

Abstract
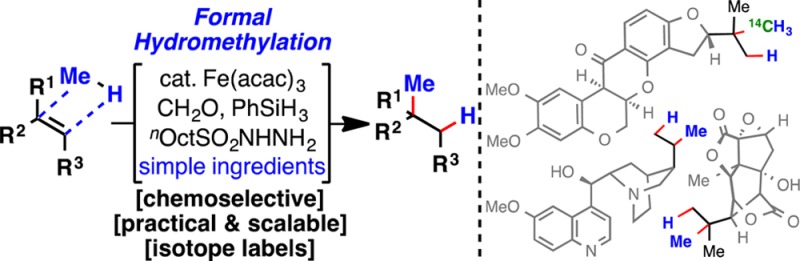
A solution to the classic unsolved problem of olefin hydromethylation is presented. This highly chemoselective method can tolerate labile and reactive chemical functionalities and uses a simple set of reagents. An array of olefins, including mono-, di-, and trisubstituted olefins, are all smoothly hydromethylated. This mild protocol can be used to simplify the synthesis of a specific target or to directly “edit” complex natural products and other advanced materials. The method is also amenable to the simple installation of radioactive and stable labeled methyl groups.
The direct, chemoselective hydromethylation of unactivated olefins is a classic unsolved problem of great importance in organic chemistry. This is clearly illustrated in the context of the total synthesis of 7-desmethyl-2-methoxycalamenene 2a (Figure 1A).1a,1b To be sure, three different indirect pathways were engineered to accomplish the seemingly simple addition of methane across olefin 1a. In the first approach, a Wacker/Wittig sequence provided intermediate A in low yield. Although the route via B gave an improvement in yield, a toxic mercury reagent was required. Finally, the most frequently used strategy for formal hydromethylation of olefins, employing a Simmons–Smith cyclopropanation and a reductive C–C bond cleavage (via C), gave an unsatisfactory yield.1b−1d This Communication reports a solution to this frequently encountered issue with the invention of a mild, scalable, and catalytic olefin hydromethylation. This simple method can be used at both the early and late stages of a synthesis, enabling access to uniquely modified natural products, medicinally relevant molecules, and even isotopically labeled structures that would be difficult or impossible to access in any other way.
Figure 1.
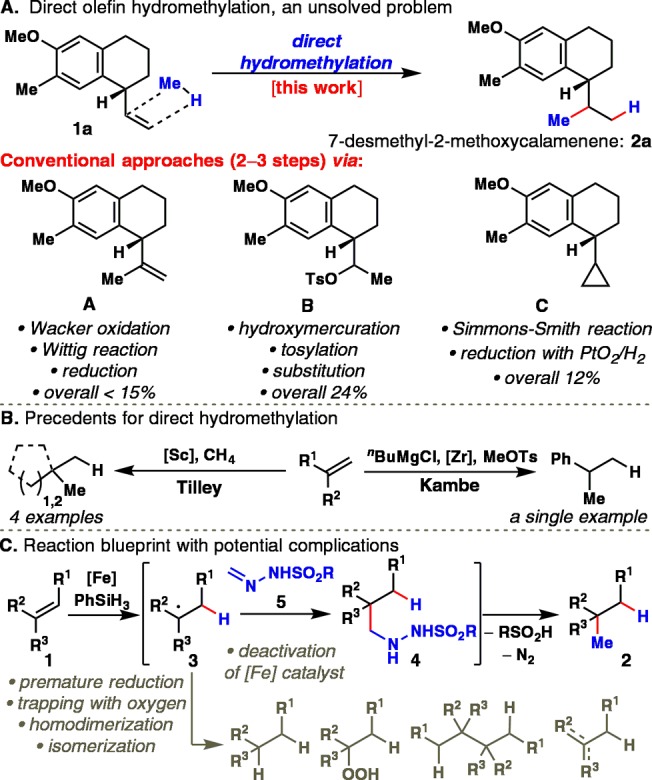
(A) Direct olefin hydromethylation, an unsolved problem in organic synthesis. (B) Precedents for hydromethylation. (C) Reaction blueprint with potential complications.
Precedents for the direct hydromethylation of olefins exist sporadically but are too substrate specific to be applicable in medicinal and natural product synthesis (Figure 1B).2 For example, Tilley described a conceptually appealing transformation for hydromethylation using methane via an in situ-formed methylscandium species.2a Only four unfunctionalized alkenes were employed due to the pyrophoric properties and the selectivity of the catalyst. In a different approach, Kambe reported a single example of direct hydromethylation of styrene employing a Cp2ZrCl2/nBuMgCl system, with methyl tosylate as the methylating reagent.2b
Following the pioneering work of Mukaiyama,3 we and others have invented a series of direct olefin functionalizations using Co-,4 Mn-,4d,5 and Fe-based systems.3d,6 Mukaiyama-type functionalizations benefit from inherently mild conditions which tolerate a variety of unprotected functionality.7 Since such reactions are thought to proceed via a nucleophilic radical 3 (Figure 1C), a variety of potential electrophilic methyl group surrogates were evaluated without success (see Supporting Information (SI)). In this context, the Kim group’s demonstration that radicals could add to hydrazones in an intramolecular context was particularly inspiring.8 In principle, capture of 3 with hydrazone 5 would form alkyl hydrazide 4, which can eliminate sulfinic acid and nitrogen to yield the net hydromethylated adduct 2. The successful execution of this plan would hinge on the ability to suppress byproducts resulting from reduction,9 oxidation,3−5 dimerization, and isomerization.10
Reducing this design to practice required extensive experimentation, the essence of which is summarized in Table 1. Initial efforts to isolate formaldehyde hydrazone 5 failed, presumably due to oligomerization. However, detection by LC-MS suggested that in situ use of 5 was feasible. To suppress the formation of oxidized byproducts, the reaction was degassed and conducted under an inert atmosphere. Systematic screening of reaction parameters led to the invention of a one-pot protocol for the transformation of 1b to 2b. Thus, Fe-mediated addition of 1b to in situ-formed hydrazone 5 yielded hydrazide 4b in THF; a solvent switch to methanol and gentle heating at 60 °C facilitated reductive C–N bond cleavage to deliver 2b in good yield (entry 1).
Table 1. Effect of Reaction Parameters on the Hydromethylation of 1b.
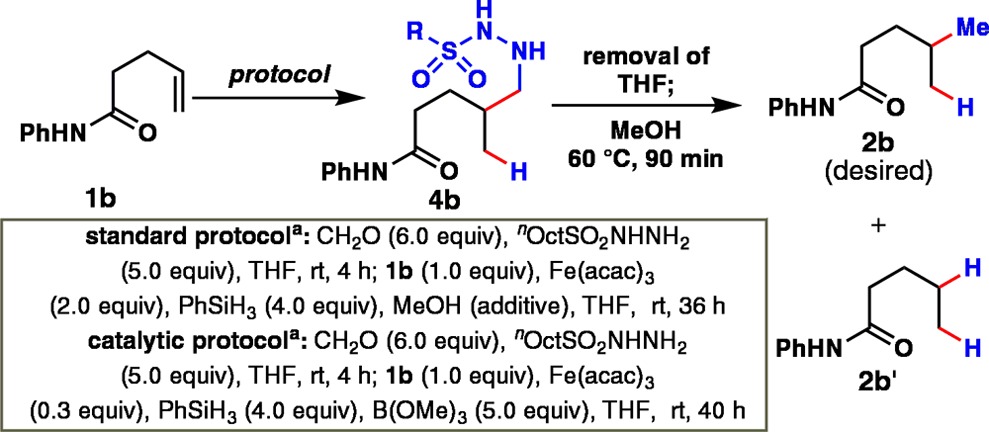
| yield (%) |
||||
|---|---|---|---|---|
| entry | deviation from standard protocol | 1bb | 2bb | 2b′b |
| 1 | none | 9 | 51 | 12 |
| 2 | nBuSO2NHNH2 instead of nOctSO2NHNH2 | 11 | 47 | 15 |
| 3 | nDecSO2NHNH2 instead of nOctSO2NHNH2 | 18 | 36 | 8 |
| 4 | iPrSO2NHNH2 instead of nOctSO2NHNH2 | 1 | 21 | 36 |
| 5 | p-MeC6H4SO2NHNH2 instead of nOctSO2NHNH2 | 27 | 19 | 22 |
| 6 | p-MeOC6H4SO2NHNH2 instead of nOctSO2NHNH2 | 2 | 31 | 20 |
| 7 | p-BrC6H4SO2NHNH2 instead of nOctSO2NHNH2 | 18 | 22 | 21 |
| 8 | Fe(dibm)3 instead of Fe(acac)3 | 19 | 31 | 18 |
| 9 | Fe2(ox)3/NaBH4 instead of Fe(acac)3/PhSiH3 | nd | 0d | nd |
| 10 | Co(II) and Co(III)c instead of Fe(acac)3 | nd | 0d | nd |
| 11 | Mn(III)e instead of Fe(acac)3 | nd | 0d | nd |
| 12 | EtOH instead of THF | 0 | 35 | 16 |
| 13 | reagents added in two portionf | 3 | 53g | 15g |
| 14 | catalytic protocolh | 0 | 56 (51g) | 4 |
Degassed and run under argon.
According to GC analysis.
Co(Sal) (1 equiv) or Co(SaltBu,tBu) (1 equiv) or Co(acac)3 (2 equiv).
According to LC-MS.
Mn(dpm)3 (2 equiv) or Jacobsen’s catalyst (0.5 equiv).
First portion: CH2O (3.0 equiv), nOctSO2NHNH2 (2.5 equiv), Fe(acac)3 (1.0 equiv), PhSiH3 (2.0 equiv), MeOH (2.0 equiv), THF, rt, 15 h. Second portion: 18 h.
Isolated yields.
This catalytic protocol requires B(OMe)3, as without it, the reaction gives <30% conversion. nd = not detected.
Entries 2–7 summarize the search for an optimal R-substituent on the hydrazone “donor” (5), eventually identified as the n-octyl group. The use of a more sterically hindered catalyst (Fe(dibm)3, entry 8) led to a slower reaction, while replacement of iron with other metal complexes did not yield any desired product (entries 9–11). These findings highlight the unique properties of iron catalysts for the current C–C bond-forming reaction. In contrast to prior studies,6e,6f THF proved superior to ethanol for the current transformation (entry 12). Ultimately, addition of the same overall quantity of reagents in two separate portions (separated by 15 h) proved to be more effective (entry 13). Finally, with the use of trimethyl borate as an additive, a catalytic reaction could be achieved (entry 14, catalytic protocol).
The scope of olefins that can be hydromethylated is illustrated in Table 2. Monosubstituted olefins bearing an aromatic triflate and even a free phenol worked smoothly (1c,d), as did a carbohydrate derivative (1e). 1,1-Disubstituted olefins (1f–m) tend to give higher yields of the hydromethylated products (2f–m), with the exception of the sterically hindered isopulegol derivative 1n. Cyclic structures such as methylenepiperidine derivative 1o gave a gem-dimethyl product in high yield, highlighting a new retrosynthetic disconnection to gem-disubstituted systems.11 Trisubstituted olefins 1p–r also delivered the desired methylated derivatives 2p–r in 41–61% yield. As Table 2 shows, the current transformation can tolerate a wide variety of acid-labile, Lewis basic, and reducible functional groups such as free alcohols, phenols, azides, alkynes, aliphatic halides, triflates, aromatic halides, and boronic esters.
Table 2. Substrate Scope of Hydromethylation Reaction.

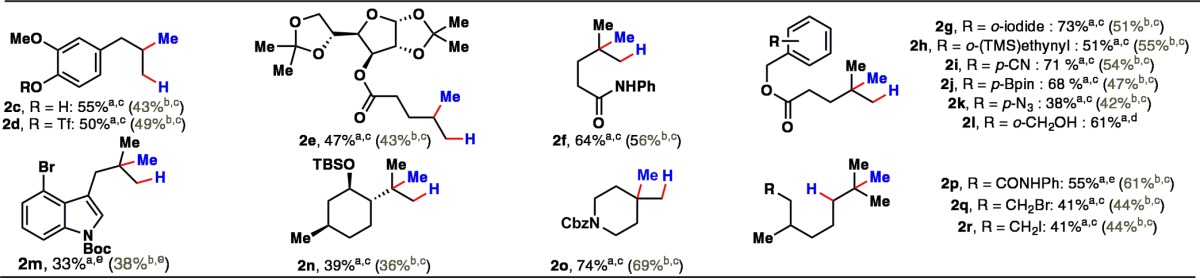
Standard protocol: first portion, CH2O (3.0 equiv), nOctSO2NHNH2 (2.5 equiv), 4 h; Fe(acac)3 (1.0 equiv), MeOH (0–2 equiv), PhSiH3 (2.0 equiv), THF, rt, 8–15 h; second portion, 8–15 h.
Catalytic protocol: CH2O (6.0 equiv), nOctSO2NHNH2 (5.0 equiv), 4 h; Fe(acac)3 (0.3–0.5 equiv), B(OMe)3 (2–5 equiv), PhSiH3 (2.0 equiv), THF, rt, 40 h.
Isolated yields.
Isolated yields, as a mixture with 2l′, ratio 2l:2l′ > 15:1.
Isolated yields, as a mixture with 2m′, ratio 2m:2m′ > 6:1.
Where the Fe-mediated hydromethylation really shines, and is likely to find widespread application, is in the context of late-stage diversification (Figure 2). For example, Apronal (1s), a hypnotic and sedative drug, could be directly transformed into hydromethylated derivative 2s in 44% yield. Complex natural products such as 1t–v could be hydromethylated, forming 2t–v in synthetically useful yields. A gram-scale transformation employing rotenone (1t), an active component of an organic insecticide known as derris, highlights the practicality of the current methodology. Next, two notoriously unstable and sensitive complex natural products were hydromethylated: picrotoxinin12a (1u) and gibberellic acid12b,12c (1v). Both of these highly congested terpenes, bearing reactive functionalities such as epoxides, free hydroxyl groups, carboxylic acids, strained lactones, and even another olefin, were chemoselectively hydromethylated (as verified by X-ray crystallography). One would be hard pressed to access these structures in any other way.
Figure 2.
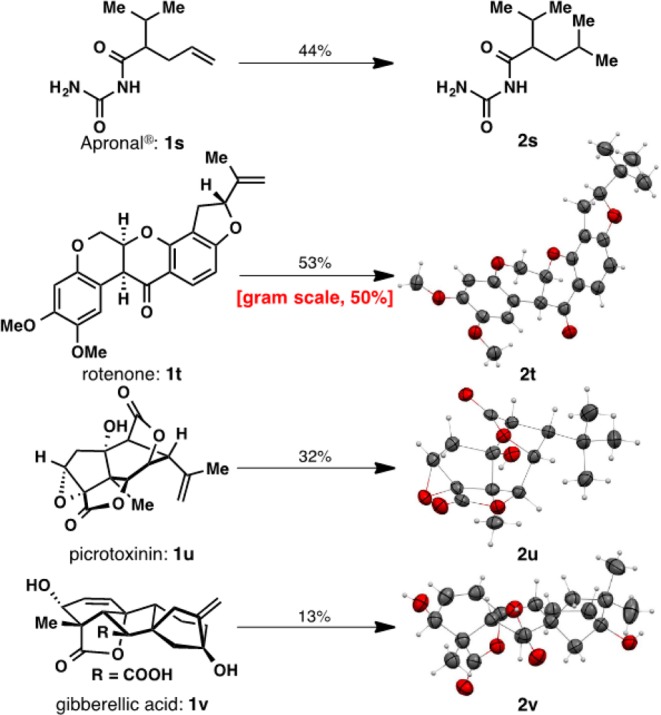
Diversification of complex molecules.
To highlight the step-economy of this newly developed C–C bond-forming methodology, the synthesis of terpene derivative 2w, alkaloid derivative 2x, and norsesquiterpene 2a was demonstrated (Figure 3). Previously, 2w was synthesized in racemic form from crotonaldehyde in a four-step route.13 It can now be synthesized in an enantiopure form from readily available citronellol (1w) in 68% yield. Hydromethylated quinine analogues such as 2x were previously accessed in a labor-intensive four-step sequence14 but can now be accessed directly in 32% yield. It is noteworthy that tertiary amine and quinoline moieties are tolerated under the mild reaction conditions. Lastly, the synthesis of 7-desmethyl-2-methoxycalamenene (2a) from the precursor 1a is a testament to the simplicity of this approach in the context of natural product synthesis. Indeed, the desired norsesquiterpene 2a was accessed in 29% yield, making this synthesis superior in yield and step count to all prior multi-step approaches (Figure 1A).1
Figure 3.
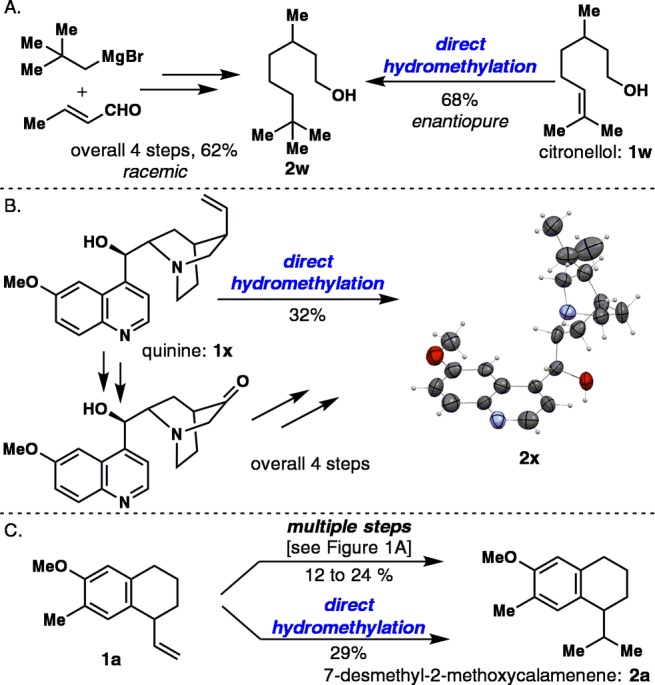
Direct hydromethylation as a step-economic method.
Another striking use of the current methodology is in the context of isotopic labeling. In theory, since formaldehyde is readily available in a myriad of radioactive and stable labeled forms, it can be used to prepare methyl groups with many permutations of 14CHn, 13CHn, 12CDn, 13CDn, and 12CTn. As a proof of concept, l-Fmoc-allylglycine-OMe (1y) was converted into l-Fmoc-leucine-OMe (CH3) and stable labeled derivatives containing CDH2 (2y-d1), CD2H (2y-d2), and CD3 (2y-d3) in >40% isolated yield without erosion of enantiopurity (Figure 4). Deuterium incorporation was >99% in the case of 2y-d2 and >75% in the other two cases; presumably this is due to incomplete deuteration of the intermediate alkylhydrazide. The divergent synthesis of leucine derivatives shown here may find use in the stereo-array isotope labeling technique15 and carbon–deuterium probes in vibrational spectroscopy.16 In an application in the synthesis of labeled drugs, (R,S)-Fmoc-pregabalin-OtBu-d2 (2z) was smoothly labeled from simple precursor 1z. As a final showcase of the utility of this method, a radioactive methyl group (14CH3) was installed to rotenone (1t) using commercially available [14C]formaldehyde solution.
Figure 4.
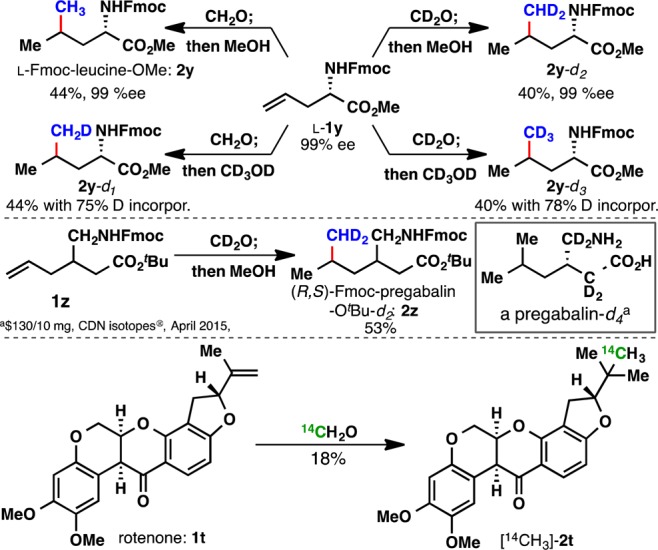
Application in synthesis of stable and radioactive labeled compounds.
Currently, this method has clearly defined limitations. For example, styrenes gave poor yields owing to the homodimerization byproducts. In the case of sterically hindered substrates (e.g., 1a, 1n, 1u, 1v, and 1x), lower yields of the desired products together with an increased amount of reduced byproducts were obtained. Procedurally, this reaction requires more experimental care than our previously reported Fe-mediated reactions6e,6f (see troubleshooting section in SI), and separation of the desired hydromethylated product from the reduced byproduct can sometimes be difficult.
A synthetically useful procedure for the formal addition of methane across a π-system has remained elusive and was heretofore only possible using multi-step sequences. This chemoselective, catalytic, one-pot method provides a solution to this problem using simple reagents and functions on a variety of substrates with mono-, di-, and trisubstitued unactivated olefins. The ability to directly “edit” complex natural products, advanced intermediates, amino acids, and pharmaceutical agents with the subtle addition of a single methyl group (in nearly any isotopic variety) represents a new strategic bond disconnection that is likely to be of broad interest and utility.
Acknowledgments
Financial support for this work was provided by NIH/NIGMS (GM-097444), Fulbright S&T Awards (predoctoral fellowship to H.T.D.), and China Scholarship Council (postdoctoral fellowship to C.L.). The authors thank A. L. Rheingold and C. E. Moore for X-ray crystallographic analysis, Dr. D.-H. Huang, Dr. L. Pasternack for assistance with NMR spectroscopy, Mr. J. T. Edwards for suggestion for the use of borate additives, and Dr. K.-J. Xiao and Dr. A. Okano for assistance with chiral and reverse phase HPLC.
Supporting Information Available
Experimental procedures and analytical data (1H and 13C NMR, MS) for all new compounds. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b05144.
Author Contributions
§ C.L. and Q.M. contributed equally to this paper.
The authors declare no competing financial interest.
Supplementary Material
References
- a Tietze L. F.; Raschke T. Synlett 1995, 597. [Google Scholar]; b Tietze L. F.; Raschke T. Eur. J. Org. Chem. 1996, 1996, 1981. [Google Scholar]; c Taber D. F.; Nakajima K.; Xu M.; Rheingold A. L. J. Org. Chem. 2002, 67, 4501. [DOI] [PubMed] [Google Scholar]; d Peng F.; Danishefsky S. J. J. Am. Chem. Soc. 2012, 134, 18860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fontaine F.-G.; Tilley T. D. Organometallics 2005, 24, 4340. [Google Scholar]; b Terao J.; Watanabe T.; Saito K.; Kambe N.; Sonoda N. Tetrahedron Lett. 1998, 39, 9201. [Google Scholar]; c Parnes Z. N.; Bolestova G. I.; Akhrem I. S. J. Chem. Soc., Chem. Commun. 1980, 748. [Google Scholar]
- a Isayama S.; Mukaiyama T. Chem. Lett. 1989, 1071. [Google Scholar]; b Mukaiyama T.; Isayama S.; Inoki S.; Kato K.; Yamada T.; Takai T. Chem. Lett. 1989, 449. [Google Scholar]; c Inoki S.; Kato K.; Takai T.; Isayama S.; Yamada T.; Mukaiyama T. Chem. Lett. 1989, 515. [Google Scholar]; d Kato K.; Mukaiyama T. Chem. Lett. 1992, 1137. [Google Scholar]
- a Zombeck A.; Hamilton D. E.; Drago R. S. J. Am. Chem. Soc. 1982, 104, 6782. [Google Scholar]; b Waser J.; Carreira E. M. J. Am. Chem. Soc. 2004, 126, 5676. [DOI] [PubMed] [Google Scholar]; c Waser J.; Nambu H.; Carreira E. M. J. Am. Chem. Soc. 2005, 127, 8294. [DOI] [PubMed] [Google Scholar]; d Waser J.; González-Gómez J. C.; Nambu H.; Huber P.; Carreira E. M. Org. Lett. 2005, 7, 4249. [DOI] [PubMed] [Google Scholar]; e Waser J.; Gaspar B.; Nambu H.; Carreira E. M. J. Am. Chem. Soc. 2006, 128, 11693. [DOI] [PubMed] [Google Scholar]; f Gaspar B.; Carreira E. M. Angew. Chem., Int. Ed. 2007, 46, 4519. [DOI] [PubMed] [Google Scholar]; g Gaspar B.; Carreira E. M. Angew. Chem., Int. Ed. 2008, 47, 5758. [DOI] [PubMed] [Google Scholar]; h Gaspar B.; Carreira E. M. J. Am. Chem. Soc. 2009, 131, 13214. [DOI] [PubMed] [Google Scholar]; i Shigehisa H.; Koseki N.; Shimizu N.; Fujisawa M.; Niitsu M.; Hiroya K. J. Am. Chem. Soc. 2014, 136, 13534. [DOI] [PubMed] [Google Scholar]
- a Magnus P.; Payne A. H.; Waring M. J.; Scott D. A.; Lynch V. Tetrahedron Lett. 2000, 41, 9725. [Google Scholar]; b Waser J.; Carreira E. M. Angew. Chem., Int. Ed. 2004, 43, 4099. [DOI] [PubMed] [Google Scholar]
- a Ishikawa H.; Colby D. A.; Seto S.; Va P.; Tam A.; Kakei H.; Rayl T. J.; Hwang I.; Boger D. L. J. Am. Chem. Soc. 2009, 131, 4904. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Leggans E. K.; Barker T. J.; Duncan K. K.; Boger D. L. Org. Lett. 2012, 14, 1428. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Barker T. J.; Boger D. L. J. Am. Chem. Soc. 2012, 134, 13588. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Taniguchi T.; Goto N.; Nishibata A.; Ishibashi H. Org. Lett. 2010, 12, 112. [DOI] [PubMed] [Google Scholar]; e Lo J. C.; Yabe Y.; Baran P. S. J. Am. Chem. Soc. 2014, 136, 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Lo J. C.; Gui J.; Yabe Y.; Pan C.-M.; Baran P. S. Nature 2014, 516, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Gui J.; Pan C.-M.; Jin Y.; Qin T.; Lo J. C.; Lee B. J.; Spergel S. H.; Mertzman M. E.; Pitts W. J.; La Cruz T. E.; Schmidt M. A.; Darvatkar N.; Natarajan S. R.; Baran P. S. Science 2015, 348, 886. [DOI] [PubMed] [Google Scholar]
- a Shi J.; Manolikakes G.; Yeh C.-H.; Guerrero C. A.; Shenvi R. A.; Shigehisa H.; Baran P. S. J. Am. Chem. Soc. 2011, 133, 8014. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hu X.; Maimone T. J. J. Am. Chem. Soc. 2014, 136, 5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kim S.; Cho J. R. Synlett 1992, 629. [Google Scholar]; b Kim S. Pure Appl. Chem. 1996, 68, 623. [Google Scholar]
- a Chung S.-K. J. Org. Chem. 1979, 44, 1014. [Google Scholar]; b Magnus P.; Waring M. J.; Scott D. A. Tetrahedron Lett. 2000, 41, 9731. [Google Scholar]; c Iwasaki K.; Wan K. K.; Oppedisano A.; Crossley S. W. M.; Shenvi R. A. J. Am. Chem. Soc. 2014, 136, 1300. [DOI] [PMC free article] [PubMed] [Google Scholar]; d King S. M.; Ma X.; Herzon S. B. J. Am. Chem. Soc. 2014, 136, 6884. [DOI] [PubMed] [Google Scholar]
- Crossley S. W. M.; Barabé F.; Shenvi R. A. J. Am. Chem. Soc. 2014, 136, 16788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebach D. Angew. Chem., Int. Ed. 2011, 50, 96. [DOI] [PubMed] [Google Scholar]
- a Porter L. A. Chem. Rev. 1967, 67, 441. [DOI] [PubMed] [Google Scholar]; b Pryce R. J. Phytochemistry 1973, 12, 507. [Google Scholar]; c Corey E. J.; Danheiser R. L.; Chandrasekaran S.; Siret P.; Keck G. E.; Gras J.-L. J. Am. Chem. Soc. 1978, 100, 8031. [Google Scholar]
- Anselmi C.; Buonocore A.; Centini M.; Facino R. M.; Hatt H. Comput. Biol. Chem. 2011, 35, 159. [DOI] [PubMed] [Google Scholar]
- Taylor E. G. Patent WO2003040083A1, 2003.
- Kainosho M.; Torizawa T.; Iwashita Y.; Terauchi T.; Mei Ono A.; Güntert P. Nature 2006, 440, 52. [DOI] [PubMed] [Google Scholar]
- Zimmermann J.; Romesberg F. E. Methods Mol. Biol. 2014, 1084, 101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.