

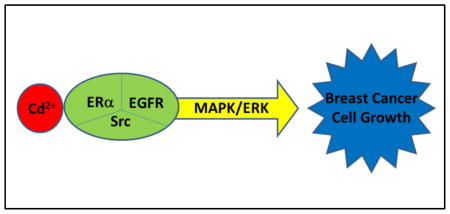
Abstract
Cadmium (Cd) is a common environmental toxicant and an established carcinogen. Epidemiological studies implicate Cd with human breast cancer. Low micromolar concentrations of Cd promote proliferation of human breast cancer cells in vitro. The growth promotion of breast cancer cells is associated with the activation of MAPK/ERK pathway. This study explores the mechanism of Cd-induced activation of MAPK/ERK pathway. Specifically, the role of cell surface receptors ERα, EGFR, and Src kinase was evaluated in human breast cancer MCF-7 cells treated with 1–3 μM Cd. The activation of ERK was studied using a serum response element (SRE) luciferase reporter assay. Receptor phosphorylation was detected by Western blot analyses. Cd treatment increased both the SRE reporter activity and ERK1/2 phosphorylation in a concentration-dependent manner. Cd treatment had no effect on reactive oxygen species (ROS) generation. Also, blocking the entry of Cd into the cells with manganese did not diminish Cd-induced activation of MAPK/ERK. These results suggest that the effect of Cd was likely not caused by intracellular ROS generation, but through interaction with the membrane receptors. While Cd did not appear to activate either EGFR or Src kinase, their inhibition completely blocked the Cd-induced activation of ERK as well as cell proliferation. Similarly, silencing ERα with siRNA or use of ERα antagonist blocked the effects of Cd. Based on these results, it is concluded that not only ERα, but also basal activities of EGFR and Src kinase are essential for Cd-induced signal transduction and activation of MAPK/ERK pathway for breast cancer cell proliferation.
Keywords: Cadmium, Breast Cancer Cells, ERα, MAPK/ERK, EGFR, Src Kinase
Graphical Abstract
Introduction
Cadmium (Cd) is a ubiquitous pollutant released into the environment from such activities as mining, electroplating, and paints, pigments, and battery manufacture. This heavy metal is a health concern because it persists in the river sediments, bioaccumulates in the ecosystems, and enters the human food chain. The dietary intake of Cd in the general population in the United States is estimated at 0.08–0.20 μg/kg body weight/day (Moschandreas et al., 2002). Due to slow elimination from the body, Cd accumulates over time primarily in the liver and kidneys, but also in testes, ovaries and breasts (Shaikh and Smith, 1980; Henson and Chedrese, 2004). In the general population, its concentration can reach 30 μg/g wet weight in kidney (Järup et al., 1998) and 160 μg/g wet weight in breast tissue (Antila et al., 1996). Among people excessively exposed to environmental Cd, kidney damage and osteoporosis are the common toxic effects (Takebayashi et al., 2000). Epidemiological studies suggest that even trace amount of Cd ingested via the diet is a cancer risk in the general population (Schwartz et al., 2000; Adams et al., 2012; Julin et al., 2012a, b). Since the recognition of Cd as a Category I carcinogen by the International Agency for Research on Cancer due to its association with lung cancer in exposed workers and evidences from animal studies (IARC, 1993), a number of epidemiological studies have implicated Cd in breast cancer development (McElroy, et al., 2006; Gallagher et al., 2010; Pan et al., 2010; Julin, et al., 2012a; Cho, et al., 2013; Nagata et al., 2013; Itoh et al., 2014). The underlying mechanism of Cd carcinogenesis is unclear, but may involve disrupted signal transduction, deregulated cell proliferation, depressed apoptosis, enhanced cell survival, and indirect genotoxic effects due to impairment of DNA repair (Waalkes, 2000).
Besides the epidemiological reports, experimental evidence also supports the role of Cd in breast cancer development. For example, Cd levels are higher in breast cancer tissue as compared to the surrounding healthy tissue (Strumylaite et al., 2011). The association of Cd with breast cancer is further supported by a number of in vivo and in vitro studies. Long-term treatment with Cd has the potential to transform normal breast epithelial cells into malignant tumor cells (Benbrahim-Tallaa et al, 2009). Although Cd as a metal is structurally unrelated to estrogen, it mimics estrogenic activity and stimulates mammary tissue growth in laboratory animals at an environmentally-relevant dose (Johnson et al., 2003). Also, when human breast cancer-derived cells are cultured in the laboratory, their growth is stimulated by treatment with low micromolar concentrations of Cd (Brama et al., 2007; Siewit et al., 2010; Yu et al., 2010). Cd may exert estrogenic effects in estrogen receptor-positive breast cancer cells through activation of ERα located in the cytosol and nucleus or on cell membrane (Stoica et al., 2000; Liu et al., 2008; Siewit et al., 2010). In estrogen receptor-negative breast cancer cells, Cd appears to promote cancer cell growth through GPR30, a membrane G-protein-coupled receptor responsive to estrogen (Yu et al., 2010).
While the exact mechanism of how Cd promotes cell growth has not been fully elucidated, studies on intracellular signaling reveal that Cd activates mitogen-activated protein kinase (MAPK) pathways in a variety of breast cancer cells (Brama et al., 2007; Liu et al, 2008; Zang et al., 2009; Casano et al., 2010; Yu et al., 2010). The MAPK pathways are pivotal signaling systems used by the cell to regulate proliferation, differentiation, survival, and apoptosis in response to diverse extracellular signals, including growth factors, hormones, and cytokines. Aberrant MAPK signaling contributes to cancer and other diseases. ERK, JNK, and P38 constitute three groups of MAPKs assigned with distinct roles. The ERK pathway is generally linked to cell proliferation and differentiation, and JNK and P38 pathways are more related to survival and apoptosis in response to cellular stresses (Seger and Krebs, 1995; Bogoyevitch et al., 1996; Pearson et al., 2001). As to which of these pathways is activated depends on the relative Cd concentration the cells are exposed to. At high concentration, Cd causes cell death and activates all three MAPK pathways by generating reactive oxygen species (Chuang, et al., 2000; Chen et al., 2008; Jing et al., 2012). However, at micromolar levels, Cd activates MAPK/ERK pathway and stimulates cell proliferation (Jiang et al., 2009; Yu et al., 2010). The MAPK/ERK signaling cascade is usually triggered by the activation of cell membrane receptors including G-protein coupled receptors GPCRs, receptor tyrosine kinases, integrins, and ion channels. Through sets of adaptor proteins Shc and GRB2, the guanine nucleotide exchange factor SOS is recruited. SOS activates small GTP binding proteins like Ras, which in turn activates a kinase cascade, the sequential phosphorylation of Raf, MEK1/2 and ERK1/2. The activated ERK1/2 translocates to the nucleus, and phosphorylates transcription factors such as ELK-1. The activated ELK-1 binds to SRF and the serum response element (SRE) located in transcription regulatory region of individual genes to form a ternary complex and activates transcription of a variety of early response genes, such as c-Fos and c-Jun, which are involved in regulation of cell proliferation (Davis et al., 2000; Roberts and Der, 2007).
EGFR is a major receptor tyrosine kinase that is involved in MAPK/ERK signaling and breast cancer development (Mendelsohn and Baselga, 2000; Arteaga, 2002). It is believed that the cross-talk between EGFR, Src kinase and ERα plays a critical role in the regulation of breast cancer development (Schiff et al., 2004). Src kinase and EGFR pathway are intertwined through reciprocal activation of each other (Simeonova et al., 2002; Bao et al., 2003). It is well-documented that ERα indirectly activates EGFR through MMP-9- and MMP-2-catalyzed release of HB-EGF (Filardo et al., 2000; Razandi et al., 2003). Since Cd activates MAPK/ERK, here we investigated the roles of EGFR, Src kinase and ERα in human breast cancer derived MCF-7 cells which are frequently used to study the involvement of Cd in breast cancer, and utilized a SRE luciferase reporter assay for detecting the MAPK/ERK signaling.
Materials and Methods
Reagents and Supplies
Dulbecco’s modified essential medium (DMEM) and phenol-red free DMEM were purchased from Lonza (Walkersville, MD). Fetal bovine serum (FBS) was from Atlanta Biologicals (Flowery Branch, GA). Charcoal-stripped FBS was from Gibco (Grand Island, NY). EGF was from Sigma Aldrich (St. Louis, MO). EGFR inhibitor (AG 1478) and Src kinase inhibitor 1, and MEK inhibitor PD 184161 were from Santa Cruz Biotechnology (Santa Cruz, CA). The antisera for EGFR (sc-03) and phospho-EGFR (Tyr 845) (sc-23420) were from Santa Cruz Biotechnology. The antisera for ERK1/2 (#9102), phospho-ERK1/2 (#4377), Src kinase (#2123), phospho-Src (Try 416) (#6943) were from Cell Signaling Technology (Beverly, MA). Dual luciferase assay kit was from Promega (Madison, WI). The luciferase reporter plasmid SRE-luc (pGL4.50[luc2/CMV/Hygro, #E1340) was purchased from Promega. The reporter plasmid 2xERE-luc was kindly provided by Dr. Ruitang Deng (Song et al., 2014). The transfection reagent, lipofectamine 2000 was from Invitrogen (Life Technologies; Grand Island, NY). The RNAi transfection reagent DharmaFECT1, siRNA-ERα (#M-003401-04-0005) and siRNA-EGFR (M-003114-03-0005) were purchased from Thermoscientificbio (Pittsburgh, PA). The plasmid dominant negative Src (K295R/Y527F) (#13657) was provided by Dr. Joan Brugge through Addgene (Cambridge, MA) (Thomas et al., 1991). All other chemicals including CdCl2, MnCl2, diphenyleneiodonium chloride (DPI), 2′,7′-dichlorofluorescin diacetate (DCFDA),17β-estradiol, and ERα inhibitor (ICI 182,780) were purchased from Sigma Aldrich. Cell Counting Kit-8 for cell proliferation assay was purchased from Dojindo Molecular Technologies (Rockville, MD).
Cell Culture Conditions for Treatment with Cd
MCF-7 human breast cancer cells were obtained from ATCC (Rockville, MD) and routinely maintained in DMEM containing 10% FBS. The cells were trypsinized and plated at a density of 4×105 cells/well in a 6-well plate in the complete medium and allowed to recover. After 2 days, the cells were washed with phosphate-buffered saline (PBS), and serum-starved in DMEM, without FBS, for 2 days. The serum-starved cells were treated with Cd in DMEM. After treatment, the medium was removed and the cells were washed with ice-cold PBS and lysed with 150 μl ice-cold RIPA buffer (150 mM sodium chloride, 100 mM Tris-HCl pH7.5, 1% deoxycholate, 0.1% SDS, 1% Triton X-100) supplemented with 2 mM phenylmethylsulphonyl fluoride, protease inhibitor cocktail, and phosphatase inhibitor cocktail. The cell lysates were inactivated at 95°C for 3 min, sonicated for 15 sec, and heated again at 95°C for 5 min in the presence of 5% mercaptoethanol. Protein concentration was determined by the micro BCA kit from Pierce, Thermoscientific (Rockford, IL).
Cell Culture Conditions for Treatment with Estrogen
The cells were cultured the same way as described above, except that instead of regular FBS cells were conditioned in DMEM containing 10% charcoal-stripped FBS (CS-FBS) for two passages.
Cell Proliferation Assay
The cells were seeded into 96-well plates at a density of 104 cells/well in DMEM containing 10% FBS. After 24 h the cells were treated with either 0.5 μM Cd alone, or with either 3 μM ERα inhibitor (ICI 182,780), 10 μM Src kinase inhibitor 1, 1 μM MEK inhibitor (PD 184161), or 4 μM EGFR inhibitor (AG 1478). After 4 days of treatment, cell proliferation was determined using the Cell Counting Kit-8. The absorbance was measured at 450 nm.
Luciferase Reporter Assay
The cells were seeded at a density of 1×105 cells/well in a 24-well plate in complete medium, and allowed to recover. After 2 days, the cells were transfected with 200 ng SRE-luc, or estrogen receptor element (ERE)-luc, and 30 ng pRL-Null by following the manufacturer’s protocol. On day-2 after transfection, the medium was replaced with DMEM without FBS, and the cells were serum-starved. After 48 h, the cells were treated with Cd in DMEM for 3 h, followed by lysis in lysis buffer. Firefly and renilla luciferase activities were measured according to the manufacturer’s protocol. The relative luciferase activity was calculated as the ratio of firefly luciferase activity to renilla luciferase activity. The results were normalized against those obtained from the control cells.
Measurement of Reactive Oxygen Species (ROS) Generation
The cells were seeded into 96 well plates at a density of 104 cells/well in DMEM containing 10% FBS for 24 h followed by serum starvation for 24 h. The cells were incubated with 20 μM DCFDA for 30 min, washed with PBS, and then treated with either 3 μM Cd alone or in the presence of 10 μM DPI for 3 h. Hydrogen peroxide (500 μM) was used as a positive control for ROS generation and its inhibition by DPI. Fluorescence intensity was measured using 485 and 525 nm as the excitation and emission wavelengths, respectively.
RNA Interference Studies
siRNAs against human EGFR and ERα were chemically synthesized by Dharmacon, Thermoscientificbio. To measure the effect of EGFR knockdown on MAPK kinase activity, MCF-7 cells were seeded at a density of 4×105 cells/well in a 6-well plate in complete medium and allowed to recover. After 2 days, the cells were transfected with 10 nM siRNA using Dharmafect1 (Dharmacon) according to the instructions of the manufacturer. The medium was replaced with DMEM without FBS 24 h post-transfection and the cells were serum-starved. After 2 days, the cells were treated with Cd or EGF for 5 min and harvested for analysis by Western blot. To measure the effect of ERα knockdown on MAPK kinase activity and on SRE luciferase reporter activity, the CS-FBS-adapted cells were used. These cells were seeded in 24-well plates at a density of 1×105 cells/well and allowed to recover for 2 days. At this point, the cells were transfected with 10 nM siRNA alone or along with 200 ng SRE-luc and 30 ng pRL-null using Dharmafect1 according to the manufacturer’s instructions. The culture medium was replaced with DMEM without FBS 24 h post-transfection and the cells were cultured for 2 days. The serum-starved cells were treated with Cd or 17β-estradiol (E2) for 3 h and proteins were harvested for measuring luciferase activity.
Western Blot Analysis
Protein expression was determined by Western blot analysis following the procedure described previously (Yu et al., 2010). Briefly, protein samples (30 μg) were loaded onto 10% SDS-PAGE gel and separated by electrophoresis. The separated proteins were transferred to the nitrocellulose membrane with transfer buffer containing 20% methanol. For EGFR, the proteins were transferred without methanol. The membranes were blocked with 5% BSA for 1 h and incubated with primary antibodies at 4°C overnight. After three 15 min washes with PBS-Tween, the membranes were incubated with Alexa Fluo® 680-conjugated anti-rabbit second antibody at a dilution of 1:10,000. The protein bands were scanned and quantified using a LI-COR Odyssey infrared imaging system.
Statistical Analysis
Each experiment was repeated at least three times and the data were analyzed with one-way ANOVA followed by the Tukey-Kramer post hoc test. P values of 0.05 or less were considered significant.
Results
Activation of MAPK/ERK pathway by Cd
In order to establish the concentration-response between Cd treatment and activation of MAPK/ERK pathway, serum-starved MCF-7 cells were treated with 0.1–3 μM Cd for 5 min. Western blot in Fig. 1A show that Cd increased ERK1/2 phosphorylation in a concentration-dependent manner to as much as 4.2-fold at 3 μM concentration, without altering total ERK1/2 levels. Similar results were obtained using a SRE luciferase reporter assay in which the MCF-7 cells were transfected with SRE-luc and then treated with 0.1–3 μM Cd for 3 h (Fig. 1B). The SRE reporter activity was increased 1.5- and 2.5-fold at 1 and 3 μM Cd, respectively.
Fig. 1.

Activation of ERK1/2 and SRE reporter by Cd. (A) MCF-7 cells were plated in 6-well plates and cultured for 48 h. The cells were serum-starved in DMEM for 24 h prior to treatment with 0.1–3 μM CdCl2 for 5 min. The cells were harvested with RIPA lysis buffer and the expression of phospho-ERK1/2 (pERK1/2) in the cell lysates was detected by Western blot analyses. The experiment was repeated three times and a representative blot is shown. (B) Cells were plated in 24-well plates with DMEM containing 10% FBS. After 48 h, the cells were transfected with 200 ng SRE-luc and 30 ng pRL-TK. The medium was replaced with serum-free DMEM 24 h post-transfection and the cells were cultured for 24 h. The serum-starved cells were treated with CdCl2 for 3 h and firefly and renilla luciferase activities were measured. The results are expressed as the ratio of firefly and renilla luciferase activities, normalized against control. The data are mean±SD of three separate experiments. *Significantly different from the untreated control cells (p<0.05). #Significantly different from each other.
MAPK/ERK pathway is a vital cell signaling system employed by cells to regulate a variety of activities, including cell proliferation, differentiation, survival, and cell death. This pathway is activated by growth factors via various cell surface receptors. One of these growth factors, EGF, plays a critical role in the regulation of breast epithelial cell development and breast cancer development by activation of MAPK via EGFR. In order to investigate whether the effect of Cd involved EGFR, the MCF-7 cells were treated for 5 min with either 100 ng/ml EGF or 3 μM Cd alone or in combination. Activation of ERK1/2 was evaluated by both Western blot analysis and luciferase activity. While both Cd and EGF alone increased ERK1/2 phosphorylation, in combination, Cd and EGF acted synergistically (Fig. 2A and B). The relative luciferase activity increased 11.5-fold over control by combination treatment as compared to 3.6- and 2.3-fold increases by EGF and Cd alone, respectively.
Fig. 2.

Synergistic enhancement of EGF-induced activation of ERK1/2 by Cd. (A) The cells were cultured and serum-starved prior to treatment as described in Fig. 1 legend. Serum-starved cells were treated with 3 μM CdCl2 for 5 min and pERK1/2 expression was detected by Western blot analyses. The experiment was repeated three times and a representative blot is shown. (B) Cells transfected with SRE-luc and pRL-TK were serum-starved for 24 h, and treated for 3 h with 3 μM CdCl2, 100 ng/ml EGF, or both Cd and EGF. Firefly and renilla luciferase activities were measured and the relative luciferase activity plotted as mean±SD of three separate experiments. *Significantly different from the untreated control cells (p<0.01). #Significantly different from cells treated with EGF or Cd alone (p<0.01).
Cd-induced Activation of MAPK is Not Due to ROS
To determine whether the observed Cd-induced activation of MAPK in MCF-7 cells was due to generation of ROS, the cells were treated with 3 μM Cd and ROS level was measured after 3 h by the DCFDA assay. As compared to the vehicle-treated control, no significant increase in ROS generation occurred at this concentration of Cd (Fig. 3A). However, the positive control, hydrogen peroxide, produced a significant increase in ROS, as expected. DPI, an inhibitor of NADPH Oxidase (Felty et al., 2005; Sim et al., 2005) was effective in significantly reducing ROS levels in all three groups. These results demonstrated that 3 μM had no effect on cellular ROS generation.
Fig. 3.

Lack of involvement of ROS in Cd-induced activation of ERK1/2. (A) MCF cells were seeded in DMEM supplemented with 10% FBS for 24 hr. The cells were serum starved for 24 h and incubated with 20 μM DCFDA for 30 min prior to treatment with 3 μM CdCl2 or 500 μM hydrogen peroxide alone or in the presence of ROS inhibitor, DPI (10 μM). Fluorescence intensity relative to vehicle control is plotted as mean±SD. *Significantly higher than the vehicle control. #Significantly lower than the respective non-DPI-treated cells. (p<0.05). (B)The cells were transfected with SRE-luc and pRL-TK, serum-starved for 24 h, and treated with 3 μM CdCl2 in the presence of serial concentrations of DPI for 3 h. Relative luciferase activity is plotted as mean±SD of three separate experiments. *Significant difference between the groups as indicated (p<0.05).
Next, the MCF-7 cells transfected with SRE reporter were treated with 3 μM Cd in the presence of 0–30 μM DPI for 3 h. As depicted in Fig. 3B, instead of decreasing the SRE reporter activity, DPI increased it about 1.5- and 2-fold at the 3 and 10 μM concentrations, respectively. The highest concentration of DPI seemed to be cytotoxic. These results further confirmed that Cd-induced activation of MAPK was not mediated through ROS.
Cd-induced activation of MAPK originates from cell membrane signaling
Next, the possibility that the activation of MAPK by Cd occurred due to its interaction with the intracellular ERα was examined. For this, the intracellular uptake of Cd was blocked by co-treatment with 20 μM manganese (Mn) (Martin et al., 2006). The cells used in this experiment were transfected with SRE reporter or estrogen receptor element (ERE) reporter prior to Cd treatment. As shown in Fig. 4, Cd treatment increased both ERE and SRE reporter activities by 2.7- and 2.8-fold, respectively. Blockage of Cd uptake by Mn limited the Cd-induced ERE reporter activity to only 1.2-fold over the Mn control. In comparison, the Cd-induced SRE reporter activity remained unaltered in the presence of Mn. This suggested that activation of MAPK in response to Cd treatment did not originate from its intracellular interaction with ERα, but likely from interaction with receptors at the cell membrane.
Fig. 4.

Differential effect of Mn on Cd-induced activation of MAPK and classical estrogen pathway. Cells transfected with either SRE-luc or ERE-luc were serum-starved for 24 h and treated with 3 μM CdCl2 alone or in the presence of 20 μM MnCl2 for 3 h. Relative luciferase activities are plotted as mean±SD of three separate experiments. *Significantly different from the untreated control cells (p<0.01). #Significantly different from the cells treated with Cd alone (p<0.01).
Role of EGFR and Src kinase in Cd-induced activation of ERK
In order to further elucidate the mechanism of how Cd might activate the MAPK pathway through interaction with the membrane receptors, MCF-7 cells were transfected with SRE reporter and then treated with Cd in the presence of EGFR and Src kinase inhibitors. While Cd alone increased SRE reporter activity by 5.3-fold, co-treatment with the EGFR inhibitor AG1478, Src inhibitor 1, or MEK1/2 inhibitor PD 184161 decreased the activity by 46, 51, and 68%, respectively (Fig. 5A). The effects of these inhibitors on the phosphorylation of ERK1/2 are shown in Fig. 5B. The MCF-7 cells were treated with Cd and the inhibitor together for 5 min, 1 h or 3 h, and p-ERK was measured by Western blot. Treatment with Cd increased ERK1/2 phosphorylation at all time-points studied, but most at 5 min. The EGFR inhibitor AG 1478, Src kinase inhibitor 1 and MEK 1/2 inhibitor PD184161 all effectively blocked the ERK1/2 phosphorylation. The consistency of the inhibitory effects EGFR and Src kinase inhibitor on both SRE reporter and ERK1/2 phosphorylation suggested that EGFR and Src kinase were involved in Cd-induced activation of MAPK.
Fig. 5.

Reduction of Cd-induced activation of ERK1/2 by EGFR and Src kinase inhibitors. (A) Cells transfected with SRE-luc were serum-starved for 24 h and treated with 3 μM CdCl2 alone or in the presence of either EGFR inhibitor AG1478 (2 μM), Src kinase inhibitor 1 (10 μM), or MEK inhibitor PD184161 (1 μM). Relative luciferase activities are plotted as mean±SD of three separate experiments. *Significantly different from the cells treated with Cd alone (p<0.05). (B) Cells were serum-starved for 24 h and treated with: (1) blank, (2) 3 μM CdCl2 alone, or along with (3) 2 μM AG1478, (4) 10 μM Src kinase inhibitor 1, or (5) 1 μM PD184161 for up to 3 h. The cell lysates were analyzed for pERK1/2 by Western blot analyses. The experiment was repeated three times and a representative blot is shown.
To further confirm that EGFR was involved in Cd-induced activation of ERK, the cells were transfected with siEGFR and then treated with Cd or EGF for 5 min prior to analyzing ERK phosphorylation. Whereas EGF treatment caused the phosphorylation of EGFR at tyr-845, treatment with Cd did not (Fig. 6A). However, transfection of cells with siEGFR to block the receptor activity resulted in 40 and 48% reduction in ERK1/2 phosphorylation in Cd and EGF treated cells, respectively (Fig. 6B). These results suggested that while Cd did not increase EGFR phosphorylation, basal activity of the receptor was required to maintain the effect of Cd on ERK phosphorylation.
Fig. 6.

Reduction of Cd-induced activation of ERK1/2 by silencing EGFR. Cells were transfected with siRNA for EGFR. Three days later, the cells were serum-starved for 24 h and treated with 3 μM CdCl2 or 100 ng/ml EGF for 5 min. (A) The levels of p-ERK1/2, and phosphorylated EGFR (pEGFR; at Tyr-845) (were analyzed by Western blot. The experiment was repeated three times and a representative blot is shown. (B) p-ERK1/2 bands were quantified and normalized values against control are plotted as mean±SD of three separate experiments. *Significantly different from the respective treatment with nonsense siRNA (p<0.05).
Next, the cells were transfected with SRE-luc and dominant negative Src (K295R/Y527F) and then treated with Cd to determine whether Src kinase was involved in Cd-induced activation of ERK. As shown in Fig. 7A, transfection with dominant negative Src-indeed decreased Cd-induced SRE reporter activity by 60%. To determine whether Cd caused the activation of Src kinase, the MCF-7 cells were treated with Cd for 5 min, and phosphorylation of Src kinase at tyr-416 was measured by Western blot analysis. Results were similar to EGFR phosphorylation in that the Cd treatment had no effect on Src kinase phosphorylation (Fig. 7B).
Fig. 7.

Reduction of Cd-induced activation of ERK1/2 by silencing Src kinase with a dominant negative mutant. (A) Cells were transfected with 200 ng SRE-luc, 30 ng pRL-TK, and 100 ng Src-K295R/Y527F (Src-DN), and serum-starved for 24 h prior to treatment with 3 μM CdCl2 for 3 h. Relative luciferase activity is plotted as mean±SD of three separate experiments. *Significantly different from the cells transfected with the vector (p<0.05). (B) Serum-starved cells were treated for 5 min with: (1) mock, (2) 3 μM CdCl2, and (3) Cd and 1 μM AG1478, or (4) Cd and 1 μM Src kinase inhibitor 1. pSrc was analyzed by Western blot. The experiment was repeated three times and a representative blot is shown.
Taken together, the above results reveal that although Cd treatment did not directly increase the phosphorylation of EGFR or Src kinase, inhibiting these proteins or silencing their expression had a negative effect on the activation of ERK. Thus both EGFR and Src kinase appeared to play active roles in Cd-induced signal transduction.
Role of ERα in Cd-induced activation of ERK
To minimize the effect of basal levels of E2 on Cd-induced activation of ERK, the MCF-7 cells were cultured in CS-FBS for two passages followed by serum-starvation in DMEM for 2 days. The role of E2 in Cd-induced activation of ERK was studied by either co-treatment or 16 h pre-treatment of the cells with the ERα antagonist ICI 182,780. In a separate experiment, the MCF-7 cells were first transfected with the SRE reporter and then treated with Cd, E2, or ICI 182,780 for 3 h. While co-treatment with ICI 182,780 did not inhibit Cd-induced ERK1/2 phosphorylation or activation of SRE reporter, pre-treatment with ICI 182,780 was highly effective (Fig. 8). Although E2 did not activate either ERK1/2 or SRE reporter, if the estrogen receptor was silenced by transfection with siRNA, the Cd-induced activation of both ERK1/2 and SRE reporter was completely blocked. These results suggest that, like EGFR and Src kinase, basal levels of ERα were not only essential, but were highly effective in signal transduction.
Fig. 8.

Role of ERα in mediating activation of ERK1/2 by Cd. (A) Cells adapted to growth in 10% charcoal-stripped FBS in DMEM for two passages were seeded in the same medium to grow for 2 more days and then serum-starved for 48 h. The cells were either treated with 10 nM E2 or 3 μM CdCl2 alone or along with 1 μM ICI 182,780 (ICI) for 5 min. In a separate experiment, the cells were pre-treated with 1 μM ICI for 16 h prior to treatment with 10 nM E2 or 3 μM CdCl2. Another batch of cells was transfected with siRNA against ERα, maintained in CS-FBS for 3 days, serum-starved for 48 h, and then treated with E2 or CdCl2. The level of pERK1/2 was analyzed by Western blot. The experiment was repeated three times and a representative blot is shown. (B) Cells were seeded in 10% CS-FBS for 2 days, transfected with 200 ng SRE-luc, 30 ng pRL-TK, and 100 nM nonsense siRNA or siERα. Three days post-transfection, the medium was replaced with serum-free DMEM and the cells were starved for 24 h and treated with Cd and ICI for 3 h. The relative luciferase activity is plotted as mean±SD of three separate experiments. *Significantly different from the vehicle-treated cells in the respective groups (p<0.05). #Significantly different from the cells treated with Cd alone (p<0.05).
The role of ERα and EGFR was further investigated in Cd-induced cell proliferation. As shown in Fig. 9, culturing the cells in the presence of 0.5 μM Cd for 4 days promoted cell growth by about 1.5-fold over control cells. Co-incubation with the inhibitors of EGFR, Src kinase, MEK, or ERα abolished this effect. Thus, these results confirmed that Cd-induced cell proliferation of MCF7 cells was dependent on EGFR, ERα and related downstream pathways.
Fig. 9.

Dependence of Cd-induced cell proliferation on EGFR, ERα, Src kinase, and MEK. Cells were seeded in DMEM containing 10% FBS for 24 h and treated with 0.5 μM CdCl2 alone or in the presence of either EGFR inhibitor AG1478 (4 μM), Src kinase inhibitor 1 (10 μM), MEK inhibitor PD184161 (1 μM), or ERα inhibitor ICI 182,780 (3 μM) in DMEM supplemented with 0.2 % BSA for 4 days. Cell proliferation was evaluated using the Cell Counting Kit-8 and plotted as mean±SD of five experiments. * Significantly higher than the control group (p<0.05).
Discussion
In agreement with a previous report from our laboratory (Liu et al., 2008), low micromolar concentrations of Cd caused the activation of ERK in MCF-7 cells. Inhibition of ROS formation did not alter the effect of Cd on ERK, suggesting that the effect of Cd is not driven by ROS. Blocking the uptake of Cd into the cells by Mn also had minimal effect on the activation of ERK, suggesting that signal for ERK activation likely originated at the cell membrane level. This conclusion was further supported by the rapid activation of ERK by Cd and its curtailment upon inhibition or silencing of EGFR. In addition, both Src kinase inhibition and silencing blocked activation of ERK indicating that not only EGFR, but also Src kinase was essential in processing the effect of Cd. Since Cd did not phosphorylate EGFR or Src, their basal activities appear to be sufficient for Cd-induced activation of ERK.
The activation of EGFR in response to Cd appears to be cell type dependent as it was reported to occur in human uterine leiomyoma and smooth muscle cells (Gao et al., 2015). In an oral carcinoma cell line Ca9-22, however, Kuribayashi et al. (2004) reported the activation of ERK by insulin like growth factor 1 (IGF-1) without EGFR phosphorylation. This effect of IGF-1, a natural ligand for EGFR, was blocked by the EGFR inhibitor AG1478. IGF-1 is believed to activate IGF-1R, which recruits IRS-1, activating Ras, and then C-Raf, and eventually the phosphorylation of ERK. This effect was shown to be dependent on the basal activity of EGFR. The basal activity of EGFR may also regulate C-Raf in the MCF-7 cells and control Cd-induced activation of ERK.
Src kinase could possibly modulate ERK pathway by interacting with EGFR, by activating Ras, or by activating c-Raf (Li et al., 1998; Biscardi et al., 1999; Xia et al., 2008). Src kinase activity is usually dependent on its phosphorylation and here it is shown that the basal activity of Src kinase is sufficient for regulating the activation of ERK.
A number of investigators have reported the activation of ERK by estrogen via non-genomic mechanisms such as through activation of membrane ERα and GPR30 (Filardo et al., 2000; Duan et al., 2001; Razandi et al., 2003). However, in the present study, estrogen did not increase the activation of ERK or SRE reporter. The potency of the same batch of estrogen on the activation of ERE reporter was validated in a separate experiment (data not shown). Similar observations were also reported by a number of other investigators (Bonapace et al., 1996; Joel et al., 1998; Lobenhofer and Marks, 2000; Lobenhofer et al., 2000; Caristi et al., 2001). Brower and colleagues attempted to address this problem by using a cell line from a laboratory which had reported positive responses (Filardo et al., 2000), but their result still turned out negative (Brower et al., 2005). Thus, it appears that the membrane ERα is non-functional in the MCF-7 cells used in the present study. Nevertheless, we reported previously that Cd activated ERK in a variety of breast cancer cell lines and that this was apparently mediated through ERα in MCF-7 cells (Liu et al., 2008). Also in the present study, short-time co-treatment with an ERα inhibitor failed to block Cd-induced activation of ERK, however, overnight pre-treatment with the inhibitor as well as ERα silencing blocked the Cd-induced activation of ERK. These data indicate that participation of ERα is required for the effect of Cd. It may be that Cd either activates ERα through a different mechanism than estrogen or it may not directly activate ERα. If membrane ERα is assumed to be functional in these MCF-7 cells, Cd may act through membrane ERα like estrogen causing sequential activation of G-protein, Src kinase, and MMP-9/MMP-2 to cleave pro-HB-EGF. The released HB-EGF could activate EGFR and MAPK/ERK cascade (Filardo et al., 2000; Razandi et al., 2003; Liu et al., 2008. However, Cd binding with ERα may be different from that of estrogen because short-term co-treatment with an ERα antagonist failed to abolish the effect of Cd in the present study. Moreover, this explanation is not fully supported since EGFR does not appear to be activated by Cd. An alternative explanation is that ERα may play an essential role in maintaining ERK activity by regulating the expression of critical components of ERK pathway, like GAB2, a docking protein which was reported to enhance EGFR-mediated ERK signaling as an ERα target gene (Daly et al., 2002). It could be that silencing of ERα by siRNA, or overnight treatment with the inhibitor, may decrease GAB2 and thus suppress MAPK/ERK activity.
Filardo et al. (2000) reported the activation of GPR30 by estrogen which led to activation of MAPK in MCF-7 cells. However, in our laboratory, both G1 and G15, the respective agonist and antagonist of GPR30, failed to activate SRE reporter (data not shown), suggesting that GRP30 is likely not involved in the effect of Cd in the MCF-7 cells used in the present study. It is also possible that Cd interacts with an unidentified membrane receptor linked to the signaling cascade of Raf-MEK-ERK, which is dependent on the basal activity of EGFR and Src kinase, similar to the IGF-1 effect in Ca9-22 cells (Kuribayashi et al., 2004). Based on these considerations, possible signaling pathways for Cd-induced activation of ERK are depicted graphically in Fig. 10.
Fig. 10.

Possible pathways of Cd-induced MAPK/ERK signaling. (1) The interaction of Cd with membrane ERα leads to EGFR-mediated activation of MAPK with the assistance of G-proteins, Src kinase, MMP-9/-2, and HB-EGF. (2) ERα regulates adaptor protein Gab2 which enhances MAPK/ERK signaling. (3) Cd interacts with an un-identified membrane receptor that sends signal to Raf whose activity is controlled by the basal activities of EGFR and Src kinase.
Our observation of the activation of SRE reporter by Cd suggests that Cd was able to activate early response genes (e.g., c-fos, egr-1) which are driven by SREs and are involved in the regulation of cell proliferation and cancer development (Matsuoka and Call, 1995; Joseph, 2001). EGFR is well known to play a critical role in breast cancer development (DiGiovanna et al., 2005; Nieto et al., 2007). Since Cd and EGF acted synergistically in the activation of ERK pathway in the present study, it underscores the potential for Cd to promote breast cancer. Considering that the effect of Cd requires only basal activities of EGFR and Src kinase, in breast cancers with high levels of EGFR and Src kinase Cd could possibly potentiate cancer development. Further studies are needed to explore this possibility.
Highlights.
Low micromolar concentrations of Cd rapidly activate ERK1/2 in MCF-7 cells.
Signal transduction and resulting cell proliferation require EGFR, ERα, and Src.
These findings implicate Cd in promotion of breast cancer.
Acknowledgments
The authors acknowledge partial salary support for XS and the use of Centralized Research Core Facility of the RI Institutional Development Award (IDeA) Network of Biomedical Research Excellence (RI-INBRE) Program supported by grant number 2P20GM103430 from the National Institute of General Medical Sciences at the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams SV, Passarelli MN, Newcomb PA. Cadmium exposure and cancer mortality in the Third National Health and Nutrition Examination Survey Cohort. Occup Environ Med. 2012;69:153–156. doi: 10.1136/oemed-2011-100111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antila E, Mussalo-Rauhamaa H, Kantola M, Atroshi F, Westermarck T. Association of cadmium with human breast cancer. Sci Total Environ. 1996;186:251–256. doi: 10.1016/0048-9697(96)05119-4. [DOI] [PubMed] [Google Scholar]
- Arteaga CL. Epidermal growth factor receptor dependence in human tumors: more than just expression? Oncologist. 2002;7(Suppl):31–39. doi: 10.1634/theoncologist.7-suppl_4-31. [DOI] [PubMed] [Google Scholar]
- Bao J, Gur G, Yarden Y. Src promotes destruction of c-Cbl: implications for oncogenic synergy between Src and growth factor receptors. Proc Natl Acad Sci USA. 2003;100:2438–2443. doi: 10.1073/pnas.0437945100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benbrahim-Tallaa L, Tokar EJ, Diwan BA, Dill AL, Coppin JF, Waalkes MP. Cadmium malignantly transforms normal human breast epithelial cells into a basal-like phenotype. Environ Health Perspect. 2009;117:1847–1852. doi: 10.1289/ehp.0900999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. C-Src-mediated phosphorylation of the epidermal growth factor receptor on tyr845 and tyr1101 is associated with modulation of receptor function. J Biol Chem. 1999;274:8335–8343. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch MA, Gillespie-Brown J, Ketterman AJ, Fuller SJ, Ben-Levy R, Ashworth A, Marshall CJ, Sugden PH. Stimulation of the stress-activated mitogen-activated protein kinase subfamilies in perfused heart. p38/RK mitogen-activated protein kinases and c-Jun N-terminal kinases are activated by ischemia/reperfusion. Circ Res. 1996;79:162–173. doi: 10.1161/01.res.79.2.162. [DOI] [PubMed] [Google Scholar]
- Bonapace IM, Addeo R, Altucci L, Cicatiello L, Bifulco M, Laezza C, Salzano S, Sica V, Bresciani F, Weisz A. 17 Beta estradiol overcomes a G1 block induced by HMG-CoA reductase inhibitors and fosters cell cycle progression without inducing ERK-1 and -2 MAP kinases activation. Oncogene. 1996;12:753–763. [PubMed] [Google Scholar]
- Brama M, Gnessi L, Basciani S, Cerulli N, Politi L, Spera G, Mariani S, Cherubini S, d’Abusco AS, Scandurra R, Migliaccio S. Cadmium induces mitogenic signaling in breast cancer cell by an ERalpha-dependent mechanism. Mol Cell Endocrinol. 2007;264:102–108. doi: 10.1016/j.mce.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Brower SL, Roberts JR, Antonini JM, Miller MR. Difficulty demonstrating estradiol-mediated Erk1/2 phosphorylation in MCF-7 cells. J Steroid Biochem Mol Biol. 2005;96:375–385. doi: 10.1016/j.jsbmb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Caristi S, Galera JL, Matarese F, Imai M, Caporali S, Cancemi M, Altucci L, Cicatiello L, Teti D, Bresciani F, Weisz A. Estrogens do not modify MAP kinase-dependent nuclear signaling during stimulation of early G1 progression in human breast cancer cells. Cancer Res. 2001;61:6360–6366. [PubMed] [Google Scholar]
- Casano C, Agnello M, Sirchia R, Luparello C. Cadmium effects on p38/MAPK isoforms in MDA-MB231 breast cancer cells. Biometals. 2010;23:83–92. doi: 10.1007/s10534-009-9268-6. [DOI] [PubMed] [Google Scholar]
- Chen L, Liu L, Huang S. Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Rad Biol Med. 2008;45:1035–1044. doi: 10.1016/j.freeradbiomed.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Cho YA, Kim J, Woo HD, Kang M. Dietary cadmium intake and the risk of cancer: a meta-analysis. PloS One. 2013;8:e75087. doi: 10.1371/journal.pone.0075087.eCollection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang SM, Wang IC, Yang JL. Roles of JNK, p38 and ERK mitogen-activated protein kinases in the growth inhibition and apoptosis induced by cadmium. Carcinogenesis. 2000;21:1423–1432. [PubMed] [Google Scholar]
- Daly RJ, Gu H, Parmar J, Malaney S, Lyons RJ, Kairouz R, Head DR, Henshall SM, Neel BG, Sutherland RL. The docking protein Gab2 is overexpressed and estrogen regulated in human breast cancer. Oncogene. 2002;21:5175–5181. doi: 10.1038/sj.onc.1205522. [DOI] [PubMed] [Google Scholar]
- Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S. The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiovanna MP, Stern DF, Edgerton SM, Whalen SG, Moore D, II, Thor AD. Relationship of epidermal growth factor receptor expression to ErbB-2 signaling activity and prognosis in breast cancer patients. J Clin Oncol. 2005;23:1152–1160. doi: 10.1200/JCO.2005.09.055. [DOI] [PubMed] [Google Scholar]
- Duan R, Xie W, Burghardt RC, Safe S. Estrogen receptor-mediated activation of the serum response element in MCF7 cells through MAPK dependent phosphorylation of Elk-1. J Biol Chem. 2001;276:11590–11598. doi: 10.1074/jbc.M005492200. [DOI] [PubMed] [Google Scholar]
- Felty Q, Xiong WC, Sun D, Sarkar S, Singh KP, Parkash J, Roy D. Estrogen-induced mitochondrial reactive oxygen species as signal-transducing messengers. Biochemistry. 2005;44:6900–6909. doi: 10.1021/bi047629p. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Bland KI, Frackelton AR. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, gpr30, and occurs via transactivation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14:1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- Gallagher CM, Chen JJ, Kovach JS. Environmental cadmium and breast cancer risk. Aging. 2010;2:804–814. doi: 10.18632/aging.100226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Yu L, Moore AB, Kissling GE, Waalkes MP, Dixon D. Cadmium and proliferation in human uterine leiomyoma cells: evidence of a role for EGFR/MAPK pathways but not classical estrogen receptor pathways. Environ Health Perspect. 2015;123:331–336. doi: 10.1289/ehp.1408234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson MC, Chedrese PJ. Endocrine disruption by cadmium, a common environmental toxicant with paradoxical effects on reproduction. Exp Biol Med. 2004;229:383–392. doi: 10.1177/153537020422900506. [DOI] [PubMed] [Google Scholar]
- IARC (International Agency for Research on Cancer) Beryllium, cadmium, mercury and exposures in the glass manufacturing industry. IARC Monogr Eval Carcinog Risk Hum. 1993;58:41–117. [PMC free article] [PubMed] [Google Scholar]
- Itoh H, Iwasaki M, Sawada N, Takachi R, Kasuga Y, Yokoyama S, Onuma H, Nishimura H, Kusama R, Yokoyama K, Tsugane S. Dietary cadmium intake and breast cancer risk in Japanese women: a case-control study. Int J Hyg Environ Health. 2014;217:70–77. doi: 10.1016/j.ijheh.2013.03.010. [DOI] [PubMed] [Google Scholar]
- Järup L, Berglund M, Elinder CG, Nordberg G, Vahter M. Health effects of cadmium exposure -- a review of the literature and a risk estimate. Scand J Work Environ Health. 1998;24(suppl):1–51. [PubMed] [Google Scholar]
- Jiang G, Duan W, Xu L, Song S, Zhu C, Wu L. Biphasic effect of cadmium on cell proliferation in human embryo lung fibroblast cells and its molecular mechanism. Toxicol In Vitro. 2009;23:973–978. doi: 10.1016/j.tiv.2009.06.029. [DOI] [PubMed] [Google Scholar]
- Jing Y, Liu LZ, Jiang Y, Zhu Y, Guo NL, Barnett J, Rojanasakul Y, Agani F, Jiang BH. Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol Sci. 2012;125:10–19. doi: 10.1093/toxsci/kfr256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joel PB, Traish AM, Lannigan DA. Estradiol-induced phosphorylation of serine 118 in the estrogen receptor is independent of p42/p44 mitogen-activated protein kinase. J Biol Chem. 1998;273:13317–13323. doi: 10.1074/jbc.273.21.13317. [DOI] [PubMed] [Google Scholar]
- Johnson MD, Kenney N, Stoica A, Hilakivi-Clake L, Singh B, Chepko G, Clarke R, Sholler PF, Lirio AA, Foss C, Reiter R, Trock B, Paik S, Martin MB. Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland. Nat Med. 2003;9:1081–1084. doi: 10.1038/nm902. [DOI] [PubMed] [Google Scholar]
- Joseph P, Muchnok TK, Klishis ML, Roberts JR, Antonini JM, Whong WZ, Ong T. Cadmium-induced cell transformation and tumorigenesis are associated with transcriptional activation of c-fos, c-jun, and c-myc proto-oncogenes: role of cellular calcium and reactive oxygen species. Toxicol Sci. 2001;61:295–303. doi: 10.1093/toxsci/61.2.295. [DOI] [PubMed] [Google Scholar]
- Julin B, Wolk A, Bergkvist L, Bottai M, Akesson A. Dietary cadmium exposure and risk of postmenopausal breast cancer: a population-based prospective cohort study. Cancer Res. 2012a;72:1459–1466. doi: 10.1158/0008-5472.CAN-11-0735. [DOI] [PubMed] [Google Scholar]
- Julin B, Wolk A, Johansson JE, Andersson SO, Andrén O, Akesson A. Dietary cadmium exposure and prostate cancer incidence: a population-based prospective cohort study. Br J Cancer. 2012b;107:895–900. doi: 10.1038/bjc.2012.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuribayashi A, Kataoka K, Kurabayashi T, Miura M. Evidence that basal activity, but not transactivation, of the epidermal growth factor receptor tyrosine kinase is required for insulin-like growth factor I-induced activation of extracellular signal-regulated kinase in oral carcinoma cells. Endocrinology. 2004;145:4976–4984. doi: 10.1210/en.2004-0713. [DOI] [PubMed] [Google Scholar]
- Li JD, Feng W, Gallup M, Kim JH, Gum J, Kim Y. Activation of NF-kB via a Src-dependent Ras-MAPK-pp90rsk pathway is required for Pseudomonas aeruginosa-induced mucin overproduction in epithelial cells. Proc Natl Acad Sci USA. 1998;95:5718–5723. doi: 10.1073/pnas.95.10.5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Yu X, Shaikh ZA. Rapid activation of ERK1/2 and AKT in human breast cancer cells by cadmium. Toxicol Appl Pharmacol. 2008;228:286–294. doi: 10.1016/j.taap.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobenhofer EK, Marks JR. Estrogen-induced mitogenesis of MCF-7 cells does not require the induction of mitogen-activated protein kinase activity. J Steroid Biochem Mol Biol. 2000;75:11–20. doi: 10.1016/s0960-0760(00)00132-1. [DOI] [PubMed] [Google Scholar]
- Lobenhofer EK, Huper G, Iglehart JD, Marks JR. Inhibition of mitogen-activated protein kinase and phosphotidylinositol 3-kinase in MCF-7 cells prevents estrogen-induced mitogenesis. Cell Growth Differ. 2000;11:99–110. [PubMed] [Google Scholar]
- Martin P, Fareh M, Poggi MC, Boulukos KE, Pognonec P. Manganese is highly effective in protecting cells from cadmium intoxication. Biochem Biophys Res Commun. 2006;351:294–299. doi: 10.1016/j.bbrc.2006.10.035. [DOI] [PubMed] [Google Scholar]
- Matsuoka M, Call KM. Cadmium-induced expression of immediate early genes in LLC-PK1 cells. Kidney Int. 1995;48:383–389. doi: 10.1038/ki.1995.306. [DOI] [PubMed] [Google Scholar]
- McElroy JA, Shafer MM, Trentham-Dietz A, Hampton JM, Newcomb PA. Cadmium exposure and breast cancer risk. J Natl Cancer Inst. 2006;98:869–73. doi: 10.1093/jnci/djj233. [DOI] [PubMed] [Google Scholar]
- Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19:6550–6565. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- Moschandreas DJ, Karuchit S, Berry MR, Ko’Rourke M, Lo D, Lebowitz MD, Robertson G. Exposure apportionment: ranking food items by their contribution to dietary exposure. J Expo Anal Environ Epidemiol. 2002;12:233–243. doi: 10.1038/sj.jea.7500230. [DOI] [PubMed] [Google Scholar]
- Nagata C, Nagao Y, Nakamura K, Wada K, Tamai Y, Tsuji M, Yamamoto S, Kashiki Y. Cadmium exposure and the risk of breast cancer in Japanese women. Breast Cancer Res Treat. 2013;138:235–239. doi: 10.1007/s10549-013-2414-4. [DOI] [PubMed] [Google Scholar]
- Nieto Y, Nawaz F, Jones RB, Shpall EJ, Nawaz S. Prognostic significance of overexpression and phosphorylation of epidermal growth factor receptor (EGFR) and the presence of truncated EGFRvIII in locoregionally advanced breast cancer. J Clin Oncol. 2007;25:4405–4413. doi: 10.1200/JCO.2006.09.8822. [DOI] [PubMed] [Google Scholar]
- Pan J, Plant JA, Voulvoulis N, Oates CJ, Ihlenfeld C. Cadmium levels in Europe: implications for human health. Environ Geochem Health. 2010;32:1–12. doi: 10.1007/s10653-009-9273-2. [DOI] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers-Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- Razandi M, Pedram A, Park ST, Levin ER. Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem. 2003;278:2701–2712. doi: 10.1074/jbc.M205692200. [DOI] [PubMed] [Google Scholar]
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- Schwartz GG, Reis IM. Is cadmium a cause of human pancreatic cancer? Cancer Epidemiol Biomarkers Prev. 2000;9:139–145. [PubMed] [Google Scholar]
- Schiff R, Massarwech SA, Shou J, Bharwani L, Mohsin SK, Osborne CK. Cross-Talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin Cancer Res. 2004;10:331–336. doi: 10.1158/1078-0432.ccr-031212. [DOI] [PubMed] [Google Scholar]
- Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- Shaikh ZA, Smith JC. Metabolism of orally ingested cadmium in humans. Dev Toxicol Environ Sci. 1980;8:569–574. [PubMed] [Google Scholar]
- Siewit CL, Gengler B, Vegas E, Puckett R, Louie MC. Cadmium promotes breast cancer cell proliferation by potentiating the interaction between ERalpha and c-Jun. Mol Endocrinol. 2010;24:981–992. doi: 10.1210/me.2009-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim S, Yong TS, Park SJ, Im KI, Kong Y, Ryu JS, Min DY, Shin MH. NADPH oxidase-derived reactive oxygen species-mediated activation of ERK1/2 is required for apoptosis of human neutrophils induced by Entamoeba histolytica. J Immunol. 2005;174:4279–4288. doi: 10.4049/jimmunol.174.7.4279. [DOI] [PubMed] [Google Scholar]
- Simeonova PP, Wang S, Hulderman T, Luster MI. c-Src-dependent activation of the epidermal growth factor receptor and mitogen-activated protein kinase pathway by arsenic. Role in carcinogenesis. J Biol Chem. 2002;277:2945–2950. doi: 10.1074/jbc.M109136200. [DOI] [PubMed] [Google Scholar]
- Song X, Vasilenko A, Chen Y, Valanejad L, Verma R, Yan B, Deng R. Transcriptional dynamics of bile salt export pump during pregnancy: Mechanisms and implications in intrahepatic cholestasis of pregnancy. Hepatology. 2014;60:1993–2007. doi: 10.1002/hep.27171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica A, Katzenellenbogen BS, Martin MB. Activation of estrogen receptor-alpha by the heavy metal cadmium. Mol Endocrinol. 2000;14:545–553. doi: 10.1210/mend.14.4.0441. [DOI] [PubMed] [Google Scholar]
- Strumylaite L, Bogusevicius A, Abdrachmanovas O, Baranauskiene D, Dregzdyte R, Pranys D, Poskiene L. Cadmium concentration in biological media of breast cancer patients. Breast Cancer Res Treat. 2011;125:511–517. doi: 10.1007/s10549-010-1007-8. [DOI] [PubMed] [Google Scholar]
- Takebayashi S, Jimi S, Segawa M, Kiyoshi Y. Cadmium induces osteomalacia mediated by proximal tubular atrophy and disturbances of phosphate reabsorption. A study of 11 autopsies. Pathol Res Pract. 2000;196:653–663. doi: 10.1016/S0344-0338(00)80010-2. [DOI] [PubMed] [Google Scholar]
- Thomas JE, Soriano P, Brugge JS. Phosphorylation of c-Src on tyrosine 527 by another protein tyrosine kinase. Science. 1991;254:568–571. doi: 10.1126/science.1719633. [DOI] [PubMed] [Google Scholar]
- Yu X, Filardo EJ, Shaikh ZA. The membrane estrogen receptor GPR30 mediates cadmium-induced proliferation of breast cancer cells. Toxicol Appl Pharmacol. 2010;245:83–90. doi: 10.1016/j.taap.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Waalkes MP. Cadmium carcinogenesis in review. J Inorg Biochem. 2000;79:241–244. doi: 10.1016/s0162-0134(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Xia F, Li J, Hickey GW, Tsurumi A, Larson K, Guo D, Yan SJ, Silver-Morse L, Li WX. Raf activation is regulated by tyrosine 510 phosphorylation in Drosophila. PLos Biol. 2008;6:1115–1129. doi: 10.1371/journal.pbio.0060128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang Y, Odwin-Dacosta S, Yager JD. Effects of cadmium on estrogen receptor mediated signaling and estrogen induced DNA synthesis in T47D human breast cancer cells. Toxicol Lett. 2009;184:134–138. doi: 10.1016/j.toxlet.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]