

Abstract
Rationale
Wnt signaling regulates key aspects of diabetic vascular disease.
Objective
We generated SM22-Cre;LRP6(fl/fl);LDLR-/- mice to determine contributions of Wnt co-receptor LRP6 in the vascular smooth muscle lineage (VSM) of male LDLR-null mice, a background susceptible to diet (HFD) - induced diabetic arteriosclerosis.
Methods and Results
As compared to LRP6(fl/fl);LDLR-/- controls, SM22-Cre;LRP6(fl/fl);LDLR-/- (LRP6-VKO) siblings exhibited increased aortic calcification on HFD without changes in fasting glucose, lipids, or body composition. Pulse wave velocity (index of arterial stiffness) was also increased. Vascular calcification paralleled enhanced aortic osteochondrogenic programs and circulating osteopontin (OPN), a matricellular regulator of arteriosclerosis. Survey of ligands and Frizzled (Fzd) receptor profiles in LRP6-VKO revealed upregulation of canonical and noncanonical Wnts alongside Fzd10. Fzd10 stimulated noncanonical signaling and OPN promoter activity via an USF-activated cognate inhibited by LRP6. RNAi revealed that USF1 but not USF2 supports OPN expression in LRP6-VKO VSM, and immunoprecipitation confirmed increased USF1 association with OPN chromatin. ML141, an antagonist of cdc42/Rac1 noncanonical signaling, inhibited USF1 activation, osteochondrogenic programs, alkaline phosphatase, and VSM calcification. Mass spectrometry identified LRP6 binding to protein arginine methyltransferase (PRMT) - 1, and nuclear asymmetric dimethylarginine modification was increased with LRP6-VKO. RNAi demonstrated that PRMT1 inhibits OPN and TNAP while PRMT4 supports expression. USF1 complexes containing the H3R17Me2a signature of PRMT4 are increased with LRP6-VKO. Jmjd6, a demethylase downregulated with LRP6 deficiency, inhibits OPN and TNAP expression, USF1:H3R17Me2a complex formation and transactivation.
Conclusions
LRP6 restrains VSM noncanonical signals that promote osteochondrogenic differentiation, mediated in part via USF1- and arginine methylation – dependent relays.
Keywords: Vascular calcification, LRP6, Wnt, USF1, protein arginine methylation, arteriosclerosis, type 2 diabetes mellitus, transcriptional regulation, signal transduction
INTRODUCTION
Hyperglycemia, hyperlipidemia, and uremia accelerate vascular aging, compromising arterial function necessary for normal blood flow, metabolism and tissue homeostasis1. Along with hypertension these dysmetabolic states induce arteriosclerotic stiffening, thereby reducing vascular compliance that underlies Windkessel physiology -- elasticity of conduit vessels that ensures smooth distal tissue perfusion throughout the cardiac cycle1. Atherosclerotic burden, mural thickening and fibrosis, medial calcification, elastin fragmentation, non-enzymatic matrix crosslinking, and endothelial dysfunction are features of arteriosclerotic aging. Multiple labs have now identified bone morphogenetic proteins (BMPs) and Wnts – polypeptides that convey paracrine cues during skeletal morphogenesis – as pathogenic signals in arteriosclerotic calcification2–7. In studies of LDLR-/- mice fed high fat diabetogenic8 diets (HFD) typical of western societies, we identified that osteogenic Msx-Wnt signaling cascades are ectopically activated in the vasculature with concomitant induction of diabetes8, obesity, and arterial calcification5. Expression of the osteoblast transcription factor Msx2 in mural myofibroblasts was shown to be activated by inflammatory signals that support arterial mineralization2, 5. Conditional deletion of Msx2 and Msx1 in the vascular smooth muscle and myofibroblast (VSM) lineage reduces arteriosclerotic calcification and vascular stiffening, with down-regulation of multiple Wnt ligands conveying canonical and noncanonical actions, including Wnt7b, Wnt5a, and Wnt2 9. In the mesenchymal lineage, Wnt7b induces osteogenesis via both pathways10, 11. Intriguingly, when expressed in the endothelial cell (EC) lineage, Wnt7b stabilizes the EC phenotype and thereby restrains mesenchymal expression of Msx210. While Wnt16 limits chondroid programing of VSM6, Wnt5a promotes osteochondral differentiation4. Thus, Wnt signals emerge as important contributors to vascular disease biology.
The panoply of Wnt receptors regulating vascular sclerosis is only beginning to be investigated. In broad terms, the 10 members of the Frizzled (Fzd) family of Wnt receptors form complexes with either (a) LDL receptor related proteins LRP5 and LRP612; or (b) other signaling proteins (ROR2, Celsr)13 to activate canonical or noncanonical signaling pathways, respectively. Additionally, LRP5 stimulates aerobic glycolysis through pathways independent of canonical β-catenin mechanisms14, and Aaronson introduced the notion that LRP6 could restrain noncanonical cascades15. Rajamannan demonstrated that LRP5 supports valve calcification in the apolipoprotein E-null mouse16. While LRP5 and LRP6 exhibit redundant roles during prenatal skeletal development17, human genetics suggest that LRP6 might play a uniquely important role in postnatal atherosclerosis and osteoporosis18 and bone-vascular interactions19.
To better understand the role of the vascular LRP6 Wnt receptor in arteriosclerosis, we utilized the SM22-Cre transgene20 to delete VSM LRP621 in LDLR-/- mice. We discover that LRP6 restrains noncanonical signals that drive VSM osteochondrogenic programs in male LDLR-/- mice fed HFD8, 9, mediated in part via USF1- and protein arginine methylation- dependent relays.
METHODS
See online Supplement.
RESULTS
Conditional deletion of LRP6 in VSM accentuates aortic calcification in LDLR-/- mice fed HFD, increases vessel stiffening and promotes ectopic arterial expression of the osteochondrogenic phenotype
LRP6 is expressed in the arterial vasculature22, primarily in mural VSM and fibrous caps of atherosclerotic lesions in the aortic sinus of LDLR-/- mice fed HFD (Figure 1A, 1B; Online Supplement Figures I–II). To better understand the role for LRP6 in the biology of arteriosclerotic calcification, we generated SM22-Cre;LRP6(fl/fl);LDLR-/- mice, conditionally depleting LRP6 in the VSM lineage in male LDLR-null mice, a background susceptible to HFD – induced diabetes and arteriosclerotic calcification23. As shown in Figure 1B, following a 3 month challenge with HFD, arterial calcification was increased in SM22-Cre;LRP6(fl/fl);LDLR-/- mice as compared to LRP6(fl/fl);LDLR-/- sibling controls, with calcium deposition observed in both medial and atherosclerotic venues (Supplement Figure III). Chow-fed animals exhibited much lower aortic calcium levels that did not differ between genotypes (Figure 1B). Aortic stiffness was increased in SM22-Cre;LRP6(fl/fl);LDLR-/- mice and by HFD as determined by echocardiography9 (Figure 1C; reduced aortic arch distensibility); insulin resistance was diet-dependent but independent of genotype (Figure 1D). RT-qPCR analysis revealed reductions in aortic LRP6 mRNA in SM22-Cre;LRP6(fl/fl);LDLR-/- mice, with concomitant increases in LRP4 and markers of osteochondrogenic programming (Figure 1E). Axin2 – a target of canonical β-catenin24 – was diminished in aortas deficient for LRP6, while Klf5 – a target of noncanonical Wnt signaling like OPN25, 26 -- was upregulated along with the progenitor marker Sca1 27(Fig. 1E). While clear trends for increased aortic osteochondrogenic programming were noted in chow-fed mice with VSM LRP6 deficiency, these differences did not reach significance (Figure IV) in the absence of HFD. However, the increases in osteochondrogenic programs were robustly elaborated in primary aortic VSM cultures from SM22-Cre;LRP6(fl/fl);LDLR-/- mice (Figure 1F; TNAP, OCN, OPN) and associated with increased mineralization in vitro (vide infra). Aspects of the contractile VSM program were diminished, indicated by down-regulation of Myh11 and myocardin (Figure 1F, and not shown). Plasma levels of osteopontin (OPN)28 were also increased in SM22-Cre;LRP6(fl/fl);LDLR-/- mice vs. LRP6(fl/fl);LDLR-/- controls following HFD challenge (Figure 1G). Measurement of medial thickness in the ascending aorta sinus revealed no significant increases in HFD-fed animals with reduced VSM LRP6 although pulse wave velocity was increased (Figure V). Aortic proliferation indices did not differ between genotypes; however, cultured VSM from SM22-Cre;LRP6(fl/fl);LDLR-/- mice exhibited 10% greater BrdU incorporation (Figure VI-VIII). Aortic lumen diameter of animal on HFD did not differ between genotypes as quantified by echocardiography (Figure IX); however, a non-significant trend (p = 0.1) for increased Mac2(+) atheroma area in the sinus was observed along with significantly increased thoracic aortic F4/80 and IL12A macrophage expression (Figures X–XI). Importantly, differences in aortic calcification and stiffness between genotypes arose in the absence of differences in HFD-induced changes in fasting blood glucose, lipids, insulin resistance, or body composition (Figures XII–XIII). Thus, absence of VSM LRP6 increases aortic calcification and vascular stiffness in diabetic LDLR-/- mice, and enhances vascular elaboration of an osteochondrogenic gene program.
Figure 1. Conditional deletion of LRP6 in VSM increases osteochondrogenic calcification and arterial stiffening in LDLR-/- mice fed high fat diabetogenic diets.







Panel A, immunohistochemistry reveals expression of LRP6 in VSM of aortic tunica media (arrows) and atherosclerotic caps (arrowheads) of the aortic sinus. Asterisks overlay atheroma. Scale bar = 50 microns. Coronary artery VSM also expresses LRP6 (Figure S1–S2). Panel B, on HFD aortic calcium content is increased in aortas of SM22-Cre;LRP6(fl/fl);LDLR-/- mice as compared to LRP6(fl/fl);LDLR-/- on HFD or chow-fed controls (11–15 weeks of age). All HFD animals were 17–20 weeks of age at aortic analysis. Numbers of animals in each group were between 12–14 as indicated. ANOVA p < 0.0001. a, p< 0.001 vs. chow-fed controls; b, p < 0.01 vs. Cre-negative mice on HFD by Holm-Sidak’s post-hoc testing corrected for multiple comparisons. Panel C, Doppler echocardiography reveals increased vascular stiffness in SM22-Cre;LRP6(fl/fl);LDLR-/- mice, reflected in reduced distensibility (% change in aortic arch diameter from diastole to systole). ANOVA p < 0.0001. ***, p < 0.001 vs LRP6(fl/fl);LDLR-/- on chow diet*, p < 0.05 vs. Cre-minus on Western Diet on post-hoc testing corrected for multiple comparisons. N = 4 to 10 per group as indicated in the X-axis labels. Panel D, HFD-induced insulin resistance did not differ between genotypes. One way ANOVA p <0.001; significant differences between the groups as indicated by Holm-Sidak post-hoc test (see also supplement Figures XXII–XXIII). a, p < 0.05 vs. chow fed of either genotype; b, p < 0.05 vs. chow fed Cre-negative animals; c, p < 0.01 between chow fed SM22-Cre;LRP6(fl/fl);LDLR-/- animals. No other differences were significant. Panel E, the aortic expression of osteochondrogenic genes is concomitantly upregulated with reductions in VSM LRP6 expression. *, p < 0.05 vs. LRP6(fl/fl);LDLR-/- control; **, p < 0.01 vs. control; ***, p < 0.001 vs. control by Student’s 2-tailed t-test; used throughout unless otherwise indicated. Panel F, cultures of primary aortic VSM also exhibit enhanced osteochondrogenic gene expression in the setting of LRP6 deficiency. *, p < 0.05 vs. LRP6(fl/fl);LDLR-/- control; **, p < 0.01 vs. control; ***, p < 0.001 vs. control by Student’s 2-tail t-test; used throughout unless otherwise indicated. Panel G, circulating OPN levels are increased in diabetic SM22-Cre;LRP6(fl/fl);LDLR-/- mice as compared to LRP6(fl/fl);LDLR-/- controls. N = 8, 7, 9, and 7, respectively as indicated. ANOVA p = 0.022. * p < 0.05 vs. all others by Fisher’s LSD post-hoc test, and p < 0.05 vs. Cre-minus control on HFD after correction for multiple comparisons.
Wnt ligands and Fzd receptors capable of activating noncanonical signals are upregulated in aortic tissues of SM22-Cre;LRP6(fl/fl);LDLR-/- mice on HFD
Skeletal biomineralization occurs via the overlapping yet distinct mechanisms of membranous (type 1 collagen-oriented) and endochondral (type 10 collagen-oriented) ossification29. Canonical Wnt signals promote initiation of the former17 and inhibit bone resorption30, while noncanonical Wnt signals promote mature tissue calcification via both mechanisms31, 32. LRP6 can restrain noncanonical signals in part by sequestering certain Fzd co-receptors15. To assess whether paracrine relays capable of noncanonical signaling were altered in aortic tissues with LRP6 deficiency, we surveyed the expression of Wnt ligands and Fzd co-receptors. Array analysis of aortic RNA from mice on HFD for 3 months (n= 5/genotype) revealed upregulation of genes encoding multiple Wnt ligands and Fzd10 (Supplement Figure XIV). This was confirmed by RT-qPCR; as compared to LRP6(fl/fl);LDLR-/- controls, SM22-Cre;LRP6(fl/fl);LDLR-/- mice on HFD exhibited elevated levels of Wnt7b, Wnt4, Wnt10a, Wnt3a, and Fzd10 (Figure 2A and Figure 2B). Increases in aortic mRNAs observed in vivo required HFD challenge (Figure XV). However, significant upregulation of ligands and Fzd10 expression was observed in primary VSM cultures (Figure XVI; and vide infra). Wnt5a -- the abundant noncanonical agonist4 -- remained unchanged. Immunohistochemistry and western blot analysis of primary VSM cultures confirmed upregulation of Wnt7b (Figure 2C), Wnt10a (Figure 2D), and Fzd10 protein with down-regulation of LRP6 (Figure 2E). Of note, while Wnt3a and Wnt10a are reported to elicit only canonical signals, Wnt433, 34 and Wnt7b11, 23 are capable of supporting both canonical and noncanonical pathways. Thus, loss of VSM LRP6 leads to the upregulation of Wnt ligands capable of supporting canonical and noncanonical signaling.
Figure 2. Wnt ligands and Fzd receptors capable of activating noncanonical signals are upregulated in aortas of SM22-Cre;LRP6(fl/fl);LDLR-/- mice fed diabetogenic diets.



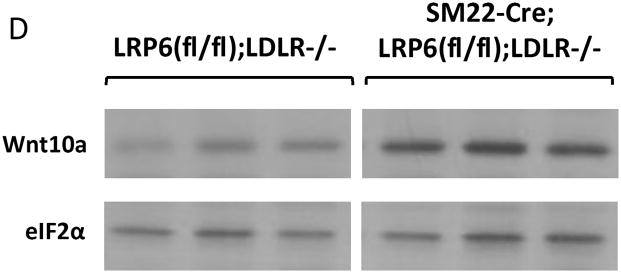

Panel A, RT-qPCR confirms upregulation of Wnt10a, Wnt7b, Wnt4, and Wnt3a in aortas of diabetic SM22-Cre;LRP6(fl/fl);LDLR-/- mice. N = 4 to 7 per group as indicated in the legend. **, p < 0.01 vs. Cre-negative control. ***, p < 0.001 vs. Cre-negative control by Student’s 2-tail t-test. Panel B, the expression of Fzd10 was selectively increased, consistent with array analyses (Figure S14). ***, p < 0.001 vs. Cre-negative control by Student’s 2-tail t-test. Panel C upper, immunohistochemistry revealed increases in aortic Wnt7b protein. Western blots using extracts from LRP6(fl/fl);LDLR-/- control and SM22-Cre;LRP6(fl/fl);LDLR-/- VSM confirmed increases in Wnt7b protein with LRP6 deficiency (lower panel). Panel D, western blot for Wnt10a protein accumulation followed by digital image analysis confirmed 3-fold upregulation of Wnt10a protein in LRP6-deficient aortic VSM (p < 0.05). Panel E, Cre-dependent reductions in LRP6 were accompanied by increased Fzd10 protein accumulation, consistent with analyses of whole aorta RNA (panel B above) and cultured VSM (Figure XVI).
Fzd10 activates noncanonical signaling that is inhibited by LRP6 expression, and promotes OPN transcription via USF protein-DNA interactions
Fzd10 activation in sarcoma elicits noncanonical signals35 similar to Fzd9 in bone31. To confirm and extend this, we transiently co-transfected Fzd10 expression vectors with NFAT-LUC36 and LEF-LUC23 reporters that register noncanonical and canonical signaling, respectively, in HEK293T cells37. As shown in Figure 3A, Fzd10 upregulated NFAT-LUC activity with modest impact on LEF-LUC, while LRP6 upregulated canonical LEF-LUC activity with little effect on NFAT-LUC. However, co-expression of LRP6 reduced Fzd10 activation of NFAT-LUC, and Fzd10 impaired LRP6 upregulation of LEF-LUC (Figure 3A). Similar responses occur with Fzd9 (Figure 3B, and data not shown), the Fzd member most closely related to Fzd10. ML141 – an inhibitor of cdc42/Rac1 signaling38 in the noncanonical Wnt planar cell polarity pathway39 – inhibited Fzd10 and Fzd9 activation of NFAT-LUC (Figure 3B). Conversely, constitutively active Cdc42(Q61L) activated NFAT-LUC, and was inhibited by LRP6 (Figure 3C). In primary cultures of aortic VSM, Fzd10 but not Fzd9 is upregulated with LRP6 deficiency (Figure XVI). Furthermore, HEK reporter cells transfected with NFAT-LUC +/- pCMV-Fzd10 exhibited Fzd10-dependent noncanonical NFAT activation when co-cultured onto VSM feeder cell layers from SM22-Cre;LRP6(fl/fl);LDLR-/- mice vs. LRP6(fl/fl);LDLR-/- controls (Figure 3D). This indicated that Fzd10 conveys responsiveness to a noncanonical agonist elaborated by SM22-Cre;LRP6(fl/fl);LDLR-/- VSM.
Figure 3. Fzd10 activation of noncanonical signaling is inhibited by LRP6 and the cdc42 antagonist ML141.




Panel A, Fzd10 activates NFAT-LUC reporter activity in HEK cells and co-expression of LRP6 inhibits induction (left). By contrast, LRP6 activates canonical TCF/LEF signaling via pathways inhibited by Fzd10 (right). ANOVA p <0.0001 with post-hoc Holm-Sidak testing. N = 3 per group. Panel B, ML141, a cdc42 antagonist, inhibits Fzd9 and Fzd10 induction of NFAT-LUC. Fzd1, Fzd2, and Fzd6 were inactive (not shown). ANOVA p <0.0001 with post-hoc Holm-Sidak testing. N = 6 per group. Panel C, constitutively active cdc42(Q61L) stimulation of noncanonical NFAT signaling is enhanced by Fzd10 and inhibited by LRP6. ANOVA p <0.0001 with post-hoc Holm-Sidak testing. N = 3 – 9 per group. Panel D, HEK cells were transfected with either pCMV vector + NFAT-LUC or pCMV-Fzd10 + NFAT-LUC, then parachuted for co-culture (Co-Cx) onto lawns of either LRP6(fl/fl);LDLR-/- VSM or SM22-Cre;LRP6(fl/fl);LDLR-/- VSM. Note that SM22-Cre;LRP6(fl/fl);LDLR-/- cells supported Fzd10-dependent activation of NFAT-LUC signaling, indicating enhanced elaboration of a noncanonical agonist with VSM LRP6 deficiency. ANOVA p < 0.0001, with post-hoc Holm-Sidak testing. N = 6 per group.
OPN is an endogenous noncanonical Wnt target40 and osteochondrogenic gene active during membranous and endochondral bone formation29, and is upregulated in SM22-Cre;LRP6(fl/fl);LDLR-/- VSM. RNAi targeting Fzd10 but not Fzd9 reduces OPN gene expression in SM22-Cre;LRP6(fl/fl);LDLR-/- VSM (Figure 4A). As with NFAT-LUC, Fzd10 upregulates OPN promoter activity and is inhibited by LRP6 expression (Figure 4B). Unlike Fzd10, Fzd7 and Fzd1 are inactive in this assay (Figure XVII and not shown). Co-expression of either Wnt7b or Wnt5a enhanced Fzd10 activation, while Wnt11 and Wnt4 did not (Figure XVIII). OPN promoter mapping identified that the glucose-responsive USF cognate41 at −80 to −72 relative to the transcriptional start site is required for Fzd10 induction (Figure 4C). Co-expression of either USF1 or USF2 activated the OPN promoter, and was inhibited by LRP6 expression (Figure 4D), indicating that OPN expression was entrained to noncanonical signals conveyed in part by USF and inhibited by LRP6. Consistent with this, siRNA targeting USF1, but not USF2, inhibited the upregulation of OPN with LRP6 deficiency (Figure 4E; Figure XIX), and chromatin immunoprecipitation revealed increased association of USF1 with OPN chromatin in LRP6-deficient VSM (Figure 4F). ML141 inhibited USF1 activation of OPNLUC (Figure 4G) as well as Fzd10 activation of NFAT-LUC (Figure 3B) and OPNLUC (Figure XX). The constitutively active variant Cdc42(Q61L) upregulated the OPN promoter in HEK cells (Figure XXI). Moreover, ML141 reduced OPN elevation in LRP6-deficient VSM (Figure 4H); by contrast, the selective Rac1 inhibitor EHT186442 exerted little if any effect on OPN expression (Figure 4H). Thus, Fzd10- and USF1- activation of the OPN promoter is inhibited by LRP6, with LRP6 actions phenocopied by ML141 treatment.
Figure 4. Fzd10 activation of OPN transcription maps to the USF cognate, and USF1 upregulation of OPN promoter activity is inhibited by LRP6 or the cdc42 antagonist ML141.








Panel A, siRNA targeting Fzd10 reduced OPN expression in SM22-Cre;LRP6(fl/fl);LDLR-/- VSM. ANOVA p = 0.001. N = 4 per group. **, p < 0.01 vs. Fzd9 siRNA and p < 0.05 vs. control siRNA by Holm-Sidak post-hoc testing. Panel B, like NFAT-LUC, Fzd10 activation of the OPN promoter is inhibited by LRP6 expression. N = 3 per group, ANOVA p < 0.001; ***, p < 0.001 vs. all other conditions by Holm-Sidak test. Panel C, Fzd10 activation of the OPN promoter maps to the proximal CCTCATGAC USF cognate. N = 3 per group, significance assessed by Student’s 2-tail t-test. Panel D, USF activation of the OPN promoter is inhibited by LRP6 expression. N =3–6 per group. ANOVA p < 0.0001. a, p < 0.05 vs. vector, b, p < 0.001 vs LRP6; p < 0.01 vs. USF1+LRP6; d, p < 0.05 vs. vector, e, p < 0.02 vs. USF2+LRP6 by Holm-Sidak post-hoc test. Panel E, USF1 siRNA, but not USF2 siRNA, significantly reduces OPN induction with LRP6 deficiency in VSM. ANOVA p < 0.0001, with post-hoc Holm-Sidak testing. N = 4 per group. Panel F, ChIP assay confirms increased association of USF1 with OPN chromatin in SM22-Cre;LRP6(fl/fl);LDLR-/- VSM. N = 4 per group; **, p ≤ 0.01 by Student’s 2-tail t-test. Panel G, USF1 activation of OPN promoter activity is inhibited by ML141. ANOVA p < 0.0001, ***, p < 0.001 by Holm-Sidak testing. N = 6 per group. Panel H, the increased OPN gene expression in SM22-Cre;LRP6(fl/fl);LDLR-/- VSM is reduced by ML141. By contrast, the Rac1 inhibitor EHT1864 had no effect. Treatments were 10 uM for 20 hours. ANOVA p < 0.0001 (post hoc Holm-Sidak). N = 4 per group.
LRP6 associates with PRMT1, and SM22-Cre;LRP6(fl/fl);LDLR-/- aortic VSM exhibits increased nuclear protein ADMA accumulation
To better understand the mechanisms whereby LRP6 regulates signaling, we expressed FLAG-epitope tagged LRP6 in HEK cells, immunoprecipitated FLAG-containing complexes under non-denaturing conditions, and began to characterize the LRP6 interactome by mass spectrometry (to be presented elsewhere). As compared to control cells, immune complexes from LRP6-FLAG expressing cells revealed enrichment of PRMT1. Co-immunoprecipitation of recombinant PRMT1 variant 1 (PRMT1v1) and LRP6-FLAG was independently demonstrated as shown in Figure 5A. PRMT1 is a protein arginine methyltransferase that catalyzes formation of asymmetric dimethylarginine (ADMA) on protein targets. Consistent with prior reports43, we identified that PRMT1v1 is present in cytoplasmic, membrane, and nuclear fractions of VSM by western blot analysis and immunohistochemistry (Figure 5B; Figure XXII). PRMT1v2, possessing a nuclear export sequence43, is only found in the non-nuclear fractions (Figure 5B). These observations prompted assessment of the cellular ADMA profile by western blot. As shown in Figure 5B, protein ADMA modification profiles were altered in the cytoplasmic, membrane, and nuclear fractions of SM22-Cre;LRP6(fl/fl);LDLR-/- VSM; the greatest changes observed were increases in a subset of ADMA-modified nuclear proteins (Figure 5B). Independent assessment of replicates demonstrated ~2- to 3 -fold increases in specific nuclear ADMA –modified proteins (Figures 5C and 5D).
Figure 5. LRP6 forms a complex with PRMT1v1, and cellular protein ADMA accumulation is perturbed in LRP6-deficient VSM.

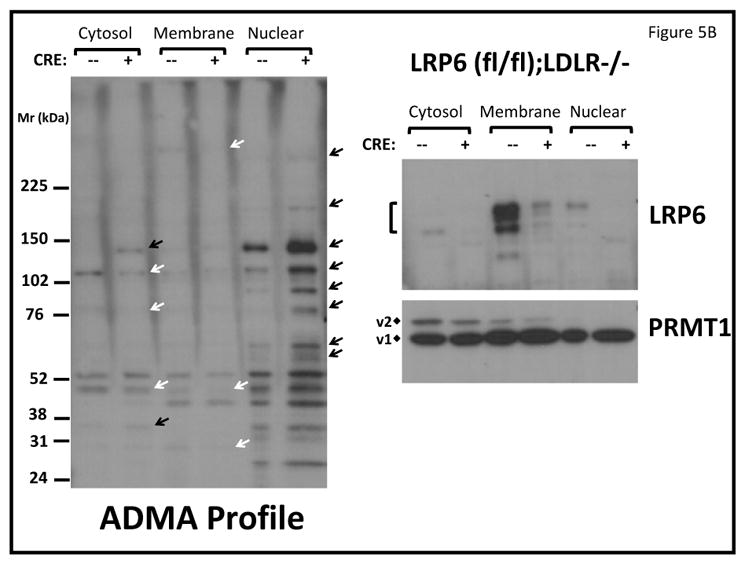
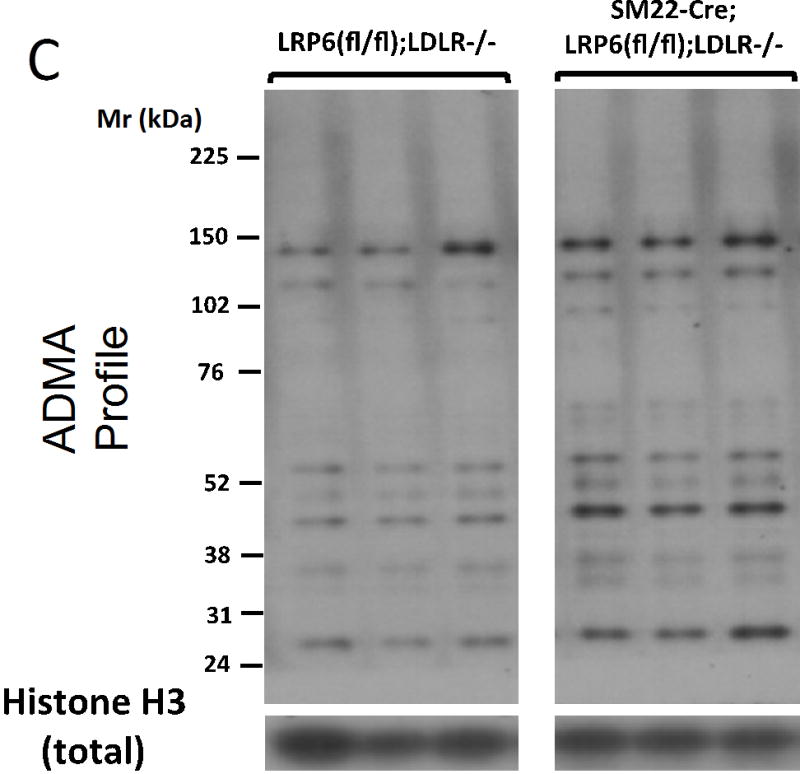

Panel A, PRMT1v1 and Flag-tagged LRP6 are co-precipitated when co-expressed in HEK cells. Panel B left, protein asymmetric dimethylarginine (ADMA) profiles are perturbed in LRP6-deficient VSM. While a few ADMA proteins are down-regulated (white arrows), the majority of those visualized are increased in SM22-Cre;LRP6(fl/fl);LDLR-/- nuclear fraction (black arrows). Right, while Cre-mediated down-regulation of LRP6 protein in the membrane fraction was readily detected (upper right), the broad distribution of PRMT1 exhibited little if any change. v2, PRMT1 variant 2 possessing the nuclear export signal (NES). v1, PRMT1 variant 1 lacking the NES. Panels C and D, digital image analysis confirmed significant 2–3 fold increases in total nuclear ADMA protein accumulation in LRP6-deficient VSM. N = 3 per group. *, p < 0.05 by Student’s t-test.
PRMT1 and PRMT4 are the two major PRMTs responsible for nuclear protein ADMA modification44. Therefore, we examined the consequence of RNAi-mediated knockdown of PRMT1 and PRMT4 in LRP6(fl/fl);LDLR-/- and SM22-Cre;LRP6(fl/fl);LDLR-/- primary VSM, emphasizing OPN expression. Knockdown of PRMT1 increased OPN expression in both genotypes (Figure 6A), indicating a suppressive role. By contrast, knockdown of PRMT4 reduced OPN upregulation in SM22-Cre;LRP6(fl/fl);LDLR-/- cultures -- indicating a sustentacular role (Figure 6B). However, while nuclear protein levels of USF1 increased in SM22-Cre;LRP6(fl/fl);LDLR-/- mice, PRMT4 levels did not (Figure 6C–6D), suggesting that ADMA accumulation might relate to alterations in arginine N-methyl group turnover.
Figure 6. RNAi targeting PRMT4 reduces, while PRMT1 siRNA increases, OPN gene induction in LRP6-deficient VSM.




Panel A, PRMT1 siRNA increases OPN gene expression. ANOVA p <0.0001, with post-hoc testing corrected for multiple comparisons. N = 4 per group. Panel B, RNAi targeting PRMT4 almost completely inhibits induction of OPN with LRP6 deficiency. ANOVA p < 0.0001, N = 4 per group. Panels C and D, LRP6 deficiency increases USF1 nuclear protein accumulation without altering PRMT4. N = 3 per group. **, p < 0.01 by Student’s t-test.
The protein arginine demethylase Jmjd6 inhibits osteochondrogenic gene expression in VSM and USF1-dependent transactivation
Because nuclear ADMA accumulation was altered in the absence of increased nuclear PRMT4, we examined the expression of Jmjd645, 46 and PADI447, genes encoding broad spectrum nuclear arginine demethylase and deiminase activities, respectively, that remove the arginine N-methyl signature. As shown in Figure 7A, the levels of both Jmjd6 and PADI4 mRNAs were reduced in SM22-Cre;LRP6(fl/fl);LDLR-/- mice, with concomitant upregulation of OPN and nuclear ADMA protein accumulation. However, only siRNA targeting Jmjd6 increased OPN and TNAP gene expression in control VSM cultures (Figure 7B; Figure XXIII); PADI4 siRNA reduced mRNA accumulation for select osteochondrogenic genes (Figure 7C). Moreover, Jmjd6 expression inhibited USF1 activation of the OPN promoter (Figure (7D) while PADI4 did not (Figure XXIV).
Figure 7. The broad specificity arginine demethylase Jmjd6 is reduced in LRP6 deficient VSM, restrains VSM osteochondrogenic gene expression, and inhibits USF1-dependent OPN transcription.







Panel A, while OPN is increased with LRP6 deficiency in VSM, Jmjd6 and PADI4 are significantly down-regulated. The relative level of PADI4 is ca. 100-fold less than Jmjd6. N = 4 per group. *, p < 0.05; ***, p < 0.001 vs. Cre-negative control by Student’s 2-tail t test. Panels B and C, Jmjd6 siRNA significantly upregulates OPN and TNAP expression in VSM, while PADI4 siRNA does not. N = 4 per group. **, p < 0.01; ***, p < 0.001 vs. control siRNA by Student’s 2-tail t test. Panel D, Jmjd6 inhibits USF1 activation of the OPN promoter in transient transfection assays. ANOVA p < 0.001. ***, p < 0.001 vs. all other treatments by Holm-Sidak post hoc test. N = 4 per group. Panels E and F, H3 co-precipitates with Flag-tagged USF1. Co-expression of Jmjd6 significantly reduces the PRMT4 H3R17Me2a signature on histone H3 co-precipitating with USF1. N = 4 per group, p < 0.01 by Student’s 2-tail t test. Panel G, USF1 immunoprecipitates from LRP6-deficient VSM cultures contain histone H3 bearing the PRMT4 signature H3R17Me2a.
In HEK cells, USF1 is not ADMA modified, but a low-molecular weight ADMA protein co-immunoprecipitated with FLAG-tagged USF1 (supplement Figure XXV). The size of this ADMA protein, 17 kDa, suggested it might be histone H3. Western blot analysis confirmed H3 co-precipitation with USF1, revealed the presence of the PRMT4-specific H3R17Me2a signature48, and demonstrated USF1-associated H3R17Me2a was reduced by co-expression of Jmjd6 (Figure 7E and 7F). Furthermore, USF1 immunoprecipitates from LRP6-deficient VSM cultures contain increased histone H3 with the H3R17Me2a signature (Figure 7G). Thus, Jmjd6 inhibits the osteochondrogenic phenotype in VSM, antagonizes USF1-dependent transcriptional activation of OPN gene expression, and reduces PRMT4-dependent histone signatures in USF1 protein complexes.
ML141 inhibits USF1 protein accumulation, osteochondrogenic gene expression, TNAP upregulation and calcification in VSM cultures from SM22-Cre;LRP6(fl/fl);LDLR-/- mice
ML141 treatment down-regulated a subset of nuclear ADMA proteins upregulated with LRP6 deficiency (Figure XXVI) -- and reversed the nuclear USF1 protein accumulation and OPN chromatin association arising in SM22-Cre;LRP6(fl/fl);LDLR-/- VSM (Figure 8A; Figure XXVII-XXVIII). Since ML141 inhibited Fzd10 activation of noncanonical Wnt signaling and USF1-dependent OPN expression (vide supra), we assessed the impact of ML141 on osteochondrogenic programs upregulated by LRP6 deficiency. Like OPN, TNAP and Col10A1 were reduced by ML141 (Figure 8B and 8C). Interestingly, Wnt ligand genes including Wnt4, Wnt5a, and Wnt5b were concomitantly down-regulated by ML141 in LRP6-deficient VSM while Jmjd6 was unaffected (Figure XXIX). TNAP enzyme activity – necessary for OPN dephosphorylation and osteochondrogenic mineralization29 -- was upregulated in SM22-Cre;LRP6(fl/fl);LDLR-/- VSM and inhibited by ML141 treatment (Figure 8D). Consistent with this, calcium deposition quantified by Alizarin red staining was significantly increased in SM22-Cre;LRP6(fl/fl);LDLR-/- VSM and inhibited by ML141 treatment (Figure 8E, 8F). Thus LRP6 restrains vascular noncanonical Wnt signals sensitive to ML141 that promote osteochondrogenic responses, mediated in part via USF1- and arginine methylation – dependent relays (Figure 8G).
Figure 8. The cdc42 antagonist ML141 down-regulates USF1 protein accumulation and osteochondrogenic mineralization of LRP6-deficient VSM without globally altering ADMA protein profiles.







Panel A, the increased VSM nuclear USF1 protein levels arising from LRP6 deficiency is reduced by ML141 treatment. Although select nuclear ADMA proteins were reduced, the global ADMA profile was largely unaffected (Figure S26 and data not shown), ANOVA p =0.015, *, p < 0.01 from others (Holm-Sidak). N = 3–4 per group as indicated. Panels B and C, like OPN, the increases in TNAP and Col10A1 arising from LRP6 deficiency are inhibited by ML141 but not by EHT1864. *, p < 0.05 vs. vehicle-treated LRP6(fl/fl);LDLR-/- control. *, p < 0.05 vs. vehicle-treated LRP6(fl/fl);LDLR-/- control by post-hoc testing. N = 4 per group. Panel D, TNAP enzyme activity is induced with LRP6 deficiency and inhibited by ML141. ANOVA p < 0.001. a, p < 0.01 vs. vehicle treated LRP6(fl/fl);LDLR-/- control. b, p < 0.0001 vs. ML141 treated LRP6(fl/fl);LDLR-/- control; c, p < 0.0001 vs. vehicle treated SM22-Cre;LRP6(fl/fl);LDLR-/- VSM by the Holm-Sidak’s multiple comparisons test. Panels E and F, Alizarin red staining of demonstrates that the increased calcification arising in SM22-Cre;LRP6(fl/fl);LDLR-/- VSM is reversed by ML141. ¶, p < 0.05 vs. vehicle SM22-Cre;LRP6(fl/fl);LDLR-/- VSM. N = 3 per group, ANOVA p = 0.011 with post-hoc Holm-Sidak’s testing. Panel G, working model. See Online Supplement Figure XXX for details.
DISCUSSION
There are three principal findings of this study that help advance our understanding of arteriosclerotic calcification. Firstly, LRP6 signaling plays a cell-autonomous role in regulating the osteochondrogenic response within the VSM lineage. The VSM functions of LRP6 in arteriosclerotic calcification have not been previously characterized. Surprisingly, in addition to conveying canonical signals, LRP6 restrains noncanonical signals that reinforce osteochondrogenic trans-differentiation and mineralization of VSM49. Based upon lineage tracing, Speer estimated that approximately 80% of mineralizing cells in the vessel wall arise from this process; the remaining 20% reflect lineage allocation of regional or circulating osteoprogenitors49. Elegant studies by Mani first identified the private mutation LRP6(R611C) as causing a precocious atherosclerosis-osteoporosis syndrome18, 22, and hypomorphic function may also extend to reduced inhibition of noncanonical signals18. Secondly, USF1 emerges as a novel mediator of noncanonical Wnt signaling alongside the Jun/ATF and Ca++/NFAT pathways. The role of USF1 as relevant to the β-globin locus control region50, glucose signaling41, and lipid homeostasis51 is well appreciated, and the genetic link between USF1 and combined hyperlipidemia has been established in parallel with the identification of USF1 within atherosclerotic plaques 51. As in bone and cartilage29, 36, cdc42-modulated NFAT signaling will also be an important component of the VSM osteochondrogenic response during arteriosclerotic calcification along with Runx2 and USF1. Given that ML141 inhibits VSM biomineralization – and cdc42’s role in vascular inflammation52 and endochondral ossification53 -- we speculate that modulators of the cdc42/Rac1 family may prove useful in treating arteriosclerosis. Thirdly, discovery that LRP6 functionally and physically interacts with the protein arginine methylation cascade reveals a new dimension in LRP biology. Our data indicate that non-nuclear PRMT1 associated with LRP6 is part of a pathway suppressing noncanonical signals, while nuclear PRMT4 supports the osteochondrogenic phenotype in collaboration with osteogenic transcription factors29. Others have noted that nuclear PRMT4 (CARM1) forms a complex with Jmjd645. Whether nuclear vs. non-nuclear pools of PRMT1 differentially impact LRP6 signaling remains to be studied.
PRMT4 plays an important role in endochondral bone formation54 and regulation of estrogen receptor methylation45 in concert with Jmjd6. Since USF1 lacks overt ADMA modification, we anticipate that proteinaceous partners of USF1 will be directed for ADMA modification. Our data indicate that histone H3 – bearing the H3R17Me2a signature of PRMT4 – is one component of the protein complexes associated with USF1. It remains to be determined whether affinity of USF1 for chromatin is enhanced by H3R17Me2a. USF1 dimerizes with other bHLH and leucine zipper transcription factors to create unique DNA binding specificities55. Furthermore, USF1 recruits nuclear PRMT1 to the β-globin locus control region to preserve euchromatin structure via H4R3Me2a formation50. It will be important to elucidate the USF1 interactome and its regulation by noncanonical signals.
Fzd9 and Fzd10 are closely related family members that convey noncanonical Wnt signals31, 35. Upregulation of noncanonical Fzd10 signaling participates in the pathobiology of synovial sarcoma35. Conversely, Fzd9-null mice exhibit reduced endochondral bone formation during fracture repair, arising from deficiency in noncanonical signals necessary for osteoblast maturation and mineralization31. It remains to be determined the relative extent to which canonical vs. noncanonical Wnt signals contribute to arterial calcification in the LDLR-/- model. As noted, osteogenic lineage allocation is promoted by Msx and canonical Wnt signals5, 29 -- but these signals need to be down-regulated to permit osteoblast maturation29. This sequence is emerging as important for the osteogenic programming of vascular progenitors9, 49. VSM trans-differentiation is responsible for a majority of the vascular osteochondrogenic cell “load”49. We propose that upregulation of noncanonical Fzd activity with LRP6 deficiency enhances VSM osteochondrogenic trans-differentiation in response to metabolic stress.
Our study has limitations. The composition of the LRP6 complex that negatively regulates noncanonical Fzd relays remains to be determined. It’s interesting to note that PRMT1 modifies G3BP family members56. Because G3BP1 localizes GTPase activating complexes56, homologs might be involved in negative regulation of cdc42. However, VSM LRP6 likely orchestrates protein-protein interactions between multiple regulatory components of the noncanonical pathway15. Furthermore, LRP6 forms heterodimers with LRP5 57. 57 Given that LRP5 activity drives pro-sclerotic canonical Wnt signaling in valve calcification16, it remains probable that heterodimeric interactions between specific LRPs and Fzds finely tune signaling via the canonical and noncanonical pathways. LRP5/6 heterodimers confer selectivity for Wnt ligand activation57; this combinatorial complexity indicates that changes in levels of a specific LRP receptor will impact canonical signaling as a function of the prevailing Wnt ligand milieu while directly modulating noncanonical tone. Even though we targeted LRP6 expression in the VSM lineage, secondary alterations in the monocyte/macrophage lineage may contribute to arteriosclerosis58. Innovative studies very recently published have identified that a subset of inflammatory mural macrophages arise from the VSM lineage (reviewed in refs.59, 60). Future studies will assess whether VSM LRP6 orchestrates phenotypic modulation and myeloid cell differentiation, recruitment and function in vascular lesions in response to metabolic stressors. As in bone31 stage-specific roles for canonical and noncanonical Wnts are emerging in the regulatory sequence that drives arterial osteochondrogenic programming. The evolving models of LRP-dependent vascular disease have yet to fully address this sequence. In vivo quantitative measures of ligand expression and signal activation are needed to temporally and spatially resolve the contributions of specific Wnt ligand-receptor engagement to disease biology. Finally, Runx2 -- the master osteochondrogenic transcriptional regulator 29 -- is post-transcriptionally activated in VSM61. How USF1 supports programs enabled by Runx2 remains to be determined. USF1 increases with osteoblast differentiation62, and USF mechanisms encompass the maintenance of chromatin structure necessary for tissue-specific enhancer function50. Nevertheless, the discovery that ML141 down-regulates noncanonical sclerotic programs restrained by LRP6 signaling indicates that strategies targeting the cdc42-related GTPases can function as LRP6 mimetics -- and might mitigate vascular disease in patients afflicted with diabetes and dyslipidemia.
Supplementary Material
Novelty and Significance.
What Is Known?
Paracrine Wnt signaling controls bone mineralization, mediated in part by LDL receptor-related protein (LRP) family heterodimers with frizzled (Fzd) co-receptors.
Rare hypomorphic mutations in human LRP6 canonical signaling proteins cause autosomal dominant osteoporosis and early cardiometabolic disease.
Multiple Wnt ligands are expressed in vessels undergoing calcification, suggesting that Wnt signaling plays an important role in arteriosclerosis.
What New Information Does This Article Contribute?
Deletion of vascular smooth muscle lineage (VSM) LRP6 worsens arteriosclerotic disease in a model of diet-induced insulin-resistant diabetes, dyslipidemia, and cardiovascular calcification.
Loss of VSM LRP6 enhances Wnt ligand expression and osteochondrogenic signaling via noncanonical Fzd co-receptors, mediated in part by USF1 and protein arginine methyltransferase (PRMT) relays.
Like LRP6, the cdc42/rac1 G-protein antagonist ML141 inhibits noncanonical Wnt signals that drive VSM mineralization, indicating that certain LRP6 mimetics may prove useful in the treatment of arteriosclerotic calcification.
In this study, we demonstrate that the Wnt co-receptor LRP6 plays a rate-limiting role in restraining VSM noncanonical Wnt signals that drive arteriosclerotic calcification and vascular stiffening with insulin-resistant hyperglycemia and hyperlipidemia. Vascular Wnt ligands, Fzd10-dependent noncanonical signals, and osteochondrogenic mineralization programs are upregulated with reduction in VSM LRP6. We show that PRMTs and the Jmjd6 demethylase are novel components of LRP6-regulated relays that control VSM drift to the osteochondrogenic phenotype. The transcription factor USF1 is identified as a new component of the VSM noncanonical response, regulated at the level of nuclear chromatin complex formation by LRP6, ML141, and Jmjd6. These results reveal novel dimensions of LRP6 vascular biology that provide insights useful for crafting new approaches to treat arteriosclerotic disease.
Acknowledgments
SOURCES OF FUNDING
Supported by NIH grants HL081138, HL069229, and HL114806 to D.A.T. and AR053292 to B.O.W.
Nonstandard Abbreviations and Acronyms
- ADMA
asymmetric dimethylarginine
- bHLH-zip
basic helix-loop-helix – leucine zipper transcription factor
- BMC/BMD
bone mineral content/bone mineral density
- BMP
bone morphogenetic protein
- BrdU
bromodeoxyuridine
- CARM1
coactivator-associated arginine methyltransferase 1, a.k.a. PRMT4
- Cdc42
cell division cycle 42, a Rac1 family member GTPase
- ChIP
chromatin immunoprecipitation
- CMV
cytomegalovirus promoter/enhancer vector
- Col10A1
type X collagen gene alpha 1 chain
- DAPI
4′,6-diamidino-2-phenylindole
- DXA
dual electron X-ray absorptiometry
- eIF
eukaryotic translation initiation factor
- FLAG
amino acid epitope D-Y-K-D-D-D-D-K
- FLAG-IP
immunoprecipation with anti-FLAG epitope antibody
- Fzd
frizzled Wnt GPCR receptor
- G3BP
Ras-GAP SH3 domain binding protein
- GAP
GTPase activating protein
- H3R17Me2a
histone H3 asymmetrically dimethylated on Arg-17, a PRMT4 product
- H4R3Me2a
histone H4 asymmetrically dimethylated on Arg-3, a PRMT1 product
- HEK
human embryonic kidney cell line
- HFD
high fat diet, Harland TD88137 Western diet
- Jmjd6
Jumonji domain containing 6 arginine demethylase
- Klf5
krüppel like factor 5
- LDLR
low density lipoprotein receptor
- LEF
lymphoid enhancing factor
- LRP
LDL-receptor related protein
- LRP6(fl/fl)
LRP6 gene floxed, e.g. flanked by lox P
- LRP6-VKO
LRP6 VSM conditional knockout
- LUC
luciferase reporter
- ML141
cdc42/Rac1 inhibitor, Chemical Abstracts Registry CAS # 71203-35-5
- MMP
matrix metalloproteinase
- Msx
muscle segment homeobox homolog
- Myh11
smooth muscle specific myosin heavy chain 11
- NFAT
nuclear factor of activated T cells
- OCN
osteocalcin
- OPN
osteopontin
- Osx
osterix Sp7 transcription factor
- PADI
peptidyl arginine deiminase
- PRMT
protein arginine N-methyltransferase
- RNAi
RNA interference
- Runx
Runt-related transcription factor
- Sca1
stem cell antigen 1
- siRNA
small interfering RNA
- SM22-Cre
transgene expressing bacterial Cre recombinase from transgelin promoter
- TCF
T cell factor
- TK
HSV Thymidine kinase promoter
- TNAP
bone alkaline phosphatase, tissue non-specific alkaline phosphatase, akp2
- TNF
tumor necrosis factor
- USF
upstream stimulatory factor
- VSM
vascular smooth muscle lineage
- Wnt
Wingless/int-1 family member
Footnotes
DISCLOSURES
D.A.T. consulted for Daiichi-Sankyo. B.O.W. consulted for Amgen, and received grant support from Genentech.
References
- 1.Thompson B, Towler DA. Arterial calcification and bone physiology: Role of the bone-vascular axis. Nature reviews Endocrinology. 2012;8:529–543. doi: 10.1038/nrendo.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:715–723. doi: 10.1161/ATVBAHA.113.302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bostrom KI, Jumabay M, Matveyenko A, Nicholas SB, Yao Y. Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circulation research. 2011;108:446–457. doi: 10.1161/CIRCRESAHA.110.236596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woldt E, Terrand J, Mlih M, Matz RL, Bruban V, Coudane F, Foppolo S, El Asmar Z, Chollet ME, Ninio E, Bednarczyk A, Thierse D, Schaeffer C, Van Dorsselaer A, Boudier C, Wahli W, Chambon P, Metzger D, Herz J, Boucher P. The nuclear hormone receptor ppargamma counteracts vascular calcification by inhibiting wnt5a signalling in vascular smooth muscle cells. Nature communications. 2012;3:1077. doi: 10.1038/ncomms2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shao JS, Cheng SL, Pingsterhaus JM, Charlton-Kachigian N, Loewy AP, Towler DA. Msx2 promotes cardiovascular calcification by activating paracrine wnt signals. The Journal of clinical investigation. 2005;115:1210–1220. doi: 10.1172/JCI24140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beazley KE, Nurminsky D, Lima F, Gandhi C, Nurminskaya MV. Wnt16 attenuates tgfbeta-induced chondrogenic transformation in vascular smooth muscle. Arteriosclerosis, thrombosis, and vascular biology. 2015 doi: 10.1161/ATVBAHA.114.304393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Derwall M, Malhotra R, Lai CS, Beppu Y, Aikawa E, Seehra JS, Zapol WM, Bloch KD, Yu PB. Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:613–622. doi: 10.1161/ATVBAHA.111.242594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schreyer SA, Vick C, Lystig TC, Mystkowski P, LeBoeuf RC. Ldl receptor but not apolipoprotein e deficiency increases diet-induced obesity and diabetes in mice. American journal of physiology Endocrinology and metabolism. 2002;282:E207–214. doi: 10.1152/ajpendo.2002.282.1.E207. [DOI] [PubMed] [Google Scholar]
- 9.Cheng SL, Behrmann A, Shao JS, Ramachandran B, Krchma K, Bello Arredondo Y, Kovacs A, Mead M, Maxson R, Towler DA. Targeted reduction of vascular msx1 and msx2 mitigates arteriosclerotic calcification and aortic stiffness in ldlr-deficient mice fed diabetogenic diets. Diabetes. 2014;63:4326–4337. doi: 10.2337/db14-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng SL, Shao JS, Behrmann A, Krchma K, Towler DA. Dkk1 and msx2-wnt7b signaling reciprocally regulate the endothelial-mesenchymal transition in aortic endothelial cells. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1679–1689. doi: 10.1161/ATVBAHA.113.300647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tu X, Joeng KS, Nakayama KI, Nakayama K, Rajagopal J, Carroll TJ, McMahon AP, Long F. Noncanonical wnt signaling through g protein-linked pkcdelta activation promotes bone formation. Developmental cell. 2007;12:113–127. doi: 10.1016/j.devcel.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joiner DM, Ke J, Zhong Z, Xu HE, Williams BO. Lrp5 and lrp6 in development and disease. Trends in endocrinology and metabolism: TEM. 2013;24:31–39. doi: 10.1016/j.tem.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rao TP, Kuhl M. An updated overview on wnt signaling pathways: A prelude for more. Circulation research. 2010;106:1798–1806. doi: 10.1161/CIRCRESAHA.110.219840. [DOI] [PubMed] [Google Scholar]
- 14.Esen E, Chen J, Karner CM, Okunade AL, Patterson BW, Long F. Wnt-lrp5 signaling induces warburg effect through mtorc2 activation during osteoblast differentiation. Cell Metab. 2013;17:745–755. doi: 10.1016/j.cmet.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grumolato L, Liu G, Mong P, Mudbhary R, Biswas R, Arroyave R, Vijayakumar S, Economides AN, Aaronson SA. Canonical and noncanonical wnts use a common mechanism to activate completely unrelated coreceptors. Genes & development. 2010;24:2517–2530. doi: 10.1101/gad.1957710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajamannan NM. The role of lrp5/6 in cardiac valve disease: Experimental hypercholesterolemia in the apoe-/-/lrp5-/- mice. Journal of cellular biochemistry. 2011;112:2987–2991. doi: 10.1002/jcb.23221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joeng KS, Schumacher CA, Zylstra-Diegel CR, Long F, Williams BO. Lrp5 and lrp6 redundantly control skeletal development in the mouse embryo. Developmental biology. 2011;359:222–229. doi: 10.1016/j.ydbio.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mani A, Radhakrishnan J, Wang H, Mani A, Mani MA, Nelson-Williams C, Carew KS, Mane S, Najmabadi H, Wu D, Lifton RP. Lrp6 mutation in a family with early coronary disease and metabolic risk factors. Science. 2007;315:1278–1282. doi: 10.1126/science.1136370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demer L, Tintut Y. The bone-vascular axis in chronic kidney disease. Current opinion in nephrology and hypertension. 2010;19:349–353. doi: 10.1097/MNH.0b013e32833a3d67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RE, Herz J, Kuhn M. Smooth muscle-selective deletion of guanylyl cyclase-a prevents the acute but not chronic effects of anp on blood pressure. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:7142–7147. doi: 10.1073/pnas.102650499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong Z, Baker JJ, Zylstra-Diegel CR, Williams BO. Lrp5 and lrp6 play compensatory roles in mouse intestinal development. Journal of cellular biochemistry. 2012;113:31–38. doi: 10.1002/jcb.23324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Go GW, Srivastava R, Hernandez-Ono A, Gang G, Smith SB, Booth CJ, Ginsberg HN, Mani A. The combined hyperlipidemia caused by impaired wnt-lrp6 signaling is reversed by wnt3a rescue. Cell Metab. 2014;19:209–220. doi: 10.1016/j.cmet.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng SL, Shao JS, Halstead LR, Distelhorst K, Sierra O, Towler DA. Activation of vascular smooth muscle parathyroid hormone receptor inhibits wnt/beta-catenin signaling and aortic fibrosis in diabetic arteriosclerosis. Circulation research. 2010;107:271–282. doi: 10.1161/CIRCRESAHA.110.219899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/tcf signaling induces the transcription of axin2, a negative regulator of the signaling pathway. Molecular and cellular biology. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ziemer LT, Pennica D, Levine AJ. Identification of a mouse homolog of the human bteb2 transcription factor as a beta-catenin-independent wnt-1-responsive gene. Molecular and cellular biology. 2001;21:562–574. doi: 10.1128/MCB.21.2.562-574.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laudes M, Oberhauser F, Schulte DM, Freude S, Bilkovski R, Mauer J, Rappl G, Abken H, Hahn M, Schulz O, Krone W. Visfatin/pbef/nampt and resistin expressions in circulating blood monocytes are differentially related to obesity and type 2 diabetes in humans. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2010;42:268–273. doi: 10.1055/s-0029-1243638. [DOI] [PubMed] [Google Scholar]
- 27.Mayr M, Zampetaki A, Sidibe A, Mayr U, Yin X, De Souza AI, Chung YL, Madhu B, Quax PH, Hu Y, Griffiths JR, Xu Q. Proteomic and metabolomic analysis of smooth muscle cells derived from the arterial media and adventitial progenitors of apolipoprotein e-deficient mice. Circulation research. 2008;102:1046–1056. doi: 10.1161/CIRCRESAHA.108.174623. [DOI] [PubMed] [Google Scholar]
- 28.Wolak T. Osteopontin - a multi-modal marker and mediator in atherosclerotic vascular disease. Atherosclerosis. 2014;236:327–337. doi: 10.1016/j.atherosclerosis.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 29.Cohen MM., Jr The new bone biology: Pathologic, molecular, and clinical correlates. American journal of medical genetics Part A. 2006;140:2646–2706. doi: 10.1002/ajmg.a.31368. [DOI] [PubMed] [Google Scholar]
- 30.Kubota T, Michigami T, Sakaguchi N, Kokubu C, Suzuki A, Namba N, Sakai N, Nakajima S, Imai K, Ozono K. Lrp6 hypomorphic mutation affects bone mass through bone resorption in mice and impairs interaction with mesd. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2008;23:1661–1671. doi: 10.1359/jbmr.080512. [DOI] [PubMed] [Google Scholar]
- 31.Heilmann A, Schinke T, Bindl R, Wehner T, Rapp A, Haffner-Luntzer M, Nemitz C, Liedert A, Amling M, Ignatius A. The wnt serpentine receptor frizzled-9 regulates new bone formation in fracture healing. PloS one. 2013;8:e84232. doi: 10.1371/journal.pone.0084232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okamoto M, Udagawa N, Uehara S, Maeda K, Yamashita T, Nakamichi Y, Kato H, Saito N, Minami Y, Takahashi N, Kobayashi Y. Noncanonical wnt5a enhances wnt/beta-catenin signaling during osteoblastogenesis. Scientific reports. 2014;4:4493. doi: 10.1038/srep04493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang J, Sonoyama W, Wang Z, Jin Q, Zhang C, Krebsbach PH, Giannobile W, Shi S, Wang CY. Noncanonical wnt-4 signaling enhances bone regeneration of mesenchymal stem cells in craniofacial defects through activation of p38 mapk. The Journal of biological chemistry. 2007;282:30938–30948. doi: 10.1074/jbc.M702391200. [DOI] [PubMed] [Google Scholar]
- 34.Lyons JP, Mueller UW, Ji H, Everett C, Fang X, Hsieh JC, Barth AM, McCrea PD. Wnt-4 activates the canonical beta-catenin-mediated wnt pathway and binds frizzled-6 crd: Functional implications of wnt/beta-catenin activity in kidney epithelial cells. Experimental cell research. 2004;298:369–387. doi: 10.1016/j.yexcr.2004.04.036. [DOI] [PubMed] [Google Scholar]
- 35.Fukukawa C, Nagayama S, Tsunoda T, Toguchida J, Nakamura Y, Katagiri T. Activation of the non-canonical dvl-rac1-jnk pathway by frizzled homologue 10 in human synovial sarcoma. Oncogene. 2009;28:1110–1120. doi: 10.1038/onc.2008.467. [DOI] [PubMed] [Google Scholar]
- 36.Bradley EW, Drissi MH. Wnt5a regulates chondrocyte differentiation through differential use of the can/nfat and ikk/nf-kappab pathways. Molecular endocrinology. 2010;24:1581–1593. doi: 10.1210/me.2010-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ring L, Perobner I, Karow M, Jochum M, Neth P, Faussner A. Reporter gene hek 293 cells and wnt/frizzled fusion proteins as tools to study wnt signaling pathways. Biological chemistry. 2011;392:1011–1020. doi: 10.1515/BC.2011.164. [DOI] [PubMed] [Google Scholar]
- 38.Surviladze Z, Waller A, Strouse JJ, Bologa C, Ursu O, Salas V, Parkinson JF, Phillips GK, Romero E, Wandinger-Ness A, Sklar LA, Schroeder C, Simpson D, Noth J, Wang J, Golden J, Aube J. Probe reports from the nih molecular libraries program. Bethesda (MD): 2010. A potent and selective inhibitor of cdc42 gtpase. [PubMed] [Google Scholar]
- 39.Yuan K, Orcholski ME, Panaroni C, Shuffle EM, Huang NF, Jiang X, Tian W, Vladar EK, Wang L, Nicolls MR, Wu JY, de Jesus Perez VA. Activation of the wnt/planar cell polarity pathway is required for pericyte recruitment during pulmonary angiogenesis. The American journal of pathology. 2015;185:69–84. doi: 10.1016/j.ajpath.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bilkovski R, Schulte DM, Oberhauser F, Gomolka M, Udelhoven M, Hettich MM, Roth B, Heidenreich A, Gutschow C, Krone W, Laudes M. Role of wnt-5a in the determination of human mesenchymal stem cells into preadipocytes. The Journal of biological chemistry. 2010;285:6170–6178. doi: 10.1074/jbc.M109.054338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bidder M, Shao JS, Charlton-Kachigian N, Loewy AP, Semenkovich CF, Towler DA. Osteopontin transcription in aortic vascular smooth muscle cells is controlled by glucose-regulated upstream stimulatory factor and activator protein-1 activities. The Journal of biological chemistry. 2002;277:44485–44496. doi: 10.1074/jbc.M206235200. [DOI] [PubMed] [Google Scholar]
- 42.Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, Der CJ. Specificity and mechanism of action of eht 1864, a novel small molecule inhibitor of rac family small gtpases. The Journal of biological chemistry. 2007;282:35666–35678. doi: 10.1074/jbc.M703571200. [DOI] [PubMed] [Google Scholar]
- 43.Goulet I, Gauvin G, Boisvenue S, Cote J. Alternative splicing yields protein arginine methyltransferase 1 isoforms with distinct activity, substrate specificity, and subcellular localization. The Journal of biological chemistry. 2007;282:33009–33021. doi: 10.1074/jbc.M704349200. [DOI] [PubMed] [Google Scholar]
- 44.Bedford MT, Clarke SG. Protein arginine methylation in mammals: Who, what, and why. Molecular cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poulard C, Rambaud J, Hussein N, Corbo L, Le Romancer M. Jmjd6 regulates eralpha methylation on arginine. PloS one. 2014;9:e87982. doi: 10.1371/journal.pone.0087982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang B, Chen Y, Zhao Y, Bruick RK. Jmjd6 is a histone arginine demethylase. Science. 2007;318:444–447. doi: 10.1126/science.1145801. [DOI] [PubMed] [Google Scholar]
- 47.Kolodziej S, Kuvardina ON, Oellerich T, Herglotz J, Backert I, Kohrs N, Buscato E, Wittmann SK, Salinas-Riester G, Bonig H, Karas M, Serve H, Proschak E, Lausen J. Padi4 acts as a coactivator of tal1 by counteracting repressive histone arginine methylation. Nature communications. 2014;5:3995. doi: 10.1038/ncomms4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gayatri S, Bedford MT. Readers of histone methylarginine marks. Biochimica et biophysica acta. 2014;1839:702–710. doi: 10.1016/j.bbagrm.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Naik V, Leaf EM, Hu JH, Yang HY, Nguyen NB, Giachelli CM, Speer MY. Sources of cells that contribute to atherosclerotic intimal calcification: An in vivo genetic fate mapping study. Cardiovascular research. 2012;94:545–554. doi: 10.1093/cvr/cvs126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang S, Li X, Yusufzai TM, Qiu Y, Felsenfeld G. Usf1 recruits histone modification complexes and is critical for maintenance of a chromatin barrier. Molecular and cellular biology. 2007;27:7991–8002. doi: 10.1128/MCB.01326-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fan YM, Hernesniemi J, Oksala N, Levula M, Raitoharju E, Collings A, Hutri-Kahonen N, Juonala M, Marniemi J, Lyytikainen LP, Seppala I, Mennander A, Tarkka M, Kangas AJ, Soininen P, Salenius JP, Klopp N, Illig T, Laitinen T, Ala-Korpela M, Laaksonen R, Viikari J, Kahonen M, Raitakari OT, Lehtimaki T. Upstream transcription factor 1 (usf1) allelic variants regulate lipoprotein metabolism in women and usf1 expression in atherosclerotic plaque. Scientific reports. 2014;4:4650. doi: 10.1038/srep04650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ito TK, Yokoyama M, Yoshida Y, Nojima A, Kassai H, Oishi K, Okada S, Kinoshita D, Kobayashi Y, Fruttiger M, Aiba A, Minamino T. A crucial role for cdc42 in senescence-associated inflammation and atherosclerosis. PloS one. 2014;9:e102186. doi: 10.1371/journal.pone.0102186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suzuki W, Yamada A, Aizawa R, Suzuki D, Kassai H, Harada T, Nakayama M, Nagahama R, Maki K, Takeda S, Yamamoto M, Aiba A, Baba K, Kamijo R. Cdc42 is critical for cartilage development during endochondral ossification. Endocrinology. 2015;156:314–322. doi: 10.1210/en.2014-1032. [DOI] [PubMed] [Google Scholar]
- 54.Ito T, Yadav N, Lee J, Furumatsu T, Yamashita S, Yoshida K, Taniguchi N, Hashimoto M, Tsuchiya M, Ozaki T, Lotz M, Bedford MT, Asahara H. Arginine methyltransferase carm1/prmt4 regulates endochondral ossification. BMC developmental biology. 2009;9:47. doi: 10.1186/1471-213X-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Massari ME, Murre C. Helix-loop-helix proteins: Regulators of transcription in eucaryotic organisms. Molecular and cellular biology. 2000;20:429–440. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bikkavilli RK, Malbon CC. Arginine methylation of g3bp1 in response to wnt3a regulates beta-catenin mrna. Journal of cell science. 2011;124:2310–2320. doi: 10.1242/jcs.084046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goel S, Chin EN, Fakhraldeen SA, Berry SM, Beebe DJ, Alexander CM. Both lrp5 and lrp6 receptors are required to respond to physiological wnt ligands in mammary epithelial cells and fibroblasts. The Journal of biological chemistry. 2012;287:16454–16466. doi: 10.1074/jbc.M112.362137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K, Aikawa E. Macrophage-derived matrix vesicles: An alternative novel mechanism for microcalcification in atherosclerotic plaques. Circulation research. 2013;113:72–77. doi: 10.1161/CIRCRESAHA.113.301036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fisher EA, Miano JM. Don’t judge books by their covers: Vascular smooth muscle cells in arterial pathologies. Circulation. 2014;129:1545–1547. doi: 10.1161/CIRCULATIONAHA.114.009075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Swirski FK, Nahrendorf M. Do vascular smooth muscle cells differentiate to macrophages in atherosclerotic lesions? Circulation research. 2014;115:605–606. doi: 10.1161/CIRCRESAHA.114.304925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heath JM, Sun Y, Yuan K, Bradley WE, Litovsky S, Dell’Italia LJ, Chatham JC, Wu H, Chen Y. Activation of akt by o-linked n-acetylglucosamine induces vascular calcification in diabetes mellitus. Circulation research. 2014;114:1094–1102. doi: 10.1161/CIRCRESAHA.114.302968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Hassan MQ, Li ZY, Stein JL, Lian JB, van Wijnen AJ, Stein GS. Intricate gene regulatory networks of helix-loop-helix (hlh) proteins support regulation of bone-tissue related genes during osteoblast differentiation. Journal of cellular biochemistry. 2008;105:487–496. doi: 10.1002/jcb.21844. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.