F. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat Med 19: 1288–1296, 2013. [DOI] [PubMed] [Google Scholar]
- 8.Caridi G, Trivelli A, Sanna-Cherchi S, Perfumo F, Ghiggeri GM. Familial forms of nephrotic syndrome. Pediatr Nephrol 25: 241–252, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clement LC, Avila-Casado C, Mace C, Soria E, Bakker WW, Kersten S, Chugh SS. Podocyte-secreted angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome. Nat Med 17: 117–122, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eddy AA, Symons JM. Nephrotic syndrome in childhood. Lancet 362: 629–639, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med 14: 931–938, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, Li J, Mattiazzi A, Ciancio G, Chen L, Zilleruelo G, Abitbol C, Chandar J, Seeherunvong W, Ricordi C, Ikehata M, Rastaldi MP, Reiser J, Burke GW 3rd.. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med 3: 85ra46, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gbadegesin R, Hinkes BG, Hoskins BE, Vlangos CN, Heeringa SF, Liu J, Loirat C, Ozaltin F, Hashmi S, Ulmer F, Cleper R, Ettenger R, Antignac C, Wiggins RC, Zenker M, Hildebrandt F. Mutations in PLCE1 are a major cause of isolated diffuse mesangial sclerosis (IDMS). Nephrol Dial Transplant 23: 1291–1297, 2008. [DOI] [PubMed] [Google Scholar]
- 14.Gbadegesin R, Lavin P, Foreman J, Winn M. Pathogenesis and therapy of focal segmental glomerulosclerosis: an update. Pediatr Nephrol 26: 1001–1015, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gbadegesin RA, Adeyemo A, Webb NJ, Greenbaum LA, Abeyagunawardena A, Thalgahagoda S, Kale A, Gipson D, Srivastava T, Lin JJ, Chand D, Hunley TE, Brophy PD, Bagga A, Sinha A, Rheault MN, Ghali J, Nicholls K, Abraham E, Janjua HS, Omoloja A, Barletta GM, Cai Y, Milford DD, O'Brien C, Awan A, Belostotsky V, Smoyer WE, Homstad A, Hall G, Wu G, Nagaraj S, Wigfall D, Foreman J, Winn MP; Members of the Mid-West Pediatric Nephrology Consortium. HLA-DQA1 and PLCG2 are candidate risk loci for childhood-onset steroid-sensitive nephrotic syndrome. J Am Soc Nephrol [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 16.Gbadegesin RA, Hall G, Adeyemo A, Hanke N, Tossidou I, Burchette J, Wu G, Homstad A, Sparks MA, Gomez J, Jiang R, Alonso A, Lavin P, Conlon P, Korstanje R, Stander MC, Shamsan G, Barua M, Spurney R, Singhal PC, Kopp JB, Haller H, Howell D, Pollak MR, Shaw AS, Schiffer M, Winn MP. Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J Am Soc Nephrol 25: 1991–2002, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gbadegesin RA, Winn MP, Smoyer WE. Genetic testing in nephrotic syndrome—challenges and opportunities. Nat Rev Nephrol 9: 179–184, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gellermann J, Stefanidis CJ, Mitsioni A, Querfeld U. Successful treatment of steroid-resistant nephrotic syndrome associated with WT1 mutations. Pediatr Nephrol 25: 1285–1289, 2010. [DOI] [PubMed] [Google Scholar]
- 19.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329: 841–845, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genovese G, Tonna SJ, Knob AU, Appel GB, Katz A, Bernhardy AJ, Needham AW, Lazarus R, Pollak MR. A risk allele for focal segmental glomerulosclerosis in African Americans is located within a region containing APOL1 and MYH9. Kidney Int 78: 698–704, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall G, Gbadegesin RA, Lavin P, Wu G, Liu Y, Oh EC, Wang L, Spurney RF, Eckel J, Lindsey T, Homstad A, Malone AF, Phelan PJ, Shaw A, Howell DN, Conlon PJ, Katsanis N, Winn MP. A novel missense mutation of Wilms' tumor 1 causes autosomal dominant FSGS. J Am Soc Nephrol 26: 831–843, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hall G, Rowell J, Farinelli F, Gbadegesin RA, Lavin P, Wu G, Homstad A, Malone A, Lindsey T, Jiang R, Spurney R, Tomaselli GF, Kass DA, Winn MP. Phosphodiesterase 5 inhibition ameliorates angiontensin II-induced podocyte dysmotility via the protein kinase G-mediated downregulation of TRPC6 activity. Am J Physiol Renal Physiol 306: F1442–F1450, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irion U, Krauss J, Nusslein-Volhard C. Precise and efficient genome editing in zebrafish using the CRISPR/Cas9 system. Development 141: 4827–4830, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K. Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell 1: 575–582, 1998. [DOI] [PubMed] [Google Scholar]
- 25.Kitamura A, Tsukaguchi H, Hiramoto R, Shono A, Doi T, Kagami S, Iijima K. A familial childhood-onset relapsing nephrotic syndrome. Kidney Int 71: 946–951, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Malone AF, Phelan PJ, Hall G, Cetincelik U, Homstad A, Alonso AS, Jiang R, Lindsey TB, Wu G, Sparks MA, Smith SR, Webb NJ, Kalra PA, Adeyemo AA, Shaw AS, Conlon PJ, Jennette JC, Howell DN, Winn MP, Gbadegesin RA. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int 86: 1253–1259, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menon MC, Chuang PY, He CJ. The glomerular filtration barrier: components and crosstalk. Int J Nephrol 2012: 749010, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mouse Genome Sequencing Consortium, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigo R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O'Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Niederhausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Mukerji N, Damodaran TV, Winn MP. TRPC6 and FSGS: the latest TRP channelopathy. Biochim Biophys Acta 1772: 859–868, 2007. [DOI] [PubMed] [Google Scholar]
- 30.National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Disease. 2013 United States Renal Data System Annual Data Report. Atlas of Chronic Kidney Disease and End-Stage Renal Disease In the United States. Bethesda, MD: National Institutes of Health, 2013. [Google Scholar]
- 31.Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev 83: 253–307, 2003. [DOI] [PubMed] [Google Scholar]
- 32.Ransom RF, Lam NG, Hallett MA, Atkinson SJ, Smoyer WE. Glucocorticoids protect and enhance recovery of cultured murine podocytes via actin filament stabilization. Kidney Int 68: 2473–2483, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Sakairi T, Abe Y, Kajiyama H, Bartlett LD, Howard LV, Jat PS, Kopp JB. Conditionally immortalized human podocyte cell lines established from urine. Am J Physiol Renal Physiol 298: F557–F567, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shankland SJ, Pippin JW, Reiser J, Mundel P. Podocytes in culture: past, present, and future. Kidney Int 72: 26–36, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Shih NY, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, Miner JH, Shaw AS. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 286: 312–315, 1999. [DOI] [PubMed] [Google Scholar]
- 36.Smokler I. NephCure Outside Witness Testimony. Senate Committee on Appropriations: Innovation Hearing. April 24, 2014. http://appropriations.gov.
- 37.Song B, Niclis JC, Alikhan MA, Sakkal S, Sylvain A, Kerr PG, Laslett AL, Bernard CA, Ricardo SD. Generation of induced pluripotent stem cells from human kidney mesangial cells. J Am Soc Nephrol 22: 1213–1220, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stanescu HC, Arcos-Burgos M, Medlar A, Bockenhauer D, Kottgen A, Dragomirescu L, Voinescu C, Patel N, Pearce K, Hubank M, Stephens HA, Laundy V, Padmanabhan S, Zawadzka A, Hofstra JM, Coenen MJ, den Heijer M, Kiemeney LA, Bacq-Daian D, Stengel B, Powis SH, Brenchley P, Feehally J, Rees AJ, Debiec H, Wetzels JF, Ronco P, Mathieson PW, Kleta R. Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med 364: 616–626, 2011. [DOI] [PubMed] [Google Scholar]
- 39.Wasilewska AM, Kuroczycka-Saniutycz E, Zoch-Zwierz W. Effect of cyclosporin A on proteinuria in the course of glomerulopathy associated with WT1 mutations. Eur J Pediatr 170: 389–391, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Westerfield M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Brachydanio rerio). Eugene, OR: Univ. of Oregon Press, 1993, p. 1 v. (unpaged). [Google Scholar]
- 41.Wingert RA, Davidson AJ. The zebrafish pronephros: a model to study nephron segmentation. Kidney Int 73: 1120–1127, 2008. [DOI] [PubMed] [Google Scholar]
- 42.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308: 1801–1804, 2005. [DOI] [PubMed] [Google Scholar]
- 43.Yamada E. The fine structure of the renal glomerulus of the mouse. J Biophys Biochem Cytol 1: 551–566, 1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yao J, Le TC, Kos CH, Henderson JM, Allen PG, Denker BM, Pollak MR. Alpha-actinin-4-mediated FSGS: an inherited kidney disease caused by an aggregated and rapidly degraded cytoskeletal protein. PLoS Biol 2: e167, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]