

Abstract
Recent studies have linked the ER stress sensor IRE1α with the RIG-I pathway, which triggers an inflammatory response upon detection of viral RNAs. In response to ER dysfunction, IRE1α cleaves mRNA into single-strand fragments that lack markers of self, which activate RIG-I. Certain microbial products from mucosal pathogens activate this pathway by binding IRE1α directly, and the discovery that IRE1 is amplified at mucosal surfaces by gene duplication suggests an important role for IRE1 in mucosal immunity. Here, we review evidence in support of this hypothesis, and propose a model wherein IRE1 surveys the integrity of the ER, acting as a guard receptor and a pattern recognition receptor, capable both of sensing cellular stress caused by microbial infection and of responding to pathogens directly.
Keywords: IRE1, RIG-I, innate immunity, mucosal immunity, inflammation, intestine
Introduction
The endoplasmic reticulum (ER) is arguably the most functionally diverse and largest of intracellular organelles, occupying all areas of the cell and affecting a wide range of cellular processes [1–3]. It is equipped to sense and accommodate problems in protein synthesis – a process termed the unfolded protein response (UPR) [2,4–8]. Almost all other forms of cell stress, including metabolic, inflammatory, and microbial, affect cellular proteostasis and can induce the UPR to effect cell survival [1,9]. In most such instances, activation of the UPR leads to a concomitant cell-autologous inflammatory response, thus linking ER homeostasis and its disruption to systemic immunity and other forms of adaptation to the environment [5,10,11].
Three ER proteins in mammalian cells – IRE1α, PERK, and ATF6 – provide for rapid adaptations to the variable demands on protein synthesis and guard against the accumulation of misfolded proteins in the ER (Box 1). Each of these sensors surveys the ER lumen for problems in protein biogenesis, and directs how the cell accommodates via the UPR. The UPR is also activated by many physiological and environmental factors that link the ER with other pathways regulating cellular homeostasis or restitution after injury (for excellent discussion see [1]).
Box 1. The UPR.
The three transmembrane proteins used by mammalian cells to sense and process ER stress are Inositol-Requiring Enzyme-1 (IRE1), Activating Transcription Factor 6 (ATF6), and the PKR-like ER-localized eIF2a Kinase (PERK) [2,4–8]. The lumenal domains of these proteins survey the ER for danger and their cytosolic domains initiate signaling to the cytosol and nucleus. Collectively, they trigger a series of responses to accommodate increased demand or problems in protein synthesis. If they are unable to resolve the stress, these proteins promote apoptosis.
The mechanisms of activation of the three sensors are not fully elucidated, but are proposed to involve receptor oligomerization, dynamic binding by the ER chaperone BIP, and direct binding by misfolded protein substrates (Figure I).
IRE1 activation initiates autophosphorylation and two distinct responses from the cytoplasmic endoribonuclease domain: (i) unique splicing of XBP-1 mRNA to produce the active transcription factor XBP-1, which activates transcription of ER chaperones; and (ii) degradation of mRNAs via RIDD. Both actions of the IRE1 RNAse domain attempt to reduce ER stress.
ATF6 activation causes the molecule to traffic to the Golgi where it is cleaved by site-1 and site-2 proteases. The cleavage product, freed from the membrane, is a transcription factor that enters the nucleus where it binds and signals expression of ER stress response elements.
PERK activation leads to phosphorylation of the alpha subunit of translation initiation factor 2 (eIF2a) to inhibit protein synthesis at the level of the ribosome. Translation of the transcription factor ATF4 however, escapes this inhibition, leading to expression of CHOP, a factor involved in apoptosis. If ER stress is not resolved, the PERK pathway strongly enhances cell-fate decisions towards apoptosis.
IRE1α is the most evolutionarily conserved of the ER stress-sensors with expression found in fission yeast [12,13]. Recent studies have revealed a pathway that connects IRE1 with the retinoic acid-inducible gene 1 (RIG-I) pathway via the generation of RNA ligands [14,15], and show that in some cases, IRE1 can be triggered by direct sensing of some pathogens [14]. We review these findings here, placing them in the context of the current understanding of the role of IRE1 in the ER stress response and associated inflammatory pathways. In light of the available evidence, we propose a model wherein IRE1 acts as both a pattern recognition receptor and a guard receptor, capable of surveying the ER and responding to pathogens directly, and indirectly via the sensing of pathogen-associated ER stress.
IRE1 and the UPR
IRE1α is a Type I membrane protein comprised of an N-terminal domain that provides surveillance of the ER lumen, followed by transmembrane and linker regions, and then two enzymatically active cytosolic domains required for signal transduction: a kinase domain and C-terminal endoribonuclease domain (Figure 1) [16]. In vitro, the core lumenal domain of yeast IRE1α oligomerizes to assume a conformation suggestive of the MHC Class I peptide-binding domain [16,17]. The human protein folds similarly, but the putative MHC Class I-like peptide-binding groove is shorter and partly occluded [18]. Both lumenal domains however, yeast and human, function to monitor for ER stress [1,19], possibly by direct and dynamic binding to misfolded proteins in the ER lumen [16,20].
Figure 1.

IRE1 domain organization and structure. (A) Schematic shows domain structures of human IRE1α and IRE1β with sequence homologies indicated. (B) Crystal structures of lumenal domain from (top) yeast IRE1 (PDB 2BEI, [17]) and (bottom) human IRE1α (PDB 2BZ6, [18]). The dotted line represents the proposed dimer interfaces observed in the crystal structures. The boxed region is enlarged in the right panels to illustrate the proposed MHC-like peptide-binding grooves. Key residues in the binding groove are indicated for one protomer of the dimer. The width of the groove in yeast IRE1 (as measured between Cα atoms of Ser 200 in each protomer) is 10.7 Å. The width of the groove in human IRE1α is reduced (6.8 Å between Cα atoms of Gln 105) and is occluded by the side chain of Gln 105.
It is thought that in vivo, IRE1α occupies the ER membrane as a monomer, likely stabilized in this conformation through binding of the chaperone protein Bip (KAR2 in yeast) [21,22]. On activation, Bip dissociates and IRE1α oligomerizes [23–30] to transduce a signal across the ER-limiting membrane and activate the kinase and ribonuclease domains. The activation of IRE1α may at times be caused by the dissociation of Bip, triggered by competing needs for Bip chaperone function in protein folding. This is proposed as one mechanism for sensing ER overload. For some forms of activating signals sensed by IRE1α, however, the core lumenal domain is dispensable. The single pass transmembrane domain on its own can sense changes in lipid composition of the ER-limiting membrane [31], and the kinase RNase domains can respond to signaling proteins and activating factors in the cytosol [32–35].
Signal transduction by IRE1α depends on the enzymatic activities of both the cytosolic kinase and endoribonuclease domains [16,26]. The second messenger in the IRE1α induced UPR response is a product of the IRE1α endoribonuclease domain: a uniquely spliced mRNA encoding the β-ZIP transcription factor XBP1 (Hac1 in Saccharomyces cerevisiae) [36–39]. XBP1/HAC1 then enters the nucleus to initiate the transcriptional responses dependent on the IRE1 branch of the UPR. The kinase domain operates to regulate oligomer formation and endonuclease activity [16], and to signal in the UPR-associated inflammatory response [40,41], as discussed further below. The specific splicing of XBP1/Hac1 was considered the only activity of IRE1α affecting the UPR, until the Weissman laboratory discovered that IRE1α could also respond to ER stress as a general endonuclease. This reaction cleaves into inactive fragments mRNAs that are targeted to the ER for translation, and that localize in proximity to IRE1α [42,43]. This activity was termed Regulated IRE1-Dependent Decay (RIDD), and thought to help relieve ER stress by diminishing protein translation (see also [44]). RIDD was soon after found to target messages with RNA motifs similar to that needed for XBP1 splicing, and to block translation of selected proteins, including by cleavage of miRNAs [45,46], thus resulting in direct physiologic effects on cell function and fate distinct from the UPR [1,2,11,47–50].
IRE1 and the inflammatory response
Each arm of the UPR (IRE1, PERK, and ATF6) can induce unique and finely tuned cell-autologous inflammatory signatures (see excellent recent reviews [5,10,11,51]). In the case of IRE1α, the cytoplasmic kinase domain can recruit the adaptor protein TRAF2 to scaffold the c-Jun amino terminal kinase (JNK) [40] and inhibitor of κB kinase (IKK) [41]; both are active in key downstream inflammatory and cell fate responses. JNK activation will induce expression of inflammatory cytokines and other factors via Activator Protein 1 (AP1) and NF-κB. IKK recruitment leads to NF-κB-dependent gene expression and a tumor necrosis factor-a autocrine loop causing enhanced susceptibility to cell death [41]. As TRAF2 has affinity for binding and can even induce clustered cell surface receptors, it is possible that the oligomerization state of IRE1α regulates (or is regulated by) IRE1α–TRAF2 interactions. This may explain in part how the cell can modify downstream inflammatory signatures in response to different causes of cellular dysfunction involving the ER.
IRE1α has also been implicated in activation of the NLR3 inflammasome. In the case of insulin-producing pancreatic β cells, the RIDD activity of IRE1α induced by prolonged ER stress will degrade the miRNA miR-17, resulting in the increased expression of thioredoxin-interacting protein TXNIP and the concomitant activation of the NLR3 inflammasome [52]. This pathway may have clinical relevance to the pathogenesis of diabetes, and in principle be operating in other forms of inflammatory signaling by cells expressing miR-17 and TXNIP.
Unrelated innate immune activation pathways can feedback on the ER stress pathways. For example, during the innate immune response to infection by Pseudomonas, the survival of Caenorhabditis elegans depends on the induction of a robust UPR [9]. Toll-like receptor (TLR) signaling in mouse macrophages activates IRE1α via the NAPDH–oxidase complex [reactive oxygen species (ROS) dependent]. Interestingly, the resulting expression of XBP1 in the case of TLR-activated macrophages does not trigger the typical ER stress transcriptional response; rather, the TLR-induced IRE1α–XBP1 cascade produces just the proinflammatory cytokines required for host defense, perhaps due to the selective activation of IRE1α by ROS mediators [53,54].
Discovery of the IRE1–RIDD–RIG-I pathway
Two recent papers have linked IRE1α with inflammatory signaling by a pathway involving RIDD and the antiviral RNA helicase RIG-I [14,15]. The discovery originated in part from our interest in the innate host response against the potent mucosal pathogen Vibrio cholera [14], and in part from the Stetson laboratory’s work on regulation of innate immunity against nucleic acids in the cytosol [15]. This newly delineated pathway, termed here IRE1–RIDD–RIG-I (Figure 2), affects adaptive immunity in humans, as evidenced by some patients with trichohepatoenteric syndrome (THES) [15]. THES is an inherited multisystem immune disease associated with interferon (IFN) signatures.
Figure 2.

The IRE1–RIDD–RIG-I pathway. The IRE1–RIDD–RIG-I pathway connects the ER with innate immune signaling in response to some forms of the microbial environment and other forms of cellular distress affecting the ER. In the resting state, IRE1α occupies the ER membrane as a monomer bound to Bip. The lumenal domain monitors the ER for dysfunction or signs of danger, which are marked at least in part by the loss of Bip binding (a putative guardee). The transmembrane domain monitors the ER membrane for perturbations in lipid structure (a second putative guardee). Activation likely also depends on IRE1α binding to signs of danger – either misfolded endogenous proteins or invading microbial products. In the case of cholera toxin, very small amounts of the A1-chain entering the ER appear to bind IRE1α, although we do not yet know in what conformation (folded or unfolded). The reaction suggests a role for IRE1α as a pattern recognition receptor. On activation IRE1α oligomerizes and the cytosolic kinase and endonuclease domains are activated to splice XBP-1 mRNA for translation into the active transcription factor, and to cleave other mRNA into fragments that lack markers of self, thus rendering them ligands for the antiviral sensor RIG-I. This general endonuclease activity is termed RIDD. Activation of RIG-I via MAVS leads to an inflammatory response. IRE1α can also induce an inflammatory response by scaffolding to TRAF2 and activating JNK- or IKK-dependent pathways.
The IRE1–RIDD–RIG-I pathway represents a new form of signal transduction elucidated by studies on bacterial toxins [14]. Cholera and Shiga toxin (CTx and STx) typify the class of AB5-subunit toxins that evolved the striking ability to enter the cytosol of host cells by coopting the machinery that normally disposes of terminally misfolded proteins in the ER, termed ER Associated Degradation (ERAD) (Box 2). IRE1α uniquely senses the portion of the toxin that engages ERAD, the A1-chain, and appears to bind the A1-chain for activation. The other toxin subunits entering the ER are not detected, and the other ER stress-sensors – PERK and ATF6 – are not activated. Splicing of XBP-1, however, is entirely dispensable for the inflammatory response [14]. Rather, it is the RIDD activity of IRE1α that signals RIG-I (Figure 2). The RIDD reaction produces single-strand mRNA fragments that lack 5′-caps or 3′ polyA-tails, which normally mark cytosolic mRNA as ‘self’; these activate RIG-I to cause a cell-autologous inflammatory response via the NF-κB and IFN pathways. The mechanism of signal transduction is analogous to how cytosolic RNase L, whose effector domain descended from IRE1α by gene duplication [55], is proposed to cleave both viral and endogenous mRNA to activate RIG-I and amplify the immune response against invading RNA-viruses [56].
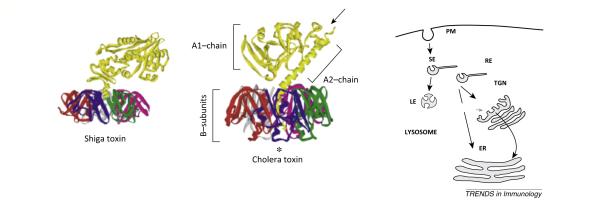
Box 2. Structure and function of the bacterial AB5 toxins.
All bacterial toxins must translocate across a cell membrane to access their cytosolic targets [103]. CTx and STx typify the AB5-subunit toxins that must enter the ER of host cells to cause disease. The related but different structures of STx and CTx are shown. They are composed of a single A-subunit and pentameric B-subunit. The B-subunit is comprised of five identical polypeptides assembled to form the highly stable pentameric rings that bind specific membrane glycosphingolipids and traffic retrogradely through the secretory pathway from the plasma membrane to the ER [104,105]. The A-subunits of both toxins are single polypeptides that are cleaved into A1- and A2-chains (arrow) before entering the cell. The chains remain linked by a single disulfide bond until they are reduced after arrival in the ER. In the case of CTx, the ER chaperones PDI and Bip are implicated in unfolding the A1-chain, separating it from the B-subunit, and releasing it into the ER lumen. The free unfolded A1-chain, perhaps stabilized by attachment to PDI or other ER chaperones, is recognized and processed by ERAD for dislocation to the cytosol; a process likely involving the HRD1 complex. ERAD is the cell’s natural quality control system for monitoring and disposing of terminally misfolded proteins in the secretory pathway. Misfolded proteins are detected within the lumen of the ER, directed to a translocation channel at the ER membrane, and shuttled (retro-translocated) into the cytoplasm where they are recognized and degraded by the ubiquitin–proteasome system (for excellent review see [106])(Figure II).
Once in the cytosol, both the STx and the CTx A1-chains refold into their enzymatically active conformation to induce disease. The CTx A1-chain is an ADP-ribosyltransferase that modifies the heterotrimeric G protein GSα to activate adenylyl cyclase [105]. The enzymatic A1-chain of Shiga toxin is an N-glycosidase that cleaves a specific adenine nucleotide in the 28S RNA of the 60S subunit of the ribosome to halt protein synthesis.
The Stetson laboratory arrived independently at the same IRE1–RIDD–RIG-I pathway discovery through studies on how the SKIV2L RNA exosome might regulate the innate inflammatory response induced during viral infection [15]. Cells devoid of the SKIV2L RNA exosome are less able to degrade cytosolic RNA and were found more sensitive to RIG-I ligands. RIG-I was also activated on induction of the UPR, thus implicating the SKIV2L RNA exosome in regulating the IRE1–RIDD–RIG-I pathway. Perhaps, most importantly, this was shown to operate in humans with the immune disease THES [15].
The products of the IRE1–endonuclease (RIDD) reaction implicated in activating RIG-I by both studies [14,15] are well delineated [37]: they are single-stranded 5′-OH and 2′-3′-cyclic phosphate mRNA fragments. They are not the double-stranded 5′-triphosphate RNA that typifies ligands activating RIG-I during viral infection [57–60], but they do activate RIG-I as first implicated in studies on the viral sensor RNase L [14,15,56]. When compared with double-stranded 5′-triphosphate RNA released by viral infection, however, activation of RIG-I by the IRE1α pathway appears less potent, suggesting that RIG-I might discern among RNA ligands, or that it is regulated by other means to induce different physiological effects, perhaps mediated by different assemblies of the RIG-I/MAVS signaling complex [61]. Similarly, the toxins were found to induce the RIDD–RIG-I pathway but not JNK [14], consistent with the capacity for differential signaling by IRE1α [1].
It is possible that such different outputs in the inflammatory response are driven by the variable degrees of IRE1α oligomerization caused by different forms of ER and cellular stress. Two groups have tested this hypothesis to explain how IRE1α modulates its enzymatic activities with respect to XBP-1- and RIDD-dependent cellular outputs [28,49,62]. The evidence so far is compelling for the dimer/oligomer model of IRE1α activation, but in one case the data implicate the higher-ordered IRE1α oligomers in amplifying the RIDD-dependent output over XBP-1 splicing [28,49], and in the other case, RIDD is favored by the smaller IRE1 dimers, and XBP-1 splicing is favored by the higher-ordered IRE1α oligomers [62]. For JNK signaling by IRE1, which is not observed when IRE1 is activated by the toxins, we note that TRAF2 binds preferentially to protein clusters. Perhaps signaling by IRE1α in the TRAF2–JNK cascade depends on higher-ordered IRE1α oligomerization, such as may occur in cases of large-scale or massive ER stress. The toxin A1-chains, which enter the ER in very low amounts, may induce only IRE1α-dimers, or small oligomers preferentially signaling via RIDD as hypothesized [62], and thus unable to activate JNK.
There are other examples of differential signaling by IRE1α [1], some caused by the association of IRE1α with other proteins or cytosolic factors [33,35]. One is by association with the proapoptotic BCL-2 family members BAX and BAK, which modify the IRE1α response to ER stress and affect cell fate decisions [32]. Another is the activation of IRE1α by flavenol-like molecules in the cytosol of S. cerevisiae [35]. An in vivo example of differential IRE1α outputs is suggested by the absence of inflammation in the liver of mice lacking XBP-1 [63]. The liver and gut cells of these mice have a strongly activated IRE1α signature, but the mice exhibit inflammation only in the intestine [63,64]. The explanation for such differential cellular outputs, we believe, will be found to depend on physiologic, cell type, and environmental context of IRE1α activation [1], as already evidenced by amplification of IRE1α signaling in macrophages exposed to activating (microbial) TLR ligands [53].
Enhanced RIDD activity at mucosal surfaces by IRE1β
The discovery of signal transduction between IRE1α and RIG-I by RIDD has important implications for immune surveillance at mucosal surfaces because this is where the endonuclease activity responsible for activating RIG-I (RIDD) is highly amplified. Epithelial cells lining the gastrointestinal and respiratory tracts uniquely express the IRE1α paralog IRE1β [65]. IRE1β has strong structural and functional homology with IRE1α, especially in the cytosolic kinase and endonuclease domains [66]; however, the enzymatic activities of the two proteins are not identical. IRE1β is tenfold more enzymatically active than IRE1α in RIDD reactions, and interestingly, less potent in splicing of XBP-1 mRNA [67]. Mucosal epithelial cells uniquely expressing IRE1β physically and functionally divide the host from the microbial laden environment [68–71], suggesting a role for IRE1β in host defense.
The notion that IRE1β might act in host defense was first suggested by earlier studies on the function of IRE1β in the intestine and lung [65,72–74]. Genetic deletion of IRE1β in mice predisposes the intestine to severe forms of chemically induced colitis, indicating a role for IRE1β in mucosal inflammation [65]. Strong evidence implicates IRE1β in the regulation of production of mucin – a component of the mucosal barrier [65] – by intestinal goblet cells [72,73]. The RIDD activity of IRE1β is considered essential for maintaining normal goblet cell function in the intestine by preventing ER stress [73]. In the lung, however, regulation of mucin secretion by IRE1β depends on XBP-1. Here, it is thought IRE1β operates to transcriptionally effect varying needs for mucin synthesis in accordance with IL13 signaling [72].
By immunocytochemistry, goblet cells appear to be the only cell type in the intestine expressing IRE1β, consistent with its activity in regulating mucin secretion [73]. However, goblet cells also have an amplified ER, typical of other secretory cell types. It is therefore likely, in our view, that detection methods of increased sensitivity will reveal expression of IRE1β in other intestinal epithelial cell lineages. This is perhaps already evidenced by the requirement for IRE1β for absorption of dietary lipids [75], which is a function of the absorptive intestinal cell type. Lipid absorption impacts the ER for synthesis and packaging of nutrient fats into chylomicrons for transport into the circulation. Thus, the evolutionary pressures responsible for the duplication of IRE1α (with amplified RIDD activity) in mucosal tissues are not fully defined, and we suggest that one of them may have been the need for IRE1 to function in innate immunity and host defense.
Such an activity for IRE1 in mucosal immunity is also suggested by the studies in mice that lack the IRE1 effector XBP-1. These mice incur spontaneous intestinal inflammation associated with proinflammatory NF-κB and JNK signaling [64]. While many factors may contribute to the onset of inflammation in these animals, the UPR and IRE1 likely play decisive roles. Such defects in the UPR leading to inflammation of the gut may apply clinically to humans as some patients with inflammatory bowel disease harbor rare variants in the XBP1 gene [64].
CT is widely acknowledged to act as a uniquely potent adjuvant for mucosal vaccinations [76–78]. The A-subunit is required for the adjuvant response in the intestine and the B-subunit is not sufficient; but mutant toxins containing enzymatically inactivated A1-chains appear to be still active as adjuvant [79,80] and the holotoxin induces T-cell responses systemically and locally [81,82]. This requirement for the A1-chain in adaptive immunity implicates the IRE1–RIDD pathway. In principle, activation of RIG-I and the IFN pathway by the toxin A1-chains could also induce T-cell responses against self, but this, so far, has not been recognized in patients infected with V. cholera.
IRE1 as both a guard receptor and a pattern recognition receptor
The available evidence indicates a general rule for IRE1α/β function in the inflammatory response, induced either by the loss of ER homeostasis or by recognition of specific microbial products. In terms of innate immune signaling, this can be understood if IRE1α/β were to operate as both a ‘Guard’ and ‘Pattern Recognition’ receptor.
In plants, the guard receptor hypothesis posits the presence of receptors that sense dysfunction in host cell processes, termed the ‘guardee’. By monitoring the guardee, the guard receptor provides a way to discern between microbes as pathogens, which cause cellular damage, or not [83,84]. The concept was extended to metazoans [84] and recently shown to explain the immune response against certain microbes by detection through their toxic effects on Rho GTPases [85,86]. In this context, IRE1α may be viewed as guarding Bip against loss of ER proteostasis, or the ER membrane against the loss of lipid structure (Figure 2) – both potential components of the cellular response to microbial infection [11,87].
The pattern recognition hypothesis on the other hand proposes recognition of conserved microbial patterns signifying non-self [88]. The idea that IRE1 may operate as a pattern recognition receptor is different from the concept that implicates the UPR in the cellular response to microbial infection [11,87], sometimes referred to as the Microbial Stress Response [89]. Many pathogens cause cellular damage or alter cellular metabolism in ways that affect the ER, and thus induce the UPR with important effects on disease or recovery [9,53]. Perhaps some of the best examples are the viral infections that were long ago recognized to induce the UPR by coopting the ER for protein synthesis [11,90,91]; the dependence on XBP-1 (IRE1α) to accommodate the innate immune response against Pseudomonas infection in C. elegans [9]; and the pathology caused by the cholera-like AB5-subunit subtilase toxins that enter the ER to induce massive damage by degrading the ER lumenal chaperone Bip [92,93]. All cause ER stress by affecting ER functions and activate IRE1α (the guard receptor). Some pathogens co-opt certain components of the UPR to amplify disease, and do so, like CTx, without activating the canonical UPR. Infection by human cytomegalovirus is one recently described example. In this case, the nascent viral membrane protein UL50 binds IRE1α and somehow causes its degradation, thus obfuscating the IRE1α branch of the UPR [94]. But this is not pattern recognition either – a term used here to denote receptor-mediated surveillance for microbes or their products [95].
Several results from our studies with cholera toxin underlie the idea that IRE1 can act as a pattern recognition receptor [14]. First, only a portion of the toxin, the A1-chain, causes activation of IRE1α, even when the A1-chain is rendered enzymatically inert. Second, the A1-chain selectively activates IRE1α; neither PERK nor ATF6 is affected, as would be expected if the A1-chain were acting as an unfolded protein to induce the UPR. Remarkably, the CTx B-subunit, which also enters the ER along with the A1-chain, does not activate IRE1α, further implicating specificity, a defining feature of receptor-mediated events. Finally, in vitro mapping of binding motifs and coimmunoprecipitation studies show the A1-chain might bind IRE1α directly.
Is this even plausible? It is possible that the cholera and Shiga toxins simply disrupt an essential ER function, such as ERAD (the guardee), to induce IRE1 activation. However, an A1-chain mutant that remains fixed to the B-subunit and cannot complete all ERAD steps still activates IRE1α [14], arguing against this possibility. Also, one of the compelling mechanistic explanations for activation of IRE1α in the UPR implicates direct binding between substrate (unfolded protein) and receptor (IRE1α) [16,17,20]. This is experimentally evidenced by binding between IRE1α and the model ERAD substrate CPY* in yeast [20] and the CTx A1-chain in mammalian cells (although it remains uncertain in which form the A1-chain binds) [14]. Like IRE1α, the lumenal domain of IRE1β displays protein–protein binding activity in vitro [96]. Arguments against the idea of IRE1α binding ER lumenal substrates most formidably include the crystal structure of human IRE1α, which folds with occlusion of the putative peptidebinding site [18,97], and in contrast to yeast IRE1α, there is no evidence for human IRE1α binding to two model-unfolded proteins when tested in vitro [30].
Another concern in the case of the toxins has to do with the nature of the activating ligand. What could be the features that mark the CTx and STx A1-chains as Pathogen Associated Microbial Patterns (PAMPs)? The two A1-chains are not structurally related, both are comprised of the same amino acids used by the host, and they have no readily apparent mark(s) signifying non-self. But such is also the case for the bacterial flagellins, which are well known to bind and activate TLR5 to induce innate immunity [98]. It is possible that CTx and STx typify a larger class of microbial products against which nature has evolved a defense. The toxin A1-chains do have some features that could mark the proteins for raising alarms. One is that they are designed to unfold in the ER, unlike all other nascent host proteins in the biosynthetic pathway. Another is that they all contain a free cysteine, which in principle may bond to exposed cysteines in the IRE1α lumenal domain or perhaps affect the regulation of IRE1α by protein disulfide isomerase 6 [99].
Concluding remarks
The discovery of the IRE1–RIDD–RIG-I pathway implicates a role for the ER in innate immunity at mucosal surfaces. This may be especially important in the intestine where epithelial cells uniquely expressing IRE1β interface directly with 100s of trillions of microbes normally colonizing the gut. The lumenal domain of IRE1β is least homologous to IRE1α, suggesting evolutionary divergence in function, perhaps driven by the need to sense the colonizing microbiome.
While we emphasize how IRE1α/β might operate as a pattern recognition receptor in immune surveillance, we fully expect IRE1α/β to signal via RIDD–RIG-I in all forms of the microbial stress response, and in other conditions that cause ER stress – essentially functioning as a guard receptor against the loss of cellular homeostasis that affects the ER. Perhaps, this is most clearly evidenced by activation of the RIDD–RIG-I pathway in cells treated with the small molecules thapsigargin and tunicamycin that massively disrupt ER function [14]. Furthermore, the relevance of such signaling at non-mucosal sites is demonstrated in some patients with THES [15], who lack regulation of the RIDD–RIG-I pathway induced by the normal UPR (Box 3).
Box 3. Outstanding questions.
Does IRE1β act like IRE1α in the RIDD–RIG-I pathway?
Does IRE1β sense invasion of the ER by the toxins or other forms of ER stress?
What is the molecular/structural mechanism by which the toxin A1-chains activate IRE1?
What effect does the IRE1–RIDD–RIG-I pathway have on mucosal immunity?
Do other microbes or microbial factors activate IRE1?
Does IRE1 operate to sense and affect microbial communities normally colonizing mucosal surfaces?
Since the ER originates protein synthesis in the secretory pathway, functioning at steps most distant from the plasma membrane, the idea that the ER evolved to sense the outside world may seem at first glance unlikely. But the ER extends throughout the cell and interacts with all other organelles including, endosomes, mitochondria, peroxisomes, autophagosomes, and the plasma membrane itself [3,100]. It is positioned and equipped to monitor all forms of cell damage or stress, and in vertebrates it is entwined in all forms of cellular signaling [1]. We believe it possible that the contact sites between ER and mitochondria and peroxisomes (MAMs) may function as synapses to amplify, or enable, signal transduction in the IRE1–RIDD–RIG-I pathway by approximating IRE1 with RIG-I and its effector MAVs located on the membranes of those organelles [101,102].
All aspects of the hypothesis are testable and clinically relevant. It will be first important to test if IRE1β acts like IRE1α in the RIDD–RIG-I pathway, and if IRE1β also senses invasion of the ER by the toxins or other forms of ER stress, including the microbial stress response. Full explanation of the reactions between IRE1 and the toxin A1-chains, in vitro and in vivo, will be required to definitively test the hypothesis for pattern recognition. If IRE1β is found to activate RIG-I, it will be important to characterize the downstream effects on the mucosal immune system. If IRE1β is found to act in pattern recognition, it will be most interesting to search for other microbial factors that induce the RIDD–RIG-I pathway. Such studies will be highly informative for the field of innate immunity, and they may explain one way that microbes colonizing mucosal tissues talk to us, and we back to them.
Highlights.
IRE1 signals in innate immunity by producing mRNA fragments that activate RIG-I.
Genetic defects in regulation of the IRE1–RIG-I pathway induce IFNs in humans.
The pathway may be uniquely amplified at mucosal surfaces by expression of IRE1β.
IRE1 may function as both a guard receptor, responding to ER stress, and a direct PRR.
Innate immune pathways may survey the ER for signals of the presence of pathogens.
Figure I.

Figure II.
Acknowledgments
The authors gratefully acknowledge all members of the Lencer laboratory for stimulating discussions and Jon Kagan for critical reading of the manuscript. The work was supported by NIH DK048106 and DK084424 to W.I.L., CCFA CDA330011 and K01 DK102828 to J.A.C., and the NIH P30 Harvard Digestive Diseases Center DK034854.
References
- 1.Rutkowski DT, Hegde RS. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J. Cell Biol. 2010;189:783–794. doi: 10.1083/jcb.201003138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 3.Friedman JR, Voeltz GK. The ER in 3D: a multifunctional dynamic membrane network. Trends Cell Biol. 2011;21:709–717. doi: 10.1016/j.tcb.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jager R, et al. The unfolded protein response at the crossroads of cellular life and death during endoplasmic reticulum stress. Biol. Cell. 2012;104:259–270. doi: 10.1111/boc.201100055. [DOI] [PubMed] [Google Scholar]
- 5.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kohno K. Stress-sensing mechanisms in the unfolded protein response: similarities and differences between yeast and mammals. J. Biochem. 2010;147:27–33. doi: 10.1093/jb/mvp196. [DOI] [PubMed] [Google Scholar]
- 7.Mori K. Signalling pathways in the unfolded protein response: development from yeast to mammals. J. Biochem. 2009;146:743–750. doi: 10.1093/jb/mvp166. [DOI] [PubMed] [Google Scholar]
- 8.Gardner BM, et al. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013;5:a013169. doi: 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richardson CE, et al. An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature. 2010;463:1092–1095. doi: 10.1038/nature08762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Todd DJ, et al. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008;8:663–674. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- 11.Janssens S, et al. Emerging functions of the unfolded protein response in immunity. Nat. Immunol. 2014;15:910–919. doi: 10.1038/ni.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cox JS, et al. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–1206. doi: 10.1016/0092-8674(93)90648-a. [DOI] [PubMed] [Google Scholar]
- 13.Mori K, et al. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell. 1993;74:743–756. doi: 10.1016/0092-8674(93)90521-q. [DOI] [PubMed] [Google Scholar]
- 14.Cho JA, et al. The unfolded protein response element IRE1α senses bacterial proteins invading the ER to activate RIG-I and innate immune signaling. Cell Host Microbe. 2013;13:558–569. doi: 10.1016/j.chom.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Eckard SC, et al. The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat. Immunol. 2014;15:839–845. doi: 10.1038/ni.2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korennykh A, Walter P. Structural basis of the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2012;28:251–277. doi: 10.1146/annurev-cellbio-101011-155826. [DOI] [PubMed] [Google Scholar]
- 17.Credle JJ, et al. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 2005;102:18773–18784. doi: 10.1073/pnas.0509487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou J, et al. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. U.S.A. 2006;103:14343–14348. doi: 10.1073/pnas.0606480103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimata Y, Kohno K. Endoplasmic reticulum stresssensing mechanisms in yeast and mammalian cells. Curr. Opin. Cell Biol. 2011;23:135–142. doi: 10.1016/j.ceb.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 20.Gardner BM, Walter P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. 2011;333:1891–1894. doi: 10.1126/science.1209126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okamura K, et al. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem. Biophys. Res. Commun. 2000;279:445–450. doi: 10.1006/bbrc.2000.3987. [DOI] [PubMed] [Google Scholar]
- 22.Bertolotti A, et al. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 23.Aragon T, et al. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature. 2009;457:736–740. doi: 10.1038/nature07641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, et al. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. U.S.A. 2010;107:16113–16118. doi: 10.1073/pnas.1010580107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korennykh AV, et al. Structural and functional basis for RNA cleavage by Ire1. BMC Biol. 2011;9:47. doi: 10.1186/1741-7007-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korennykh AV, et al. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimata Y, et al. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J. Cell Biol. 2007;179:75–86. doi: 10.1083/jcb.200704166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghosh R, et al. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell. 2014;158:534–548. doi: 10.1016/j.cell.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimata Y, et al. A role for BiP as an adjustor for the endoplasmic reticulum stress-sensing protein Ire1. J. Cell Biol. 2004;167:445–456. doi: 10.1083/jcb.200405153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oikawa D, et al. Activation of mammalian IRE1α upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp. Cell Res. 2009;315:2496–2504. doi: 10.1016/j.yexcr.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 31.Volmer R, et al. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. U.S.A. 2013;110:4628–4633. doi: 10.1073/pnas.1217611110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hetz C, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1α. Science. 2006;312:572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- 33.Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: assembling the IRE1α interactome. Mol. Cell. 2009;35:551–561. doi: 10.1016/j.molcel.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korennykh AV, et al. Cofactor-mediated conformational control in the bifunctional kinase/RNase Ire1. BMC Biol. 2011;9:48. doi: 10.1186/1741-7007-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiseman RL, et al. Flavonol activation defines an unanticipated ligand-binding site in the kinase–RNase domain of IRE1. Mol. Cell. 2010;38:291–304. doi: 10.1016/j.molcel.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calfon M, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 37.Gonzalez TN, et al. Mechanism of non-spliceosomal mRNA splicing in the unfolded protein response pathway. EMBO J. 1999;18:3119–3132. doi: 10.1093/emboj/18.11.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawahara T, et al. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/ Ern4p that activates the unfolded protein response. Mol. Biol. Cell. 1997;8:1845–1862. doi: 10.1091/mbc.8.10.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sidrauski C, Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90:1031–1039. doi: 10.1016/s0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- 40.Urano F, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 41.Hu P, et al. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α-mediated NF-κB activation and downregulation of TRAF2 expression. Mol. Cell. Biol. 2006;26:3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 43.Hollien J, et al. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reid DW, et al. The unfolded protein response triggers selective mRNA release from the endoplasmic reticulum. Cell. 2014;158:1362–1374. doi: 10.1016/j.cell.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Upton JP, et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic caspase-2. Science. 2012;338:818–822. doi: 10.1126/science.1226191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oslowski CM, et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012;16:265–273. doi: 10.1016/j.cmet.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osorio F, et al. The unfolded-protein-response sensor IRE-1α regulates the function of CD8α+ dendritic cells. Nat. Immunol. 2014;15:248–257. doi: 10.1038/ni.2808. [DOI] [PubMed] [Google Scholar]
- 48.So JS, et al. Silencing of lipid metabolism genes through IRE1α-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012;16:487–499. doi: 10.1016/j.cmet.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han D, et al. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee AH, et al. Dual and opposing roles of the unfolded protein response regulated by IRE1α and XBP1 in proinsulin processing and insulin secretion. Proc. Natl. Acad. Sci. U.S.A. 2011;108:8885–8890. doi: 10.1073/pnas.1105564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hasnain SZ, et al. The interplay between endoplasmic reticulum stress and inflammation. Immunol. Cell Biol. 2012;90:260–270. doi: 10.1038/icb.2011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lerner AG, et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012;16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martinon F, et al. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. immunol. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinon F, Glimcher LH. Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Curr. Opin. Immunol. 2011;23:35–40. doi: 10.1016/j.coi.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bork P, Sander C. A hybrid protein kinase–RNase in an interferon-induced pathway? FEBS Lett. 1993;334:149–152. doi: 10.1016/0014-5793(93)81701-z. [DOI] [PubMed] [Google Scholar]
- 56.Malathi K, et al. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007;448:816–819. doi: 10.1038/nature06042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bowzard JB, et al. PAMPer and tRIGer: ligand-induced activation of RIG-I. Trends Biochem. Sci. 2011;36:314–319. doi: 10.1016/j.tibs.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 58.Hornung V, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 59.Pichlmair A, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 60.Zheng C, Wu H. RIG-I ‘sees’ the 5′-triphosphate. Structure. 2010;18:894–896. doi: 10.1016/j.str.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Poeck H, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat. Immunol. 2010;11:63–69. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- 62.Tam AB, et al. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. 2014;9:850–858. doi: 10.1016/j.celrep.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee AH, et al. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaser A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bertolotti A, et al. Increased sensitivity to dextran sodium sulfate colitis in IRE1β-deficient mice. J. Clin. Invest. 2001;107:585–593. doi: 10.1172/JCI11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang XZ, et al. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998;17:5708–5717. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Imagawa Y, et al. RNase domains determine the functional difference between IRE1α and IRE1β. FEBS Lett. 2008;582:656–660. doi: 10.1016/j.febslet.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 68.Duerkop BA, et al. Immune responses to the microbiota at the intestinal mucosal surface. Immunity. 2009;31:368–376. doi: 10.1016/j.immuni.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 69.Huttenhower C. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ley RE, et al. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 2008;6:776–788. doi: 10.1038/nrmicro1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Backhed F, et al. Host–bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 72.Martino MB, et al. The ER stress transducer IRE1β is required for airway epithelial mucin production. Mucosal Immunol. 2013;6:639–654. doi: 10.1038/mi.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsuru A, et al. Negative feedback by IRE1β optimizes mucin production in goblet cells. Proc. Natl. Acad. Sci. U.S.A. 2013;110:2864–2869. doi: 10.1073/pnas.1212484110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Iwawaki T, et al. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat. Cell Biol. 2001;3:158–164. doi: 10.1038/35055065. [DOI] [PubMed] [Google Scholar]
- 75.Iqbal J, et al. IRE1β inhibits chylomicron production by selectively degrading MTP mRNA. Cell Metab. 2008;7:445–455. doi: 10.1016/j.cmet.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Elson CO, Ealding W. Generalized systemic and mucosal immunity in mice after mucosal stimulation with cholera toxin. J. Immunol. 1984;132:2736–2741. [PubMed] [Google Scholar]
- 77.Dertzbaugh MT, Elson CO. Cholera toxin as a mucosal adjuvant. In: Spriggs DR, Koff WC, editors. Topics in Vaccine Adjuvant Research. CRC Press; 1990. pp. 119–131. [Google Scholar]
- 78.Elson CO, Ealing W. Genetic control of the murine immune response to cholera toxin. J. Immunol. 1985;135:930–932. [PubMed] [Google Scholar]
- 79.Anosova NG, et al. Cholera toxin, E. coli heat-labile toxin, and non-toxic derivatives induce dendritic cell migration into the follicleassociated epithelium of Peyer’s patches. Mucosal Immunol. 2008;1:59–67. doi: 10.1038/mi.2007.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dickinson BL, Clements JD. Dissociation of Escherichia coli heat-labile enterotoxin adjuvanticity from ADP-ribosyltransferase activity. Infect. Immun. 1995;63:1617–1623. doi: 10.1128/iai.63.5.1617-1623.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bhuiyan TR, et al. Cholera caused by Vibrio cholerae O1 induces T-cell responses in the circulation. Infect. Immun. 2009;77:1888–1893. doi: 10.1128/IAI.01101-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kuchta A, et al. Vibrio cholerae O1 infection induces proinflammatory CD4+ T-cell responses in blood and intestinal mucosa of infected humans. Clin. Vaccine Immunol. 2011;18:1371–1377. doi: 10.1128/CVI.05088-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dangl JL, Jones JD. Plant pathogens and integrated defence responses to infection. Nature. 2001;411:826–833. doi: 10.1038/35081161. [DOI] [PubMed] [Google Scholar]
- 84.Vance RE, et al. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe. 2009;6:10–21. doi: 10.1016/j.chom.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Keestra AM, et al. Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1. Nature. 2013;496:233–237. doi: 10.1038/nature12025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu H, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513:237–241. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 87.Celli J, Tsolis RM. Bacteria, the endoplasmic reticulum and the unfolded protein response: friends or foes? Nat. Rev. Microbiol. 2015;13:71–82. doi: 10.1038/nrmicro3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Medzhitov R, Janeway C.A., Jr. Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 89.Claudio N, et al. Mapping the crossroads of immune activation and cellular stress response pathways. EMBO J. 2013;32:1214–1224. doi: 10.1038/emboj.2013.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pahl HL, Baeuerle PA. Expression of influenza virus hemagglutinin activates transcription factor NF-kappa B. J. Virol. 1995;69:1480–1484. doi: 10.1128/jvi.69.3.1480-1484.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pahl HL, et al. Activation of transcription factor NF-kappaB by the adenovirus E3/19K protein requires its ER retention. J. Cell Biol. 1996;132:511–522. doi: 10.1083/jcb.132.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Paton AW, et al. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature. 2006;443:548–552. doi: 10.1038/nature05124. [DOI] [PubMed] [Google Scholar]
- 93.Wolfson JJ, et al. Subtilase cytotoxin activates PERK. IRE1 and ATF6 endoplasmic reticulum stress-signalling pathways. Cell. Microbiol. 2008:1775–1786. doi: 10.1111/j.1462-5822.2008.01164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stahl S, et al. Cytomegalovirus downregulates IRE1 to repress the unfolded protein response. PLoS Pathog. 2013;9:e1003544. doi: 10.1371/journal.ppat.1003544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu. Rev. Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 96.Oikawa D, et al. Direct association of unfolded proteins with mammalian ER stress sensor. IRE1β. PLoS One. 2012;7:e51290. doi: 10.1371/journal.pone.0051290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dufey E, et al. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 1. An overview. Am. J. Physiol. Cell Physiol. 2014;307:C582–C594. doi: 10.1152/ajpcell.00258.2014. [DOI] [PubMed] [Google Scholar]
- 98.Hayashi F, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 99.Eletto D, et al. Protein disulfide isomerase A6 controls the decay of IRE1α signaling via disulfide-dependent association. Mol. Cell. 2014;53:562–576. doi: 10.1016/j.molcel.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.English AR, Voeltz GK. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb. Perspect. Biol. 2013;5:a013227. doi: 10.1101/cshperspect.a013227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dixit E, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Horner SM, et al. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 2011;108:14590–14595. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]