

Abstract
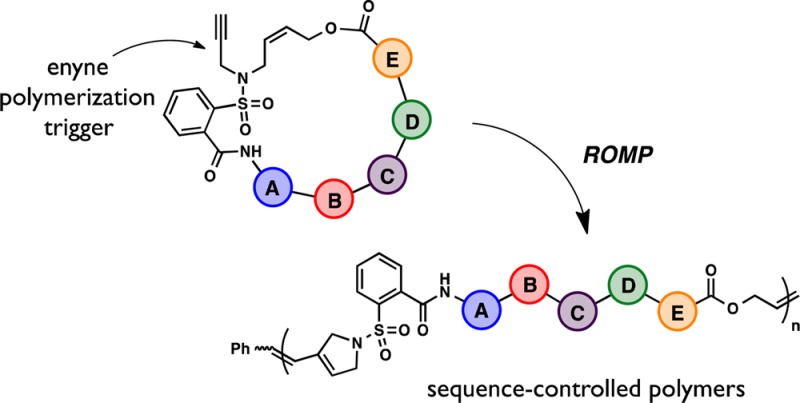
A new and general strategy for the synthesis of sequence-defined polymers is described that employs relay metathesis to promote the ring opening polymerization of unstrained macrocyclic structures. Central to this approach is the development of a small molecule “polymerization trigger” which when coupled with a diverse range of sequence-defined units allows for the controlled, directional synthesis of sequence controlled polymers.
Biopolymers, such as proteins and DNA, clearly illustrate the power of defined polymer sequences, with remarkable functions such as catalysis, molecular recognition, and data storage emerging. Inspired by these natural, sequence-defined materials, strategies to control the primary structure of synthetic polymers have become an active area of research with the aim to develop materials with next-generation performance and functions.1 Synthetic methods for sequence-controlled polymerization fall into three general categories: iterative, step growth, and chain growth. Iterative strategies employ the classic Merrifield approach in which each monomer is appended to the chain end sequentially.2 This leads to highly controlled microstructures, but is unable to produce high molecular weights and is generally time intensive. Step-growth approaches, such as click polymerization3 or ADMET,4 can generate elaborate periodic polymers from preformed sequences, but lack control over polymer molecular weight and dispersity. In contrast, chain-growth strategies have controlled polymerization characteristics, but lack a high level of sequence precision when compared to the previous methods. Recent advances in chain-growth chemistry include the timed addition of kinetically fast monomers,5 templated polymerizations,6 cyclooctene ring opening metathesis polymerization (ROMP),7 and multiblock copolymers.8 In order to combine the sequence precision of step-growth methods with the functional group tolerance and chain-growth characteristics of ROMP, the living polymerization of sequence-defined macrocycles is reported herein.
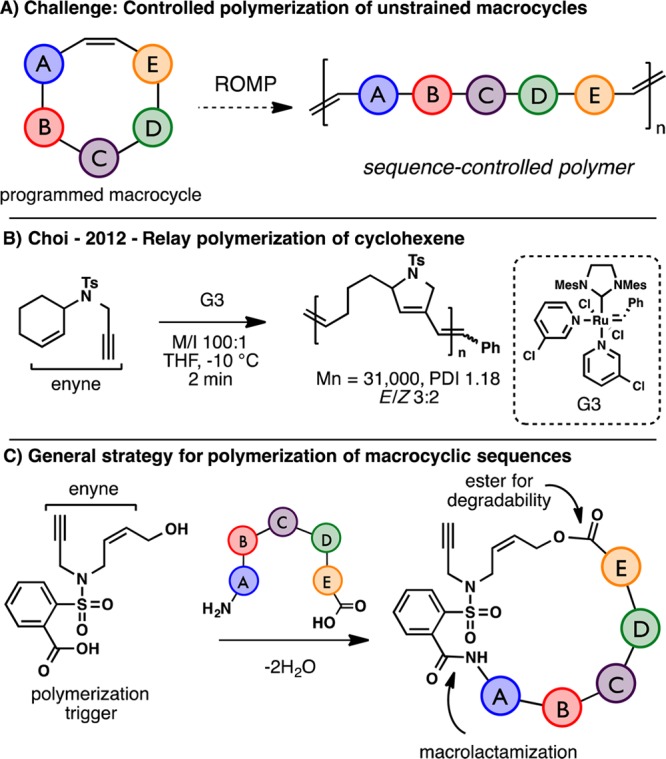
The challenge inherent to this approach is the lack of ring strain for macrocycles when compared with traditional ROMP monomers (Scheme 1A). Inspiration for this new strategy originates from Hoye’s seminal work on relay-ring closing metathesis.9 A tandem reaction was therefore desired that could “relay” a ruthenium carbene to affect a kinetically driven, intramolecular ring opening of the macrocyclic olefin. Further encouragement derives from Choi’s polymerization of an unstrained cyclohexene using an alkyne unit as the relay (Scheme 1B).10 While NHC-ligated Grubbs catalysts are highly reactive toward terminal alkynes,11 the resulting vinyl ruthenium carbenes are usually resistant to further metathesis chemistry. As a result, the key to this process is the close proximity of the cyclohexene olefin to the alkyne, permitting fast intramolecular cyclization and rapid polymerization. Given this precedent, it was reasoned that an unstrained macrocyclic enyne should also be a competent monomer. With this idea in mind, a small molecule “polymerization trigger” was envisioned that contains the critical enyne motif and also functional handles for incorporation of arbitrary macrocyclic sequences (Scheme 1C).
Scheme 1. Development of Macrocyclic ROMP.
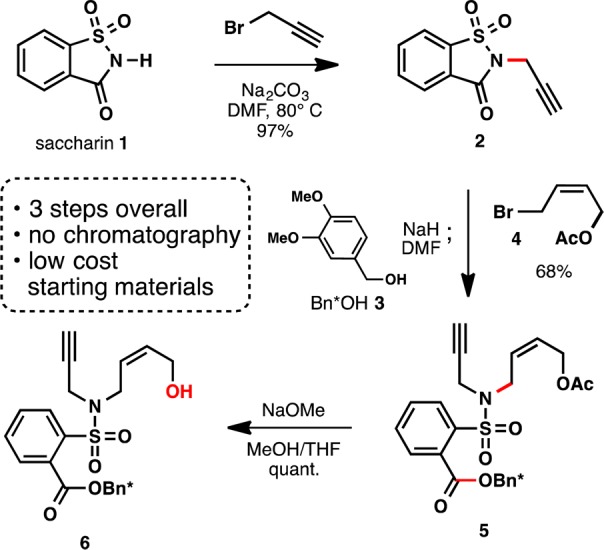
In order to test this concept, an efficient and scalable synthesis of the polymerization trigger was devised starting from readily available saccharin 1, a commercial artificial sweetener. N-Alkylation of saccharin with propargyl bromide gives 2, which was then subjected to a one-pot difunctionalization sequence (Scheme 2). First, the saccharin heterocycle was regioselectively ring-opened with veratryl alcohol 3, an acid labile protecting group, to generate an intermediate sulfonamide anion. Addition of acetoxy allyl bromide 4 to this anion in the same pot furnished the desired enyne system 5 as a crystalline solid in 68% yield. Finally, the acetate was quantitavely cleaved with sodium methoxide to give the monoprotected polymerization trigger 6. Notably, this short sequence employs inexpensive, readily available reagents and does not require column chromatography, as each intermediate is isolated via precipitation or recrystallization.
Scheme 2. Polymerization Trigger Synthesis.
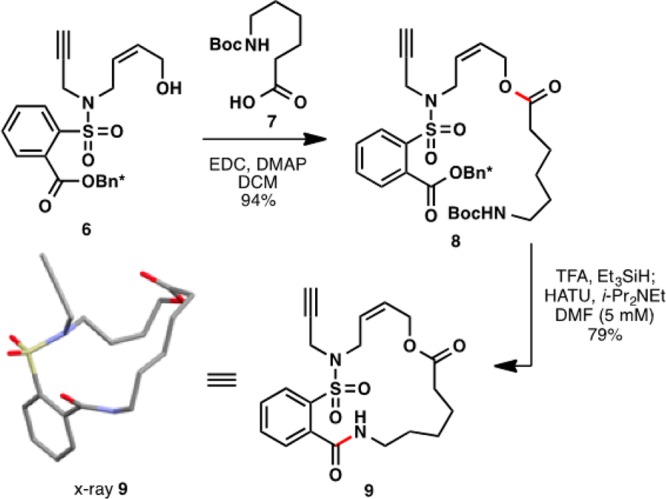
In order to evaluate the competency of the polymerization trigger, a simple aliphatic amino acid “sequence” was investigated. The macrocyclic monomer 9 was prepared as shown in Scheme 3, involving EDC coupling, deprotection with TFA, and HATU mediated macrocyclization. Significantly, the macrocyclization gave the crystalline, 17-membered monomer 9 in a very reasonable 79% yield.
Scheme 3. Synthesis and X-ray Crystal Structure of Macrocyclic Monomer 9.
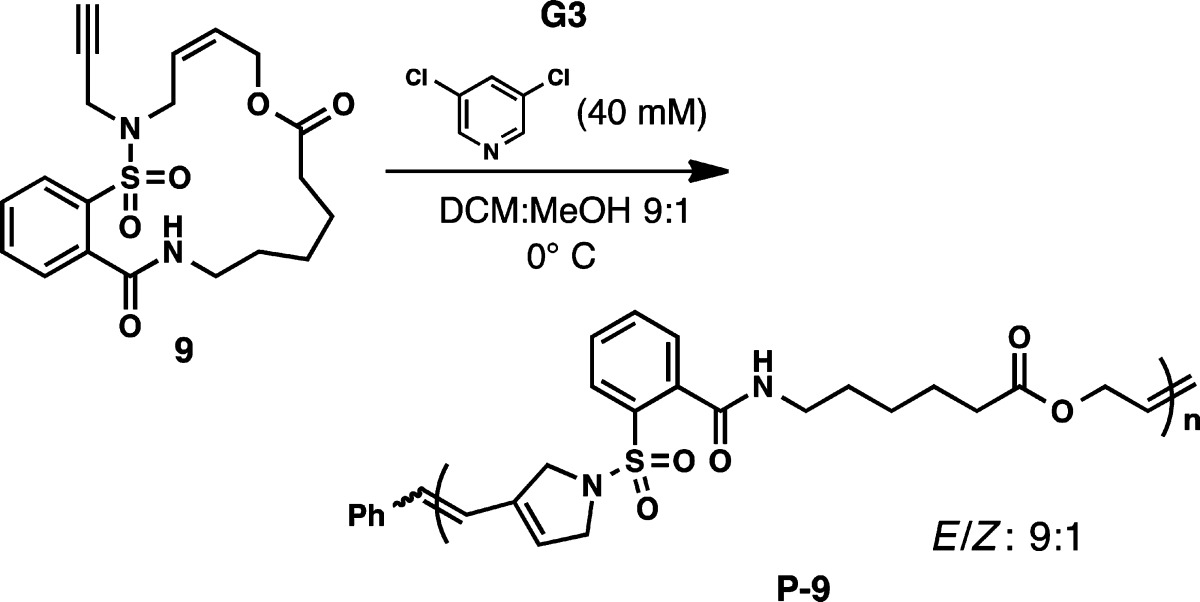
Initial attempts to polymerize macrocycle 9 with Grubbs third generation catalyst (G3) revealed solubility issues in THF, DCM, and other traditional solvents. While a DCM/methanol solvent system resulted in homogeneous polymerization, rapid chain transfer caused considerable broadening of the PDIs. However, adding 3,5-dichloropyridine as an additional ligand resulted in controlled polymerization of 9 to P-9 with excellent olefin stereoselectivity (9:1 E/Z), as shown in Table 1.12 When the polymerization was performed with a monomer to initiator ratio (M/I) of 50:1 (entries 1–4), conversion of the monomer increased with molecular weight over time, with full conversion reached in 15 min at 0 °C.13 Further support for the chain growth and controlled nature of this polymerization is the ability to target different molecular weights by varying the M/I, with high conversions being obtained in each case (entries 5–8). This demonstrates the synthetic utility of this novel macrocyclic ROMP as a variety of polymers can be prepared with excellent control over molecular weight and molecular weight distribution.
Table 1. Polymerization of Macrocyclic Monomer 9.
| entry | time (min) | M/I | Mn (g/mol)a | Đa | conversionb |
|---|---|---|---|---|---|
| 1 | 1 | 50 | 11 500 | 1.07 | 38% |
| 2 | 2 | 50 | 17 100 | 1.12 | 48% |
| 3 | 5 | 50 | 22 100 | 1.20 | 79% |
| 4 | 15 | 50 | 24 900 | 1.30 | 98% |
| 5 | 3 | 12.5 | 8500 | 1.18 | 90% |
| 6 | 5 | 25 | 12 800 | 1.19 | 91% |
| 7 | 10 | 50 | 21 000 | 1.26 | 89% |
| 8 | 15 | 75 | 32 900 | 1.51 | 95% |
Determined using DMF size exclusion chromatography calibrated with polystyrene standards.
Determined by crude 1H NMR.
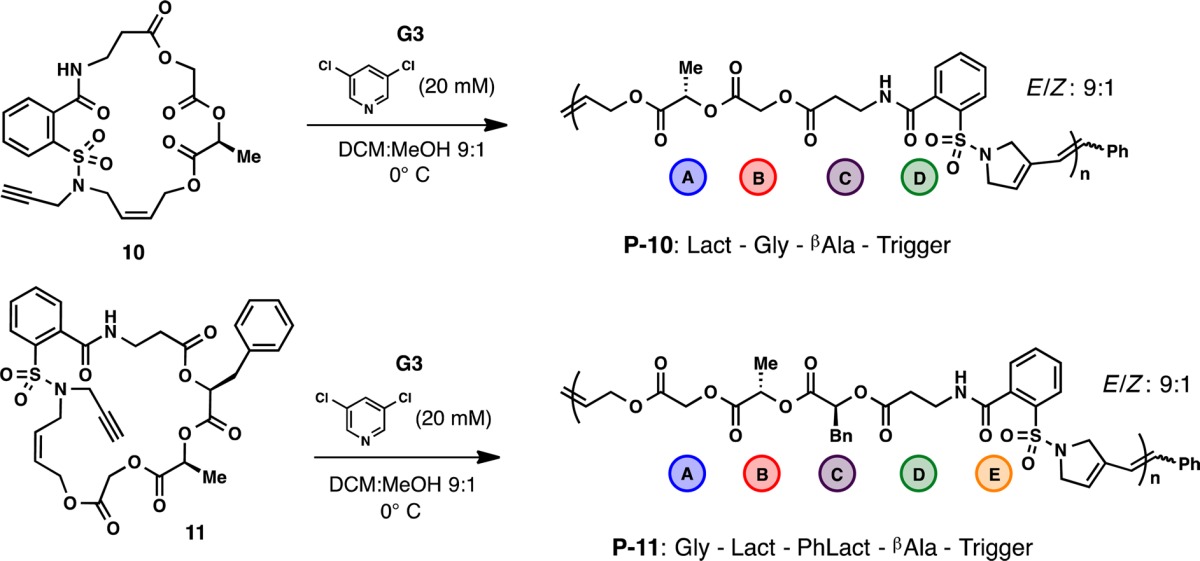
With this proof of concept established and general conditions developed, more elaborate sequences were developed following Meyer’s work on sequenced polyesters.14 The hybrid-polyester monomers 10 and 11 were readily prepared using iterative coupling methods15 (see SI for details), resulting in 20- and 23-membered macrocycles, respectively (Table 2). The monomers are comprised of the polymerization trigger attached to a series of glycolate (Gly), (S)-lactate (Lact), (S)-phenyllactate (PhLact), and β-alanine (βAla) units to give ABCD and/or E sequences that will be displayed in the backbone after ROMP. These monomers proved to polymerize slower than 9 using the developed reaction conditions, but reducing the amount of ligand to 20 mM led to comparable reaction rates. Under these conditions, both monomer 10 (entries 1 and 2) and monomer 11 (entries 3–6) produced sequence-defined polymers with accurate control over molecular weight and low polydispersity. For example, entry 4 gave P-11 with an Mn of 32 600 and a PDI of 1.26, which corresponds to a polymer chain consisting of ∼250 perfectly sequenced units. This readily illustrates the precision and wide range of chemical diversity that can be incorporated into these synthetic polymers with functionality ranging from chiral esters to sulfonamides to amides.
Table 2. Polymerization of Sequenced Macrocycles 10 and 11.
| entry | monomer | time (min) | M/I | Mn (g/mol)a | Đa | conversionb |
|---|---|---|---|---|---|---|
| 1 | 10 | 5 | 25 | 10 800 | 1.18 | 81% |
| 2 | 10 | 10 | 50 | 19 300 | 1.27 | 92% |
| 3 | 11 | 3 | 12.5 | 9400 | 1.16 | 77% |
| 4 | 11 | 5 | 25 | 16 300 | 1.15 | 81% |
| 5 | 11 | 10 | 50 | 32 600 | 1.26 | 92% |
| 6 | 11 | 15 | 75 | 41 400 | 1.39 | 84% |
Determined using DMF size exclusion chromatography calibrated with polystyrene standards.
Determined by crude 1H NMR.
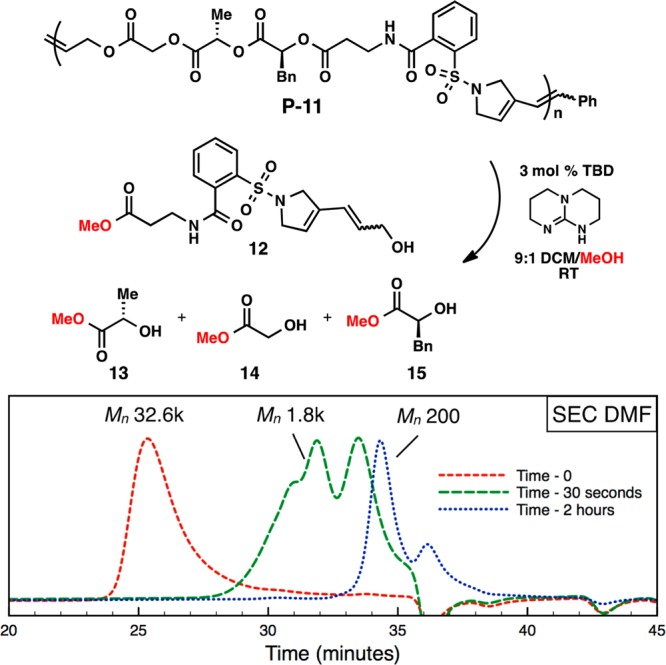
A further unique aspect of this methodology compared to traditional ROMP is the ability to control functionality in the backbone of the polymer. Traditional ROMP polymers are not hydrolytically degradable, a quality that limits their use in a number of applications.16 Since P-10 and P-11 are polyesters, they can break down into their small molecule constituents. To demonstrate this reactivity, P-11 was degraded with triazabicyclodecene (TBD) using conditions developed by Leibfarth and co-workers.17 The reaction was readily followed by GPC analysis with a dramatic molecular weight reduction observed after only 30 s and complete conversion to small molecule methyl esters (12–15) after 2 h (Scheme 4). These degradation experiments highlight the enabling nature of this macrocyclic ROMP strategy for accessing sequence-defined polymers, as well as for introducing specific properties into these materials.
Scheme 4. Degradation of P-11 with TBD.
In summary, a small molecule polymerization trigger 6 has been developed that allows for the facile synthesis and polymerization of unstrained macrocyclic sequences. For the first time, polymers with arbitrary functionality (ester, amide, sulfonamide, aliphatic, aromatic, heterocyclic, etc.) within the backbone can be produced while still providing control over molecular weight and molecular weight distribution. Future work will examine the general nature of this platform and its application to the synthesis copolymers with traditional ROMP monomers for the creation of new functional materials with applications in fields ranging from catalysis to self-assembly to drug delivery.
Acknowledgments
We thank the MRSEC program of the National Science Foundation (DMR 1121053, W.R.G. and C.J.H.) for funding. W.R.G. thanks the NIH for a postdoctoral fellowship (F32GM108323). We also thank Dr. Guang Wu (UCSB) for X-ray analysis.
Supporting Information Available
Experimental procedures for preparation of all compounds and characterization data for all compounds. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b04940.
The authors declare no competing financial interest.
Supplementary Material
References
- a Lutz J.-F. Polym. Chem. 2010, 1, 55–62. [Google Scholar]; b Badi N.; Lutz J.-F. Chem. Soc. Rev. 2009, 38, 3383. [DOI] [PubMed] [Google Scholar]; c Lutz J.-F.; Ouchi M.; Liu D. R.; Sawamoto M. Science 2013, 341, 1238149. [DOI] [PubMed] [Google Scholar]
- a Merrifield R. B. J. Am. Chem. Soc. 1963, 85, 2149. [Google Scholar]; b Espeel P.; Carrette L. L. G.; Bury K.; Capenberghs S.; Martins J. C.; Du Prez F. E.; Madder A. Angew. Chem., Int. Ed. 2013, 52, 13261. [DOI] [PubMed] [Google Scholar]; c Pfeifer S.; Zarafshani Z.; Badi N.; Lutz J.-F. J. Am. Chem. Soc. 2009, 131, 9195. [DOI] [PubMed] [Google Scholar]; d Rosales A. M.; Segalman R. A.; Zuckermann R. N. Soft Matter 2013, 9, 8400. [Google Scholar]
- Chen Y.; Guan Z. J. Am. Chem. Soc. 2010, 132, 4577. [DOI] [PubMed] [Google Scholar]
- a Atallah P.; Wagener K. B.; Schulz M. D. Macromolecules 2013, 46, 4735. [Google Scholar]; b Li Z.-L.; Lv A.; Du F.-S.; Li Z.-C. Macromolecules 2014, 47, 5942. [Google Scholar]
- a Pfeifer S.; Lutz J.-F. J. Am. Chem. Soc. 2007, 129, 9542. [DOI] [PubMed] [Google Scholar]; b Schmidt B. V. K. J.; Fechler N.; Falkenhagen J.; Lutz J.-F. Nat. Chem. 2011, 3, 234. [DOI] [PubMed] [Google Scholar]; c Zamfir M.; Lutz J.-F. Nat. Commun. 2012, 3, 1138. [Google Scholar]; d Moatsou D.; Hansell C. F.; O’Reilly R. K. Chem. Sci. 2014, 5, 2246. [Google Scholar]
- a Hibi Y.; Ouchi M.; Sawamoto M. Angew. Chem. 2011, 123, 7572. [DOI] [PubMed] [Google Scholar]; b Niu J.; Hili R.; Liu D. R. Nat. Chem. 2013, 5, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang J.; Matta M. E.; Hillmyer M. A. ACS Macro Lett. 2012, 1, 1383. [DOI] [PubMed] [Google Scholar]; b Kobayashi S.; Pitet L. M.; Hillmyer M. A. J. Am. Chem. Soc. 2011, 133, 5794. [DOI] [PubMed] [Google Scholar]
- a Gody G.; Maschmeyer T.; Zetterlund P. B.; Perrier S. Macromolecules 2014, 47, 3451. [Google Scholar]; b Anastasaki A.; Nikolaou V.; Pappas G. S.; Zhang Q.; Wan C.; Wilson P.; Davis T. P.; Whittaker M. R.; Haddleton D. M. Chem. Sci. 2014, 5, 3536. [Google Scholar]
- Hoye T. R.; Jeffrey C. S.; Tennakoon M. A.; Wang J.; Zhao H. J. Am. Chem. Soc. 2004, 126, 10210. [DOI] [PubMed] [Google Scholar]
- a Park H.; Choi T.-L. J. Am. Chem. Soc. 2012, 134, 7270. [DOI] [PubMed] [Google Scholar]; b Park H.; Lee H.-K.; Choi T.-L. J. Am. Chem. Soc. 2013, 135, 10769. [DOI] [PubMed] [Google Scholar]
- Lee O. S.; Kim K. H.; Kim J.; Kwon K.; Ok T.; Ihee H.; Lee H.-Y.; Sohn J.-H. J. Org. Chem. 2013, 78, 8242. [DOI] [PubMed] [Google Scholar]
- Kang E.-H.; Yu S. Y.; Lee I. S.; Park S. E.; Choi T.-L. J. Am. Chem. Soc. 2014, 136, 10508. [DOI] [PubMed] [Google Scholar]
- During polymerization a monomer isomerization process is observed (15–20%); see SI for discussion.
- a Li J.; Rothstein S. N.; Little S. R.; Edenborn H. M.; Meyer T. Y. J. Am. Chem. Soc. 2012, 134, 16352. [DOI] [PubMed] [Google Scholar]; b Li J.; Stayshich R. M.; Meyer T. Y. J. Am. Chem. Soc. 2011, 133, 6910. [DOI] [PubMed] [Google Scholar]
- a Takizawa K.; Nulwala H.; Hu J.; Yoshinaga K.; Hawker C. J. J. Poly. Sci., Part A: Polym. Chem. 2008, 46, 5977. [Google Scholar]; b Takizawa K.; Tang C.; Hawker C. J. J. Am. Chem. Soc. 2008, 130, 1718. [DOI] [PubMed] [Google Scholar]
- Fishman J. M.; Kiessling L. L. Angew. Chem., Int. Ed. 2013, 52, 5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibfarth F. A.; Moreno N.; Hawker A. P.; Shand J. D. J. Poly. Sci., Part A: Polym. Chem. 2012, 50, 4814. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.