

Abstract
Methylation of histone H3 lysine-4 (H3K4) is an important, regulatory, epigenetic post-translational modification associated with actively transcribed genes. In humans, the principal mediators of this modification are part of the MLL/SET1 family of methyltransferases, which comprises six members, MLLs1–4 and SET1A/SET1B. Aberrations in the structure, expression, and regulation of these enzymes are implicated in various disease states, making them important potential targets for drug discovery, particularly for oncology indications. The MLL/SET1 family members are most enzymatically active when part of a “core complex,” the catalytic SET-domain-containing subunits bound to a subcomplex consisting of the proteins WDR5, RbBP5, Ash2L and a homodimer of DPY-30 (WRAD2). The necessity of MLL/SET1 members to bind WRAD2 for full activity is the basis of a particular drug development strategy, which seeks to disrupt the interaction between the MLL/SET1 subunits and WDR5. This strategy is not without its theoretical and practical drawbacks, some of which relate to the ease with which complexes of Escherichia coli-expressed MLL/SET1 and WRAD2 fall apart. As an alternative strategy, we explore ways to stabilize the complex, focusing on the use of an excess of WRAD2 to drive the binding equilibria toward complex formation while maintaining low concentrations of the catalytic subunits. The purpose of this approach is to seek inhibitors that bind the SET domain, an approach proven successful with the related, but inherently more stable, enhancer of zeste homolog 2 (EZH2) complex.
Introduction
The methylation of the ɛ-amino group of lysine-4 in histone H3 (H3K4) of chromatin is an epigenetic mark both correlated with, and a regulator of, the activation of gene transcription in eukaryotes.1–5 H3K4 can accept up to three methyl groups (H3K4me1/2/3) and the mono-, di-, and trimethylated forms play differing roles in the control of chromatin structure and function.3–6 For example, it has recently been shown that, for osmotic stress responsive genes in yeast, the presence or absence of the H3K4 monomethyl form (H3K4me1) regulates the participation of alternative chromatin remodeling modeling complexes, and hence, the nature of remodeling events, such as the exchange of histone H2A for histone H2A.Z.7 H3K4me3 levels are typically elevated near the transcription start site of active genes, whereas H3K4me2 and H3K4me1 are distributed across active genes and in their enhancer elements.3–6
Lysine methytransferases (KMTs) transfer the methyl group from S-adenosylmethionine (SAM) to the side chain, ɛ-amino group of protein lysyl residues, functioning predominately, but not exclusively, as histone methyltransferases (HMTs).8 Major histone sites of regulatory lysine modifications include H3K4, H3K9, H3K27, H3K36, H3K79, and H4K20. With the exception of the H3K79 methyltransferase Dot1L,9 and another possible case to be discussed below, the human KMTs contain a catalytic SET domain, named for three Drosophila methyltransferases, Su(var)3–9, enhancer of zeste, and trithorax.10
There is a single H3K4 methyltransferase, SET1, in the budding yeast Saccharomyces cerevisiae,11 while the human proteome comprises two orthologs of SET1, SET1A and SET1B, plus an additional four homologs, the MLL proteins 1 to 4.12–17 Human SET1-containing complexes (SET1A and SET1B) appear to be responsible for maintaining global levels of H3K4me3.17,18 In contrast, the H3K4-methylating activities of other MLL/SET1 family members are thought to be more narrowly focused, as for example, in the case of MLL1 and MLL2 and the HOX genes.9 With the possible exception of SET1A, the human SET1 homologs display a weak, primarily monomethylation activity on H3K4,20 but catalysis is dramatically stimulated by the binding of a protein complex, here termed WRAD2, consisting of the proteins WDR5, RBBP5, ASH2L, and DPY-30 (2 copies, homodimer).20–27 For example, addition of the WRAD2 subunits in vitro to recombinant MLL1 stimulates methyltransferase activity about 600-fold.28 Interaction between MLL1 and WRAD2 is required for sequential mono- and dimethylation of H3K428 and for methylation of nucleosomal histone H329 and, generally, the interaction of WRAD2 with each of the six MLL/SET1 family members appears to dictate their specificity in terms of mono-, di-, and trimethylating activities.20 Thus, WRAD2 is considered an essential subcomplex of SET1 family H3K4 methyltransferases, necessary for complex assembly and full activity.20,22,26–28 Moreover, recent work has shown that the WRAD2 complex by itself has the H3K4 methyltransferase activity, making it, along with Dot1L, one of the only two non-SET domain lysine methyltransferases to be identified.28,29
The four MLL enzymes are considered promising targets for drug development, particularly for various oncology indications. MLL1 gene rearrangements resulting in chimeric proteins, usually with the N-terminal MLL1 sequence, but not the C-terminal, catalytic SET domain, are a feature of aggressive leukemias (see review Hess30). However, MLL1 rearrangements that include the SET domain (partial tandem duplications) occur in 5%–10% of acute myeloid leukemias,31 and wild-type MLL1 can also be required for the leukemic transformation driven by the MLL1 fusion protein derived from the other rearranged allele.32,33 Thus, inhibition of MLL1 methyltransferase activity may represent a strategy for antileukemia therapy.32 MLL1 may also act more generally in the progression of various cancers, including roles in cell cycle progression and angiogenesis.34 MLL2 expression is elevated in breast and colon cancer cell lines and in both breast and colon tumor tissue.35 Possible mechanistic links between the MLL2 complex activity and breast carcinogenesis include its association with Pygo2 and consequent activation of Wnt target genes36 and its association with the JMJD2B H3K9 demethylase in a complex required for estrogen receptor α-activated transcription.37 The MLL2 complex may therefore represent a therapeutic target for breast and colon cancer or possibly for solid tumors in general. Furthermore, knockdown of MLLs3 and 4 diminishes estrogen-induced expression of HOXC10, which is overexpressed in breast cancer.38
The WRAD2 subunit WDR5 is critical for assembly of the full MLL/SET1 core complexes, and interactions between the MLL/SET1 subunits and WDR5 are mediated by the Win motif (WDR5 interaction motif), a six-residue sequence located just N-terminal to the overall C-terminal location of the SET domains in this family.27,39 The last four residues of the Win motif (AR-S/A-E) have homology to the four N-terminal residues of histone H3 (ARTK) and, for two of the family members, MLL1 and MLL4, this homology extends beyond the Win motif to include downstream residues identical or similar to H3 residues 8–10 (RKS).39 In fact, the Win motif interacts with WDR5 at a site also implicated in binding the N-terminus of histone H3, and peptides based on both Win and H3 sequence have been found to disrupt the MLL1-WDR5 interaction and inhibit methyltransferase activity.29,40
To date, most of the reported effort to pharmacologically target the MLL1 catalytic activity has centered on attempts to disrupt the MLL1-WDR5 interaction by means of Win-motif-mimicking peptides and small-molecule peptidomimetics.39–43 A parallel strategy aimed at disrupting the Menin-MLL1 interaction of oncogenic MLL1 fusion proteins and/or wild-type MLL144–46 appears to be bearing fruit in the form of efficacy in mouse models of MLL and primary samples from MLL patients47 and in mouse xenograft models of castration-resistant prostate cancer.48 However, for reasons that we will discuss, we have chosen to pursue an assay development strategy that instead focuses on optimizing conditions for the discovery and characterization of inhibitors of MLL/SET1 catalysis per se, that is, inhibitors more likely to bind and affect the SET domain. We describe those efforts here as well as some preliminary results from overlapping proof-of-principle screens of the enhancer of zeste homolog 2 (EZH2) and MLL1 methyltransferase complexes.
Materials and Methods
Materials
S-adenosyl-l-[methyl-3H]-methionine (3H-SAM), 96-well, glass fiber filter plates (UniFilter GF/B), MicroScint-O scintillation fluid, and vacuum filtration rig were obtained from PerkinElmer. The control methyltransferase inhibitor S-adenosyl-l-homocysteine was from Sigma. The RBC Clinical compound library is composed of compounds from the NIH Clinical Collection (NIHCC; www.nihclinicalcollection.com/product_info.php), the LOPAC® 1280 library (Sigma), and the Screen-Well® FDA-approved drug library (Enzo). In addition, a chemically diverse ∼30,000 compound library was assembled from multiple commercial sources. Chromatography media were from GE Life Sciences, except for the Strep-Tactin resin, which was from Qiagen. Chicken core histones were purchased from Millipore. HeLa nucleosomes were prepared from HeLa nuclei (Biovest International) according to Schnitzler.49 Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels were 12% acrylamide and from Pierce.
Recombinant Protein Expression and Complex Assembly
Human recombinant MLL1 (residues 3745–3969; Accession No. NM_005933), MLL2 (residues 5319–5537; Accession No. NM_003482), MLL3 (residues 4689–4911; Accession No. NM_170606), MLL4 (residues 2490–2715; Accession No. NM_014727), WDR5 (residues 22–334; Accession No. NM_017588), RbBP5 (residues 1–538; Accession No. NM_005057), Ash2L (residues 2–534; Accession No. NM_001105214), and DPY-30 (residues 1–99; Accession No. NM_0325742) were expressed in Escherichia coli with N-terminal His-tags. Human recombinant SET1A (residues 1418–1707; Accession No. NM_014712) and SET1B (residues 1629–1923 [C-term.]; Accession No. NM_015048) were expressed in E. coli as N-terminal glutathione S-transferase (GST) fusions. After purification by standard methods, culminating in gel filtration into the assembly and storage buffer (20 mM Tris-HCl, pH 7.5, 300 mM NaCl, 1 mM TCEP, 10% [w/v] glycerol, 1 μM ZnCl2), protein concentrations were determined by measurement of absorbance at 280 nm using calculated extinction coefficients based on the protein sequences. Complexes comprising one each of MLL(1–4) or SET1(A/B), with WDR5, RbBP5, ASH2L, and DPY-30 (WRAD2) in respective molar ratios of 1:1:1:1:2, were assembled by simply mixing the individual component subunits.39 Similarly, the WRAD2 complex was produced separately as a 1:1:1:2 mixture of the latter four subunits.
Recombinant human histone H3.3 (residues 1–136; Accession No. NM_002107) was expressed in E. coli, isolated from inclusion bodies and solubilized, purified, and refolded by standard techniques.50
A baculovirus/insect cell-expressed human recombinant MLL4-WRAD2 complex was produced by coinfection with five viruses encoding MLL4 (residues 2063–2715; Accession No. NM_014727), WDR5 (residues 2–334; Accession No. NM_017588), RbBP5 (residues 1–538; Accession No. NM_005057), Ash2L (residues 2–534; Accession No. NM_001105214), and DPY-30 (residues 1–99; Accession No. NM_0325742). MLL4, RbBP5, ASH2L, and DPY-30 were expressed with N-terminal His-tags and WDR5 with an N-terminal Strep-tag™ II. Purification of the full complex was achieved by Strep-Tactin® chromatography, and a gel filtration step was used to exchange the Strep-Tactin elution buffer for the same storage buffer used for the E. coli-expressed complexes.
The baculovirus/insect cell expression of the human recombinant five-member complex of EZH2 with AEBP2, EED, RbAp48, and SUZ12 was as described previously as was the expression or sources of other enzymes in the HMT profiling panel.51
Methyltransferase Assays
Screening assays were performed with the miniaturized radiolabeled HotSpotSM format, in duplicate, as previously described,51 except that compound concentrations were 50 μM (MLL1, EZH2 RBC Clinical Library screen) or 10 μM (EZH2 HTS Library screen) rather than 20 μM. Briefly, for MLL1, reactions were 1 h, 30°C, with 1 μM 3H-SAM, 0.05 mg/mL chicken core histones (histones H3, H4, H2A, and H2B, ∼1 μM H3) in 50 mM Tris-HCl, pH 8.5, 50 mM NaCl, 5 mM MgCl2, 1 mM DTT, 1 mM PMSF, 1% (v/v) DMSO, with 150 nM MLL1-WRAD2 complex plus an additional 100 nM WRAD2 complex. EZH2 reactions were done under the same conditions except that the EZH2 five-member complex was 50 nM and the reaction buffer was 50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, 1% (v/v) DMSO. Assay statistics for the two enzymes were as follows: MLL1-WRAD2, Z′-factor=0.80, signal/background (S/B)=10.3, coefficient of variation (CV) (%)=5.5; EZH2 complex, Z′-factor=0.76, S/B=5.2, CV (%)=4.1.
Determination of 10-dose compound IC50s was performed in the HotSpot format, as described,51 with a four-parameter curve fitting done with GraphPad Prism. The top compound concentration in all cases was 100 μM. Substrate and other conditions for MLL1 and EZH2 were as described above for screening. For MLL2, MLL3, MLL4, and SET1B, conditions for compound IC50 determinations were as for MLL1, except for enzyme concentrations, which were as follows: MLL2, 50 nM MLL2-WRAD2 complex plus an additional 500 nM WRAD2 complex; MLL3, 50 nM MLL3-WRAD2 complex plus an additional 250 nM WRAD2 complex; MLL4, 50 nM MLL4-WRAD2 complex plus an additional 1,000 nM WRAD2 complex; SET1B, 50 nM SET1B-WRAD2. For selected compounds, MLL1-WRAD2 IC50s were determined with the conditions given above, except that chicken core histones were replaced with HeLa oligonucleosomes (0.05 mg/mL as DNA; ∼0.8 μM histone H3). Conditions for other enzymes in the 15 or 20 HMT profiling panels were as previously described.51
For certain experiments, the methyltransferase activity was determined as TCA-precipitable counts from 25 or 50 μL reactions, as single determinations, in a scintillation/filter plate assay. Assay conditions were the same as described above, but in addition to chicken core histones, HeLa mono/di-nucleosomes were also used (0.05 mg/mL as DNA; ∼0.8 μM histone H3). Twenty-five microliters of reactions was terminated by the addition of 6.3 μL of 50% (w/v) TCA and transferred; after the addition of 31.2 μL of 10% (w/v), 80% of the reaction (50 of 62.5 μL) was transferred to the wells of a 96-well glass fiber filtration plate mounted on a vacuum filtration manifold and suction applied. The preceding TCA addition volumes were doubled for 50 μL reactions. Successive filter washes with 100 μL 10% TCA (3×) and 100 μL 95% ethanol (1×) were accomplished by repeating the vacuum filtration process. Measurement of the methyl group transfer from 3H-SAM to protein substrates was done in a TopCount™ scintillation counter after addition of the scintillation fluid MicroScint-O to the wells of the washed, dried, and bottom-sealed glass fiber filter plate.
Cell-Based Assays
Assays to determine cellular H3K4me2 levels were performed using the AlphaLISA® H3K4Me2 Cell Based Assay for Methyl Modifications, a kit from PerkinElmer (Cat. No. AL716C), with other procedures as described previously for determining cellular H3K27me3,52 except that compound treatments were for 1 day, rather than 3 days. The HeLa H3K4me2 AlphaLISA signal was abolished by competition with an H3K4me2 peptide at 1 μM or higher (IC50=9 nM). In the HeLa cell/H3K4me2 10-dose IC50 experiments performed with the inhibitor compounds RBC-1 to RBC-4, the bottoms of the four-parameter fits were 10% or less of the signal from vehicle-treated cells. Cell viability was determined with the CellTiter-Glo® Luminescent Cell Viability Assay (Promega), also after 1 day of compound treatment.
Gel Densitometry
To quantitate the MLL4 content of fractions from the size exclusion chromatography (SEC) of MLL4-WRAD2, SDS-PAGE gels were stained with GelCode Blue (Thermo), washed with water, scanned with a Typhoon 8610 imager and the bands quantitated using the ImageQuant 5.2 software (Molecular Dynamics).
Results
Properties of the In Vitro-Assembled MLL/SET1-WRAD2 Complexes
The full-length MLL/SET1 family proteins are large, ranging from 1,707 amino acids (SET1A) to 5,537 (MLL2), making recombinant expression in the most economical system, E. coli, unfeasible. Therefore, essentially following the procedures developed for MLL1 in Michael Cosgrove's Laboratory,28 we produced E. coli expression constructs incorporating the C-terminally located, catalytic SET domains and the N-SET region, including the Win-motif, of all six human MLL/SET1 family members. These were then assembled in vitro with the components of the WRAD2 subcomplex, also expressed in E. coli. With the exceptions of SET1A and SET1B, which required N-terminal GST-tags for soluble expression, all eight other proteins were expressed with N-terminal His-tags.
Enzyme titrations of all six MLL/SET1-WRAD2 complexes, using either HeLa mono/di-nucleosomes or core histones as protein substrate, were performed with a radiolabeled filter plate assay (Fig. 1). Several points emerged from this study. First, there is broad variation in the relative activity of the six complexes with their respective preferred substrates (MLL4>MLL1, MLL2>SET1B>MLL3>>SET1A). Second, the three most active enzyme complexes, MLL1, MLL2, and MLL4, preferred the nucleosomal substrate, whereas MLL3, SET1A, and SET1B preferred the core histones. Finally, with the exception of the SET1A/B complexes, the titration curves display a distinctly sigmoidal shape at lower enzyme concentrations (<25 nM).
Fig. 1.

Enzyme titrations of the six MLL/SET1-WRAD2 complexes comparing the activity of HeLa mono/di-nucleosomes (0.05 mg/mL, as DNA) to core histones (0.05 mg/mL) as substrates. Reactions were 1 h, 30°C, with 1 μM 3H-SAM, and the extents of methylation were determined with the scintillation/filter plate assay. (A–F) HeLa mono/di-nucleosomes (■); chicken core histone (□).
Since the full activity of these complexes requires the interaction of the MLL/SET1 subunits with the WRAD2 subcomplex, this latter point suggested that, at lower concentrations, the subunits of the complex may be dissociating, thus reducing the observed methyltransferase activities. Consistent with this occurring in the concentration range of 25 nM, the reported pairwise dissociation constants of the MLL1-WRAD2 complex all fall in the range of 100 nM [Ash2L-(DPY-30)2] to 2.4 μM (WDR5-RbBP5).28 Using the most active of the six complexes, MLL4-WRAD2, we undertook a verification and visualization of this dissociation process, and its effect on activity, by subjecting it to SEC (Superdex 200). We then characterized various portions of the elution profile for subunit content by SDS-PAGE and for activity by the radiolabeled filtration plate assay with HeLa mono/di-nucleosomes as substrate (Fig. 2). Broadly speaking, the elution/activity profile comports with the predictions made by the pairwise interactions reported by Patel et al.28 (MLL-W; W-R; R-A; A-D2) and their relative strength (A-D2, MLL-W>R-A>W-R). As might be expected, the greatest activity is observed in the fractions (B12, C1), which were collected at an elution volume consistent with the molecular weight of the intact, MLL4-WRAD2 complex (212 kDa). Furthermore, fractions C3 and C7/C8 appear to be enriched in subunits consistent with the presence of an RbBP5-Ash2L-(DPY-30)2 subcomplex (∼150 kDa) and an MLL4-WDR5 subcomplex (62 kDa), possibly reflecting breakdown of the weakest pairwise interaction (KD=2.4 μM28), between WDR5 and RbBP5. It is also notable that the greatest of the estimated specific activities, based on densitometry of the MLL4 bands (numbers above the graph columns, lower panel), was in fraction C2. Conceivably, this may reflect an excess of the WRAD2 complex, and/or other subcomplexes, relative to the MLL4 content of that fraction.
Fig. 2.

Analytical size exclusion chromatography (SEC) and activity assays of the fractionated MLL4-WRAD2 complex. MLL4-WRAD2 (225 μL, 10 μM) was loaded on a 24-mL Superdex 200 column (10/300; GE Life Sciences) and eluted, in storage buffer, at 0.5 mL/min. Fractions (0.5 mL) were concentrated 5× (10 kDa MWCO, Ultra 4; Amicon) before analysis by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; 20 μL/lane; 12% acrylamide gel) and methyltransferase assays (25 μL reactions, HeLa oligonucleosomes, 60 min, 30°C, scintillation/filter plate assay). Gel loads of both the 10 μM unfractionated complex (5 μL) and the concentrated fractions (20 μL) were 8× the volume assayed. The stained gel was scanned and the MLL4 bands quantitated by densitometry (Typhoon 8610, ImageQuant software, v. 5.2). The numbers above each column in the lower panel represent estimates of specific activity relative to MLL4 subunit content and were obtained by dividing the activity numbers (CPM; the blue columns) by the arbitrary units from the densitometry of each corresponding gel lane/fraction. Upper panel: UV-absorbance elution profile of the MLL4-WRAD2 subjected to SEC. Middle panel: SDS-PAGE of the column load (left lane) and selected fractions. Lower panel: Methyltransferase activity data for the fraction loaded in the gel lane directly above the column/axis label.
It would be desirable, in terms of the ability to characterize potent inhibitors for purposes of structure/activity studies to inform medicinal chemistry efforts, to be able to run assays with complex concentrations in the nanomolar range. We therefore decided to express a different set of MLL4-WRAD2 constructs, by coinfection in the insect cell/baculovirus system. The MLL4 subunit in this system contains an additional sequence N-terminal to the N-SET region (residues 2063–2715 as opposed to 2490–2715) with the N-terminus placed at the presumptive Taspase1 cleavage site.53 In addition, the N-terminal His-tag of WDR5 is replaced by a Strep-tag II. A similar system, with a FLAG-tagged WDR5, enabled purification of a stable MLL1-WRA complex from insect cells.22 Intriguingly, when assayed with HeLa mono/di- (Fig. 3A) or oligonucleosomes (Fig. 3B) or chicken core histones (Fig. 3C), the insect cell-expressed complex, purified through the Strep-tag II on WDR5, does not display a sigmoidal titration curve, but does when assayed with recombinant histone H3.3 (Fig. 3C). This might suggest that another of the core histones (H4, H2A, H2B) and/or a post-translational modification present on HeLa and chicken H3, but not on E. coli-expressed recombinant H3.3, interacts with the insect cell-expressed complex and stabilizes it. Moreover, while the insect cell-expressed complex has a similar or slightly greater activity with H3.3 and core histones (Fig. 3C), it is only weakly active with nucleosomal substrates (Fig. 3A, B).
Fig. 3.
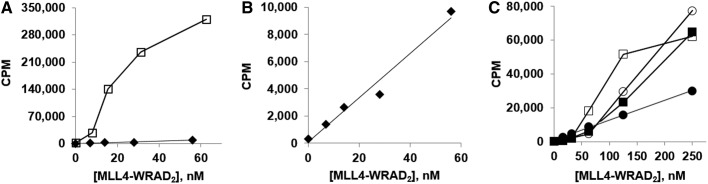
Enzyme titrations comparing the methyltransferase activities of insect cell-expressed and Escherichia coli-expressed MLL4-WRAD2 complexes in the scintillation/filter plate assay. (A) The E. coli-expressed (□) and insect cell-expressed (♦) preparations assayed with HeLa mono/di-nucleosomes. (B) Insect cell-expressed complex assayed with HeLa oligonucleosomes (same data as [A], expanded scale). Linear regression line is y=162.6x+113.6, with R2=0.9717. (C) E. coli-expressed enzyme (□, ■) was assayed with recombinant histone H3.3 (□) or core histones (■), as was the insect cell-expressed preparation (◯, H3.3), (●, core histones). The linear fit to the insect/core histones data (●) is y=117.9x+783.0 with R2=0.9986.
Relative Merits of the Win-Motif Mimetic and SET-Targeted Approaches to MLL/SET1 Drug Discovery
One means to enable assays at low concentrations of the MLL/SET1 component of these complexes would be to perform them in the presence of an excess of WRAD2, thereby driving the subunit binding equilibria toward formation of the fully active complex. This would have the disadvantage, however, of being unsuitable for assays that serve the aim of discovery, characterization, and development of drugs that target the MLL/SET1-WDR5 protein–protein interaction; that is, to say small-molecule mimics of the Win-motif. Almost by definition, such drugs would be specific to the MLL/SET1 family.
Win-motif mimetics, by outcompeting the native Win-motifs of the MLL/SET1 SET-domain-containing subunits, would be expected to displace WDR5 from its interaction with those subunits. To gain some insight into the maximum potential inhibitory effects of such a displacement on the complexes of the MLL/SET1 family, we assembled complexes of three of the family members, MLL1, SET1A, and SET1B, in the presence or absence of the WDR5 subunit and measured the activities of each complex as a function of complex concentration (Fig. 4). Seemingly, in some measure of conflict with some other reported results,43 we find that the omission of WDR5 from assembled MLL/SET1 complexes is by no means completely inhibitory, relative to the activities of each of the full MLL/SET1-WRAD2 complexes (Fig. 4A–C). Furthermore, such diminution of activity as does occur is not specific to MLL1 (compare Fig. 4A, B, and C and see also Zhang et al.54). These radiolabeled assay results with HeLa oligonucleosomes (MLL1) or recombinant histone H3.3 (SET1A/B) are in substantial agreement with the peptide-based results recently reported by Shinsky et al.20 For MLL1, as opposed to SET1B, the loss of activity in the absence of WDR5 is concentration dependent (Fig. 4D), a point that may be related to the enhancement of Win-motif mimetic inhibition of MLL1 at lower complex concentrations.55 This raises the question of how effective this mode of inhibition can be in the absence of an additional factor, such as the dilution-induced disruption of the WDR5-RbBP5 interaction, acting to destabilize other binding interactions in the complex. In contrast to these results with omission of WDR5, Shinsky et al.20 recently reported that the absence of either RbBP5 or Ash2L completely eliminates peptide H3K4 methyltransferase activity for all six SET1/MLL family members.
Fig. 4.

The methyltransferase activities of MLL1 (A), SET1A (B), and SET1B (C) complexes assembled with (■) or without (□) WDR5 are compared. Activities were determined with the scintillation/filter plate assay using HeLa oligonucleosomes as substrate for MLL1 and recombinant histone H3.3 for SET1A and SET1B. In (D), the activity ratios of the −WDR5 to the +WDR5 complex preparations [V(−WDR5)/V(+WDR5)] are compared for SET1B (■) and MLL1 (×) as a function of complex concentration.
Finally, it should be noted that the sigmoidal shape of, for example, the MLL1-WRAD2 titration curve is not a universal feature of the KMTs (Fig. 5A), and in the case of MLL1, it can be eliminated by addition of excess WRAD2 (Fig. 5B).
Fig. 5.
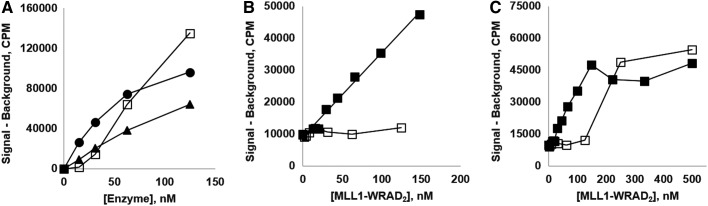
(A) Single-subunit methyltransferases NSD2 (▲) and DOT1L (●) do not display the sigmoidal titration curve that is evident for MLL1-WRAD2 (□). All three were assayed in 1 h, 25 μL reactions with HeLa oligonucleosomes as substrate. (B, C) Addition of an excess of WRAD2 enhances activity and linearizes the lower portion of the MLL1-WRAD2 enzyme titration curve. MLL1-WRAD2 was titrated in the presence (■) or absence (□) of an added 500 nM WRAD2 (HotSpotSM assays with core histones as substrate, 15-min reaction time, each point the mean of duplicate). The linear best fit regression to the 0–150 nM data (■) in (B) is y=263.9x+9045, R2=0.9901.
Compound Screening and Profiling
As proof-of-principle that the approach of compound screening with assay conditions incorporating an added excess of WRAD2 can yield inhibitors with some degree of specificity for MLL1, we undertook screens of the EZH2 and the MLL1-WRAD2 complexes (Fig. 6). Twenty-eight compounds producing 55% or greater inhibition at 10 μM in the EZH2 screen against the HTS Library (hit rate: 0.1%) were advanced to confirmatory IC50 determinations. Fifteen of these compounds with the lowest IC50s were selected for profiling against a panel of 15 HMTs by determination of IC50s. In addition, both the EZH2 and MLL1 complexes were screened against the RBC Clinical Library. IC50s for EZH2 were determined for compounds producing 50% or greater inhibition in the screen (31 compounds; screened at 50 μM; hit rate: 2.3%). The three compounds with the lowest EZH2 IC50s had, in the MLL1 screen, produced inhibitions of 100%, 94%, and 92% at 50 μM and were added to the aforementioned 15 compounds for profiling against the HMT panel. The screen of the MLL1 complex itself against the RBC Clinical Library at 50 μM compound concentrations had yielded, including those three compounds, a total of 32 compounds producing 70% or greater inhibition (hit rate: 2.3%).
Fig. 6.

Outline of the process leading from the screening of enhancer of zeste homolog 2 (EZH2) and MLL1-WRAD2 to the profiling of RBC compounds 1–4.
Included in the HMT panel, along with MLL1-WRAD2, were the MLL2, MLL3, MLL4, and SET1B complexes with WRAD2. For the IC50 determination with these enzymes as well as the initial screening of the EZH2 complex and MLL1-WRAD2, a single substrate was used, chicken core histones. Titrations were performed to determine optimal concentrations of these five MLL/SET1-WRAD2 complexes and the optimal concentration of additional WRAD2. These are listed in the Methyltransferase Assays section. Note that SET1B-WRAD2 was not stimulated by additional WRAD2 and that SET1A-WRAD2, although assayable in the filter plate format, has activity too weak to reliably measure in the HotSpot format. After profiling the 18 hit compounds against the panel of 15 HMTs, 4 compounds (RBC-1 to RBC-4) were chosen for further study by the following: (i) IC50 determinations of HeLa cellular H3K4me2 levels, (ii) IC50 determinations in HeLa cell viability assays, (iii) determination of in vitro MLL1-WRAD2 IC50s with an additional substrate (nucleosomes), and (iv) profiling against five additional HMTs, enlarging the total panel to 20. Two of these four compounds (RBC-1, RBC-2; Table 1) were chosen for their relative specificity for the MLL1 complex and derive from EZH2 hits of the HTS library. The two others (RBC-3, RBC-4) were from the three RBC Clinical Library hits and were chosen for their broad inhibition of the MLL/SET1 family complexes. A flow chart of the overall screening and profiling scheme is depicted in Figure 6. The profiling data for the four compounds with the 20 HMT panel are presented in Table 1 along with IC50s for cellular H3K4me2 and cell viability.
Table 1.
Profiling of Four Screening Hit Compounds by In Vitro Methyltransferase Assays and Assays for Cellular H3K4me2 and Cell Viability
| Methyltransferase or cell-based assay | Compound IC50s, μMb (IC50s<25 μM) | |||||
|---|---|---|---|---|---|---|
| Substratea | RBC-1 | RBC-2 | RBC-3 | RBC-4 | SAH | |
| Assay for cellular H3K4me2c | N/A | 3.4f | 7.2 | 24.3 | 3.9 | ND |
| 9.2 | 23.4 | 1.7 | ||||
| 7.7 | 20.5 | 2.0 | ||||
| Cell viability assayd | N/A | 9.8 | >200 | 85.1 | 30.6 | ND |
| MLL1-WRAD2 | Core histones | 3.7 | 8.4 | 17.6 | 3.0 | 1.3 |
| 3.4 | 11.4 | 17.8 | 3.2 | 0.69 | ||
| MLL1-WRAD2 | Nucleosomes | 2.6 | 33.4 | 10 | 1.8 | 4.7 |
| SET1B-WRAD2 | Core histones | 43.8 | 64.4 | 9.3 | 5.2 | 16 |
| 49.3 | 87 | 9.7 | 5.7 | 7.1 | ||
| MLL2-WRAD2 | Core histones | 132.0 | 179.0 | 11.1 | 13.9 | 14.5 |
| MLL3-WRAD2 | Core histones | 109.0 | >200 | 11.5 | 16.8 | 13.1 |
| MLL4-WRAD2 | Core histones | 84.6 | 75.0 | 8.8 | 5.7 | 9.8 |
| DOT1L | Nucleosomes | >200 | 186.0 | 8.4 | 6.3 | 0.8 |
| G9a | Histone H3 | >200 | >200 | >200 | >200 | ND |
| GLP | Histone H3 | 96.9 | >200 | 24.7 | 12.1 | 0.4 |
| SET7/9 | Core histones | 179.0 | 136.0 | 39.8 | 17.9 | 110.0 |
| SMYD2 | Histone H3 | >200 | 64.5 | 11.6 | 10.7 | 0.7 |
| SUV39H2 | Histone H3 | >200 | >200 | 49.4 | 26.5 | 27.5 |
| SET8 | Histone H4 | >200 | >200 | 27.2 | 12.9 | ND |
| NSD2 | Nucleosomes | >200 | >200 | 3.0 | 4.1 | 3.6 |
| EZH1 complexe | Core histones | >200 | >200 | >200 | 28.8 | 75.4 |
| EZH2 complexe | Core histones | 16.7 | 22.8 | 9.1 | 7.7 | 7.7 |
| 18.4 | 26.6 | 10.4 | 6.5 | 14 | ||
| PRMT1 | Histone H4 | 184.0 | >200 | >200 | >200 | 0.3 |
| PRMT3 | Histone H3 | 17.4 | 185.0 | >200 | >200 | 0.4 |
| PRMT4 | Histone H3 | 117.0 | 188.0 | >200 | >200 | 0.1 |
| PRMT5 | Histone H4 | 14.4 | >200 | >200 | >200 | 0.4 |
| PRMT6 | Histone H4 | 12.6 | 86.5 | >200 | >200 | 0.1 |
Substrate concentrations: Histone H3, Histone H4, and H3 Peptide—5 μM; core histones—0.05 mg/mL; nucleosomes (HeLa oligonucleosomes)—0.05 mg/mL as DNA.
All IC50s represent 10 dose studies (100 μM top concentration) with singlicate data points, 60-min reaction times. Curves were fitted to a four-parameter model with GraphPad Prism.
AlphLISA® H3K4 H3K4Me2 Cell Based Assay for Methyl Modifications.
CellTiter-Glo®Luminescent Cell Viability Assay
Includes AEBP2, EED, RbAp48, and SUZ12 subunits.
IC50 values less than 25 μM are italicized.
EZH, enhancer of zeste homolog; N/A, not applicable; ND, not determined.
Discussion
The tendency of these recombinant MLL/SET1-WRAD2 complexes to dissociate in the tens of nanomolar concentration range somewhat weighs against the practical advantages of the ease of expression and complex assembly afforded by the E. coli-expressed subunits, including the N-SET (Win-motif)-SET subunit constructs. Unfortunately, the insect cell expression system, the major alternative for producing reasonable quantities of the complexes for screening purposes, would seem to be of little practical benefit in terms of lowering the range of MLL4-WRAD2 concentrations at which it would be feasible to conduct assays. Note that although the activity of the insect cell-expressed complex is linear with enzyme concentration into the nanomolar range (Fig. 3B), its activity with the preferred nucleosome substrate is extremely weak compared with that of the E. coli-expressed complex (Fig. 3A). Furthermore, the data of Figure 3 were obtained with the scintillation/filter plate assay which, due to the much larger sample volume, is far more sensitive than assays performed in the HotSpot format used for HTS. Moreover, it is also far more expensive and time-consuming to produce the complex in the insect cell system, and maintaining consistency for the sake of comparisons among the MLL/SET1 family members would have entailed using that system for all six complexes.
Because the E. coli-expressed MLL4-WRAD2 has the greatest activity of the six complexes, we chose it for the SEC/dissociation studies, since this would better allow for activity measurements of partially dissociated complexes than with the other family members that have a weaker activity. The MLL1-WRAD2 complex, however, is currently of greater interest as a drug development target,43,55 so primary emphasis was placed on it in the course of the screening and profiling work.
As noted previously, because of the unique dependence of the MLL/SET1 family complexes on the Win-motif-mediated interaction between MLL/SET1 subunits and WDR5, small-molecule peptidomimetics that blocked this interaction would almost by definition be specific for this family. In contrast, assay conditions that incorporate an excess of WRAD2 might be limited to seeking conventional KMT inhibitors; that is, ones that target the SET catalytic domain, since it would be difficult to outcompete the MLL/SET1-WDR5 binding interaction in the presence of a large excess of WDR5 and the other three subunits of the WRAD2 complex. However, for reasons we discuss below, we would argue that targeting the MLL/SET1 SET domains would, on the one hand, be a worthwhile complement to the Win peptidomimetic approach and, on the other, may in fact have better overall prospects for success.
Based on the results with the Win-mimetic small molecule, MM-410, and on experiments in which WDR5 was omitted from assembled MLL/SET1 family complexes, Cao et al. have argued that the specificity of such inhibitors may be greater still and that only MLL1 activity depends on WDR5.43 We would note that, however, MM-401 inhibition experiments by Cao et al. showing specificity for MLL1 as well as the WDR5 omission experiments showing a lack of effect on the activity of all MLL/SET1 family complexes other than MLL1 were all performed at a single relatively high complex concentration (500 nM)43 and thus might have missed a concentration-dependent effect on WDR5 dependence, such as that observed for MLL1 (Fig. 4D). However, we would also point out that at a 500 nM complex concentration for MLL1 and SET1A, the results reported herein (Fig. 4A, B) are strikingly different from those reported by Cao et al., in which omission of WDR5 has essentially no effect on the SET1A activity, while the MLL1 activity is nearly completely eliminated.43 This might be attributable to the use of different substrates in the respective assays, 50 μM of a 10-mer H3 peptide by Cao et al.43 as opposed to 0.05 mg/mL nucleosomes (∼0.8 μM in histone H3) for MLL1 or 1 μM recombinant histone H3.3 for SET1A/B as herein. On the other hand, Shinsky et al., employing peptide substrates (100 μM) with differing H3K4 methylation states, report, for example, a strong dependence of SET1A complex activity on WDR5 with H3K4me1 or H3K4me2 peptides and actual inhibition of MLL3 complex activity by WDR5 with an unmethylated peptide substrate (H3K4me0).20 Given the conflicting results just discussed, the question of the potential efficacy of a Win-peptidomimetic strategy for either MLL1 specificity or complete inhibition of any MLL/SET1 family member would have to be rated as debatable and open. That said, Cao et al. do report promising cell-based results with MM-401, which inhibited the growth of human mixed-lineage leukemia cell lines and patient-derived cells at concentrations in the tens of micromolar range. Nevertheless, given the doubts that remain, a strategy of developing SET-domain-targeted inhibitors, as enabled by the assay conditions we have described, would seem worthwhile, at the very least, as a complementary approach.
With regard to the points just discussed, it is worth noting the recent spate of successes in developing potent, SET-targeted, SAM-competitive inhibitors of EZH256–62 and to also note that the MLL/SET1 family presents a number of features that parallel those of the EZHs. First, despite one group's catalysis of a gene repressive modification (EZH, H3K27 methylation) and the others catalysis of an activating one (MLL/SET1, H3K4 methylation), the two groups share the same branch of the KMT phylogeny tree.63 In addition, the members of each of the two groups are most active as the five subunit complexes and their subunits share structural features—catalytic subunits with C-terminal SET domains, two subunits each with WD40 domains, one subunit each with zinc fingers.64,65 Moreover, the recent report of a somatic cancer-associated mutation in MLL3 that shifts substrate and product specificity to favor H3K4 trimethylation66 echoes the functional shift described earlier for mutations of EZH2.67,68 Overall, it might be argued that assaying MLL/SET1 family members in the presence of an excess of WRAD2 might be effectively equivalent to assaying a somewhat related, but dilution-stable, complex such as that of EZH2.
Of the four inhibitor compounds profiled in some detail, RBC-2, displayed the highest degree of specificity toward MLL1-WRAD2 (Table 1). At the other end of the specificity spectrum, RBC-4 appears to be a modest and nonspecific lysine methyltransferase inhibitor, while RBC-3 has some selectivity for the entire MLL/SET1 family, relative to most other HMTs. While SET1B and SET1A are thought to contribute the bulk of global H3K4 methylation,17,18 there seems to be a better correlation between the in vitro inhibitory potency toward MLL1-WRAD2 and the decrease in cellular H3Kme2 than there is between SET1B inhibition and declines in H3K4me2. On the one hand, this could be an artifact of following that particular degree of H3K4 methylation. Recent results would seem to indicate that MLL1-WRAD2 can methylate both H3K4me0 and H3K4me1, but is poorly active on H3K4me2, while the SET1A/B-WRAD2s are active on the me0, me1, and me2 forms.20 In that case, inhibition of SET1A/B-WRAD2 could act, in part, to prevent H3K4me2 conversion to H3K4me3 and thereby somewhat counteract the effect of inhibiting the methylation of H3K4me1 to me2. On the other hand, and this need not conflict with the idea just mentioned, MLL1 may play a more widespread role in global H3K4 methylation than is generally suggested.69,70
An ongoing concern for drug discovery efforts involving high-throughput screening of large compound collections are promiscuous inhibitors or nuisance compounds,71,72 which by various mechanisms, such as chemical assay interference73 or compound aggregation-based effects,74 show up repeatedly, across multiple target classes, as false-positive hits.75 A different but related concern is that hit compounds exhibit lead-like properties; that is, they have physical and chemical properties suitable for the downstream analytical and chemical modification procedures employed in the further steps of drug development.76 Since compounds are no longer present at the time of signal measurement, the radiolabeled HotSpot methyltransferase assay51 used in our screening and IC50-based profiling eliminates many, but not all types of assay interferences. A leading cause of compound inhibitory promiscuity, and likely the most significant among the potential interferences not eliminated by the HotSpot assay, is nonspecific inhibition due to compound aggregation.74
We have only screened the 30,000 compound HTS Library against several of the methyltransferases, so any direct assessment of the possible promiscuity of the two hits originating in that library (RBC-1, RBC-2) will necessarily come from examination of the HMT profiling data (Table 1). In this regard, it is noteworthy that of the 20 methyltransferases profiled, RBC-1 and RBC-2 did not inhibit 7 and 9, respectively, (IC50>200 μM) and only weakly inhibited a further 4 and 5 (100 μM<IC50<200 μM), respectively. In addition, although this is merely consistent with a lack of promiscuity due to aggregation,74 the Hill slopes for each compound with EZH2 (RBC-1: ∼0.8; RBC-2: ∼0.5) and MLL1 (RBC-1: ∼1; RBC-2: ∼1) are relatively low. Moreover, each of these compounds has reasonably lead-like properties (e.g., RBC-1: MW<300; RBC-2: MW<400; both compounds 4<logP<5).
In contrast, the case for the nonpromiscuity and nonaggregating character of RBC-3 and RBC-4 is less strong, since the number of HMTs they did not inhibit was fewer, 7 and 6, respectively (IC50>200 μM), and each inhibited none with IC50s in the 100–200 μM range. The Hill slopes of RBC-3 and RBC-4 were also higher with both EZH2 (RBC-3: ∼1.5; RBC-4: ∼2.8) and MLL1 (RBC-3: ∼2.3; RBC-4: ∼2.1). These two compounds also had less lead-like properties (e.g., RBC-3: MW>500, logP>5; RBC-4: MW>400, logP>7). Furthermore, the RBC Clinical Library has been screened against one other target class, the BET bromodomains (BRD2-2, BRD3-1, BRD3-2, BRD4-1, BRD4-2, and BRDT-1 assayed for binding of tetraacetylated histone H4 peptide by AlphaLISA), and RBC-3 and RBC-4 inhibited all of these with IC50s ranging from 10 to 70 μM.
In the course of constructing our screening and profiling strategy for MLL1 and relatives, we became interested in the analogy between the MLL/SET1 family of complex-forming methyltransferases and its similarities to the phylogenetically related group, the EZHs. As noted above, the two displaying the greatest specificity for MLL1-WRAD2, RBC-1, and RBC-2 were actually chosen from the hits identified in the screen of EZH2 with the ∼30,000 compound HTS Library (Fig. 6). Data on cell-based inhibition of H3K27me3 by RBC-2 (designated as “RBC-124”; IC50=13.0 μM in AlphaLISA) have been previously reported.52 We take this as some measure of confirmation of the idea that assay of the MLL/SET1 family in the presence of excess WRAD2 can provide a condition equivalent to that which is feasible, without a molar excess of ancillary non-SET-domain subunits, for a stably assembled preparation such as the EZH2 complex.
In light of the screening and profiling strategy just mentioned, we sought to simplify our procedures and data interpretation by maintaining a uniform assay condition across the MLL/SET1 family and the EZHs. We therefore used core histones as the screening and primary profiling substrate for the EZH2 complex and all of the MLL/SET1 family complexes. For MLL1 in particular, this necessitated the use of a relatively high MLL1-WRAD2 concentration (150 nM) plus an additional 100 nM of WRAD2. However, MLL1-WRAD2 is tremendously more active with nucleosomal substrates (Fig. 1A) and our current routine screening and profiling condition with HeLa oligonucleosomes as substrate is 20 nM MLL1-WRAD2 plus an addition 100 nM WRAD2. By combining the addition of an excess of WRAD2 with a detection technique of greater sensitivity, such as biotinylated nucleosomes in conjunction with streptavidin-coated FlashPlates®, it should prove possible to drop the MLL1 subunit concentration into the low nanomolar range and thereby enable the characterization of highly potent inhibitors that target the SET domain.
Abbreviations Used
- AEBP2
adipocyte enhancer-binding protein 2
- ASH2L
absent small homeotic 2-like, and 2 copies (homodimer) of DPY-30, dumpy-30
- CV
coefficient of variation
- EED
embryonic ectoderm development (polycomb protein EED)
- EZH2
enhancer of zeste homolog 2
- GST-tag
glutathione S-transferase-tag
- H3K4
histone H3 lysine-4
- H3K4me1, 2, or 3
mono-, di-, or trimethylated histone H3 lysine-4
- MLL1/2/3/4
mixed-lineage leukemia protein 1/2/3/4
- RbAp48
retinoblastoma-binding protein p48
- RbBP5
retinoblastoma-binding protein 5
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- S/B
signal/background
- SDS-PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- SEC
size exclusion chromatography
- SET1A/B
SET-domain-containing protein 1A/B
- SET7/9
SET-domain-containing protein 7/9
- SET-domain
su(var)3-9, enhancer of zeste, trithorax-domain
- SUZ12
suppressor of zeste 12 protein homolog
- TCEP
tris(2-carboxyethyl)phosphine HCl
- Win motif
WDR5 interaction motif
- WRAD2
protein complex assembled with one copy each of WDR5, WD-40 repeat protein 5
Disclosure Statement
No competing financial interests exist.
References
- 1.Ruthenburg AJ, Allis CD, Wysocka J: Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell 2007;25:15–30 [DOI] [PubMed] [Google Scholar]
- 2.Shilatifard A: Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol 2008;20:341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernstein BE, Kamal M, Lindblad-Toh K, et al. : Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 2005;120:169–181 [DOI] [PubMed] [Google Scholar]
- 4.Barski A, Cuddapah S, Cui K, et al. : High-resolution profiling of histone methylations in the human genome. Cell 2007;129:823–837 [DOI] [PubMed] [Google Scholar]
- 5.Santos-Rosa H, Schneider R, Bannister AJ, et al. : Active genes are tri-methylated at K4 of histone H3. Nature 2002;419:407–411 [DOI] [PubMed] [Google Scholar]
- 6.Heintzman ND, Stuart RK, Hon G, et al. : Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 2007;39:311–318 [DOI] [PubMed] [Google Scholar]
- 7.Nadal-Ribelles M, Mas G, Millan-Zambrano G, et al. : H3K4 monomethylation dictates nucleosome dynamics and chromatin remodeling at stress-responsive genes. Nucleic Acids Res 2015;43:4937–4949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greer EL, Shi Y: Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 2012;13:343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng Q, Wang H, Ng HH, et al. : Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol 2002;12:1052–1058 [DOI] [PubMed] [Google Scholar]
- 10.Eissenberg JC, Shilatifard A: Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol 2010;339:240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Briggs SD, Bryk M, Strahl BD, et al. : Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev 2001;15:3286–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakamura T, Mori T, Tada S, et al. : ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell 2002;10:1119–1128 [DOI] [PubMed] [Google Scholar]
- 13.Dou Y, Milne TA, Tackett AJ, et al. : Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 2005;121:873–885 [DOI] [PubMed] [Google Scholar]
- 14.Hughes CM, Rozenblatt-Rosen O, Milne TA, et al. : Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell 2004;13:587–597 [DOI] [PubMed] [Google Scholar]
- 15.Wysocka J, Myers MP, Laherty CD, Eisenman RN, Herr W: Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev 2003;17:896–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JH, Skalnik DG: CpG-binding protein (CXXC finger protein 1) is a component of the mammalian Set1 histone H3-Lys4 methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J Biol Chem 2005;280:41725–41731 [DOI] [PubMed] [Google Scholar]
- 17.Lee JH, Tate CM, You JS, Skalnik DG: Identification and characterization of the human Set1B histone H3-Lys4 methyltransferase complex. J Biol Chem 2007;282:13419–13428 [DOI] [PubMed] [Google Scholar]
- 18.Wu M, Wang PF, Lee JS, et al. : Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol Cell Biol 2008;28:7337–7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang P, Lin C, Smith ER, et al. : Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol Cell Biol 2009;29:6074–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shinsky SA, Monteith KE, Viggiano S, Cosgrove MS: Biochemical reconstitution and phylogenetic comparison of human SET1 family core complexes involved in histone methylation. J Biol Chem 2015;290:6361–6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yokoyama A, Wang Z, Wysocka J, et al. : Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol 2004;24:5639–5649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dou Y, Milne TA, Ruthenburg AJ, et al. : Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol 2006;13:713–719 [DOI] [PubMed] [Google Scholar]
- 23.Steward MM, Lee JS, O'Donovan A, Wyatt M, Bernstein BE, Shilatifard A: Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol 2006;13:852–854 [DOI] [PubMed] [Google Scholar]
- 24.Mo R, Rao SM, Zhu YJ: Identification of the MLL2 complex as a coactivator for estrogen receptor alpha. J Biol Chem 2006;281:15714–15720 [DOI] [PubMed] [Google Scholar]
- 25.Lee S, Lee DK, Dou Y, et al. : Coactivator as a target gene specificity determinant for histone H3 lysine 4 methyltransferases. Proc Natl Acad Sci U S A 2006;103:15392–15397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho YW, Hong T, Hong S, et al. : PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem 2007;282:20395–20406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ernst P, Vakoc CR: WRAD: enabler of the SET1-family of H3K4 methyltransferases. Brief Funct Genomics 2012;11:217–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel A, Dharmarajan V, Vought VE, Cosgrove MS: On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J Biol Chem 2009;284:24242–24256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel A, Vought VE, Dharmarajan V, Cosgrove MS: A novel non-SET domain multi-subunit methyltransferase required for sequential nucleosomal histone H3 methylation by the mixed lineage leukemia protein-1 (MLL1) core complex. J Biol Chem 2011;286:3359–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hess JL: MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med 2004;10:500–507 [DOI] [PubMed] [Google Scholar]
- 31.Dorrance AM, Liu S, Chong A, et al. : The Mll partial tandem duplication: differential, tissue-specific activity in the presence or absence of the wild-type allele. Blood 2008;112:2508–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thiel AT, Blessington P, Zou T, et al. : MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell 2010;17:148–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milne TA, Kim J, Wang GG, et al. : Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol Cell 2010;38:853–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ansari KI, Kasiri S, Mandal SS: Histone methylase MLL1 has critical roles in tumor growth and angiogenesis and its knockdown suppresses tumor growth in vivo. Oncogene 2013;32:3359–3370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Natarajan TG, Kallakury BV, Sheehan CE, et al. : Epigenetic regulator MLL2 shows altered expression in cancer cell lines and tumors from human breast and colon. Cancer Cell Int 2010;10:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen J, Luo Q, Yuan Y, et al. : Pygo2 associates with MLL2 histone methyltransferase and GCN5 histone acetyltransferase complexes to augment Wnt target gene expression and breast cancer stem-like cell expansion. Mol Cell Biol 2010;30:5621–5635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi L, Sun L, Li Q, et al. : Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc Natl Acad Sci U S A 2011;108:7541–7546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ansari KI, Hussain I, Kasiri S, Mandal SS: HOXC10 is overexpressed in breast cancer and transcriptionally regulated by estrogen via involvement of histone methylases MLL3 and MLL4. J Mol Endocrinol 2012;48:61–75 [DOI] [PubMed] [Google Scholar]
- 39.Patel A, Vought VE, Dharmarajan V, Cosgrove MS: A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J Biol Chem 2008;283:32162–32175 [DOI] [PubMed] [Google Scholar]
- 40.Karatas H, Townsend EC, Bernard D, Dou Y, Wang S: Analysis of the binding of mixed lineage leukemia 1 (MLL1) and histone 3 peptides to WD repeat domain 5 (WDR5) for the design of inhibitors of the MLL1-WDR5 interaction. J Med Chem 2010;53:5179–5185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patel A, Dharmarajan V, Cosgrove MS: Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J Biol Chem 2008;283:32158–32161 [DOI] [PubMed] [Google Scholar]
- 42.Karatas H, Townsend EC, Cao F, et al. : High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein-protein interaction. J Am Chem Soc 2013;135:669–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cao F, Townsend EC, Karatas H, et al. : Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell 2014;53:247–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grembecka J, He S, Shi A, et al. : Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol 2012;8:277–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li L, Zhou R, Geng H, et al. : Discovery of two aminoglycoside antibiotics as inhibitors targeting the menin-mixed lineage leukaemia interface. Bioorg Med Chem Lett 2014;24:2090–2093 [DOI] [PubMed] [Google Scholar]
- 46.Cierpicki T, Grembecka J: Challenges and opportunities in targeting the menin-MLL interaction. Future Med Chem 2014;6:447–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borkin D, He S, Miao H, et al. : Pharmacologic inhibition of the menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015;27:589–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Malik R, Khan AP, Asangani IA, et al. : Targeting the MLL complex in castration-resistant prostate cancer. Nat Med 2015;21:344–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schnitzler GR: Isolation of histones and nucleosome cores from mammalian cells. Curr Protoc Mol Biol 2001;Chapter 21:Unit 21.5; DOI: 10.1002/0471142727.mb2105s50 [DOI] [PubMed] [Google Scholar]
- 50.Luger K, Rechsteiner TJ, Richmond TJ: Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol Biol 1999;119:1–16 [DOI] [PubMed] [Google Scholar]
- 51.Horiuchi KY, Eason MM, Ferry JJ, et al. : Assay development for histone methyltransferases. Assay Drug Dev Technol 2013;11:227–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qian J, Lu L, Wu J, Ma H: Development of multiple cell-based assays for the detection of histone H3 Lys27 trimethylation (H3K27me3). Assay Drug Dev Technol 2013;11:449–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bier C, Knauer SK, Klapthor A, et al. : Cell-based analysis of structure-function activity of threonine aspartase 1. J Biol Chem 2011;286:3007–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang P, Lee H, Brunzelle JS, Couture JF: The plasticity of WDR5 peptide-binding cleft enables the binding of the SET1 family of histone methyltransferases. Nucleic Acids Res 2012;40:4237–4246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Senisterra G, Wu H, Allali-Hassani A, et al. : Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5. Biochem J 2013;449:151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Knutson SK, Wigle TJ, Warholic NM, et al. : A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 2012;8:890–896 [DOI] [PubMed] [Google Scholar]
- 57.McCabe MT, Ott HM, Ganji G, et al. : EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012;492:108–112 [DOI] [PubMed] [Google Scholar]
- 58.Konze KD, Ma A, Li F, et al. : An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem Biol 2013;8:1324–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knutson SK, Kawano S, Minoshima Y, et al. : Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther 2014;13:842–854 [DOI] [PubMed] [Google Scholar]
- 60.Knutson SK, Warholic NM, Johnston LD, et al. : Synergistic anti-tumor activity of EZH2 inhibitors and glucocorticoid receptor agonists in models of germinal center non-Hodgkin lymphomas. PLoS One 2014;9:e111840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Aller GS, Pappalardi MB, Ott HM, et al. : Long residence time inhibition of EZH2 in activated polycomb repressive complex 2. ACS Chem Biol 2014;9:622–629 [DOI] [PubMed] [Google Scholar]
- 62.Kung PP, Huang B, Zehnder L, et al. : SAH derived potent and selective EZH2 inhibitors. Bioorg Med Chem Lett 2015;25:1532–1537 [DOI] [PubMed] [Google Scholar]
- 63.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M: Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov 2012;11:384–400 [DOI] [PubMed] [Google Scholar]
- 64.Takahashi YH, Westfield GH, Oleskie AN, Trievel RC, Shilatifard A, Skiniotis G: Structural analysis of the core COMPASS family of histone H3K4 methylases from yeast to human. Proc Natl Acad Sci U S A 2011;108:20526–20531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ciferri C, Lander GC, Maiolica A, Herzog F, Aebersold R, Nogales E: Molecular architecture of human polycomb repressive complex 2. Elife 2012;1:e00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weirich S, Kudithipudi S, Kycia I, Jeltsch A: Somatic cancer mutations in the MLL3-SET domain alter the catalytic properties of the enzyme. Clin Epigenetics 2015;7:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sneeringer CJ, Scott MP, Kuntz KW, et al. : Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A 2010;107:20980–20985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McCabe MT, Graves AP, Ganji G, et al. : Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A 2012;109:2989–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guenther MG, Jenner RG, Chevalier B, et al. : Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci U S A 2005;102:8603–8608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bina M, Wyss P, Novorolsky E, et al. : Discovery of MLL1 binding units, their localization to CpG Islands, and their potential function in mitotic chromatin. BMC Genomics 2013;14:927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shoichet BK: Screening in a spirit haunted world. Drug Discov Today 2006;11:607–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sink R, Gobec S, Pecar S, Zega A: False positives in the early stages of drug discovery. Curr Med Chem 2010;17:4231–4255 [DOI] [PubMed] [Google Scholar]
- 73.Dahlin JL, Nissink JW, Strasser JM, et al. : PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J Med Chem 2015;58:2091–2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Feng BY, Simeonov A, Jadhav A, et al. : A high-throughput screen for aggregation-based inhibition in a large compound library. J Med Chem 2007;50:2385–2390 [DOI] [PubMed] [Google Scholar]
- 75.Baell JB, Holloway GA: New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 2010;53:2719–2740 [DOI] [PubMed] [Google Scholar]
- 76.Rishton GM: Molecular diversity in the context of leadlikeness: compound properties that enable effective biochemical screening. Curr Opin Chem Biol 2008;12:340–351 [DOI] [PubMed] [Google Scholar]