

Abstract
Although disruption of DNA repair capacity is unquestionably associated with cancer susceptibility in humans and model organisms, it remains unclear if the inherent tumor phenotypes of DNA repair deficiency syndromes can be regulated by manipulating DNA repair pathways. Loss-of-function mutations in BLM, a member of the RecQ helicase family, cause Bloom's syndrome (BS), a rare, recessive genetic disorder that predisposes to many types of cancer. BLM functions in many aspects of DNA homeostasis, including the suppression of homologous recombination (HR) in somatic cells. We investigated whether BLM overexpression, in contrast to loss-of-function mutations, attenuated the intestinal tumor phenotypes of ApcMin/+ and ApcMin/+;Msh2-/- mice, animal models of familial adenomatous polyposis coli (FAP). We constructed a transgenic mouse line expressing human BLM (BLM-Tg) and crossed it onto both backgrounds. BLM-Tg decreased adenoma incidence in a dose-dependent manner in our ApcMin/+ model of FAP, although levels of GIN were unaffected, and concomitantly increased animal survival over 50%. It did not reduce intestinal tumorigenesis in ApcMin/+;Msh2-/- mice. We used the pink-eyed unstable (pun) mouse model to demonstrate that increasing BLM dosage in vivo lowered endogenous levels of HR by two-fold. Our data suggests that attenuation of the Min phenotype is achieved through a direct effect of BLM-Tg on the HR repair pathway. These findings demonstrate that HR can be manipulated in vivo to modulate tumor formation at the organismal level. Our data suggests that lowering HR frequencies may have positive therapeutic outcomes in the context of specific hereditary cancer predisposition syndromes, exemplified by FAP.
Keywords: homologous recombination, intestinal tumorigenesis, mouse models, RecQ helicase
Introduction
The RecQ-like helicase family members WRN, BLM, and RECQL4 are linked to human genetic diseases characterized by genome instability, premature aging and cancer predisposition (1, 2). BLM is a structure-specific helicase with 3′-5′ directionality which is involved in DNA double-strand break (DSB) repair (3). BLM functions in many aspects of DNA homeostasis, including the restart/repair of stalled and collapsed replication forks during DNA replication, repair of inter-strand cross-links, and resolution of Holliday junctions (4-6). While it is accepted that BLM promotes resolution of Holliday intermediates by dissolution, thus suppressing crossovers, the role of BLM in homologous recombination (HR) is more complex than merely this late stage role. BLM also disrupts formation of RAD51-ssDNA filaments, leading to disruption of D-loops and thus suppression of HR at earlier stages (7). BLM-deficient cells have an approximate tenfold increase in the number of sister chromatid exchanges (SCE) caused by inappropriate HR between sister chromatids at the S- or G2-phases of the cell cycle (8). Bloom's syndrome (BS) is a rare, recessive genetic disorder which is caused by loss-of-function mutations in the BLM gene (9). BS patients have a predisposition to develop many types of cancer, presenting with a mean age of 24 years at diagnosis.
Several lines of evidence indicate that Blm dosage is critical for controlling the onset of tumorigenesis in mice. Mouse models demonstrate that chromosomal instability directly correlates with the levels of Blm; as Blm decreases, genomic instability and tumor burden increase (10-12). In addition, haploinsufficiency for Blm on the C57Bl-6J ApcMin/+ background increases spontaneous adenoma formation and dysplasia (11). Genomic analyses of ApcMin/+;BlmCin/+ mice indicate that increased adenoma formation is a direct consequence of reduced Blm levels and an increase in somatic recombination. This, in turn, facilitates loss of the wild-type Apc allele by inter-chromosomal recombination and leads to increased loss-of-heterozygosity (LOH). In humans, similar conclusions have been reached about carriers of specific BLM mutations and their resulting susceptibilities to colorectal cancer (13).
Familial adenomatous polyposis coli (FAP) is a hereditary human cancer predisposition syndrome characterized by the growth of hundreds to thousands of small adenomatous polyps throughout the colon (reviewed in 14). FAP requires the inheritance of a mutated allele of the adenomatous polyposis coli (APC) gene (15). Depending on the nature of the inherited germline allele, second-hit inactivation of the wild-type allele is achieved either by LOH of the APC locus or intragenic mutation of the APC gene (16). APC is also inactivated by intragenic mutation in 70-80% of individuals with sporadic colorectal cancer (14).
Given the demonstrated relationship between low or absent expression levels of BLM and cancer, we investigated whether constitutive overexpression of BLM modulated adenoma formation in the ApcMin/+ mouse model of FAP (17). We hypothesized that if halving Blm gene dosage increased predisposition to tumorigenesis, over-expression would conversely decrease tumor susceptibility. Understanding the mechanism by which BLM attenuates tumor susceptibility will aid our fundamental understanding of its roles in maintaining genomic stability and suggest new strategies for cancer prevention involving direct regulation of DNA repair pathways. Our data suggests that levels of specific DNA repair proteins may be titrated to achieve positive therapeutic outcomes in the context of specific hereditary cancer syndromes, exemplified by FAP.
Materials and Methods
Generation of the transgenic mouse line expressing BLM
The procedure is outlined below. The human BLM cDNA was amplified from plasmid pJK1 and cloned into the TA-vector (Promega). The construct was sequenced and verified. A 0.44 kb fragment, corresponding to the phosphoglycerate kinase (PGK) promoter was cloned in to the 5′ end of the BLM cDNA. The PGK-BLM cDNA fragment was then cloned into the vector pOPRSVICat, containing a synthetic intron and the HSV thymidine kinase (TK) polyadenylation signal. The PGK-BLMcDNA-p(A) fragment was removed by restriction digestion from the vector, purified and introduced into C57Bl-6J oocytes by pronuclear injection. Founder lines were generated and initially screened for the presence of the transgene by Southern blotting. A probe corresponding to the 3′ end of the BLM cDNA was used. Once germline transmission had been established, transgenic animals were routinely identified using PCR.
Generation of mice lines and genotyping
ApcMin/+ mice were originally obtained from The Jackson Laboratories (stock: 002020; strain: C57BL/6J-ApcMin/J). BlmCin/+ mice have been previously reported (11). The pink-eyed unstable mouse model (18) was a gift from Dr. A.J.R. Bishop, University of Texas Health Science Center at San Antonio. Heterozygous Msh2+/- mice (19) were obtained from the laboratory of Dr. Winfried Edelmann, Albert Einstein College of Medicine, New York. ApcMin/+, BlmCin/+, Msh2+/- and BLM-Tg lines were intercrossed to generate mice of the required genotypes, all on congenic C57BL/6J backgrounds. Animals were bred in a barrier facility and were maintained according to the NIH animal care and use guidelines. Both male and female mice were included in experimental study groups for subsequent analyses. All experiments involving animals received prior approval from the OSU Institutional Animal Care and Use Committee. ApcMin/+, BlmCin/+ and Msh2+/- mice were genotyped as described previously (11, 19).
Genotyping BLM-Tg mice
Mice were genotyped as follows: primers hBE3F (5′- TAT GCA CTA CCC AAA ACA CAC C -3′; forward) and hBE3R (5′- TCA GTC AAA TCT ATT TGC TCG C -3′; reverse), were used in a PCR reaction to amplify a 310 bp product from exon 3 of human BLM. Primers HMGAPF (5′- GAC ATC AAG AAG GTG GTG AAG -3′; forward) and HMGAPR (5′- CCA GGA AAT GAG CTT GAC AAA G -3′; reverse) were used to amplify a 171 bp product from mouse Gapdh as an internal positive control. PCR reactions were performed with standard Taq polymerase.
qPCR to determine allelic status of BLM-Tg
Primers BLM09F (5′- TGG TGC GGA AGT GAT TTC AGT A -3′; forward) and BLM12R (5′- TTT ATA GGC TTC GGT GGA GC -3′) were used to amplify a 396 bp amplicon from the 3′ end of the BLM cDNA. The SYBR Green PCR Master Mix (Invitrogen) was used for all qPCR reactions. Purified DNAs from BLM-Tg mice were used as templates. Reactions were performed with a series of dilutions of each template. Cycle threshold (Ct) was plotted against log10[DNA] and used to identify hemizygous (BLM+/T) and homozygous (BLMT/T) transgenic mice. All qPCR reactions were run in triplicate and repeated at least twice.
Animal dissection
Mice were euthanized at 16 weeks by CO2 inhalation, followed by cervical dislocation. Intestines were removed, rinsed in PBS and cut into sections corresponding to the duodenum, jejunum, ileum, cecum and colon (large intestine). Tissues were opened longitudinally, washed twice in PBS and examined under a dissecting microscope. Gross numbers of adenomas/intestinal polyps were counted. Tissues were fixed in 10% formalin overnight, blocked in paraffin, sectioned and stained with hematoxylin and eosin. Slides were evaluated to confirm tumors and determine gastrointestinal neoplasia (GIN). The criteria for GIN are as described previously (20).
Ethics statement
All experiments involving mice received prior approval from The Ohio State University Institutional Animal Care and Use Committee (IACUC), OLAW Assurance #A3261-01. Animal work was conducted in accordance with the established criteria of our animal use protocol, #2012A00000021, approved by IACUC. Mice were observed on a daily basis for predetermined criteria necessitating removal and euthanasia. Decisions to remove animals were made in conjunction with the veterinarian staff of our animal facility. Mice were euthanized by CO2 inhalation, followed by cervical dislocation.
Dissection of the RPE/scoring reversion events
These have been described previously (18).
Statistical methods
All statistical analyses were performed with Prism 6.1.
Results
BLM expression rescues the embryonic lethality of th BlmCin knockout mouse
A transgenic mouse was generated that expressed human BLM on a congenic C57Bl-6J background, hereafter designated BLM-Tg. qPCR was used to establish the allelic status of the BLM transgene in sibling mice bred from the established colony (Supplementary Fig. S1). Protein expression levels correlated with the allelic status of the transgene; homozygous (BLMT/T) mice expressed approximately twice as much BLM as hemizygous (BLM+/T) mice. The BLM-Tg also rescued the embryonic lethality of the conventional BlmCin/Cin knockout mouse (11). Mating of BLM+/T;BlmCin/+ animals yielded BLM+/T;BlmCin/Cin mice at normal Mendelian ratios. Long-term expression of BLM for over 24 months had no apparent deleterious effects on animal health. BLM-Tg mice (n=3) were subjected to full necropsy and phenotypic analyses. No overt phenotypic abnormalities were detected; only age- or strain-related lesions were observed (data not shown). Tumors were not apparent in any tissue at necropsy and there was no evidence of architectural destruction or invasion which would suggest neoplastic transformation.
Transgenic expression of BLM reduces adenoma numbers in ApcMin/+ mice
The BLM-Tg line was crossed to ApcMin/+ mice, also on a C57Bl-6J background. Animals were maintained in a barrier environment and four different groups of litter mates were generated: (i) ApcMin/+, (ii) ApcMin/+;BLM+/T, (iii) ApcMin/+;BLMT/T, and (iv) BLM+/T and/or wild-type mice. Mice were euthanized after 16 weeks and gross intestinal adenomas were counted with a dissecting microscope to differentiate adenomas in the duodenum, jejunum, ileum, cecum and colon. Pathology studies confirmed and categorized adenomas as low- or high-grade and scored GIN according to the criteria of Boivin et al. as an early marker for neoplasia (20).
BLM-Tg significantly reduced gross numbers of intestinal adenomas in ApcMin/+ litter mates (Fig. 1A, Table 1). ApcMin/+ mice developed a mean of 46.44 ± 11.42 adenomas, compared to 24.44 ± 8.22 for ApcMin/+;BLM+/T and 17.24 ± 8.26 for ApcMin/+;BLMT/T mice (P<0.0001 for both groups; Mann-Whitney U test). Tumor attenuation of the Min phenotype was dose-dependent; ApcMin/+;BLMT/T mice developed significantly less adenomas than ApcMin/+;BLM+/T mice (P=0.016; Mann-Whitney U test). Suppression of adenoma formation by BLM-Tg was most evident in the jejunal and ileac segments of the gastrointestinal tract (Fig. 1B) which is not surprising, as these regions comprise the predominant site of adenoma formation in the ApcMin/+ mode (17). There was no difference in mean adenoma numbers between male and female mice. Adenomas were not observed in control groups of BLM-Tg or wild-type mice.
Figure 1.

Transgenic BLM suppresses adenoma formation in the ApcMin/+ mouse model of intestinal tumorigenesis. A, Total adenoma numbers and total levels of GIN for ApcMin/+, ApcMin/+;BLM+/T, ApcMin/+;BLMT/T and BLM-Tg/wild-type mice. Mean adenoma number ± standard deviation and mean GIN ± standard deviation was calculated for each genotype. Significant values are indicated above each bracket: *P<0.02; ****P<0.0001. B, The intestines of ApcMin/+, ApcMin/+;BLM+/T and ApcMin/+;BLMT/T mice were dissected and divided into each intestinal compartment shown. Mean adenoma number ± standard deviation was calculated for each tissue. Statistical analyses were performed using the Mann-Whitney U test. Significant values are indicated above each bracket: * P≤0.02; ** P<0.002; **** P<0.0001.
Table 1.
Mean number of intestinal adenomas per region in ApcMin/+, ApcMin/+;BLM+/T and ApcMin/+;BLMT/T mice
| genotype | n | intestinal region1 |
all tumors | ||||
|---|---|---|---|---|---|---|---|
| du. | je. | il. | ce. | co. | |||
| ApcMin/+ | 27 | 7.84 | 13.56 | 23.11 | 0.44 | 1.37 | 46.44 |
| ApcMin/+; BLM+/T | 18 | 4.00 | 7.44 | 11.83 | 0.28 | 0.94 | 24.44 |
| ApcMin/+; BLMT/T | 17 | 3.30 | 4.12 | 8.47 | 0.29 | 1.06 | 17.24 |
| BLM-Tg & wild-type | 22 | 0 | 0 | 0 | 0 | 0 | 0 |
du.=duodenum; je.=jejunum; il.=ileum; ce.=cecum; co.=colon
Despite the reduction in total adenomas by BLM-Tg, there was no significant reduction in the levels of GIN between groups (Fig. 1A), although the trend was suggestive. Compare means of 4.69 ± 4.22 for ApcMin/+ to 2.71 ± 2.05 for ApcMin/+;BLM+/T, and 3.00 ± 3.33 for ApcMin/+;BLMT/T mice. However, these sample sizes confer only a 30% power to detect differences between means of 1.56 with a significance level (α) of 0.05 (two-tailed). We would require 80 or more animals in each group to have 80% power to detect a difference between means of 1.34 with a significance level (α) of 0.05 (two-tailed).
BLM-Tg increases survival in the ApcMin/+ mouse model of intestinal tumorigenesis
Survival of ApcMin/+ mice was increased by the BLM transgene (Fig. 2). Median survival for ApcMin/+ mice was 137 days compared to 196 days for ApcMin/+;BLM+/T mice and 221 days for ApcMin/+;BLMT/T mice (P<0.0001; log-rank test for both groups). Survival times between ApcMin/+;BLM+/T and ApcMin/+;BLMT/T mice were also significantly different (P<0.0053; log-rank test), indicating a dose-dependent effect of the transgene. Although BLM-Tg significantly extended survival of ApcMin/+ mice, most likely due to reduced intestinal tumor burden, they died earlier than BLM-Tg litter mate controls, which are all still alive after 350 days (Fig. 2). It is not surprising that BLM overexpression was unable to mitigate the persistent intestinal tumorigenesis that is characteristic of the Min phenotype.
Figure 2.

Transgenic BLM increases survival in ApcMin/+ mice. Groups of BLM+/T and/or BLMT/T, (n=12), ApcMin/+ (n=26), ApcMin/+;BLM+/T (n=28) and ApcMin/+;BLMT/T (n=19) mice were aged. Mice were observed on a daily basis for pre-determined criteria necessitating removal and euthanasia. Kaplan-Meier plots were generated and median survival times of each group were calculated. The log-rank test was used to determine the significance of differences in survival.
Transgenic BLM does not affect tumor etiology
While genetic background modifies tumor penetrance in the ApcMin/+ model (21), adenomas rarely progress to carcinoma (22). Comparative histopathological evaluation of intestinal lesions from ApcMin/+;BLM+/T and ApcMin/+;BLTT/T mice was consistent with the well-characterized etiology of tumor development in the ApcMin/+ model (Fig. 3A-F), (17, 20). Most adenomas were classified as low-grade; no adenocarcinomas were observed. Although more high-grade adenomas developed in ApcMin/+ (10/282) compared to ApcMin/+;BLM+/T (1/148) or ApcMin/+;BLMT/T (1/101) mice, the numbers of adenomas assessed were insufficient to determine significance.
Figure 3.
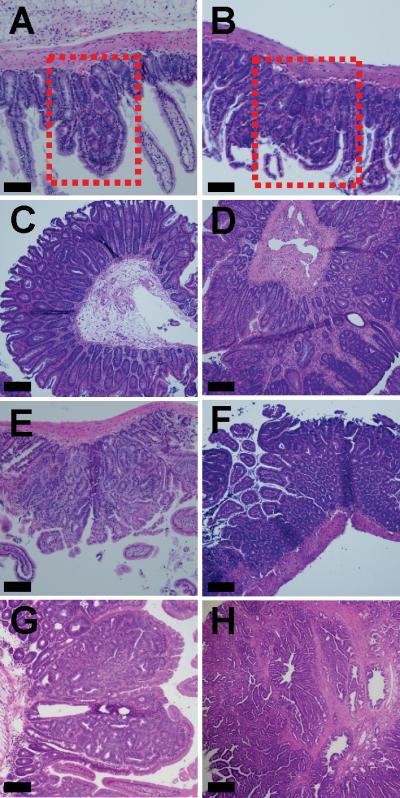
Histopathology of intestinal tumor development associated with Blm/BLM in ApcMin/+ mice. A-F, Although BLM-Tg reduces adenoma numbers on the ApcMin/+ background, it does not alter the underlying histopathology of emerging adenomas: A, ApcMin/+ jejunum, boxed area highlights GIN; B, ApcMin/+;BLMT/T jejunum, boxed area highlights GIN; C, ApcMin/+;BLMT/T cecum, adenoma; D, ApcMin/+;BLM+/T colon, adenoma; E, ApcMin/+;BLM+/T jejunum, adenoma; F, ApcMin/+;BLMT/T ileum, adenoma. G-H, Blm haplo-insufficiency drives tumor development and accelerates the development of intestinal adenomas with high-grade dysplasia and invasive carcinoma: G, ApcMin/+;BlmCin/+, adenoma with high-grade dysplasia; H, ApcMin/+;BlmCin/+, invasive adenocarcinoma. All sections were stained with hematoxylin and eosin. Scale bars each represent 100 μm.
In order to more fully understand the nuanced effects of Blm/BLM dosage on tumor initiation versus progression in the ApcMin/+ intestine, we employed the BlmCin/+ knockout mouse model (11). We examined tumors from aged ApcMin/+;BlmCin/+ mice (n=20) to investigate how Blm haploinsufficiency affected tumor progression within the same intestinal model. Adenomas with high-grade dysplasia (Fig. 3G) were observed in 15 ApcMin/+;BlmCin/+ mice and 4 mice also developed adenocarcinomas that invaded the serosa (Fig. 3H). There was a statistically significant increase in both carcinomas and high-grade dysplasia in the ApcMin/+;BlmCin/+ mice (P<0.0001). These data suggest that Blm/BLM dosage can modulate tumor burden and progression in the mouse. Although BLM-Tg most likely suppresses adenoma formation by inhibiting progression from GIN to intestinal adenoma, it may also have subtle effects on the initiation of dysplasia that this study is not sufficiently powered to reveal.
Transgenic BLM does not attenuate intestinal tumor numbers in mismatch repair-deficient ApcMin/+ mice
A well-documented aspect of the ApcMin/+ phenotype on the C57Bl-6J background is that inactivation of the wild-type Apc locus by LOH is essential for subsequent intestinal adenoma formation (17). Given the known roles of Blm/BLM in HR, we investigated if our transgenic BLM could likewise reduce numbers of intestinal adenomas in an ApcMin/+ model that was not dependent on LOH as a second hit mechanism of inactivation. It has been observed that when ApcMin/+ is combined with mismatch repair- (MMR-) null mouse models, either Mlh1-/- or Msh2-/-, the mechanism of Apc inactivation changes from that of LOH to intragenic mutation. Analyses of adenomas from Mlh1-/-;ApcMin/+ and Msh2-/-;ApcMin/+ mice demonstrated intragenic (point) mutation of the wild-type Apc allele in 81% and 85% of cases, respectively (23, 24). This shift is most likely due to the characteristic mutator phenotypes inherent to these specific models of MMR-deficiency. Analyses of respective control groups of ApcMin/+ mice from both of the above models confirmed LOH in all of the adenomas examined (23, 24).
We combined our ApcMin/+;BLM-Tg model with the Msh2 null allele, Msh2Δ7N (19), and generated all possible combinations of ApcMin/+;BLMT;Msh2+/- animals. Mice transgenic for BLM are collectively represented as BLMT; they were not stratified as BLM+/T and BLMT/T for this analysis. The increased intestinal tumor burdens that developed in cohorts of ApcMin/+;BLMT;Msh2-/- and ApcMin/+;Msh2-/- mice resulted in a severe decline in animal survival, compelling necropsy of these groups at 11-12 weeks. Control groups of ApcMin/+, ApcMin/+;BLMT and ApcMin/+;BLMT;Msh2+/- mice were analyzed at the standard 16-17 week time point. Intestinal adenoma counts for the control groups were: ApcMin/+, 49.5 ± 12.0; ApcMin/+;BLMT, 23.7 ± 8.9 and ApcMin/+;BLMT;Msh2+/-, 24.1 ± 10.3 (Fig. 4). Adenoma numbers for ApcMin/+ and ApcMin/+;BLMT groups are similar to those of Fig.1, indicating that introduction of the Msh2Δ7N allele onto the ApcMin/+;BLMT background did not alter tumor susceptibility in the intestine. It is also evident that heterozygosity for Msh2 does not perturb adenoma development in the intestines of ApcMin/+ mice which is consistent with published data (24, 25). The ApcMin/+;BLMT;Msh2-/- and ApcMin/+;Msh2-/- groups presented comparable adenoma counts of 272.0 ± 28.5 and 288.2 ± 32.3, respectively (Fig. 4). This striking increase in intestinal adenomas is a characteristic feature of MMR-deficient ApcMin/+ mice phenotypes (23-26). Our data suggests that when Apc is inactivated by intragenic mutation in this model, rather than by LOH, transgenic BLM has no significant effect on the outcome of intestinal adenoma development.
Figure 4.

Transgenic BLM does not attenuate intestinal tumor numbers in mismatch repair-deficient ApcMin/+ mice. Box and whiskers plot showing total adenoma numbers arising in ApcMin/+ and ApcMin/+;BLMT mice on Msh2-deficient backgrounds. Significant values are indicated above each bracket: **** P<0.0001; NS, not significant. Statistical analyses were performed using the Mann-Whitney U test.
Overexpression of BLM modulates DNA repair by down-regulating homologous recombination
Given the known role of BLM in maintaining genomic integrity (1, 2), we hypothesized that BLM-Tg ameliorated tumorigenesis in ApcMin/+ mice by suppressing HR. To investigate this possible mechanism, BLM-Tg mice were crossed to the pink-eyed unstable (pun) mouse model which measures in vivo HR levels. In this model, a somatic intra-chromosomal deletion within the mouse p gene restores melanin production in the otherwise transparent cells of the retinal pigment epithelium (RPE), generating a clone of brown cells, or eye-spot (18). This deletion event occurs spontaneously and is dependent on HR. Thus, the number of RPE eye-spots represents an in vivo read-out for HR. Cohorts of pun/un and pun/un;BLMT mice were euthanized after 20 days and RPE eye-spots were counted. Representative examples are shown in Fig. 5A and B. In contrast to ApcMin/+ mice, BLM-Tg dosage does not appear to be a critical modifier in the pun/un model; differences in eye-spot numbers for pun/un;BLM+/T and pun/un;BLMT/T mice were not significant. Mitotic cell division is essentially complete in the RPE by P20 (27), so it is possible that the restricted developmental window of this tissue is insufficient to highlight subtle differences in HR between pun/un;BLM+/T and pun/un;BLMT/T genotypes. Therefore, these mice were combined and analyzed as one group (Fig. 5C). The number of eye-spots in control pun/un mice of 6.9 ± 3.2 is comparable to previous reports (18, 28), whereas BLM overexpression reduces HR two-fold, resulting in 3.4 ± 1.9 eye-spots per RPE in pun/un;BLMT mice (P<0.0001; Mann-Whitney U test). Although it was not possible to directly measure HR in the intestinal epithelial compartment of our intestinal model, our data suggests that the observed reduction in adenoma numbers is also due to modulation of HR by BLM-Tg.
Figure 5.

Expression of BLM-Tg down-regulates homologous recombination in the pun mouse RPE. Eye-spots consist of pigmented clones of revertant RPE cells in which melanin production has been restored by an intra-chromosomal recombination event. They are easily visible on the transparent RPE background. A and B, Representative examples of eye-spots in pun/un;BLMT mice. These are comprised of: A, single or simple multiples of revertant RPE cells although B, sometimes rarer, more complex configurations are observed. Eye-spots separated by a single non-revertant (clear) cell are scored as a single unit. C, BLM-Tg suppresses HR in pun RPE cells. pun/un;BLM+/T and pun/un;BLMT/T mice have been combined and represented as a single group, pun/un;BLMT, because analyses demonstrated both genotypes are similarly effective in this model. Mean ± standard deviation are shown for each group. pun/un mice have 6.9 ± 3.2 eye-spots, n=277, 40 RPE; pun/un;BLMT mice have 3.4 ± 1.9 eye-spots, n=192, RPE=56. Statistical analyses were performed using the Mann-Whitney U test; **** P<0.0001. Scale bars each represent 50 μm.
Discussion
Rescue of the BlmCin/Cin embryonic lethal phenotype by BLM-Tg indicates that expression of this human ortholog is sufficiently regulated, within the physiological context of our model, to direct normal development in Blm null mice. Our findings that BLM-Tg reduces adenoma numbers in the ApcMin/+ mouse model of intestinal tumorigenesis (Fig. 1) are consistent with the known role of BLM in HR and its requirement for maintaining genomic integrity (2, 11). Moreover, BLM-Tg expression suppressed adenoma formation in the ApcMin/+ model by a dose-dependent mechanism (Fig. 1), suggesting that augmentation of the HR pathway may be a viable objective for attenuating tumor suppression in specific in vivo milieus, notably the intestinal compartment. Reduction of intestinal tumor burden results in accompanying dose-dependent increases in median survival times for ApcMin/+;BLM+/T and ApcMin/+;BLMT/T mice (Fig. 2).
Adenomas that developed in ApcMin/+;BLM+/T mice were pathologically identical to those from ApcMin/+ animals (Fig. 3), indicating that the BLM-Tg did not affect tumor origin or skew tumor spectrum in this intestinal model. Furthermore, long-term expression does not adversely alter the wild-type phenotype of BLM+/T and BLMT/T mice. Mice (n=12) have now been aged for over 24 months without deleterious effects on survival or overt signs of tumorigenesis. This is somewhat surprising because data from the pun model indicate that, mechanistically, BLM-Tg overexpression down-regulates HR two-fold (Fig. 5). However, given the crucial function of HR in mediating error-free repair of DNA damage, it is possible that aging BLM-Tg mice may ultimately prove more susceptible to spontaneous tumorigenesis. Perhaps if aged mice were challenged with radiomimetic agents, the inflicted DNA damage might exceed their (lowered) threshold for HR repair, consequently resulting in an increased susceptibility to tumorigenesis. Transgenic mouse lines have also been generated for Wrn, another member of the RecQ helicase family (29). Although the wild-type Wrn transgene has yet to be tested in models of tumorigenesis, it has no effect on HR in RPE cells of the pun mouse (30).
The elevated BLM levels observed in our ApcMin/+;BLM-Tg model most likely reduce adenoma formation through suppression of HR, thus maintaining heterozygosity of the wild-type Apc allele. We used the pun and MMR-deficient Msh2-/- models to further investigate the mechanism of tumor reduction in the ApcMin/+;BLMT mice, rather than attempting to correlate a reduction in surrogate markers of HR, such as Rad51 foci, with reduction in adenoma burden. These genetic models presented a more relevant system for assessing the effects of the BLM-Tg on HR in vivo. A two-fold reduction of eyespots in RPE cells of pun/un;BLMT mice (Fig. 5) suggests that BLM-Tg directly modulates HR in this tissue. Our interpretation of the observed reduction in adenoma numbers in the ApcMin/+;BLMT model is that it is caused by the effect of BLM-Tg on HR in the intestinal epithelia, thus suppressing LOH of the wildtype Apc allele. This conclusion is supported by the data from the ApcMin/+;BLMT;Msh2-/- mice. When Apc is activated by point mutation, due to innate MMR deficiency, thus precluding the requirement for inactivation of the wild-type Apc allele by LOH, there are no observable differences in intestinal adenoma numbers between ApcMin/+;BLMT;Msh2-/- and ApcMin/+;Msh2-/- mice (Fig. 4). If BLM-Tg was affecting adenoma formation through other mechanisms unrelated to HR, one would predict that adenoma numbers should still differ between these two models. This is not the case.
Consistent with this model, levels of GIN are also reduced between ApcMin/+ and ApcMin/+;BLM-Tg genotypes, although they do not meet statistical significance (Fig. 1A). If correct, BLM-Tg would act before the potential onset of GIN, since GIN pathologically precedes adenomas in the ApcMin/+ model and since LOH of the wild-type Apc allele is a fundamental requirement for GIN development on a C57Bl-6J background (31). In addition to perturbing LOH, and hence subsequent levels of GIN and adenomas, it is possible that BLM-Tg may selectively target neoplastic cells after they have emerged as larger lesions on the ApcMin/+ background. It remains unclear whether elevating BLM levels could eliminate, perhaps through apoptosis, single or small populations of nascent cells that have acquired two mutant alleles of Apc.
Data from our ApcMin/+;BLMT models suggest that levels of specific DNA repair proteins may be titrated to achieve positive therapeutic outcomes in the context of specific hereditary cancer syndromes, exemplified by FAP. There are many inhibitors readily available that target the homologous recombination repair pathway and down-regulate HR (reviewed 32, 33). However, we are unaware of any small molecule inhibitors, or other reagents, that effect up-regulation of endogenous BLM and thus, might prove more suitable for therapeutic applications. With this in mind, we are investigating expression profiles of our BLM-Tg model to determine if there are other molecular targets that are more amenable to therapeutic modulation.
Our study establishes that BLM expression can be effectively manipulated in a mouse model of intestinal tumorigenesis to successfully attenuate the tumor phenotype. We show that overexpression of human BLM reduces intestinal adenoma formation in the ApcMin/+ mouse model and propose that the mechanism is through down-regulation of the homologous recombination repair pathway. This presents the potential to explore new avenues for intestinal tumor prevention by controlling levels of BLM expression or other genes of the DNA repair pathways. Our data demonstrates the therapeutic potential of titrating levels of specific DNA repair proteins that may be protective against tumor formation and suggests that this approach of modulating fundamental DNA repair pathways may be a viable pharmacological strategy for cancer prevention.
Supplementary Material
Acknowledgments
We would like to acknowledge support from the Bloom's Syndrome Foundation and The Solid Tumor Biology Program in the OSU Comprehensive Cancer Center (J. Groden). K. Murnan was supported by the Pelotonia Fellowship Program. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Advancing Translational Sciences or the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Grant Support
This work was supported by NIH Grants CA117898 (J. Groden), CA63507 (J. Groden), CA98013 (J. Groden), CA152758 (C.M. Croce) and CA166905 (C.M. Croce). The project was supported by awards 8UL1TR000090-05, 8KL2TR000112-05, and 8TL1TR000091-05 from the National Center for Advancing Translational Sciences (J. Groden).
Footnotes
Disclosure of Potential Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- 1.Chu WK, Hickson ID. RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer. 2009;9:644–54. doi: 10.1038/nrc2682. [DOI] [PubMed] [Google Scholar]
- 2.Risinger MA, Groden J. Crosslinks and crosstalk: human cancer syndromes and DNA repair defects. Cancer Cell. 2004;6:539–45. doi: 10.1016/j.ccr.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Karow JK, Chakraverty RK, Hickson ID. The Bloom's syndrome gene product is a 3’-5’ DNA helicase. J Biol Chem. 1997;272:30611–14. doi: 10.1074/jbc.272.49.30611. [DOI] [PubMed] [Google Scholar]
- 4.Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, et al. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–66. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- 5.German J, Archibald R, Bloom D. Chromosomal breakage in a rare and probably genetically determined syndrome of Man. Science. 1965;148:506–7. doi: 10.1126/science.148.3669.506. [DOI] [PubMed] [Google Scholar]
- 6.Braybrooke JP, Li J-L, Wu L, Caple F, Benson FE, Hickson ID. Functional interaction between the Bloom's syndrome helicase and the RAD51 paralog, RAD51L3 (RAD51D). J Biol Chem. 2003;278:48357–66. doi: 10.1074/jbc.M308838200. [DOI] [PubMed] [Google Scholar]
- 7.Bugreev DV, Yu X, Egelman EH, Mazin AV. Novel pro- and anti-recombination activities of the Bloom's syndrome helicase. Genes Dev. 2007;21:3085–94. doi: 10.1101/gad.1609007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaganti RS, Schonberg S, German JA. A manyfold increase in sister chromatid exchanges in Bloom's syndrome lymphocytes. Proc Natl Acad Sci U S A. 1974;71:4508–12. doi: 10.1073/pnas.71.11.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.German J, Sanz MM, Ciocci S, Ye TZ, Ellis NA. Syndrome-causing mutations of the BLM gene in persons in the Bloom's Syndrome Registry. Hum Mutat. 2007;28:743–53. doi: 10.1002/humu.20501. [DOI] [PubMed] [Google Scholar]
- 10.Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, Youssoufian H, et al. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet. 2000;26:424–9. doi: 10.1038/82548. [DOI] [PubMed] [Google Scholar]
- 11.Goss KH, Risinger MA, Kordich JJ, Sanz MM, Straughen JE, Slovek LE, et al. Enhanced tumor formation in mice heterozygous for Blm mutation. Science. 2002;297:2051–3. doi: 10.1126/science.1074340. [DOI] [PubMed] [Google Scholar]
- 12.McDaniel LD, Chester N, Watson M, Borowsky AD, Leder P, Schultz RA. Chromosome instability and tumor predisposition inversely correlate with BLM protein levels. DNA Repair. 2003;2:1387–1404. doi: 10.1016/j.dnarep.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Gruber SB, Ellis NA, Scott KK, lmog R, Kolachana P, Bonner JD. BLM heterozygosity and the risk of colorectal cancer. Science. 2002;297:2013. doi: 10.1126/science.1074399. [DOI] [PubMed] [Google Scholar]
- 14.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 15.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 16.Albuquerque C, Breukel C, van der Luijt R, Fidalgo P, Lage P, Slors FJ, et al. The 'just-right' signaling model: APC somatic mutations are selected based on a specific level of activation of the beta-catenin signaling cascade. Hum Mol Genet. 2002;11:1549–60. doi: 10.1093/hmg/11.13.1549. [DOI] [PubMed] [Google Scholar]
- 17.Shoemaker AR, Gould KA, Luongo C, Moser AR, Dove WF. Studies of neoplasia in the Min mouse. Biochim Biophys Acta. 1997;1332:F25–48. doi: 10.1016/s0304-419x(96)00041-8. [DOI] [PubMed] [Google Scholar]
- 18.Brown AD, Claybon AB, Bishop AJ. A conditional mouse model for measuring the frequency of homologous recombination events in vivo in the absence of essential genes. Mol Cell Biol. 2011;31:3593–602. doi: 10.1128/MCB.00848-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smits R, Hofland N, Edelmann W, Geugien M, Jagmohan-Changur S, Albuquerque C, et al. Somatic Apc mutations are selected upon their capacity to inactivate the beta-catenin downregulating activity. Genes Chrom Can. 2000;29:229–39. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1033>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 20.Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology. 2003;124:762–77. doi: 10.1053/gast.2003.50094. [DOI] [PubMed] [Google Scholar]
- 21.Shoemaker AR, Moser AR, Midgley CA, Clipson L, Newton MA, Dove WF. A resistant genetic background leading to incomplete penetrance of intestinal neoplasia and reduced loss of heterozygosity in ApcMin/+ mice. Proc Natl Acad Sci U S A. 1998;95:10826–31. doi: 10.1073/pnas.95.18.10826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halberg RB, Waggoner J, Rasmussen K, White A, Clipson L, Prunuske AJ, et al. Long-lived Min mice develop advanced intestinal cancers through a genetically conservative pathway. Cancer Res. 2009;69:5768–75. doi: 10.1158/0008-5472.CAN-09-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shoemaker AR, Haigis KM, Baker SM, Dudley S, Liskay RM, Dove WF. Mlh1 deficiency enhances several phenotypes of Apc(Min)/+ mice. Oncogene. 2000;19:2774–9. doi: 10.1038/sj.onc.1203574. [DOI] [PubMed] [Google Scholar]
- 24.Reitmair AH, Cai JC, Bjerknes M, Redston M, Cheng H, Pind MT, et al. MSH2 deficiency contributes to accelerated APC-mediated intestinal tumorigenesis. Cancer Res. 1996;56:2922–6. [PubMed] [Google Scholar]
- 25.Lal G, Ash C, Hay K, Redston M, Kwong E, Hancock B, et al. Suppression of intestinal polyps in Msh2-deficient and non-Msh2-deficient multiple intestinal neoplasia mice by a specific cyclooxygenase-2 inhibitor and by a dual cyclooxygenase-1/2 inhibitor. Cancer Res. 2001;61:6131–6. [PubMed] [Google Scholar]
- 26.Belcheva A, Green B, Weiss A, Streutker C, Martin A. Elevated incidence of polyp formation in APCMin/+ Msh2−/− mice is independent of nitric oxide-induced DNA mutations. PLoS One. 2013;8:e65204. doi: 10.1371/journal.pone.0065204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bodenstein L, Sidman RL. Growth and development of the mouse retinal pigment epithelium. I. Cell and tissue morphometrics and topography of mitotic activity. Dev Biol. 1987;121:192–204. doi: 10.1016/0012-1606(87)90152-7. [DOI] [PubMed] [Google Scholar]
- 28.Bishop AJ, Hollander MC, Kosaras B, Sidman RL, Fornace AJ, Jr, Schiestl RH. Atm-, p53- and Gadd45a-deficient mice show an increased frequency of homologous recombination at different stages during development. Cancer Res. 2003;63:5335–43. [PubMed] [Google Scholar]
- 29.Wang L, Ogburn CE, Ware CB, Ladiges WC, Youssoufian H, Martin GM, et al. Cellular Werner phenotypes in mice expressing a putative dominant-negative human WRN gene. Genetics. 2000;154:357–62. doi: 10.1093/genetics/154.1.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamamoto ML, Reliene R, Oshima J, Schiestl RH. Effects of human Werner helicase on intrachromosomal homologous recombination mediated DNA deletions in mice. Mutat Res. 2008;644:11–6. doi: 10.1016/j.mrfmmm.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Paulsen JE, Steffensen IL, Løberg EM, Husøy T, Namork E, Alexander J. Qualitative and quantitative relationship between dysplastic aberrant crypt foci and tumorigenesis in the Min/+ mouse colon. Cancer Res. 2001;61:5010–5. [PubMed] [Google Scholar]
- 32.Carvalho JF, Kanaar R. Targeting homologous recombination-mediated DNA repair in cancer. Expert Opin Ther Targets. 2014;18:427–58. doi: 10.1517/14728222.2014.882900. [DOI] [PubMed] [Google Scholar]
- 33.Huang F, Mazin AV. Targeting the homologous recombination pathway by small molecule modulators. Bioorg Med Chem Lett. 2014;24:3006–13. doi: 10.1016/j.bmcl.2014.04.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.