

Abstract
The consumption of ethanol by pregnant women may cause neurological abnormalities, affecting learning and memory processes in children, and are collectively described as fetal alcohol spectrum disorders (FASDs). However, the molecular mechanisms underlying these changes are still poorly understood. In our previous studies, we found that ethanol treatment of postnatal day 7 (P7) mice significantly enhances anandamide (AEA) levels but not 2-arachidonylglycerol (2-AG) levels and induces widespread neurodegeneration, but the reason for the lack of significant effects of ethanol on the 2-AG level is unknown. In this study, we examined developmental changes in diacylglycerol lipase-α, β (DAGL-α and β) and monoacylglycerol lipase (MAGL). We found that the levels of these proteins were significantly higher in adult brains compared to those detected early in brain development. Next, we examined the influence of P7 ethanol treatment on these enzymes, finding that it differentially altered the DAGL-α protein and mRNA levels but consistently enhanced those of the DAGL-β. Interestingly, the ethanol treatment enhanced MAGL protein and mRNA levels. Inhibition of MAGL with KML29 failed to induce neurodegeneration in P7 mice. Collectively, these findings suggest that ethanol significantly activates DAGL-β and MAGL in the neonatal brain, resulting in no net change in 2-AG levels.
Keywords: neurodegeneration, DAGL-α/β, MAGL, anandamide, cannabinoids, development, CB1R, fetal alcohol spectrum disorder (FASD)
Introduction
Fetal alcohol spectrum disorder (FASD) is an umbrella term used to describe a range of neurological defects that can occur in an individual whose mother abused alcohol during pregnancy (Feldman et al. 2012, Lewis et al. 2012, Paintner et al. 2012a, Paintner et al. 2012b). A recent survey has suggested that the present prevalence of FASD in the United States and several Western European countries may be as high as 2-5% (May et al. 2009). The increase in the incidence of FASD is a major factor underlying the rises in the number of children and adults with learning disabilities and cases of non-genetic intellectual disability in Western nations (Morleo et al. 2011). FASD is associated with widespread neuropsychological manifestations (Mattson & Riley 1998, Mattson et al. 1998), such as reduced intellectual ability and behavioral problems (Goodman et al. 1999, Harris et al. 1995, Mattson et al. 2011, Mattson et al. 1999, Rasmussen et al. 2006). In mice, a significant proportion of third trimester-equivalent (Bayer et al. 1993) brain development takes place following birth (Cronise et al. 2001, Tran et al. 2000), and rapid synaptic growth occurs during postnatal days 4-10 (P4-10). Therefore, in binge ethanol models, ethanol has been directly administered to neonatal pups to examine the effects of ethanol consumption during the third trimester of fetal development in humans (Gil-Mohapel et al. 2010). The major immediate effect of single-day ethanol intoxication in P7 mice has been found to be the widespread activation of caspase-3 (Ikonomidou et al. 2000) in many brain regions including hippocampus (HP) and neocortex (NC), which are important for learning and memory (Kelly et al. 2009). This ethanol paradigm has been shown to cause persistent neurobehavioral abnormalities in adult mice (Noel et al. 2011, Sadrian et al. 2012, Subbanna & Basavarajappa 2014, Subbanna et al. 2015, Subbanna et al. 2013a, Wilson et al. 2011).
The effects of ethanol are mediated through several signal transduction pathways involving many neurotransmitters and ion channels in various brain regions, one of which is the endocannabinoid system (EC), which is comprised of endogenous cannabinoids, their receptors (cannabinoid receptors type 1 and 2; CB1R and CB2R), and the enzymes involved in their metabolism (Basavarajappa & Arancio 2008, Mechoulam & Parker 2013). The EC system regulates synaptic events via endocannabinoids, such as anandamide (AEA) and 2-arachidonylglycerol (2-AG) (Kreitzer & Regehr 2001, Ohno-Shosaku et al. 2001, Wilson & Nicoll 2001), in developing (Hansen et al. 2008, Harkany et al. 2008, Subbanna et al. 2015, Subbanna et al. 2013a) and adult brains (Mechoulam & Parker 2013). AEA, 2-AG and their G-protein-coupled receptor, CB1R, have been shown to be prime targets of ethanol sensitivity, tolerance and dependence in adult animals (Basavarajappa 2007, Basavarajappa et al. 2008, Basavarajappa et al. 2006, Hungund et al. 2003, Rubio et al. 2009), including humans (Ceccarini et al. 2014, Hirvonen et al. 2013, Marcos et al. 2012). There is strong evidence that ECs and CB1R have decisive functions in neuronal maturation during brain development (Bisogno et al. 2003, Fernandez-Ruiz et al. 2000).
In our previous studies, we have found enhanced CB1R expression and increased AEA but not 2-AG levels which lead to neurodegeneration in ethanol-treated P7 mice. Enhanced AEA levels in ethanol-treated P7 mice were achieved via transcriptional activation of enzymes involved in AEA biosynthesis. Blockade with CB1R antagonist or genetic deletion of CB1R provided protection against ethanol-induced neurodegeneration, suggesting that the brain response involves the specific activation of the AEA-CB1R pathway and the maintenance of the 2-AG mediated effects under normal conditions (Subbanna et al 2013a). The reason for the lack of ethanol effects on 2-AG levels is not known. Unlike the AEA level, the 2-AG level is two hundred-fold higher in the brain (Sugiura et al. 2006), and its biosynthesis is catalyzed by two diacylglycerol lipase isoforms (diacylglycerol lipase-α/β and DAGLα/β) (Bisogno et al. 2003). DAGL-α is expressed throughout the brain, but the expression pattern of DAGL-β has not been well characterized (Basavarajappa 2014, Oudin et al. 2011). In vivo, 2-AG biosynthesis appears to be carried out by these two DAGLs, but the isoforms that are actually functioning in normal brains and in those that are diseased still remains to be established (Murataeva et al. 2014). It is well accepted that monoacylglycerol lipase (MAGL) is the predominant enzyme responsible for degrading 2-AG during synaptic activities, but at least four other enzymes, including alpha-beta-hydrolase domain 6 and 12 (ABHD6 and 12), fatty acid amide hydrolase (FAAH) and cyclooxygenase-2 (COX-2), have shown to be involved in 2-AG degradation, depending on the cell type and tissue-specific conditions (Basavarajappa 2014, Murataeva et al. 2014). Based on this interesting fact, in the current study, we further assessed the means by which 2-AG is maintained in the steady state in ethanol-treated P7 mice by examining the transcription and translation of enzymes involved in 2-AG metabolism. Postnatal ethanol treatment differentially regulated the DAGL-α/β and MAGL enzymes, resulting in steady-state 2-AG levels in the hippocampus and neocortex of the P7 mice. Inhibition of MAGL with KML29 failed to induce neurodegeneration in P7 mice.
Methods
Animals and ethanol treatment
C57BL/6J (Jackson Laboratory, Bar Harbor, ME, USA) mice were generated from breeding colony at NKI and housed in groups under standard laboratory conditions (12 h light/12 h dark cycle), with food and water available ad libitum. Animal care and handling procedures followed the Institutional (NKI IACUC) and National Institutes of Health guidelines. An ethanol treatment paradigm, which has been previously shown to induce robust apoptotic neurodegeneration in P7 mice (Olney et al. 2002), was used in the current study. Half of the male and female pups in each litter were treated subcutaneously (s.c.) with saline, and the other half were treated with ethanol at P7 (based on the day of birth) (2.5 g/kg s.c. at 0 h and again at 2 h) as described previously by our laboratory (Subbanna & Basavarajappa 2014, Subbanna et al. 2014, Subbanna et al. 2013b). The mice were kept with the dams until the pups were sacrificed, and their brains were removed at 4–24 h after the first saline/ethanol injection.
Analysis of AEA and 2-AG levels by LC/MS
Hippocampus and neocortex tissues were dissected at 8 h after first dose of ethanol or saline injection, flash frozen and stored at -80°C. Hippocampus and neocortex tissue homogenates were subjected to LC–MS using the isotopic dilution procedure to measure AEA and 2-AG levels as described previously (Subbanna et al. 2013a). The standard curve was fitted with a quadratic equation, with the curve encompassing ranges of 0.5–50 ng for AEA and 50-2500 ng for 2-AG, and was processed similar to quality controls prepared with brain tissue extracts.
KML29 treatment
For the KML29 experiments, KML29 (Cayman Chemicals, Ann Arbor, Michigan) was dissolved in 10μl of ethanol followed by a few drops of Tween 80 (10 μl) and the volume was made up with sterile saline solution. Half of the male and female C57BL/6J mice pups in each litter were treated subcutaneously (s.c.) with vehicle and the other half were treated with KML29 solution (0-20 mg/kg) by s. c. injection at a volume of 5 μl/g body weight as described previously by our laboratory (Subbanna & Basavarajappa 2014, Subbanna et al. 2014, Subbanna et al. 2013b). The KML29 vehicle solution was injected as a control. The mice were kept with the dams until the pups were sacrificed, and their brains were removed at 4–24 h after the KML29 injection.
Protein extraction, electrophoresis and immunoblotting
Four to 24 h after the first saline or ethanol injection, pups were sacrificed by decapitation, and the cortex and hippocampus were dissected, flash frozen and stored at -80°C. Hippocampus and neocortex tissue homogenates containing a freshly added 1% protease inhibitor mixture (Roche, Indianapolis, IN, USA) and phosphatase inhibitors were centrifuged at 7700 g for 1 min, and the supernatant (total extract) was aspirated. The membrane and cytosolic fractions were prepared from the total extract as described previously (Basavarajappa & Hungund 2001) and stored at -80°C until use. The samples for electrophoresis were prepared in a sample buffer as previously described by our laboratory (Basavarajappa et al. 2008). Blots were stained with Ponceau S to confirm equal loading in each lane and were incubated with a primary antibody, such as anti-rabbit MAGL (polyclonal #ab24701, 1:1000, Abcam, Cambridge, MA, USA), anti-goat DAGL-α (polyclonal #46-829, 1:1000), anti-rabbit-DAGL-β (1:1000; Kind gift from Dr. Ken Mackie, Indian University, Bloomington, IN), or anti-mouse-β-actin (monoclonal, catalog #3700, Cell Signaling), for 3 h at room temperature (22°C) or overnight at 4°C and processed as previously described by our laboratory (Basavarajappa et al. 2008). The incubation of the blots with a secondary antibody (goat anti-mouse peroxidase conjugate, #AP 124P, 1:5000 or goat anti-rabbit, #AP132P, 1:5000, Millipore, Billerica, MA, USA) alone resulted in no protein bands.
Real-time quantitative polymerase chain reaction (qPCR)
Four to 24 h after the first saline or ethanol injection, total RNA from the hippocampus and cortex samples was isolated using an RNeasy Mini Kit (Qiagen, Valencia, CA). mRNA was reverse-transcribed into complementary deoxyribonucleic acid (cDNA) using a Maxima First Strand cDNA Synthesis Kit from Fermentas in a programmable thermal cycler (PCR-Sprint, Thermo Electron, Milford, MA). qPCR for the amplification of Faah, Dagl-α, β, Magl, Abdh4, Abdh6 and Abdh12 was performed with an integrated thermocycler and fluorescence detector (ABI PRISM 7900HT Sequence Detector; Applied Biosystems) using TaqMan® Gene Expression Assays Mm00813830_m1 (Dagl-α), Mm00523381_m1 (Dagl-β), Mm00449274_m1 (Magl), Mm00506368_m1 (Abdh4), Mm00481199_m1 (Abdh6), Mm00470489_m1 (Abdh12) and 4352932 (Gapdh) (Applied Biosystems). Glyceraldehyde3-phosphate dehydrogenase (GAPDH) was used as an endogenous mRNA control. For each run, triplicate reactions were carried out for each sample. Three independent experiments were carried out for each set of samples. Data were analyzed using SDS2.4 software (Applied Biosystems). The amount of target (Faah, Dagl-α, Dagl-β, Magl, Abdh4, Abdh6 and Abdh12), normalized to the endogenous reference (Gapdh) and relative to a calibrator was calculated with the equation -2ΔΔCt (Subbanna et al. 2014, Subbanna et al. 2013a, Subbanna et al. 2013b).
Statistical analysis
All of the data are presented as the mean ± SEM. A statistical comparison of the data was performed by one-way analysis of variance (ANOVA) with Bonferroni's post hoc test. For all comparisons, a p < 0.05 was considered statistically significant. Statistical analyses were performed using Prism software (GraphPad, San Diego, CA).
Results
P7 ethanol treatment induces caspase-3 activation and specifically enhances AEA levels without altering 2-AG levels in hippocampus and neocortex tissues
The exposure of P7 mice to the ethanol (2.5 g/kg, s.c. at 0 h and again at 2 h) treatment resulted in a BEL of 0.43 ± 0.34 g/dl at 3 h that was gradually reduced to 0.23 ± 0.06 g/dl at 9 h after the first ethanol injection. This treatment resulted in the widespread activation of capsase-3, as measured using a cleaved caspase-3 (CC3) antibody throughout the forebrain. Measurements were made at 8 h after the first dose of ethanol or saline [hippocampus (F1, 11 = 55, p < 0.05) and cortex (F1, 11 = 120, p < 0.05) regions] in the ethanol-exposed P7 brains (one-way ANOVA with Bonferroni's post hoc test) (Fig. 1a). We also measured CC3 protein levels by Western blot in hippocampal and neocortical protein cytosolic extracts. The results suggested that at 8 and 24 h after the first dose of ethanol, CC3 protein levels were significantly enhanced in both the hippocampus (F3, 28 = 66, p < 0.05) and neocortex (F3, 28 = 55, p < 0.05) (Fig. 1b) regions compared to those detected at 0 h (saline control) (one-way ANOVA with Bonferroni's post hoc test). The ethanol treatment significantly enhanced AEA but not 2-AG levels (p > 0.05) in both the hippocampus (F3, 28 = 26, p < 0.05) and neocortex (F3, 28 = 25, p < 0.05) tissues, as measured at 8 h after the first dose of ethanol or saline (Table 1). These observations suggest that ethanol-treatment of P7 mice recapitulated the previous findings on endocannabinoid levels and neurodegenerative conditions.
Fig. 1.
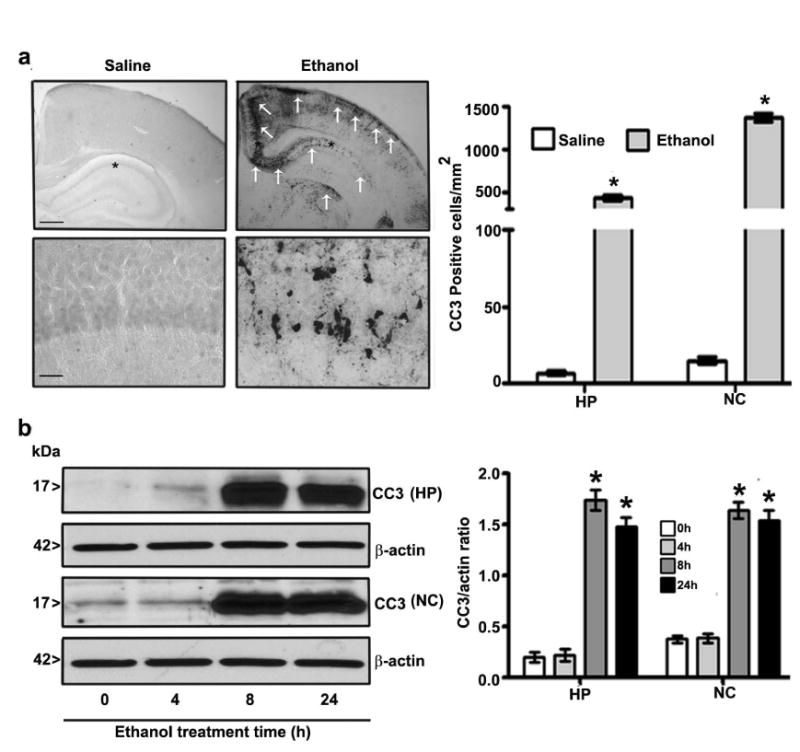
Ethanol exposure activates caspase-3 and alters endocannabinoid levels in the P7 mouse brain. (a) Coronal brain sections with hippocampus and retrosplenial cortex regions from saline- and ethanol-treated P7 mice were immunostained with an anti-rabbit CC3 antibody. CC3-positive neurons in the hippocampus and retrosplenial cortex are indicated by white arrows. Scale bars = 200 μm. Hippocampus and retrosplenial cortex images are enlarged to indicate CC3-positive cells (*). The scale bars represent 50 μm. CC3-positive neurons were counted in the hippocampus and the retrosplenial cortex (n = 8 pups/group). (b) CC3 levels were measured in cytosolic extracts of hippocampal and neocortical samples from the saline and ethanol groups (n = 15 pups/group) by Western blot analysis. β-actin was used as a loading control. HP, hippocampus; NC, neocortex.
Table 1.
Ethanol-treatment of P7 mice enhances AEA but not 2-AG contents in hippocampus and neocortex tissues.
| Endocannabinoids | Hippocampus | Neocortex | ||
|---|---|---|---|---|
| Saline | Ethanol | Saline | Ethanol | |
| AEA (pmol/g) | 6.54 ± 0.38 | 8.37 ± 0.17* | 4.51 ± 0.11 | 6.87 ± 0.24* |
| 2-AG (nmol/g) | 32.84 ± 1.93 | 32.92 ± 1.63 | 31.67 ± 1.24 | 31.94 ±` 0.58 |
AEA and 2-AG levels in hippocampal and neocortical total extracts were analyzed by LC-MS (n = 10 pups/group)
(p < 0.05 vs. saline-treated group).
All statistical analyses were conducted using one-way ANOVA with Bonferroni's post hoc tests. The error bars represent the SEM.
Ethanol exposure of P7 mice affects transcription and translation of 2-AG biosynthetic enzymes
Prior to the examination of the events responsible for the lack of effects of ethanol on 2-AG formation in P7 mice, we determined the developmental patterns of the DAGL-α and β proteins using cortical extracts. The levels of these proteins dramatically increased from the early postnatal to adult stages (F7, 40 = 102, p < 0.05) (Fig. 2). The levels of the housekeeping protein actin did not change significantly during the various stages of brain development, as shown previously (Jacob et al. 2011, Subbanna et al. 2013a, Subbanna et al. 2013b). We then determined whether ethanol affects the two major enzymes responsible for 2-AG biosynthesis (Basavarajappa 2014, Murataeva et al. 2014) and examined DAGL-α and β protein levels by Western blot analysis. Our results suggested that ethanol significantly reduced DAGL-α protein levels in the hippocampus (F3, 28 = 15, p < 0.05) but not in the neocortex (F3, 28 = 1, p > 0.05) (Fig. 3a) (one-way ANOVA with Bonferroni's post hoc test). We also measured DAGL-α mRNA levels. The results indicated mRNA levels were similar to the observed protein levels in the hippocampus (F3, 28 = 15, p < 0.05)) and neocortex (Fig. 3b) (F3, 28 = 1.2, p > 0.05) (one-way ANOVA with Bonferroni's post hoc test). Next, we measured DAGL-β protein levels. Our results suggested that ethanol significantly enhanced the levels of this protein in the hippocampus (F3, 20 = 19, p < 0.001) and neocortex (F3, 20 = 27, p < 0.001) (Fig. 4a) (one-way ANOVA with Bonferroni's post hoc test). Further, we determined DAGL-β mRNA levels to examine the transcriptional activation of the DAGL-β gene by the P7 ethanol treatment. Consistent with the protein levels, the mRNA levels were also significantly increased by ethanol in the hippocampus (F3, 28 = 13, p < 0.05) and neocortex (F3, 28 = 12, p < 0.05) at 8 and 24 h (Fig. 4b) (one-way ANOVA with Bonferroni's post hoc test). Together, these findings imply that although ethanol significantly enhanced 2-AG biosynthetic enzymes, especially DAGL-β, no significant increase in the 2-AG level occurred, which suggests that this endocannabinoid may be undergoing degradation.
Fig. 2.
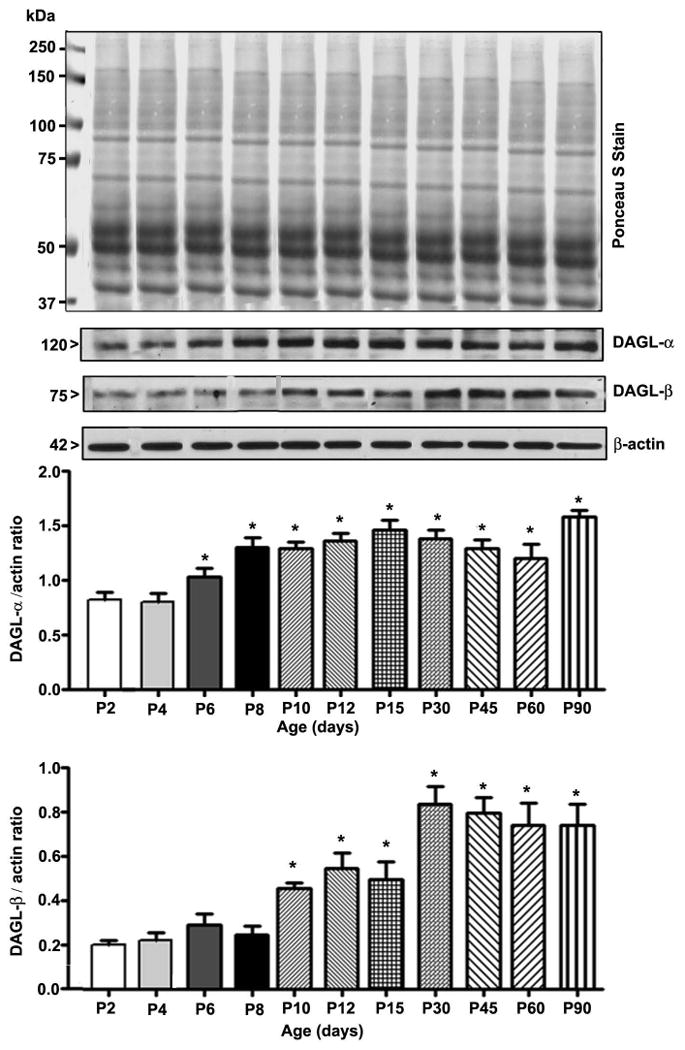
Developmental patterns of 2-AG biosynthetic enzymes. Western blot analysis of DAGL-α and β protein expression in neocortical total extracts. Ponceau S Stain confirmed equal loading. The representative blot shows the developmental patterns of DAGL-α and β protein expression (n = 8 pups/group; *p < 0.05 compared with the P2 group). Statistical analysis was conducted using one-way ANOVA with Bonferroni's post hoc tests. The error bars represent the SEM. HP, hippocampus; NC, neocortex.
Fig. 3.
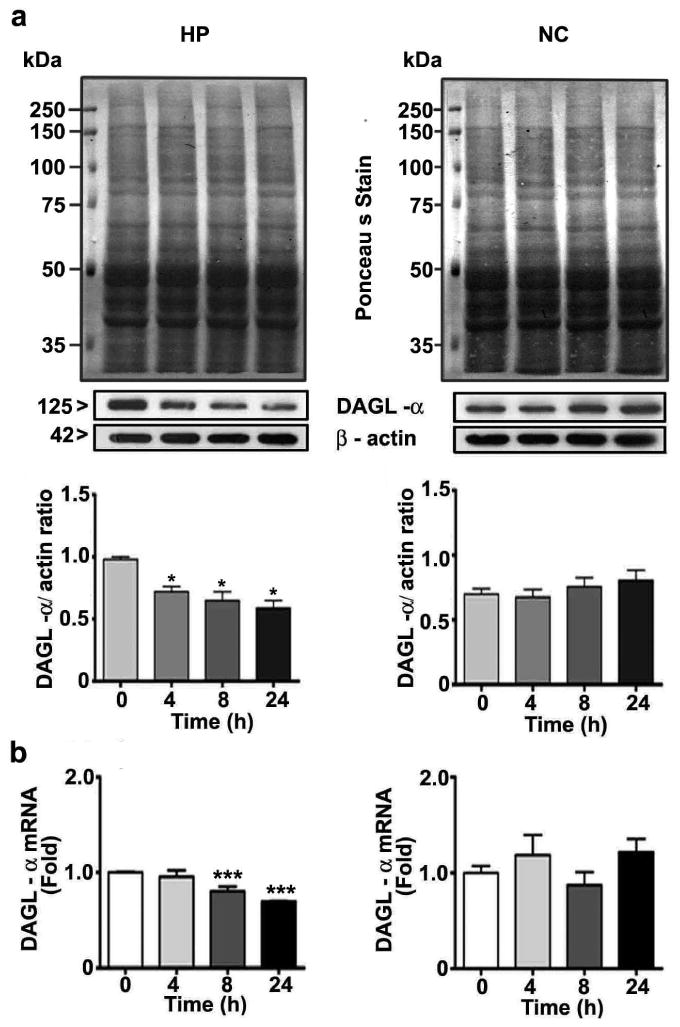
The ethanol treatment of P7 mice reduces the DAGL-α level. (a) DAGL-α levels in hippocampal and neocortical cytosolic extracts were determined by Western blot analysis. Blots were stained with Ponceau S to confirm equal loading in each lane, and β-actin was used as a loading control (n = 15 pups/group). (b) DAGL-α mRNA levels in hippocampal and cortical total extracts from the saline- and ethanol-treated groups (n = 15 pups/group) were measured by qPCR (*p < 0.05; ***p < 0.01). Statistical analysis was conducted with one-way ANOVA with Bonferroni's post hoc tests. The error bars represent the SEM. HP, hippocampus; NC, neocortex.
Fig. 4.
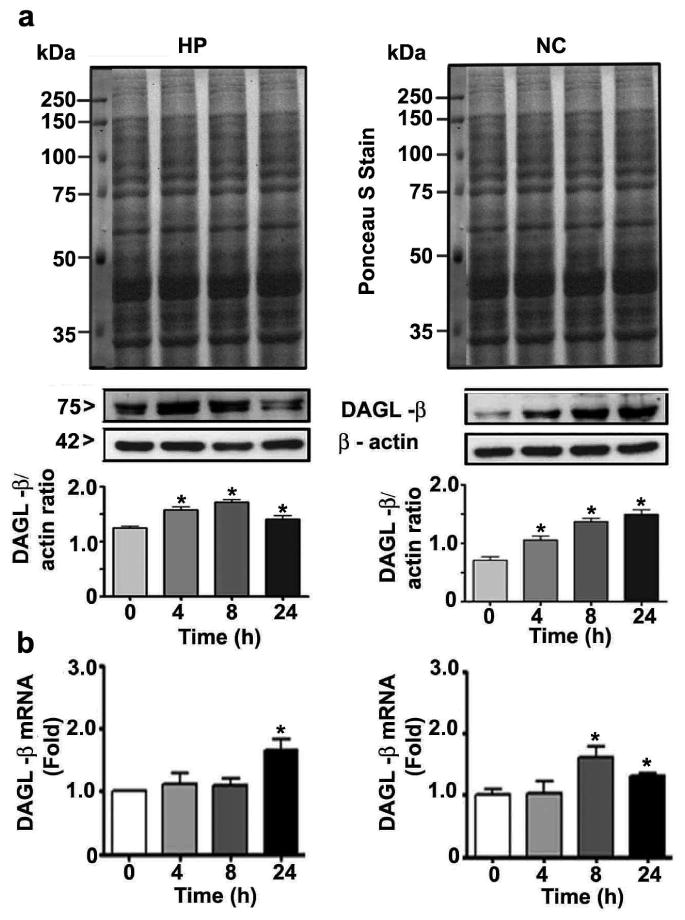
DAGL-β levels were enhanced in P7 mice treated with ethanol. (a) Hippocampal and neocortical levels of DAGL-β were determined in cytosolic extracts by Western blot analysis. Blots were stained with Ponceau S to confirm equal loading in each lane, and β-actin was used as a loading control (n = 15 pups/group). (b) qPCR was used to determine DAGL-β mRNA levels in hippocampal and cortical total extracts from the saline- and ethanol-treated groups (n = 15 pups/group; *p < 0.05). Statistical analysis was conducted using one-way ANOVA with Bonferroni's post hoc tests. The error bars represent the SEM. HP, hippocampus; NC, neocortex.
Ethanol treatment of P7 mice specifically enhances transcription and translation of MAGL enzyme
As discussed earlier, 2-AG degradation is carried out by more than one degradative enzyme (Basavarajappa 2014, Murataeva et al. 2014). Thus, we screened the mRNA levels of the following enzymes: ABHD4, ABHD6, ABHD12, FAAH and MAGL. The results suggested that there were no significant increases in the ABHD4, ABHD6, ABHD12 or FAAH mRNA levels in the hippocampus (Fig. 5a) or neocortex (Fig. 5b) tissues (p > 0.05), as measured at 8 h after the first dose of ethanol or saline. However, MAGL mRNA levels were significantly increased in the hippocampus (F3, 28 = 23, p < 0.05) and neocortex (F3, 28 = 10, p < 0.05) tissues (one-way ANOVA with Bonferroni's post hoc test) (Fig. 5a and b). Prior to analysis of the effects of ethanol on MAGL protein levels, we determined the developmental pattern of the MAGL protein using cortical extracts. The levels of this protein dramatically increased from the early postnatal to adult stages (F7, 40 = 287, p<0.05) (Fig. 6a). The levels of the housekeeping protein actin did not change significantly during the various stages of brain development, as shown previously (Jacob et al. 2011, Subbanna et al. 2013a, Subbanna et al. 2013b). We then examined whether ethanol treatment affects MAGL protein levels in P7 mice. Our Western blot analyses suggested that ethanol significantly enhanced the levels of this protein in both the hippocampus (F3, 28 = 14, p < 0.05) and neocortex (F3, 28 = 45, p < 0.05) (Fig. 6b) tissues. We also examined MAGL mRNA levels to determine whether ethanol affected the transcriptional activity of the MAGL gene in a time-dependent manner in P7 mice. Consistent with the protein levels, ethanol also enhanced the mRNA levels of this protein in the hippocampal (F3, 28 = 58, p < 0.05) and cortical (F3, 28 = 12, p < 0.05) (Fig. 6c) regions. Thus, although ethanol persistently enhanced the transcription and translation of DAGL-β, the simultaneous transcriptional activation of MAGL led to the normalization of the 2-AG levels in the P7 mice.
Fig. 5.
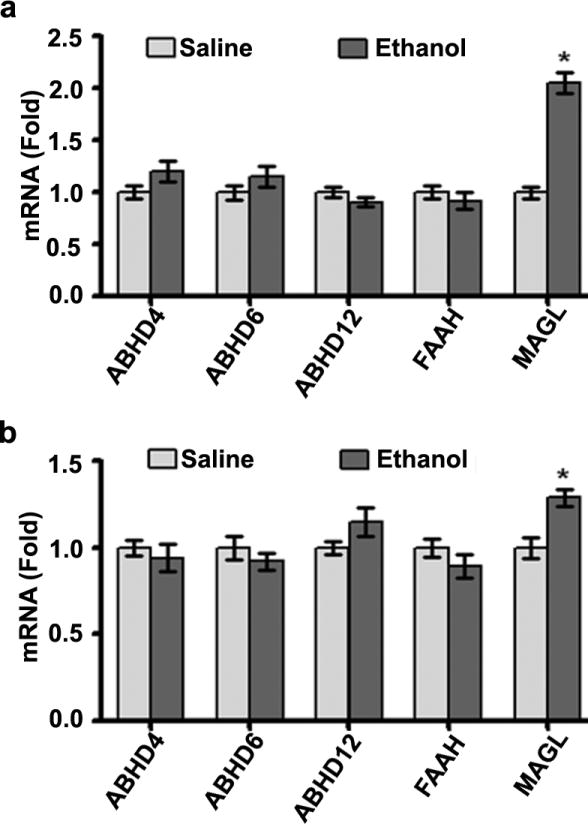
Influence of ethanol treatment of P7 mice on the mRNA levels of endocannabinoid-metabolizing enzymes. The mRNA levels of FAAH, ABHD4, ABHD6, ABHD12 and MAGL were determined in (a) hippocampal and (b) neocortex total extracts from the saline- and ethanol-treated groups (at 8 h after the first dose of ethanol or saline) by qPCR (n = 10 pups/group; *p < 0.05). Statistical analysis was conducted using one-way ANOVA with Bonferroni's post hoc tests. The error bars represent the SEM. HP, hippocampus; NC, neocortex.
Fig. 6.
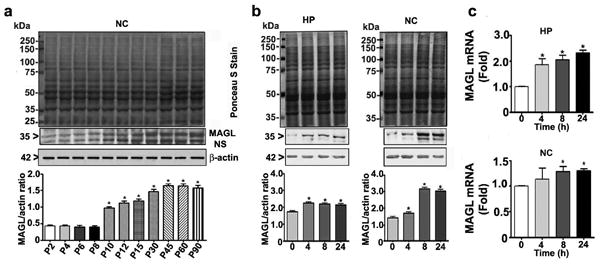
Developmental pattern and influence of ethanol treatment of P7 mice on MAGL expression. (a) Western blot analysis of MAGL expression in neocortical total extracts. Blots were stained with Ponceaus s Stain to confirm equal loading. The representative blot shows the developmental patterns of MAGL protein expression (n = 8 pups/group; *p < 0.05 compared with the P2 group). (b) Western blot analysis of MAGL in hippocampal and neocortex extracts from the saline and ethanol-treated groups (*p < 0.05). (c) MAGL mRNA levels in hippocampal and cortical total extracts from the saline- and ethanol-treated groups were measured by qPCR (n = 8 pups/group compared with the 0.0 control group; *p < 0.05). Statistical analysis was conducted using one-way ANOVA with Bonferroni's post hoc tests. The error bars represent the SEM. HP, hippocampus; NC, neocortex.
Inhibition of MAGL by KML29 failed to activate caspase-3 in P7 mice
To explore whether the inhibition of MAGL activity in vivo induces activation of caspase-3, we injected P7 mice with low to high dose of KML29 for up to 24 h. The results suggested that there were no significant increases in the cleaved caspase-3 levels in neocortex (Fig. 7) tissue (p > 0.05), as measured at 4- 24 h after the KML29 or vehicle treatment. In previous studies, KML29 has been shown to enhance 2-AG levels in mice at the doses used (Ignatowska-Jankowska et al. 2014, Kinsey et al. 2013, Schlosburg et al. 2010). Thus, inhibition of MAGL by KML29 which was shown to elevate endogenous levels of 2-AG failed to induce neurodegeneration in P7 mice.
Fig. 7.
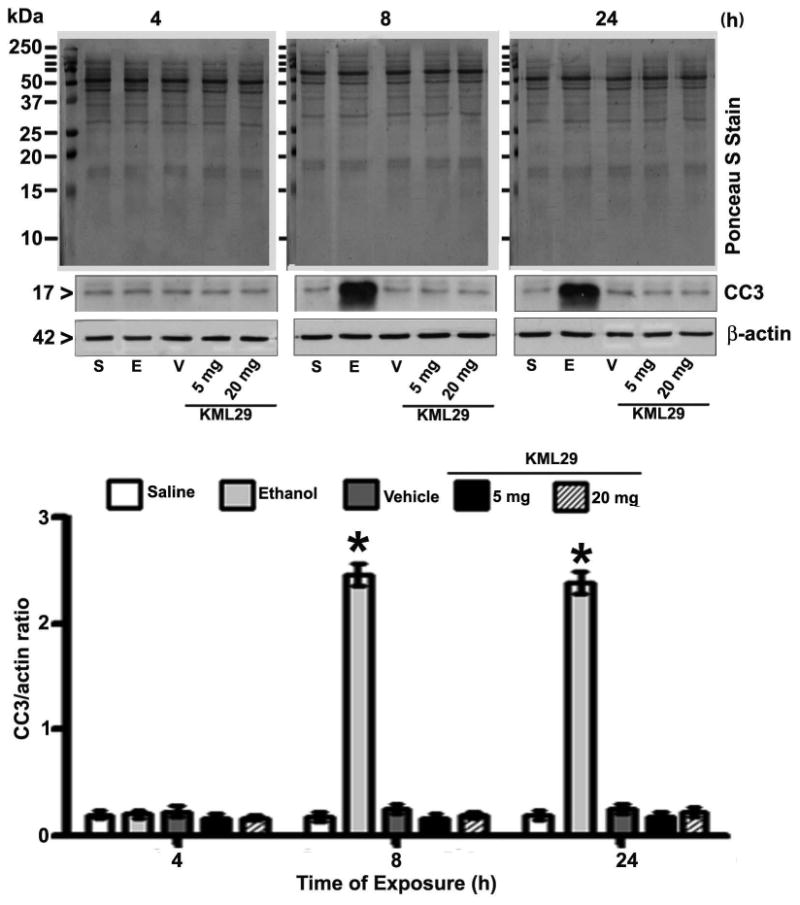
Pharmacological inhibition of MAGL by KML29 failed to induce caspase-3 activation in P7 mice. Mice were treated for 4-24 h with two doses of KML29 (5 and 20 mg/kg, s.c.) or vehicle and CC3 levels were determined in neocortex tissue by a Western blot analysis (p > 0.05). P7 were also treated with ethanol and used as positive controls. The error bars represent the SEM. S, saline; E, ethanol; V, vehicle.
Discussion
Here, we showed the means by which the postnatal ethanol treatment achieved enhanced AEA over 2-AG levels in the neonatal mouse brain. This precise on-demand local increase in AEA over 2-AG is attained through transcriptional and translational activation of well-characterized biosynthetic and degradative enzymes. In P7 ethanol-treated mice, AEA biosynthesis was enhanced through the transcriptional and translational upregulation of the N-arachidonoyl phosphatidylethanolamine-phospholipase D (NAPE-PLD) and glycerophosphodiesterase (GDE1) enzymes, but the catabolizing enzymes (ABHD4 and FAAH) were not altered (Subbanna et al. 2013a). In addition, although the P7 ethanol treatment consistently enhanced 2-AG biosynthetic enzymes, such as DAGL-β, also enhanced 2-AG catabolizing enzymes, such as MAGL, resulting in no significant change in the 2-AG levels. While these events are not economical to cells overall function, these observations suggest that the enhancement of AEA over 2-AG-CB1R pathway constitutes an example of the generation of unique cellular responses depending on the environment of the cell. Although our current and previous observations are in agreement with the suggested roles of endocannabinoids as modulators of neural cell fate (Berrendero et al. 1999, Fernandez-Ruiz et al. 2000, Rodriguez de Fonseca et al. 1993, Romero et al. 1997, Subbanna et al. 2015, Subbanna et al. 2013a) and with their ability to modulate signal transduction pathways that are essential for the regulation of cell fate in general (Fernandez-Ruiz et al. 2000, Mato et al. 2003, Rodriguez de Fonseca et al. 1993, Subbanna et al. 2015, Subbanna et al. 2013a), our present data provide the transcriptional and translational means through which postnatal ethanol was able to achieve specific AEA-CB1R-mediated ERK1/2/pCREB/Arc signaling (Subbanna et al. 2015, Subbanna et al. 2013a), rather than 2-AG mediated events in ethanol-induced neurodegeneration in the neonatal mice.
Although both AEA and 2-AG have been detected from the fetal period, the concentration of AEA has been found to be 1000-fold lower than that of 2-AG (Fernandez-Ruiz et al. 2000). Likewise, the developmental patterns significantly differ between the two endocannabinoids. AEA concentrations gradually increase throughout development to the adult stage (Berrendero et al. 1999), whereas 2-AG levels remain same throughout ontogeny, with a remarkably distinct peak on the first day after birth in rats. However, DAGL-α, β and MAGL expression levels are lower during early development and gradually increase throughout development to adulthood. Although the 2-AG concentration in the rodent brain is in the nanomolar range, which is one hundred times higher than that of AEA, it is 20 times less potent than AEA (Mechoulam et al. 1995). DAGL-α and β both contribute substantially to the regulation of steady-state levels of 2-AG in the brain and other tissues. However, in mice lacking DAGL-α, 2-AG levels are reduced by up to 80% in the brain and up to 50% in the DAGL-β null mouse brain (Gao et al. 2010). The manner by which ethanol preferably activates DAGL-β over DAGL-α even though both DAGL- α and β are less abundant in the developing brain compared to the adult brain (Bisogno et al. 2003) are unknown and warrant further investigations. Nonetheless, this work is still in its infancy, and future studies should address the mechanism by which AEA and 2-AG signaling is regulated in the developing brain.
While ethanol in the developing brain led to significantly increased levels of DAGL-β compared with those of DAGL-α, the net increase in 2-AG levels was not altered due to the ethanol-induced increase in the 2-AG-degrading enzyme MAGL. MAGL is a serine hydrolase that preferentially hydrolyzes 2-AG into glycerol and fatty acids, and it is the most highly expressed in the brain (Dinh et al. 2002). In our present study, we observed that the MAGL protein level was significantly lower during early brain development compared to that in the adult mouse brain. MAGL null mice or those with the pharmacological inhibition of MAGL exhibit elevated levels of 2-AG in the brain, and MAGL has been shown to be the primary enzyme responsible for 2-AG degradation in the brain (Chanda et al. 2010, Long et al. 2009, Pan et al. 2011, Saario et al. 2005, Schlosburg et al. 2010). However, ABHD6 and ABHD12, which show lower brain 2-AG serine hydrolase activity (approximately 5 and 9%, respectively) (Blankman et al. 2007), are not altered in the hippocampus or neocortex tissues of P7 mice exposed to ethanol. The ethanol treatment of P7 mice significantly enhanced MAGL protein levels in both the hippocampus and neocortex, which might have caused the immediate degradation of the excess 2-AG. These findings may also represent one of the reasons for the unaltered levels of 2-AG in the ethanol-treated P7 mouse brain. Therefore, ethanol reorganizes 2-AG metabolism through the transcriptional activation of synthesizing and degrading enzymes in the developing brain, facilitating the activation of AEA-CB1R over 2-AG/CB1R events, leading to neurodegeneration in the P7 mouse brain (Subbanna et al. 2015, Subbanna et al. 2013a). As DAGL and MAGL enzymes are distinctly localized at the neuroanatomical levels (Katona et al. 2006, Yoshida et al. 2006), local alterations of 2-AG and their influence on CB1R pathway cannot be ruled out from the current findings.
Several lines of evidence suggest that the origin of neurodevelopmental psychiatric disorders caused by stress or other environmental insults involve alterations in endocannabinoid metabolizing enzymes in the brain. In contrast with our observations of the ethanol-exposed P7 mice, the exposure of neonatal P9 rats to a single prolonged 24-h episode of maternal deprivation significantly enhances DAGL-α without affecting DAGL-β immunoreactivity in the P13 hippocampus. In the same model, MAGL mRNA levels are reduced (Suarez et al. 2010). Another study has revealed that the exposure of P10 mice to a low dose of chlorpyrifos for 7 days significantly enhances the accumulation of AEA in the forebrain through the inhibition of FAAH. This treatment has no effect on 2-AG or MAGL (Carr et al. 2014). An additional study has found the accumulation of AEA and its associated lipid families, such as N-acylethanolamines (NAEs), in P7 rats exposed to an NMDA or NMDA antagonist or head trauma (Hansen et al. 2001). However, 2-AG and its associated lipid families, such as 2-monoacylglycerols (2-MAGs), are virtually unaffected. These three models have been well characterized to induce widespread caspase-3 activation in neonatal rodents (Hansen et al. 2001, Ikonomidou et al. 2000, Ikonomidou et al. 1999, Subbanna et al. 2013a). Although no biosynthetic enzymes have been evaluated in these studies, their findings suggest that even neurotoxic insults occurring during brain development significantly activate AEA/CB1R over 2-AG/CB1R events by regulating specific metabolic pathways that are required for normal brain development (Alpar et al. 2014, Berghuis et al. 2007, Keimpema et al. 2013a, Keimpema et al. 2013b, Keimpema et al. 2011, Keimpema et al. 2013c, Tortoriello et al. 2014). Further, DAGL-α and MAGL expression are reduced in the hippocampus of adolescent mice treated with valproic acid on gestational day 12.5. 2-AG levels are not altered by this regimen, but the animals exhibit autistic behavior (Kerr et al. 2013). Another study has shown that the expression levels of both DAGL-α and MAGL are be enhanced in FMR null mice (fragile X mental retardation syndrome 1 homolog) (Jung et al. 2012, Maccarrone et al. 2010), a model of fragile X syndrome, which is the most common genetic form of autism. In our previous studies, the pre-administration of a CB1R antagonist before P7 ethanol treatment or the use of CB1R null mice has been shown to prevent P7 ethanol-induced long-lasting neurobehavioral abnormalities, including spatial and social interaction memories, in adult mice (Subbanna et al. 2015, Subbanna et al. 2013a). Together, these observations suggest that alterations in the metabolism of specific endocannabinoids and their signaling pathways during the critical period of brain development cause long-lasting behavioral abnormalities in adulthood. In our study, treatment of P7 mice with MAGL inhibitor (KML29) failed to induce activation of caspase-3. JZL184 and its analog KML29 show good selectivity (>100-fold) for MAGL over FAAH (Fowler 2012, King et al. 2009, Mulvihill & Nomura 2013) and most other serine hydrolases (Chang et al. 2012, Long et al. 2009). The doses used were shown to inhibit MAGL and enhance brain levels of 2-AG in animals (Ignatowska-Jankowska et al. 2014, Kinsey et al. 2013, Schlosburg et al. 2010). Therefore, these observations suggest that 2-AG may not be involved in P7 ethanol-induced neurodegeneration but our study does not rule out the possible additional impact of 2-AG (both DAGL and MAGL enzymes) on the CB1R-mediated long-lasting effect on the brain function.
In summary, the ethanol treatment of P7 mice causes the specific up-regulation of AEA-CB1R signaling over the 2-AG-CB1R pathway by the specific reorganization of the enzymes that synthesize (DAGL-α/β) and degrade (MAGL) 2-AG. Further investigations of the roles of 2-AG during early brain development will be useful for understanding and providing a novel pharmacological target for the treatment of behavioral traits associated with the neurodevelopmental origin of several brain diseases, including FASD.
Acknowledgments
This work was supported by NIH/NIAAA grant AA019443 (BSB). DP was supported by NIDA grant F32DA021977. We thank Neha Balapal for editing the final version of the manuscript.
Abbreviations
- AEA
anandamide
- 2-AG
2-arachidonylglycerol
- CB
cannabinoid receptor
- FASD
fetal alcohol spectrum disorder
- DAGL
diacylglycerol lipase
- MAGL
monoacylglycerol lipase
Footnotes
Conflicts of interest disclosure: The authors declare no competing financial interests. All experiments were conducted in compliance with the ARRIVE guidelines.
References
- Alpar A, Tortoriello G, Calvigioni D, et al. Endocannabinoids modulate cortical development by configuring Slit2/Robo1 signalling. Nature communications. 2014;5:4421. doi: 10.1038/ncomms5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavarajappa BS. The endocannabinoid signaling system: a potential target for next-generation therapeutics for alcoholism. Mini-Rev Med Chem. 2007;7:769–779. doi: 10.2174/138955707781387920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavarajappa BS. Major Enzymes of Endocannabinoid Metabolism. In: Dunn Ben M., editor. Frontiers in Protein and Peptide Sciences. Vol. 1. Bentham Science Publishers; Oak Park, IL, USA: 2014. pp. 31–62. [Google Scholar]
- Basavarajappa BS, Arancio O. Synaptic Plasticity: Emerging Role for Endocannabinoid system. In: Kaiser TF, Peters FJ, editors. Synaptic Plasticity: New Research. Nova Science Publishers Inc.; NY, USA: 2008. pp. 77–112. [Google Scholar]
- Basavarajappa BS, Hungund BL. Cannabinoid receptor agonist-stimulated [35S]Guanosine TriphosphategS binding in the brain of C57BL/6 and DBA/2 mice. J Neurosci Res. 2001;64:429–436. doi: 10.1002/jnr.1094. [DOI] [PubMed] [Google Scholar]
- Basavarajappa BS, Ninan I, Arancio O. Acute Ethanol Suppresses Glutamatergic Neurotransmission through Endocannabinoids in Hippocampal Neurons. J Neurochem. 2008;107:1001–1013. doi: 10.1111/j.1471-4159.2008.05685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavarajappa BS, Yalamanchili R, Cravatt BF, Cooper TB, Hungund BL. Increased ethanol consumption and preference and decreased ethanol sensitivity in female FAAH knockout mice. Neuropharmacology. 2006;50:834–844. doi: 10.1016/j.neuropharm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Bayer SA, Altman J, Russo RJ, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology. 1993;14:83–144. [PubMed] [Google Scholar]
- Berghuis P, Rajnicek AM, Morozov YM, et al. Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science. 2007;316:1212–1216. doi: 10.1126/science.1137406. [DOI] [PubMed] [Google Scholar]
- Berrendero F, Sepe N, Ramos JA, Di Marzo V, Fernandez-Ruiz JJ. Analysis of cannabinoid receptor binding and mRNA expression and endogenous cannabinoid contents in the developing rat brain during late gestation and early postnatal period. Synapse. 1999;33:181–191. doi: 10.1002/(SICI)1098-2396(19990901)33:3<181::AID-SYN3>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, et al. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr RL, Graves CA, Mangum LC, Nail CA, Ross MK. Low level chlorpyrifos exposure increases anandamide accumulation in juvenile rat brain in the absence of brain cholinesterase inhibition. Neurotoxicology. 2014;43:82–89. doi: 10.1016/j.neuro.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarini J, Hompes T, Verhaeghen A, Casteels C, Peuskens H, Bormans G, Claes S, Van Laere K. Changes in cerebral CB1 receptor availability after acute and chronic alcohol abuse and monitored abstinence. J Neurosci. 2014;34:2822–2831. doi: 10.1523/JNEUROSCI.0849-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda PK, Gao Y, Mark L, et al. Monoacylglycerol lipase activity is a critical modulator of the tone and integrity of the endocannabinoid system. Mol Pharmacol. 2010;78:996–1003. doi: 10.1124/mol.110.068304. [DOI] [PubMed] [Google Scholar]
- Chang JW, Niphakis MJ, Lum KM, et al. Highly selective inhibitors of monoacylglycerol lipase bearing a reactive group that is bioisosteric with endocannabinoid substrates. Chem Biol. 2012;19:579–588. doi: 10.1016/j.chembiol.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronise K, Marino MD, Tran TD, Kelly SJ. Critical periods for the effects of alcohol exposure on learning in rats. Behav Neurosci. 2001;115:138–145. doi: 10.1037/0735-7044.115.1.138. [DOI] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci U S A. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman HS, Jones KL, Lindsay S, Slymen D, Klonoff-Cohen H, Kao K, Rao S, Chambers C. Prenatal alcohol exposure patterns and alcohol-related birth defects and growth deficiencies: a prospective study. Alcohol Clin Exp Res. 2012;36:670–676. doi: 10.1111/j.1530-0277.2011.01664.x. [DOI] [PubMed] [Google Scholar]
- Fernandez-Ruiz J, Berrendero F, Hernandez ML, Ramos JA. The endogenous cannabinoid system and brain development. Trends Neurosci. 2000;23:14–20. doi: 10.1016/s0166-2236(99)01491-5. [DOI] [PubMed] [Google Scholar]
- Fowler CJ. Monoacylglycerol lipase - a target for drug development. Br J Pharmacol. 2012;166:1568–1585. doi: 10.1111/j.1476-5381.2012.01950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Vasilyev DV, Goncalves MB, et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci. 2010;30:2017–2024. doi: 10.1523/JNEUROSCI.5693-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Mohapel J, Boehme F, Kainer L, Christie BR. Hippocampal cell loss and neurogenesis after fetal alcohol exposure: insights from different rodent models. Brain Res Rev. 2010;64:283–303. doi: 10.1016/j.brainresrev.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Goodman AM, Delis DC, Mattson SN. Normative data for 4-year-old children on the California Verbal Learning Test-Children's Version. The Clinical neuropsychologist. 1999;13:274–282. doi: 10.1076/clin.13.3.274.1748. [DOI] [PubMed] [Google Scholar]
- Hansen HH, Krutz B, Sifringer M, Stefovska V, Bittigau P, Pragst F, Marsicano G, Lutz B, Ikonomidou C. Cannabinoids enhance susceptibility of immature brain to ethanol neurotoxicity. Ann Neurol. 2008;64:42–52. doi: 10.1002/ana.21287. [DOI] [PubMed] [Google Scholar]
- Hansen HH, Schmid PC, Bittigau P, et al. Anandamide, but not 2-arachidonoylglycerol, accumulates during in vivo neurodegeneration. J Neurochem. 2001;78:1415–1427. doi: 10.1046/j.1471-4159.2001.00542.x. [DOI] [PubMed] [Google Scholar]
- Harkany T, Keimpema E, Barabas K, Mulder J. Endocannabinoid functions controlling neuronal specification during brain development. Mol Cell Endocrinol. 2008;286:S84–90. doi: 10.1016/j.mce.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Harris SR, MacKay LL, Osborn JA. Autistic behaviors in offspring of mothers abusing alcohol and other drugs: a series of case reports. Alcohol Clin Exp Res. 1995;19:660–665. doi: 10.1111/j.1530-0277.1995.tb01564.x. [DOI] [PubMed] [Google Scholar]
- Hirvonen J, Zanotti-Fregonara P, Umhau JC, et al. Reduced cannabinoid CB1 receptor binding in alcohol dependence measured with positron emission tomography. Mol Psychiatry. 2013;18:916–921. doi: 10.1038/mp.2012.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hungund BL, Szakall I, Adam A, Basavarajappa BS, Vadasz C. Cannabinoid CB1 Receptor Knockout Mice Exhibit Markedly Reduced Voluntary Alcohol Consumption and Lack Alcohol-Induced Dopamine Release in the Nucleus Accumbens. J Neurochem. 2003;84:698–704. doi: 10.1046/j.1471-4159.2003.01576.x. [DOI] [PubMed] [Google Scholar]
- Ignatowska-Jankowska BM, Ghosh S, Crowe MS, et al. In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: antinociceptive activity without cannabimimetic side effects. Br J Pharmacol. 2014;171:1392–1407. doi: 10.1111/bph.12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Jacob C, Christen CN, Pereira JA, et al. HDAC1 and HDAC2 control the transcriptional program of myelination and the survival of Schwann cells. Nat Neurosci. 2011;14:429–436. doi: 10.1038/nn.2762. [DOI] [PubMed] [Google Scholar]
- Jung KM, Sepers M, Henstridge CM, et al. Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nature communications. 2012;3:1080. doi: 10.1038/ncomms2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Urban GM, Wallace M, Ledent C, Jung KM, Piomelli D, Mackie K, Freund TF. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci. 2006;26:5628–5637. doi: 10.1523/JNEUROSCI.0309-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keimpema E, Alpar A, Howell F, et al. Diacylglycerol lipase alpha manipulation reveals developmental roles for intercellular endocannabinoid signaling. Scientific reports. 2013a;3:2093. doi: 10.1038/srep02093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keimpema E, Calvigioni D, Harkany T. Endocannabinoid signals in the developmental programming of delayed-onset neuropsychiatric and metabolic illnesses. Biochem Soc Trans. 2013b;41:1569–1576. doi: 10.1042/BST20130117. [DOI] [PubMed] [Google Scholar]
- Keimpema E, Mackie K, Harkany T. Molecular model of cannabis sensitivity in developing neuronal circuits. Trends Pharmacol Sci. 2011;32:551–561. doi: 10.1016/j.tips.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keimpema E, Tortoriello G, Alpar A, et al. Nerve growth factor scales endocannabinoid signaling by regulating monoacylglycerol lipase turnover in developing cholinergic neurons. Proc Natl Acad Sci U S A. 2013c;110:1935–1940. doi: 10.1073/pnas.1212563110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MP, Stein JM, Vecsey CG, et al. Developmental etiology for neuroanatomical and cognitive deficits in mice overexpressing Galphas, a G-protein subunit genetically linked to schizophrenia. Mol Psychiatry. 2009;14:398–415. 347. doi: 10.1038/mp.2008.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr DM, Downey L, Conboy M, Finn DP, Roche M. Alterations in the endocannabinoid system in the rat valproic acid model of autism. Behav Brain Res. 2013;249:124–132. doi: 10.1016/j.bbr.2013.04.043. [DOI] [PubMed] [Google Scholar]
- King AR, Dotsey EY, Lodola A, Jung KM, Ghomian A, Qiu Y, Fu J, Mor M, Piomelli D. Discovery of potent and reversible monoacylglycerol lipase inhibitors. Chem Biol. 2009;16:1045–1052. doi: 10.1016/j.chembiol.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Wise LE, Ramesh D, Abdullah R, Selley DE, Cravatt BF, Lichtman AH. Repeated low-dose administration of the monoacylglycerol lipase inhibitor JZL184 retains cannabinoid receptor type 1-mediated antinociceptive and gastroprotective effects. J Pharmacol Exp Ther. 2013;345:492–501. doi: 10.1124/jpet.112.201426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Lewis SJ, Zuccolo L, Davey Smith G, et al. Fetal alcohol exposure and IQ at age 8: evidence from a population-based birth-cohort study. PloS one. 2012;7:e49407. doi: 10.1371/journal.pone.0049407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M, Rossi S, Bari M, De Chiara V, Rapino C, Musella A, Bernardi G, Bagni C, Centonze D. Abnormal mGlu 5 receptor/endocannabinoid coupling in mice lacking FMRP and BC1 RNA. Neuropsychopharmacology. 2010;35:1500–1509. doi: 10.1038/npp.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos M, Pastor I, de la Calle C, Barrio-Real L, Laso FJ, Gonzalez-Sarmiento R. Cannabinoid receptor 1 gene is associated with alcohol dependence. Alcohol Clin Exp Res. 2012;36:267–271. doi: 10.1111/j.1530-0277.2011.01623.x. [DOI] [PubMed] [Google Scholar]
- Mato S, Del Olmo E, Pazos A. Ontogenetic development of cannabinoid receptor expression and signal transduction functionality in the human brain. Eur J Neurosci. 2003;17:1747–1754. doi: 10.1046/j.1460-9568.2003.02599.x. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Crocker N, Nguyen TT. Fetal alcohol spectrum disorders: neuropsychological and behavioral features. Neuropsychol Rev. 2011;21:81–101. doi: 10.1007/s11065-011-9167-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson SN, Goodman AM, Caine C, Delis DC, Riley EP. Executive functioning in children with heavy prenatal alcohol exposure. Alcohol Clin Exp Res. 1999;23:1808–1815. [PubMed] [Google Scholar]
- Mattson SN, Riley EP. A review of the neurobehavioral deficits in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcohol Clin Exp Res. 1998;22:279–294. doi: 10.1111/j.1530-0277.1998.tb03651.x. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Riley EP, Gramling L, Delis DC, Jones KL. Neuropsychological comparison of alcohol-exposed children with or without physical features of fetal alcohol syndrome. Neuropsychology. 1998;12:146–153. doi: 10.1037//0894-4105.12.1.146. [DOI] [PubMed] [Google Scholar]
- May PA, Gossage JP, Kalberg WO, Robinson LK, Buckley D, Manning M, Hoyme HE. Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies. Developmental disabilities research reviews. 2009;15:176–192. doi: 10.1002/ddrr.68. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Parker LA. The endocannabinoid system and the brain. Annu Rev Psychol. 2013;64:21–47. doi: 10.1146/annurev-psych-113011-143739. [DOI] [PubMed] [Google Scholar]
- Morleo M, Woolfall K, Dedman D, Mukherjee R, Bellis MA, Cook PA. Under-reporting of foetal alcohol spectrum disorders: an analysis of hospital episode statistics. BMC pediatrics. 2011;11:14. doi: 10.1186/1471-2431-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvihill MM, Nomura DK. Therapeutic potential of monoacylglycerol lipase inhibitors. Life Sci. 2013;92:492–497. doi: 10.1016/j.lfs.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murataeva N, Straiker A, Mackie K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br J Pharmacol. 2014;171:1379–1391. doi: 10.1111/bph.12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel M, Norris EH, Strickland S. Tissue plasminogen activator is required for the development of fetal alcohol syndrome in mice. Proc Natl Acad Sci U S A. 2011;108:5069–5074. doi: 10.1073/pnas.1017608108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminal. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Olney JW, Tenkova T, Dikranian K, Qin YQ, Labruyere J, Ikonomidou C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res Dev Brain Res. 2002;133:115–126. doi: 10.1016/s0165-3806(02)00279-1. [DOI] [PubMed] [Google Scholar]
- Oudin MJ, Hobbs C, Doherty P. DAGL-dependent endocannabinoid signalling: roles in axonal pathfinding, synaptic plasticity and adult neurogenesis. Eur J Neurosci. 2011;34:1634–1646. doi: 10.1111/j.1460-9568.2011.07831.x. [DOI] [PubMed] [Google Scholar]
- Paintner A, Williams AD, Burd L. Fetal alcohol spectrum disorders-- implications for child neurology, part 1: prenatal exposure and dosimetry. J Child Neurol. 2012a;27:258–263. doi: 10.1177/0883073811428376. [DOI] [PubMed] [Google Scholar]
- Paintner A, Williams AD, Burd L. Fetal alcohol spectrum disorders--implications for child neurology, part 2: diagnosis and management. J Child Neurol. 2012b;27:355–362. doi: 10.1177/0883073811428377. [DOI] [PubMed] [Google Scholar]
- Pan B, Wang W, Zhong P, Blankman JL, Cravatt BF, Liu QS. Alterations of endocannabinoid signaling, synaptic plasticity, learning, and memory in monoacylglycerol lipase knock-out mice. J Neurosci. 2011;31:13420–13430. doi: 10.1523/JNEUROSCI.2075-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen C, Horne K, Witol A. Neurobehavioral functioning in children with fetal alcohol spectrum disorder. Child neuropsychology : a journal on normal and abnormal development in childhood and adolescence. 2006;12:453–468. doi: 10.1080/09297040600646854. [DOI] [PubMed] [Google Scholar]
- Rodriguez de Fonseca F, Ramos JA, Bonnin A, Fernandez-Ruiz JJ. Presence of cannabinoid binding sites in the brain from early postnatal ages. Neuroreport. 1993;4:135–138. doi: 10.1097/00001756-199302000-00005. [DOI] [PubMed] [Google Scholar]
- Romero J, Garcia-Palomero E, Berrendero F, Garcia-Gil L, Hernandez ML, Ramos JA, Fernandez-Ruiz JJ. Atypical location of cannabinoid receptors in white matter areas during rat brain development. Synapse. 1997;26:317–323. doi: 10.1002/(SICI)1098-2396(199707)26:3<317::AID-SYN12>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Rubio M, de Miguel R, Fernandez-Ruiz J, Gutierrez-Lopez D, Carai MA, Ramos JA. Effects of a short-term exposure to alcohol in rats on FAAH enzyme and CB1 receptor in different brain areas. Drug Alcohol Depend. 2009;99:354–358. doi: 10.1016/j.drugalcdep.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Saario SM, Salo OM, Nevalainen T, Poso A, Laitinen JT, Jarvinen T, Niemi R. Characterization of the sulfhydryl-sensitive site in the enzyme responsible for hydrolysis of 2-arachidonoyl-glycerol in rat cerebellar membranes. Chem Biol. 2005;12:649–656. doi: 10.1016/j.chembiol.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Sadrian B, Subbanna S, Wilson DA, Basavarajappa BS, Saito M. Lithium prevents long-term neural and behavioral pathology induced by early alcohol exposure. Neuroscience. 2012;206:122–135. doi: 10.1016/j.neuroscience.2011.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez J, Rivera P, Llorente R, Romero-Zerbo SY, Bermudez-Silva FJ, de Fonseca FR, Viveros MP. Early maternal deprivation induces changes on the expression of 2-AG biosynthesis and degradation enzymes in neonatal rat hippocampus. Brain Res. 2010;1349:162–173. doi: 10.1016/j.brainres.2010.06.042. [DOI] [PubMed] [Google Scholar]
- Subbanna S, Basavarajappa BS. Pre-administration of G9a/GLP inhibitor during Synaptogenesis Prevents Postnatal Ethanol-induced LTP Deficits and Neurobehavioral Abnormalities in Adult Mice. Exp Neurol. 2014;261:34–43. doi: 10.1016/j.expneurol.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbanna S, Nagaraja NN, Umapathy NS, Pace BS, Basavarajappa BS. Ethanol Exposure Induces Neonatal Neurodegeneration by Enhancing CB1R Exon1 Histone H4K8 Acetylation and Up-regulating CB1R Function causing Neurobehavioral Abnormalities in Adult Mice. International Journal of Neuropsychopharmacology. 2015 doi: 10.1093/ijnp/pyu028. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbanna S, Nagre NN, Shivakumar M, Umapathy NS, Psychoyos D, Basavarajappa BS. Ethanol Induced Acetylation of Histone at G9a Exon1 and G9a-Mediated Histone H3 Dimethylation leads to Neurodegeneration in Neonatal Mice. Neuroscience. 2014;258C:422–432. doi: 10.1016/j.neuroscience.2013.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbanna S, Shivakumar M, Psychoyos D, Xie S, Basavarajappa BS. Anandamide-CB1 Receptor Signaling Contributes to Postnatal Ethanol-Induced Neonatal Neurodegeneration, Adult Synaptic and Memory Deficits. Journal of neuoscience. 2013a;33:6350–6366. doi: 10.1523/JNEUROSCI.3786-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbanna S, Shivakumar M, Umapathy NS, et al. G9a-Mediated Histone Methylation Regulates Ethanol-Induced Neurodegeneration in the Neonatal Mouse Brain. Neurobiol Dis. 2013b;54:475–485. doi: 10.1016/j.nbd.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T, Kishimoto S, Oka S, Gokoh M. Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog Lipid Res. 2006;45:405–446. doi: 10.1016/j.plipres.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Tortoriello G, Morris CV, Alpar A, et al. Miswiring the brain: Delta9-tetrahydrocannabinol disrupts cortical development by inducing an SCG10/stathmin-2 degradation pathway. EMBO J. 2014;33:668–685. doi: 10.1002/embj.201386035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TD, Cronise K, Marino MD, Jenkins WJ, Kelly SJ. Critical periods for the effects of alcohol exposure on brain weight, body weight, activity and investigation. Behav Brain Res. 2000;116:99–110. doi: 10.1016/s0166-4328(00)00263-1. [DOI] [PubMed] [Google Scholar]
- Wilson DA, Peterson J, Basavaraj BS, Saito M. Local and regional network function in behaviorally relevant cortical circuits of adult mice following postnatal alcohol exposure. Alcoholism Clin and Exp Res. 2011;35:1974–1984. doi: 10.1111/j.1530-0277.2011.01549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Fukaya M, Uchigashima M, Miura E, Kamiya H, Kano M, Watanabe M. Localization of diacylglycerol lipase-alpha around postsynaptic spine suggests close proximity between production site of an endocannabinoid, 2-arachidonoyl-glycerol, and presynaptic cannabinoid CB1 receptor. J Neurosci. 2006;26:4740–4751. doi: 10.1523/JNEUROSCI.0054-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]