

Abstract
Background
Mutations in heat shock 27 kDa protein 1 (HSP27 or HSPB1) cause distal hereditary motor neuropathy (dHMN) or Charcot-Marie-Tooth disease type 2 F (CMT2F) according to unknown factors. Mutant HSP27 proteins affect axonal transport by reducing acetylated tubulin.
Results
We generated a transgenic mouse model overexpressing HSP27-S135F mutant protein driven by Cytomegalovirus (CMV) immediate early promoter. The mouse phenotype was similar to dHMN patients in that they exhibit motor neuropathy. To determine the phenotypic aberration of transgenic mice, behavior test, magnetic resonance imaging (MRI), electrophysiological study, and pathology were performed. Rotarod test showed that founder mice exhibited lowered motor performance. MRI also revealed marked fatty infiltration in the anterior and posterior compartments at calf level. Electrophysiologically, compound muscle action potential (CMAP) but not motor nerve conduction velocity (MNCV) was reduced in the transgenic mice. Toluidine staining with semi-thin section of sciatic nerve showed the ratio of large myelinated axon fiber was reduced, which might cause reduced locomotion in the transgenic mice. Electron microscopy also revealed abundant aberrant myelination. Immunohistochemically, neuronal dysfunctions included elevated level of phosphorylated neurofilament and reduced level of acetylated tubulin in the sural nerve of transgenic mice. There was no additional phenotype besides motor neuronal defects.
Conclusions
Overexpression of HSP27-S135F protein causes peripheral neuropathy. The mouse model can be applied to future development of therapeutic strategies for dHMN or CMT2F.
Electronic supplementary material
The online version of this article (doi:10.1186/s12929-015-0154-y) contains supplementary material, which is available to authorized users.
Keywords: Heat shock 27 kDa protein 1 (HSP27 or HSPB1), Charcot-Marie-Tooth disease (CMT), Distal hereditary motor neuropathy (dHMN), Axonopathy, Mouse model, Magnetic resonance image (MRI)
Background
Mutations in heat shock 27 kDa protein 1 (HSP27, also called HSPB1) are associated with hereditary peripheral neuropathy [1–4]. The clinical aspects of the patients are heterogeneous and include motor neuropathy or sensory neuropathy with various onset ages, resulting in distal hereditary motor neuropathy (dHMN) type II (OMIM 608634) or Charcot-Marie-Tooth disease (CMT) type 2 F (OMIM 606595) [1–4].
HSP27 is located in chromosome 7q11-q21 and encodes a 27 kDa protein possessing an α-crystallin domain [5]. HSP27 protects cells from stresses and is associated with cell motility and cytoskeletal stabilization [6–10]. The expression of HSP27 ameliorates neurodegenerative diseases, such as Alzheimer’s disease [11, 12], Parkinson disease [13], and amyotrophic lateral sclerosis [14]. On the other hand, mutation in HSP27 deteriorates axonal transport in the peripheral nervous system [15]. Mutant HSP27 protein reduces neuronal cell viability and impairs neurofilament assembly [1, 16]. In addition, the expression of mutant HSP27 reduces acetylated α-tubulin in dorsal root ganglion cell (DRG) in mouse model, resulting in reduced mitochondrial movement and dysfunction of axon cytoskeleton and axonal transport [17].
So far, eight mutations in HSP27 are reported: L99M, R127W, S135F, R136W, R140G, T151I, P182S, and P182L. Intriguingly, the Ser135Phe mutation, which is the best characterized mutation, is associated with both CMT2F and dHMN [1]. This implies that various clinical phenotypes such as involvement of sensory neuron, onset age, and severity caused by the same mutation, might be dependent on the genetic background.
Currently, several mouse models (FVB/N strain) have been developed for HSP27 mutations including Pro182Leu [18], Ser135Phe [18], and Arg136Trp [19]. The phenotypes of these mice are similar to patients who develop late-onset motor neuropathy. Since the clinical symptoms of patients are heterogeneous according to genetic background, various models might be required for further study.
In this study, we generated a transgenic mouse (C57BL6/J strain) expressing S135F mutation in HSP27 to further investigate the clinical aspects and development of therapeutic strategies.
Methods
Preparation of HSP27 construct
To obtain human HSP27 gene, total mRNA from HEK 293 cells was used as the template for cDNA synthesis and PCR amplification. The amplified gene was cloned into the pcDNA3.1(+) vector, where CMV promoter drives the expression of the gene, for recombination and expression in mammalian cells. To generate the S135F mutant gene, site-directed mutagenesis was performed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). All the sequences were confirmed by capillary sequencing.
Generation of transgenic mouse model
To establish a mouse model for HSP27-S135F mutation, pcDNA3.1-HSP27-S135F was injected into fertilized eggs (C57BL6/J strain). The eggs were implanted into surrogate female mice. Seventy-two mice were generated and 12 harbored the HSP27-S135F construct. All experiments were conducted according to protocols approved by the Institutional Animal Care and Use Committees of Samsung Medical Center.
Rotarod test
To evaluate motor coordination and balance of HSP27-S135F transgenic mice, the rotarod test was performed on a 3 cm horizontal rotating rod (2 m/min). To adapt to the test, the mice were pre-trained for one week. Testing was for a maximum of 7 min.
Electrophysiological study
Ten control and transgenic mice, aged 7 months and weighing 25–30 g, were used for the electrophysiological study. The mice were anesthetized with 50 mg/kg Zoletil (Virbac, Seoul, Korea) intraperitoneally and the fur from the distal back and the hind limbs was completely removed. Nerve conduction study (NCS) was performed using Nicolet VikingQuest (Natus Medical, San Carlos, CA) [20]. The compound motor action potential (CMAP) amplitudes and motor nerve conduction velocity (MNCV) were determined.
Magnetic resonance imaging (MRI)
In vivo monitoring of mouse hind limbs and muscle damage was performed using a Biospec 7.0 Tesla 30 cm horizontal bore scanner with Paravision 5.1 software (Bruker Biospin MRI GmbH, Germany) as detailed [21]. Briefly, Bruker four-element 1H volume coil array and Bruker 72 mm linear-volume coil were used as the receiver and the transmitter, respectively. Aaxial, mid-sagittal, and coronal scout rapid acquisition with fast low angle shot imaging was used to localize the leg, and high resolution T2-weighted images in the cross-sectional view were acquired with TR/TE (repetition time/echo time) = 5000/32 ms, field of view = 30 × 30 mm2, matrix size = 250 × 250, slice thickness = 0. 5 mm without a gap.
Electron microscopy
The distal sural nerve was biopsied from mice at 10 months, and pathological examinations of affected individuals included light and electron microscopic analyses. Specimens were individually fixed in 2 % glutaraldehyde in 25 mM cacodylate buffer. Semithin sections were stained with toluidine blue and ultra-thin cut samples were contrasted with uranyl acetate and lead citrate.
Immunohistochemistry and western blotting
Tissues from sciatic and median/ulnar nerve of the mice were analyzed for tubulin acetylation and neurofilament phosphorylation. Formalin-fixed sections were stained with hematoxylin and eosin (H&E). Anti-phospho-neurofilament and acetylated-α-tubulin antibody (Abcan, Cambridge, UK) were used. Standard Western blotting was performed using anti-tubulin (Abcam), anti-β-actin antibody, anti-mouse secondary antibody, and anti-rabbit secondary antibody (Sigma-Aldrich, St. Louis, MO) as well as ECL plus Western blotting substrate (Thermo Scientific, Rockford, IL). Band intensities were calculated by Image J program (www.imagej.nih.gov) then the ratio of the band intensity of acetylated-α-tubulin over α-tubulin and phospho-neurofilament over neurofilament was calculated.
Statistical analysis
All animals were studied with a blind test. Comparison between normal and HSP27-S135F mice were made by Student’s t-test. P < 0.05 was considered statistically significant.
Results
HSP27-S135F transgenic mice exhibit reduced motor function
Wild type and S135F mutant HSP27 gene was cloned as described in Materials and Methods. Expression of the genes was confirmed in HEK293 and NSC34 cells (Additional file 1: Figure S1). To generate HSP27-S135F transgenic mice, pcDNA3.1-HSP27-S135F was injected into fertilized eggs. Twelve transgenic mice were confirmed to have the HSP27-S135F construct. From 5 months, motor function was assessed using the rotarod test. Compared with age matched control mice group (n = 10), several transgenic mice exhibited reduced motor performance (Fig. 1a). Upon completion of the tests, two mice (#11 and #26) were chosen as founders for further studies. Rotarod testing of the founders’ siblings also showed the same results (Fig. 1b). Measurement of grip strength on the progeny (F1) also demonstrated reduced motor function in the transgenic mice (Additional file 2: Figure S2). Besides the motor function, all the mice exhibited normal phenotype, except that #60 had white fur on chest, which was transmitted to its siblings.
Fig. 1.
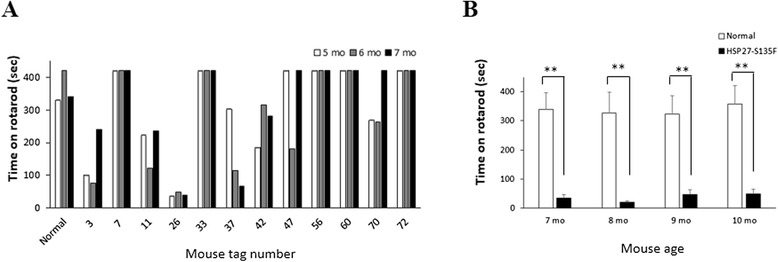
Reduced motor function in HSP27-S135F mouse. Rotarod test were applied to measure motor function of mice. Reduced performance was observed from both the founders from 5 months of age (a) and siblings of #11 and #26 from 7 months of age (b). **, p < 0.01
MRI demonstration of fatty infiltration in lower extremities
T2-weighted imaging and fat-suppressed imaging of leg and thigh muscles were performed on 7 month-old mice. Heterogeneous areas of elevated intensity were visible in all HSP27-S135F transgenic mice (Fig. 2a) in contrast to the more uniform and dark signal for healthy control muscle tissue (Fig. 2b). Transgenic mice showed marked fatty infiltration in the anterior and posterior compartments at the calf level.
Fig. 2.
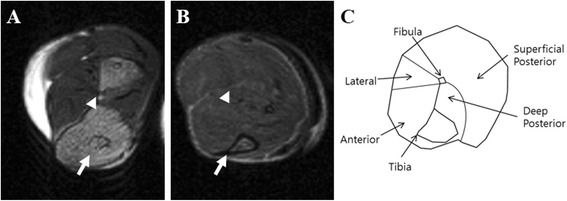
Fatty replacement of calf muscles in HSP27 transgenic mice on T2-weighted MRI. a. Fatty infiltration of leg muscles was observed in transgenic mice with HSP27 mutations on MRI. b. Normal muscular findings of control mice were shown on MRI. c. Orientation and anatomy of calf cross sections. Anterior muscle groups (anterior) include tibialis anterior, extensor halluces longus, and extensor digitorum longus. Lateral muscle groups (lateral) include peroneus longus and peroneus brevis. Deep posterior groups (deep posterior) include tibialis posterior and flexor digitorum longus. Superficial posterior groups (superficial posterior) include gastrocnemius and soleus. The tibial and fibular bones are marked as arrow and arrowhead, respectively
HSP27-S135F transgenic mice exhibit reduced CMAP
To determine the functional defect in hind limbs, nerve conduction was examined in both sides of sciatic nerve of five F1 mice of the #11 founder at 7 months of age. Five age-matched normal mice were used as control. As shown in Fig. 3a-c and Table 1, transgenic mice showed significantly reduced CMAP than control mice. However, there was no difference in MNCV between normal and HSP27-S135F mice (Table 1 and Fig. 3). These data suggest that HSP27-S135F transgenic mice have abnormal motor function and the phenotype is consistent with CMT2 or dHMN, rather than CMT1, which is caused by Schwann cell malfunction and shows reduced MNCV.
Fig. 3.
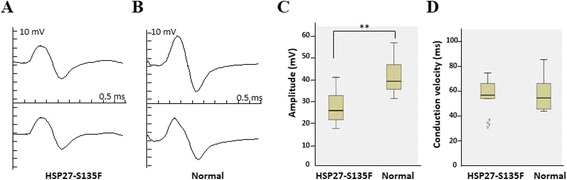
Reduced CMAP was observed from NCS. Representative sciatic CMAP responses from HSP27-S135F (a) and normal mice (b). Sweep speed, 0.5 ms per division; Amplitude, 10 mV per division. Mean sciatic CMAP amplitudes (c) and conduction velocity (d) for each group revealed that HSP27-S135F exhibit abnormality in only CMAP. Five mice of each group were used and data from both right and left were pooled (n = 10). **, p < 0.01
Table 1.
Summary of nerve conduction study
| Control | HSP27-S135F | p-value | |
|---|---|---|---|
| na | 10 | 10 | - |
| CMAP | 41.27. ± 7.51 | 27.48 ± 8.21 | 0.004 |
| MNCV | 57.92 ± 14.28 | 56.50 ± 13.95 | 0.912 |
a n = sum of left and right sciatic nerve; data expressed as mean ± standard deviation
Electron microscopy demonstration of reduced motor neuron
To analyze the ultra-structure of the sciatic nerve of HSP27-S135F mice, sciatic nerves of the founder and their progeny mice were collected. Semi-thin sections of sciatic nerve displayed reduced number of large myelinated fibers in HSP27-S135F mice compared to control mice (Fig. 4a and 4b). Direct counts also revealed that the ratio of large myelinated fibers (>15 μm) was reduced in transgenic mice compared to control mice (Fig. 4c). Since large myelinated fibers are motor neurons, this result is consistent with previously demonstrated impaired motor function and muscle integrity. Electron microscopic analysis revealed mixed forms of demyelinated or dysmyelinated fibers compared to normal fibers (Fig. 4d-g).
Fig. 4.
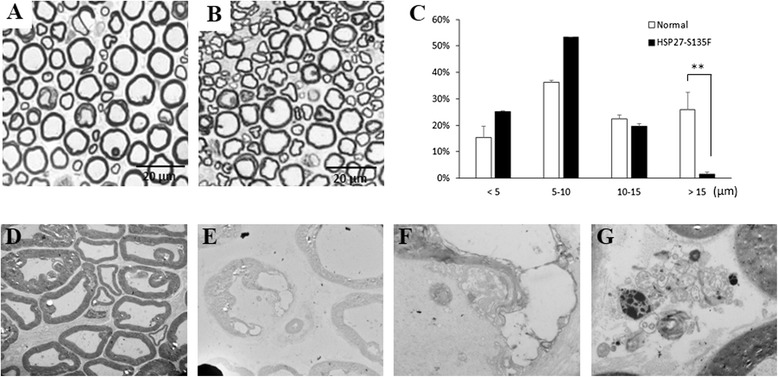
Aberration in myelination was observed in HSP27-S135F mice. Toluidine blue staining showed normal myelination in normal mice (a) and demyelination in HSP27-S135F mice (b). Scale bar = 20 μM. c. Distribution of diameter of myelinated axon demonstrated that the proportion of large (>15 μm) myelinated fibers are reduced in HSP27-S135F mice. **, p < 0.01. Electron microscopy demonstrated normal pattern in normal mice (d) and abnormal myelination in transgenic mice (e-g). Magnifications: (d) and (e), x 1000; (f) and (g), x 4000
Decreased level of acetyl-tubulin in sciatic nerve
Since phosphorylated neurofilament and acetylated tubulin have been reported as molecular pathological markers for HSP27 mutation mediated axonopathy, we determined their levels in the sciatic nerve. As expected, tubulin acetylation was reduced and phosphorylated neurofilament was elevated in sciatic nerve of HSP27-S135F mice compared to control mice (Fig. 5a-d). Western blotting from each of the three mice (Fig. 5e) and quantitation from the data (Fig. 5f) were also consistent with the immunohistochemical data.
Fig. 5.
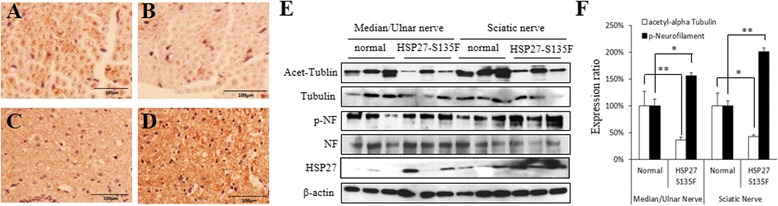
Reduced acetylated tubulin and increased phosphorylated neurofilament. a-d. Immunohistochemical analyses revealed the reduced level of acetylated tubulin in HSP27-S135F mice (b) compared to normal mice (a) and elevated level of phosphorylated neurofilament in HSP27-S135F mice (d) compared to normal mice (c). Scale bar = 100 μM. e. Western blotting from 3 mice from each group demonstrated the changes of the proteins level. f. The Western blotting image were quantitated. Data are mean ± standard deviation. *, p < 0.05; and **, p < 0.01
Discussion
Transgenic mice overexpressing HSP27-S135F were generated and phenotypic characteristics were assessed. The mice exhibited axonal neuropathy as expected. Electron microscopy and toluidine blue staining revealed deteriorated integrity of Schwann cells and axons in transgenic mice. However, the major pathogenesis occurred in the axon because only CMAP rather than MNCV was affected. MNCV is lowered by the aberrant myelination of Schwann cell, which is a typical pattern of CMT1, whereas CMAP is affected by axonal neuropathy, such as CMT2. These axonal defects increase fatty infiltration in the anterior and posterior compartments of the calf, which eventually led to reduced locomotion function. Since HSP27-S135F mutation cause both CMT2F and dHMN, we tried to examine whether the mice exhibit sensory neuropathy. From hot plate test, we could observe that HSP27-S135F mice show normal sensory function on temperature (Additional file 3: Figure S3). Thus phenotypic characteristics of our new mice might be close to dHMN.
Compared to previously reported mouse models, the newly developed model exhibited early onset and more severe phenotype. In a prior study, rotarod performance deteriorated from 6 months of age [18]; however, the newly developed mice showed poor performance from 5 months. Moreover, the performance in two founders was only 55 % and 23 % of control mice, respectively. These observations imply that the phenotype of the mice with the same mutation can be diverse according to the genetic background and driving promoter. In addition, phenotypic diversity could be derived by the position or copy number of the transgene. The phenotypic variation might also be dependent on the expression level of the transgene. We tried to correlate the expression level and the phenotypic severity (Additional file 4: Figure S4), however, further study is needed to provide clear conclusion.
MRI was used to assess muscle integrity in the CMT mouse model. Since the pathological findings from MRI are consistent with previous reports [4], MRI can be adopted as a new assessment tool for CMT research. In CMT, huge gaps still remain between preclinical study and clinical outcomes. Although the administration of ascorbic acid can dramatically reduce the mouse phenotype [22], the effects remain unclear even after phase III clinical trial [23].
The HSP27-S135F missense mutation is located in the HSP20-α-crystallin domain. This domain is related to mitochondrial movement, which is important to maintain the integrity of cellular cytoskeleton transport [18, 24]. HSP27-S135F mutation induces hyper-phosphorylation of neurofilament light protein, resulting in axonal transport defect by aggregation of phosphorylated neurofilament proteins, which is mediated by Cyclin-dependent kinase (Cdk5) [25]. Since elevated level of phospho-NEFL was reduced by inhibitor or by the Cdk5-specific shRNA and the reduced mitochondrial movement, which is mediated by reduced acetylated-α-tubulin, was recovered by the utilization of HDAC inhibitor [18], targeting Cdk5 or HDAC could be the therapeutic strategies for the treatment of axonal neuropathy caused by the mutation in HSP27.
Conclusion
In conclusion, the phenotype of the newly developed mouse model is consistent with CMT2 or dHMN patients. The model is a potentially useful tool for future assessment of therapeutic candidates for CMT2 or dHMN.
Acknowledgements
This study was supported by the Korean Health Technology R&D Project, Ministry of Health & Welfare (HI12C0135 and HI14C3484) and by the National Research Foundation of Korea (NRF) grants funded by the Korea governments, MSIP (NRF-2014R1A2A2A01004240) and MOE (NRF-2013R1A1A2007873).
Abbreviations
- HSP27
Heat shock 27 kDa protein 1
- CMT2F
Charcot-Marie-Tooth disease type2F
- dHMN
Distal hereditary motor neuropathy
- MRI
Magnetic resonance image
- CMAP
Compound motor action potential
- MNCV
Motor nerve conduction velocity
- DRG
Dorsal root ganglion
Additional files
Generation and expression of HSP27-S135F. (A) Chromatograms of the sequences of wild type and S135F mutants of HSP27. Arrows indicate the mutation site. (B) Western blotting for the determination of the expression wild type and S135F mutant HSP27 in HEK293 cells or NSC34 cells.
Grip strength test for offspring of #11 and #26. Grip strength was performed using all four limbs of the mice. *, p < 0.05.
Hot plate test for sensory nerve function. Mice were place on a preheated (52 °C) acrylic box. Latency of paw withdrawal, shaking, or licking was calculated and compared (n = 7 per each group). Trembler J mice is well-known CMT1 mouse model, which naturally carries PMP22-L16P mutation in one allele. **, p < 0.01.
Expression of HSP27 in the siblings. (A) CMAP were performed to divide the sibling into moderate and severe phenotype. #201, #243, and #293 mice showed relatively mild phenotype, and #217, #240, and #280 mice showed relatively severe phenotype. (B) Western blotting for determination of HSP27 expression level in the sciatic nerve of the mice. (C) Determination of HSP27 expression in the various organs including sciatic nerve, spinal cord, spleen, and liver from normal and HSP27-S135F transgenic mice.
Supplementary Methods.
Footnotes
Jinho Lee and Sung-Chul Jung contributed equally to this work.
Competing interest
The authors declare that they have no competing interest.
Authors’ contributions
SCJ, YBH and BOC: study concept and design; JL, JJ, YRC, JSL, HJA, NJ, HWM, HKY, GK and SH: acquisition of data; JL, JR, SYW and JYK: analysis and interpretation of data; JL, SCJ and YBH: drafting of the manuscript; JR, SYW and JYK: critical revision of the manuscript for important intellectual content; SCJ, YBH and BOC: provision of funding; YBH and BOC: study supervision. All authors read and approved the final manuscript.
Contributor Information
Young Bin Hong, Phone: +82-2-2148-9497, Email: youngbinhong@gmail.com.
Byung-Ok Choi, Phone: +82-2-3410-1296, Email: bochoi77@hanmail.net.
References
- 1.Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL, Schagina O, Verpoorten N, Van Impe K, Fedotov V, Dadali E, Auer-Grumbach M, Windpassinger C, Wagner K, Mitrovic Z, Hilton-Jones D, Talbot K, Martin JJ, Vasserman N, Tverskaya S, Polyakov A, Liem RK, Gettemans J, Robberecht W, De Jonghe P, Timmerman V. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004;36:602–606. doi: 10.1038/ng1354. [DOI] [PubMed] [Google Scholar]
- 2.Kijima K, Numakura C, Goto T, Takahashi T, Otagiri T, Umetsu K, Hayasaka K. Small heat shock protein 27 mutation in a Japanese patient with distal hereditary motor neuropathy. J Hum Genet. 2005;50:473–476. doi: 10.1007/s10038-005-0280-6. [DOI] [PubMed] [Google Scholar]
- 3.Tang B, Liu X, Zhao G, Luo W, Xia K, Pan Q, Cai F, Hu Z, Zhang C, Chen B, Zhang F, Shen L, Zhang R, Jiang H. Mutation analysis of the small heat shock protein 27 gene in Chinese patients with Charcot-Marie-Tooth disease. Arch Neurol. 2005;62:1201–1207. doi: 10.1001/archneur.62.8.1201. [DOI] [PubMed] [Google Scholar]
- 4.Chung KW, Kim SB, Cho SY, Hwang SJ, Park SW, Kang SH, Kim J, Yoo JH, Choi BO. Distal hereditary motor neuropathy in Korean patients with a small heat shock protein 27 mutation. Exp Mol Med. 2008;40:304–312. doi: 10.3858/emm.2008.40.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plumier JC, Hopkins DA, Robertson HA, Currie RW. Constitutive expression of the 27-kDa heat shock protein (HSP27) in sensory and motor neurons of the rat nervous system. J Comp Neurol. 1997;384:409–428. doi: 10.1002/(SICI)1096-9861(19970804)384:3<409::AID-CNE7>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 6.Wagstaff MJ, Collaco-Moraes Y, Smith J, de Belleroche JS, Coffin RS, Latchman DS. Protection of neuronal cells from apoptosis by HSP27 delivered with a herpes simplex virus-based vector. J Biol Chem. 1999;274:5061–5069. doi: 10.1074/jbc.274.8.5061. [DOI] [PubMed] [Google Scholar]
- 7.Hirano S, Shelden EA, Gilmont RR. HSP27 regulates fibroblast adhesion, motility, and matrix contraction. Cell Stress Chaperones. 2004;9:29–37. doi: 10.1379/471.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heo JI, Lee MS, Kim JH, Lee JS, Kim J, Park JB, Lee JY, Han JA, Kim JI. The role of tonicity responsive enhancer sites in the transcriptional regulation of human hsp70-2 in response to hypertonic stress. Exp Mol Med. 2006;38:295–301. doi: 10.1038/emm.2006.35. [DOI] [PubMed] [Google Scholar]
- 9.Williams KL, Rahimtula M, Mearow KM. Heat shock protein 27 is involved in neurite extension and branching of dorsal root ganglion neurons in vitro. J Neurosci Res. 2006;84:716–723. doi: 10.1002/jnr.20983. [DOI] [PubMed] [Google Scholar]
- 10.Arrigo AP, Simon S, Gibert B, Kretz-Remy C, Nivon M, Czekalla A, Guillet D, Moulin M, Diaz-Latoud C, Vicart P. HSP27 (HspB1) and &B-crystallin (HspB5) as therapeutic targets. FEBS Lett. 2007;581:3665–3674. doi: 10.1016/j.febslet.2007.04.033. [DOI] [PubMed] [Google Scholar]
- 11.Shimura H, Miura-Shimura Y, Kosik KS. Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J Biol Chem. 2004;279:17957–17962. doi: 10.1074/jbc.M400351200. [DOI] [PubMed] [Google Scholar]
- 12.Tóth ME, Szegedi V, Varga E, Juhász G, Horváth J, Borbély E, Csibrány B, Alföldi R, Lénárt N, Penke B, Sántha M. Overexpression of Hsp27 ameliorates symptoms of Alzheimer’s disease in APP/PS1 mice. Cell Stress Chaperones. 2013;18:759–771. doi: 10.1007/s12192-013-0428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Renkawek K, Stege GJ, Bosman GJ. Dementia, gliosis and expression of the small heat shock proteins HSP27 and alpha B-crystallin in Parkinsons’ disease. Neuroreport. 1999;10:2273–2276. doi: 10.1097/00001756-199908020-00009. [DOI] [PubMed] [Google Scholar]
- 14.Vleminckx V, Van Damme P, Goffin K, Delye H, Van Den Bosch L, Robberecht W. Upregulation of HSP27 in a transgenic model of ALS. J Neuropathol Exp Neurol. 2002;61:968–974. doi: 10.1093/jnen/61.11.968. [DOI] [PubMed] [Google Scholar]
- 15.Almeida-Souza L, Asselbergh B, d’Ydewalle C, Moonens K, Goethals S, de Winter V, Azmi A, Irobi J, Timmermans JP, Gevaert K, Remaut H, Van Den Bosch L, Timmerman V, Janssens S. Small heat-shock protein HSPB1 mutants stabilize microtubules in Charcot-Marie-Tooth neuropathy. J Neurosci. 2011;31:15320–15328. doi: 10.1523/JNEUROSCI.3266-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ackerley S, James PA, Kalli A, French S, Davies KE, Talbot K. A mutation in the small heat-shock protein HSPB1 leading to distal hereditary motor neuronopathy disrupts neurofilament assembly and the axonal transport of specific cellular cargoes. Hum Mol Genet. 2006;15:347–354. doi: 10.1093/hmg/ddi452. [DOI] [PubMed] [Google Scholar]
- 17.Irobi J, De Jonghe P, Timmerman V. Molecular genetics of distal hereditary motor neuropathies. Hum Mol Genet. 2004;13:R195–202. doi: 10.1093/hmg/ddh226. [DOI] [PubMed] [Google Scholar]
- 18.d’Ydewalle C, Krishnan J, Chiheb DM, Van Damme P, Irobi J, Kozikowski AP, Vanden Berghe P, Timmerman V, Robberecht W, Van Den Bosch L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med. 2011;17:968–974. doi: 10.1038/nm.2396. [DOI] [PubMed] [Google Scholar]
- 19.Srivastava AK, Renusch SR, Naiman NE, Gu S, Sneh A, Arnold WD, Sahenk Z, Kolb SJ. Mutant HSPB1 overexpression in neurons is sufficient to cause age-related motor neuronopathy in mice. Neurobiol Dis. 2012;47:163–173. doi: 10.1016/j.nbd.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xia RH, Yosef N, Ubogu EE. Dorsal caudal tail and sciatic motor nerve conduction studies in adult mice: technical aspects and normative data. Muscle Nerve. 2010;41:850–856. doi: 10.1002/mus.21588. [DOI] [PubMed] [Google Scholar]
- 21.Pratt SJ, Xu S, Mullins RJ, Lovering RM. Temporal changes in magnetic resonance imaging in the mdx mouse. BMC Res Notes. 2013;6:262. doi: 10.1186/1756-0500-6-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Passage E, Norreel JC, Noack-Fraissignes P, Sanguedolce V, Pizant J, Thirion X, Robaglia-Schlupp A, Pellissier JF, Fontés M. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med. 2004;10:396–401. doi: 10.1038/nm1023. [DOI] [PubMed] [Google Scholar]
- 23.Lewis RA, McDermott MP, Herrmann DN, Hoke A, Clawson LL, Siskind C, Feely SM, Miller LJ, Barohn RJ, Smith P, Luebbe E, Wu X, Shy ME, Muscle Study Group High-dosage ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A: results of a randomized, double-masked, controlled trial. JAMA Neurol. 2013;70:981–987. doi: 10.1001/jamaneurol.2013.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, Gurbuxani S, Arrigo AP, Kroemer G, Solary E, Garrido C. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol. 2000;2:645–652. doi: 10.1038/35023595. [DOI] [PubMed] [Google Scholar]
- 25.Holmgren A, Bouhy D, De Winter V, Asselbergh B, Timmermans JP, Irobi J, Timmerman V. Charcot-Marie-Tooth causing HSPB1 mutations increase Cdk5-mediated phosphorylation of neurofilaments. Acta Neuropathol. 2013;126:93–108. doi: 10.1007/s00401-013-1133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]