

Abstract
Chemotherapeutic resistance, particularly to doxorubicin (Dox), represents a major impediment to successfully treating breast cancer and is linked to elevated tumor metabolism and tumor over-expression and/or activation of various families of receptor- and non-receptor-associated tyrosine kinases. Disruption of circadian time structure and suppression of nocturnal melatonin production by dim light exposure at night (dLEN), as occurs with shift work, and/or disturbed sleep-wake cycles, is associated with a significantly increased risk of an array of diseases, including breast cancer. Melatonin inhibits human breast cancer growth via mechanisms that include the suppression of tumor metabolism and inhibition of expression or phospho-activation of the receptor kinases AKT and ERK1/2 and various other kinases and transcription factors. We demonstrate in tissue-isolated estrogen receptor alpha-positive (ERα+) MCF-7 human breast cancer xenografts, grown in nude rats maintained on a light/dark cycle of LD 12:12 in which dLEN is present during the dark phase (suppressed endogenous nocturnal melatonin), a significant shortening of tumor latency-to-onset, increased tumor metabolism and growth, and complete intrinsic resistance to Dox therapy. Conversely, a LD 12:12dLEN environment incorporating nocturnal melatonin replacement resulted in significantly lengthened tumor latency-to-onset, tumor regression, suppression of nighttime tumor metabolism, and kinase and transcription factor phosphorylation, while Dox sensitivity was completely restored. Melatonin acts as both a tumor metabolic inhibitor and circadian-regulated kinase inhibitor to reestablish the sensitivity of breast tumors to Dox and drive tumor regression indicating that dLEN-induced circadian disruption of nocturnal melatonin production contributes to a complete loss of tumor sensitivity to Dox chemotherapy.
Keywords: Melatonin, Doxorubicin, Circadian, Breast, Warburg
Introduction
Breast cancer is a leading cause of death in women around the world despite recent advances in therapeutic options. Although the goal of chemotherapy is to kill disseminated cancer cells and prevent metastatic progression, many cancers are intrinsically resistant to conventional chemotherapeutic agents while others that initially respond develop resistance (acquired resistance) during treatment [1]. The anthracycline, doxorubicin (Dox), is a commonly used chemotherapeutic for the treatment of patients whose breast tumors are resistant to endocrine therapy or are metastatic [2]. The antitumor activity of Dox results from the initiation of DNA damage via the inhibition of topoisomerase II and lipid peroxidation resulting in the generation of free radicals [3, 4]. Although Dox is still considered to be one of the most effective agents in the treatment of breast cancer, particularly following tamoxifen failure, its efficacy as a curative agent is compromised by cumulative dose-related development of resistance and cardiomyopathy [5].
Resistance to Dox may occur through a variety of mechanisms [6–10] including the activation of key-signaling pathways including the phosphatidylinositol 3-kinase/protein B (PI3K/AKT), the epidermal growth factor (EGFR), human epidermal growth factor receptor (HER), their downstream mitogen-activated protein kinase/extracellular signal-related kinases (MAPK/ERK1/2), and other downstream signaling pathways all of which can drive cancer cell progression and drug resistance [10–13]. Other signaling pathways involved in Dox-resistant breast tumors include signal transducer and activator of transcription 3 (STAT3), nuclear factor-kappa B (NF-kB), and protein kinase C alpha and delta (PKCα and δ) [14–16].
Activation of signaling pathways including ERK1/2, PI3K/AKT, NF-kB, and STAT3 are reported to induce the expression of the ABC transporters ABCB1 (MDR1), ABCC1 (MRP1), and ABCG2 (BCRP) and metabolizing enzymes such as carbonyl reductase 1 (CBR1) and aldo-keto reductase 1C3 (AKR1C3) which are involved in the development of both intrinsic and acquired drug resistance [17–22]. In addition, elevated expression of CBR1 and AKR1C3 induce the metabolism of Dox to doxurubicinol, the primary Dox metabolite, which is reported to be a million times less active than Dox at inducing DNA damage and promoting breast tumor cell apoptosis [23] but significantly more cardiotoxic than Dox [23]. Thus, individual variations in conversion of Dox to doxorubicinol may confer individualized variation in its antitumor and cardiotoxic effects.
Disruption of circadian time structure by light at night as encountered during shift work and/or disturbed sleep-wake cycle leads to a significantly increased risk of an array of diseases, including breast cancer [24]. The circadian hormone melatonin, produced by the pineal gland at night, inhibits the growth of breast cancer through activation of the MT1 G protein coupled melatonin receptor [25]. Our most recent studies [26] demonstrate that circadian/melatonin disruption by exposure of tumor-bearing female nude rats to dim light at night (dLEN) promotes tumor growth and drives intrinsic resistance to the anti-estrogen tamoxifen in tissue-isolated ERα-positive (ERα+) human breast tumor xenografts and that melatonin via inhibition of tumor metabolism and kinase activity re-establishes tumor sensitivity to tamoxifen and acts synergistically with tamoxifen to drive tumor regression.
In the present study, we again utilize our circadian-complete (e.g., intact nocturnal melatonin production), tissue-isolated breast tumor xenograft nude rat model to address the hypothesis that disruption of the nocturnal circadian melatonin signal, by exposure of tumor-bearing animals to dLEN, induces rapid breast tumor growth and resistance to chemotherapy with Dox. In addition, we tested the postulate that the administration of melatonin during dLEN would attenuate tumor metabolism, inhibit the expression and/or phospho-activation of key kinases and transcription factors as well as Dox metabolizing enzymes and ABC-transporters that efflux Dox to block the development of dLEN-induced intrinsic resistance to Dox.
Materials and methods
Chemicals and reagents
All chemicals including Dox and tissue culture reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Cell culture medium, RPMI 1640 and fetal bovine serum (FBS) were purchased from Invitrogen Corporation (Carlsbad, CA). High-performance liquid chromatography (HPLC)-grade reagents were purchased from Fisher Chemical (Pittsburgh, PA). Free fatty acid, rapeseed oil methyl ester standards, as well as boron trifluoride-methanol, potassium chloride, sodium chloride, perchloric and trichloroacetic acids were purchased from Sigma-Aldrich. The HPLC standards, (+/−) 5-HETE and 13(S)-HODE were purchased from Cayman Chemical Co. (Ann Arbor, MI).
Cell line and cell culture
The ERα+ MCF-7 human breast cancer cell line used in these studies was obtained from American Tissue Culture Collection (Manassas, VA). These cells were tested and authenticated by ATCC and upon receipt were immediately expanded and frozen-down as stock for future studies. Cells from low passage frozen stocks (passage numbers 18–20) were used in these studies. Briefly, cells were cultured in RPMI 1640 medium supplemented with 10% FBS, 50 mM minimum essential medium non-essential amino acids, 1 mM sodium pyruvate, 2 mM glutamine, 10 mM basal medium eagle, 100 mg/mL streptomycin, and 100 U/mL penicillin, and maintained at 37° C in a humidified atmosphere of 5% CO2 and 95% air.
Animals, housing conditions, and diet
Female athymic, inbred nude rats (Crl:NIH- Foxn1rnu) used in this study were purchased from Charles River Laboratories (Wilmington, MA) at 1–2 weeks of age. Upon arrival, animals were maintained in environmentally controlled rooms (25°C; 50–55% humidity) with controlled diurnal lighting schedule of 12 h light:12 h dark at subjective night (LD, 12:12) [304 lux; 123 μW/cm2; lights on, 0600 hr and off at 1800 hr]. Animal rooms were completely devoid of light contamination during the dark phase [27–29]. One week prior to tumor implantation the animals were switched to a 12 h light:12 h dim light at night (dLEN), subjective night cycle [0.2 lux, with lights on at 0600 hr and off at 1800 hr, and dLEN on at 1800 hr and off at 0600 hr] in an AAALAC-accredited facility and in accordance with The Guide. Animals were given free access to food (Purina 5053 Irradiated Laboratory Rodent Diet, Richmond, IN), and acidified water as previously described [27–29]. All procedures employed for animal studies were approved by the Tulane University Institutional Animal Care and Use Committee.
Arterial blood collection
Diurnal plasma melatonin levels (pg/mL; mean ± 1 SD) of naïve, female nude rats (n = 12) maintained initially in the control LD 12:12 cycle or in the dLEN cycle were measured as previously described [26, 28]. In experimental animals during the course of these studies blood was collected over time at six circadian time points (0400-, 0800-, 1200-, 1600-, 2000, and 2400-hr) and melatonin levels measured using a radioimmunoassay kit (Alpco, Salem, NH), as previously described [26].
MCF-7 tumor xenografts development in nude mice
Ovariectomized athymic nude mice (4–5 week old females) were obtained from Charles River (Indianapolis, IN) and maintained in pathogen-free aseptic conditions with phytoestrogen-free food and water ad libitum. All mice were supplemented with estrogen pellets (0.72 mg of 17β-estradiol 60-day release from Innovative Research of America (Sarasota, FL) and estradiol-dependent MCF-7 xenografts were propagated. Exponentially growing MCF-7 cells (Passage numbers 18–20) were harvested and approximately 5x106 MCF-7 cells in 150 μl of PBS-matrigel mixture were orthotopically and bilaterally implanted into the mammary fat pads of female nude mice, as previously described [26].
Tumor transplantation into athymic nude rats
After one week of photoperiod acclimation (dLEN or dLEN plus nighttime melatonin supplementation), nude rats were implanted in a tissue-isolated fashion with MCF-7 human tumor xenografts obtained from the tumor xenografts initially developed in mice, as described previously [26–28]. Once implanted, tissue-isolated tumor xenografts reached a palpable size (approximately pea size) in the nude rats they were measured every day for estimated tumor weights, as previously described [26]. All animals were maintained in the dLEN lighting schedule and were randomized so that half received either vehicle or a supplement of melatonin in their nighttime drinking water (0.1 μg/ml) such that they received approximately 2.5 μg melatonin daily intake, based on a daily water intake of about 25 ml; this simulates the high normal nighttime physiologic levels of melatonin. When tumor weights reached approximately 2.5 g estimated weight, one-half of the animals (n = 3/group) maintained in either the dLEN lighting or dLEN supplemented with melatonin were treated daily at 1600 hr with either 0.1 ml of Dox (6 mg/kg/day) or diluent by i.p. injection. Thus this study consisted of 4 groups: Group I (dLEN treated with vehicle), Group II (dLEN treated with Dox), Group III (dLEN supplemented with melatonin and treated with vehicle), and Group IV (dLEN supplemented with melatonin and treated with Dox). When tumors reached an estimated weight of 8 g or regressed to 1.6 g following treatment, tumors were prepared for A-V difference measurements between 2400 and 0200 hr; animal preparation and blood collection, blood and plasma analysis of constituents were conducted as previously described [26–28]. Twenty minutes prior to end of blood collection [3H]-thymidine (0.05 μCi/g of est. tumor weight) was injected intravenously via carotid catheter. Details for the these procedures and the kits used for measurement of glucose, lactate, fatty acids, LA, 13-HODE, cAMP, O2, CO2, and melatonin have been described previously [26–28].
Tumor and heart lysate extraction and Western blot analysis
Tumor xenografts and heart samples harvested and snap frozen at 2400 hr (mid-dLEN phase) were pulverized and homogenized in RIPA buffer (1 × PBS, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS) containing protease and phosphatase inhibitor cocktails (Sigma). Total tumor protein was isolated from tumor lysates as previously described [26, 28] and aliquots were stored at −80° C. One hundred twenty micrograms of protein from each sample were separated on a Criterion™ precast gel (Bio-Rad, Richmond, CA) and transferred onto nitrocellulose membranes (Bio-Rad). After incubation with 5% nonfat milk in Tris-buffered saline containing 0.1% Tween 20, Western blots were probed with various antibodies including phospho- (p)-HER2 Tyr1121/1222, (t)-HER2, p-HER3 Tyr1222, t-HER3 p-ERK1/2 Thr202/Tyr204, total t-ERK1/2, p-AKT Ser473, t-AKT, p-nuclear factor-kappa beta (NF-kB) Ser536, t-NF-KB, p-cAMP response element binding protein (CREB) Ser133, t-CREB, p-pyruvate dehydrogenase kinase 1 (PDK-1) Ser241 and t-PDK-1, p-p21 activating kinase 1 (PAK1) Tyr423 and t-PAK1, p-signal transducer and activator of transcription 3 (STAT3) Tyr707 and t-STAT3, t-protein kinase C alpha/delta (PKCα, t-PKCδ), t-CBR1, t-AKR1C1, t-AKR1C2, and t-AKR1C3, t-superoxide dismustase 2 (SOD2), and t-proliferating cell nuclear antigen (PCNA) from Cell Signaling (Danvers, MA). The blots were stripped and re-probed with anti-β-actin antibody (Sigma, St. Louis, MO) to evaluate loading. Quantitation of Western blots and differences in expression of total and phosphorylated proteins were determined by digital quantitation of phosphorylated and total protein levels and normalizing phosphorylated levels to the levels of the total protein of interest and comparing the levels of dLEN, and dLEN + melatonin in the presence or absence of Dox, to the dLEN control to determine the percent or fold change.
Statistical analysis
Data are represented as the mean ± standard error of the mean. Statistical differences between mean values in the LEN-exposed group versus the control group at circadian time points were assessed by the Student’s t-test. Statistical differences among group means in tumor perfusion studies were determined by a one-way ANOVA followed by Bonferroni’s multiple comparison test. Differences in the tumor growth rates among groups were determined by regression analyses and tests for parallelism (Student’s t-test). Differences were considered to be statistically significant at p < 0.05.
Results
We previously reported that exposure to dLEN represses the nighttime rise of melatonin in female nude rats [26]. Figure 1A shows plasma levels of melatonin of female nude rats housed under LD 12:12 conditions increased and remained high during the dark phase reaching a peak at 2400 hr that was more than 70-fold higher than during the light phase. Figure 1B shows that melatonin levels were low to undetectable under the dLEN lighting schedule prior to the administration of exogenous melatonin. After supplementation with melatonin in the drinking water (2.73 ± 0.34 μg/day) plasma melatonin levels at 2400 hr (mid-dLEN/subjective night) were 142-fold higher (352.5 ± 17.74 pg/ml) than at 1200 hr in control (dLEN) rats (2.48 ± 0.67 pg/ml), respectively.
Figure 1.

Effect of dLEN versus LD 12:12 lighting schedules or administration of exogenous melatonin in the dLEN lighting schedule on the serum melatonin profile in female nude rats. Female nude rats with (ERα+) tissue-isolated breast tumor xenografts were housed under control (LD 12:12) or experimental, dLEN (with light at 0.2 lux) lighting schedules, or dLEN and supplemented with nighttime melatonin (MLT), and treated with diluent or doxorubicin (Dox). (A) Diurnal plasma melatonin levels (pg/ml; mean ± 1 SD) of female nude rats maintained in a controlled LD 12:12 or experimental dLEN lighting cycle (n=6/group) were measured as described in “Materials and Methods”. Data are double-plotted to better visualize rhythmicity (n = 6/group). Asterisks (*) denote significant differences (p < 0.05) in serum melatonin levels in rats under the different lighting schedules. (B) Plasma melatonin levels from dLEN at 1200 hr (red bars) and 2400 hr (blue bars) from animals maintained in dLEN and treated with vehicle (dLEN Cntl) or Dox (dLEN + Dox), or from animals on dLEN but supplemented with melatonin (in nighttime drinking water) and treated with vehicle (dLEN+MLT) or Dox (dLEN+MLT+Dox), as described in “Materials and Methods”. Significant differences (p < 0.05) in serum melatonin levels in rats (n=3/group) under the different lighting schedules are denoted by an asterisk (*).
Given that dLEN promotes tumor growth and intrinsic resistance to tamoxifen in MCF-7 tissue-isolated breast tumor xenografts, we asked if reduction of nighttime melatonin by exposure to dLEN could also drive resistance to Dox. Figure 2 shows that tissue-isolated ERα+ MCF-7 breast tumor xenografts from rats housed in a dLEN lighting schedule had a significantly shorter (p < 0.001) latency-to-tumor-onset and increased growth rate (2.3-fold) compared to tumors in rats in dLEN that received supplemental melatonin during the subjective dark phase. Tumor xenografts from rats in dLEN showed complete intrinsic resistance to Dox growing at the same rate as vehicle-treated dLEN xenografts. However, xenografts in dLEN rats supplemented with melatonin showed a significant response to Dox administered in the late light phase at 1600 hr, regressing at a rate of −0.18 g/day. Figure 2B–E shows the visually dramatic difference in tumor growth and regression between the various groups with or without melatonin supplementation and Dox.
Figure 2.

Differential effects of Dox on the growth and regression of (ERα+) MCF-7 tissue-isolated breast tumor xenografts in female nude rats housed in a lighting schedule of dLEN or dLEN supplemented with melatonin during dLEN. (A) Estimated tumor weight [g/day] of MCF-7 human breast tumor xenografts from nude rats exposed to a dLEN lighting schedule and treated with diluent (red triangles) or Dox (blue diamonds) [6 mg/kg/day] or a dLEN lighting schedule and supplemented with melatonin during dLEN (black triangles) or dLEN + melatonin + Dox (green inverted triangles). Tumor weights were estimated daily as described in “Materials and Methods.” Images of tumor-bearing nude rats in maintained in either experimental (LD 12:12dLEN) or (LD 12:12dLEN and supplemented with nighttime melatonin) lighting schedules, as described in “Materials and Methods.” Panel B and C represent xengografts from experimental animals 28 days after tumor implant in dLEN (B) and dLEN receiving melatonin supplementation (C). Panels D and E show tumor xenografts from experimental animals 45 days after tumor implant in dLEN supplemented with melatonin and administered a diluent (D) and dLEN supplemented with melatonin and treated with DOX beginning on day 28 (E). Asterisks (*) indicate location of tumor xenografts.
Because tumor metabolism has been reported play an important role in drug resistance, we felt it necessary to evaluate key aspects of tumor metabolism including FFA and LA uptake and glucose metabolism. Administration of Dox alone to rats on dLEN did not suppress tumor LA uptake, its metabolism to 13-HODE, or the incorporation of [3H]-thymidine into DNA as compared with dLEN vehicle-treated controls. Tumor LA uptake and 13-HODE formation were completely suppressed in both vehicle- and Dox-treated rats on dLEN when melatonin was supplemented (Table 1). Incorporation of [3H]-thymidine into DNA in xenografts from vehicle and Dox-treated dLEN rats was elevated by 10-fold at the mid-dLEN phase (2400 hr) compared with vehicle- and Dox-treated dLEN tumors receiving melatonin supplementation.
Table 1.
Tumor cAMP levels, arteriovenous difference measurements in tumor total fatty acid (TFA) and linoleic acid (LA) utilization, 13-HODE production rates, and 3H-thymidine incorporation into tumor DNA and tumor DNA content in vivo during dark phase (2400 hrs) in MCF-7 (ER+) dLEN-Diluent Controls, –dox (6 mg/kg), –melatonin (2.5μg/d), and –dox + melatonin in human breast tumor xenografts. Values are means ± SD (n = 3/group).
| Treatmenta | cAMP (nmol/g tissue) | TFA uptake (μg/min/g) | LA uptake (μg/min/g) | 13-HODE (ng/min/g) | 3H-Thymidine Incorporation (dpms/μg DNA) | DNA Content (mg/gm) | |
|---|---|---|---|---|---|---|---|
| Arterial Supply | Venous Output | ||||||
| dLEN Vehicle | 1.74 ± 0.18 | 8.57 ± 0.85 (32.5 ± 2.7%) | 2.62 ± 0.75 (36.1 ± 4.3%) | NDb | 7.05 ± 0.42 | 69.4 ± 2.8 | 3.46 ± 0.17 |
| dLEN dox | 1.76 ± 0.25 | 8.50 ± 1.19 (32.5 ± 2.7%c) | 2.25 ± 0.33 (31.6 ± 0.7%) | NDb | 6.90 ± 0.59 | 67.4 ± 4.7 | 3.43 ± 0.16 |
| dLEN melatonin | 0.15 ± 0.06c | −0.01 ± 0.02c (−0.4 ± 0.1%c) | 0.03 ± 0.03c (0.4 ± 0.3%c) | NDb | NDb,c | 6.7 ± 1.7c | 2.00 ± 0.10c |
| dLEN dox + melatonin | 0.06 ± 0.03c | −0.11 ± 0.11c (−0.1 ± 0.1%c) | −0.05 ± 0.05c (−0.2 ± 0.1%c) | NDb | NDb,c | 6.1 ± 1.4 c | 2.01 ± 0.10c |
Three animals (tumors)/group; (+ SD) tumor weights, MCF-7 (ER+) dLEN Group, vehicle treatment = 8.30 ± 0.27 g; dox treatment = 8.08 ± 0.17 g; melatonin treatment = 7.93 ± 0.37 g; and, dox + melatonin treatment = 1.60 ± 0.18 g, respectively. All tumors were harvested at 2400 hrs.
ND, not detectable.
P< 0.05 vs. vehicle and dLEN+dox+melatonin group.
In rats treated with Dox alone, no suppressive effect of the drug on the high rates of tumor glucose and O2 uptake were observed at 2400 hr in dLEN xenografts versus those receiving vehicle (Table 2). In dLEN rats receiving melatonin without Dox, uptake of tumor glucose and O2 were significantly reduced by 66% and 29%, respectively, compared to vehicle treated dLEN xenografts. In melatonin supplemented Dox-treated xenografts tumor glucose uptake was 89% lower than in either vehicle or Dox-treated dLEN tumors, and 67% lower than in melatonin-supplemented xenografts. Tumor O2 uptake in dLEN xenografts receiving melatonin and Dox was 72% lower than in vehicle or Dox-treated dLEN group, and 66% lower than in melatonin-treated tumors (Table 2).
Table 2.
Tumor uptake of glucose and O2 and production of lactate and CO2 production in vivo (Warburg effect) during the mid-dark phase (2400 hrs) in tissue-isolated MCF-7 (ER+) human breast cancer xenografts in nude female rats exposed to dLEN and treated with either vehicle, dox (6 mg/kg), melatonin (2.5μg/d), or dox + melatonin. Values are means ± SD (n =3/group).
| Treatmenta | Glucose uptake (μg/min/g) | Lactate release (nmol/min/g) | O2 uptake (% of supply) | CO2 production (% of original value) |
|---|---|---|---|---|
| dLEN Vehicle | 4.5 ± 0.6 (34.9 ± 1.3%)b | 26.2 ± 1.9 | 78.9 ± 0.4 | 132.9 ± 10.8 |
| dLEN dox | 4.2 ± 0.3 (33.8 ± 3.4%)b | 25.2 ± 1.1 | 78.7 ± 2.3 | 136.9 ± 4.6 |
| dLEN melatonin | 1.5 ± 0.2c (12.0 ± 1.5%)b,c | 10.5 ± 0.9c | 64.3 ± 3.9c | 74.0 ± 6.3c |
| dLEN dox + melatonin | 2.1 ± 0.5e (3.7 ± 0.5%)b,d | 2.9 ± 0.6d | 22.4 ± 1.9d | 49.1 ± 1.7d |
Three animals (tumors)/group; (± SD) tumor weights, in MCF-7 (ER+) dLEN groups, vehicle treatment = 8.30 ± 0.27 g; dox treatment = 8.08 ± 0.17 g; melatonin treatment = 7.93 ± 0.37 g; dox + melatonin treatment = 1.60 ± 0.18 g, respectively. All tumors were harvested at 2400 hrs.
Values expressed as % of arterial glucose supply.
p< 0.05 vs. vehicle;
p < 0.05 vs. dox and melatonin;
p < 0.05 vs. dox.
In rats treated with Dox alone, there was no suppressive effect on the high rates of tumor lactate release and CO2 production observed at 2400 hr in dLEN xenografts receiving vehicle or Dox (Table 2). In dLEN rats receiving melatonin alone, tumor lactate release and CO2 production were significantly reduced by 58% and 44%, respectively, as compared with dLEN vehicle treated animals. In melatonin + Dox-treated rats, tumor lactate release was 88% lower than in either vehicle or Dox-treated rats, and 73% lower than in melatonin-treated animals (Table 2). In melatonin + Dox-treated rats, tumor CO2 production was 64% lower than in either vehicle or Dox-treated rats, and 33% lower than animals receiving only melatonin (Table 2).
We next asked if melatonin could repress the stimulatory effect of dLEN on breast cancer cAMP production. In rats treated with Dox alone, no suppression of tumor cAMP levels was observed, compared with vehicle-treated control animals. Tumor cAMP levels were 30-fold higher at the mid-dLEN phase in rats in vehicle- and Dox-treated dLEN rats respectively, compared with the corresponding groups supplemented with melatonin (Table 1). Administration of Dox to rats in dLEN did not affect tumor cAMP levels, however; it diminished tumor cAMP levels by 2.8-fold in dLEN supplemented with melatonin as compared to vehicle treated animals.
As the expression and/or phospho-activation of numerous kinases and transcription factors in proliferative and survival signaling pathways are known to be elevated in drug-resistant breast cancer [6–13], we asked if such signaling nodes were modulated by dLEN or melatonin in the presence or absence of Dox. In our current study, tumors from the dLEN group treated with vehicle or Dox showed high levels of p-ERK1/2 expression, while those in dLEN but supplemented with melatonin showed almost complete suppression of p-ERK1/2 expression (Fig. 3). Elevated expression of t-AKT and t-NF-kB, but not their phosphorylated forms, were observed in dLEN xenografts treated with either vehicle or Dox, but were suppressed in dLEN xenografts supplemented with melatonin. Tumors from vehicle or Dox-treated rats in dLEN also showed elevated expression of p-PDK1, p-RSK2, t-PKCα, and t-PKCδ, respectively; however, dramatic suppression of the total and phosphorylated forms of these kinases was seen in melatonin supplemented dLEN xenografts with or without Dox treatment (Fig. 3). A high level of p-STAT3 expression was also evident in vehicle or Dox treated dLEN tumors, whereas its expression was greatly suppressed in melatonin supplemented dLEN xenografts with or without Dox therapy.
Figure 3.

Modulation of signaling kinases and transcription factors in tissue-isolated (ERα+) MCF-7 human breast tumor xenografts from female nude rats exposed to control dLEN treated with vehicle or Dox or dLEN supplemented with nighttime melatonin and treated with vehicle or Dox. Total-(t) and phospho-(p)Western blot analysis of total tissue lysates from breast tumor xenografts harvested from rats in a dLEN lighting schedule and treated with vehicle (Cntl) or Dox (Dox) or in dLEN but supplemented with melatonin during dLEN and treated with vehicle (MLT) or Dox (MLT+Dox). All tumors were harvested at 2400 hr (mid-dLEN phase) from 3 animals in each group. Total cell lysates (120 μg of protein per sample) from each tumor were analyzed by Western blot for expression of total and/or phosphorylated forms of HER2, HER3, ERK1/2, PDK1, AKT, NF-kB, CREB, STAT3, PKCα, PKCδ, RAS, RSK2, and PCNA. β-actin was used as a control for equal loading. This figure was generated by re-probing the same Western blot with all the different antibodies, thus β–actin loading is representative for all samples.
The transcription factor CREB is phospho-activated by the cAMP/PKA pathway and up regulated in many breast tumors [29]. In dLEN xenografts CREB is highly phospho-activated in the presence or absence of Dox, but supplementation with melatonin suppressed the levels of p-CREB in both the absence and presence of Dox. Elevated expression of t-HER2, t-HER3, and t-PCNA (marker of cell proliferation) was also seen in dLEN tumors treated with either vehicle or Dox, all of which were suppressed in response to melatonin supplementation during dLEN.
We next examine the expression of known Do metabolizing enzymes and ABC transporters which are known to be involved in the development of both intrinsic and acquired drug resistance [17–21]. dLEN breast tumor xenografts treated with either vehicle or Dox showed high levels of the reductases CBR1 and AKR1C3 while dLEN tumors supplemented with melatonin and treated with vehicle or Dox showed suppressed levels of CBR1 and AKR1C3 (Fig. 4A). AKR1C1 and AKR1C2 were also elevated in dLEN but only modestly inhibited by melatonin (data not shown).
Figure 4.
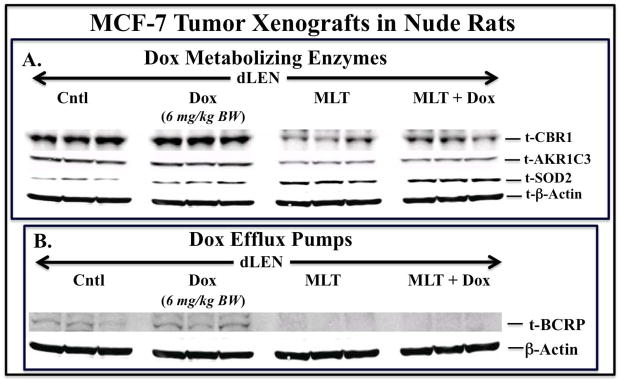
Modulation of Dox metabolizing enzymes, the antioxidant SOD2, and the ABC efflux pump protein ABCG2/BCRP in tissue-isolated (ER+) MCF-7 human breast tumor xenografts from female nude rats exposed to control dLEN treated with vehicle or Dox or dLEN supplemented with nighttime melatonin and treated with vehicle or Dox. Western blot analysis of total tissue lysates from breast tumor xenografts harvested from rats in dLEN lighting schedule and treated with vehicle (Cntl) or Dox (Dox) or in dLEN supplemented with melatonin during dLEN and treated with vehicle (MLT) or melatonin and Dox (MLT+Dox). All tumors were harvested at 2400 hr (mid-dLEN phase) from 3 animals in each group. Total cell lysates (120 μg of protein per sample) from each tumor were analyzed by Western blot for expression of total forms of (A) CBR1, AKR1C3, SOD2, and (B) BCRP. β-actin was used as a control for equal loading. This figure was generated by re-probing the same Western blot with all the different antibodies, thus β–actin loading is representative for all samples.
Expression of ABC transporters is a key event in the development of multidrug resistance in cancer cells [17]. Breast tumor xenografts from the dLEN group treated with vehicle or Dox showed elevated levels of ABCG2/BCRP. However, tumors from rats in dLEN receiving melatonin supplementation and treated with vehicle or Dox showed greatly reduced BCRP levels (Fig. 4B). The levels of ABCB1/MDR1 or ABCC1/MRP1 were undetectable in all the groups (data not shown).
Given the reported ability of melatonin to relieve the cardotoxicity of Dox [30], we examined expression of several factors associated with this action of melatonin. In heart tissue from rats in the dLEN lighting schedule, the expression of total and phospho-active ERK1/2 and AKT was evident in both the presence and absence of Dox (Fig. 5). Unlike tumor xenografts, cardiomyocytes did not show suppressed expression or phosphorylation of ERK1/2 and AKT in dLEN rats when supplemented with melatonin in any treatment group. No significant change in cardiomyocyte expression of the reductase CBR1 was observed between the different treatment groups, but a significant (p<0.05) 38% increase in the anti-oxidant enzyme SOD2 was evident in cardiomyocytes from Dox-treated dLEN rats supplemented with melatonin vs. those from Dox-treated rats in dLEN.
Figure 5.
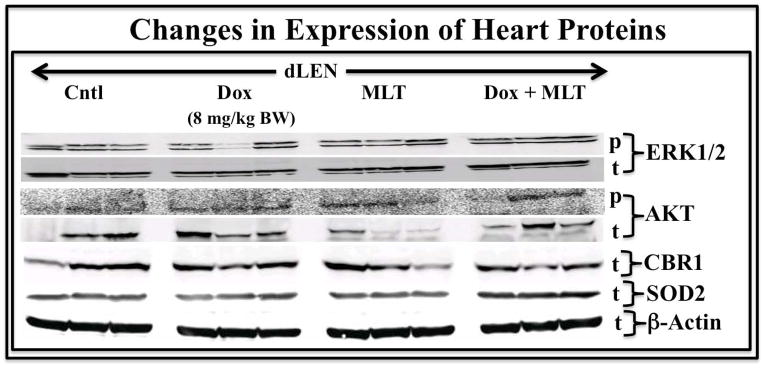
Modulation of heart ERK1/2 and AKT, the Dox metabolizing enzyme CBR1, and the anti-oxidant enzyme SOD2 in female nude rats bearing tissue-isolated (ERα+) MCF-7 human breast tumor xenografts and exposed to dLEN treated with vehicle or Dox or dLEN supplemented with nighttime melatonin and treated with vehicle or Dox. Expression of total (t)- or phospho (p)- ERK1/2, AKT, CBR1, and SOD2 in heart tissue from nude rats in dLEN and treated with vehicle (Cntl) or Dox (Dox) or in a dLEN lighting schedule but supplemented with exogenous melatonin and treated with vehicle (MLT) or Dox (MLT+TAM). All tumors were harvested at 2400 hr (mid-dLEN phase) from 3 animals in each group. Total cell lysates (120 μg of protein/sample) from tumors were analyzed by Western blotting for expression of total or phospho- ERK1/2 and AKT, and total levels of CBR1 and SOD2. β-actin was used as a control for equal loading. This figure was generated by re-probing the same Western blot with all the different antibodies, thus β–actin loading is representative for all samples.
Discussion
Enhancement of tumor metabolism and cell signaling by various kinase and transcription factor pathways is acknowledged as key for the development of drug resistance in human malignancies [1–8, 31]. We have recently demonstrated that disruption of the endogenous circadian melatonin signal in female nude rats in response to dLEN drives human breast tumor xenografts to develop intrinsic resistance to tamoxifen [26]. Unlike bright LEN, which disrupts the activity of the central circadian clock and the circadian melatonin signal, dLEN suppresses only melatonin production without affecting overall central circadian regulatory activities [25, 28]. In the present study, we extended our previous findings on dLEN-induced tamoxifen resistance to examine if circadian disruption may also cause a similar intrinsic resistance to Dox chemotherapy via common mechanistic pathways.
Doxorubicin in addition to other chemotherapeutic agents is commonly used as a first-line treatment for patients with endocrine-resistant or metastatic breast cancer [32]. As with all chemotherapeutic agents many patients develop intrinsic and/or acquired resistance to Dox [2]. Our studies clearly show that host exposure to dLEN drives breast tumor xenografts to intrinsic resistance to Dox indicating that drug resistance induced by dLEN is a broader event impacting both endocrine and drug therapies. To begin to explore the mechanisms involved in dLEN induction and melatonin inhibition of Dox resistance in breast cancer, we focused on three key events involved in cancer chemo-resistance including: (1) tumor metabolism, (2) tumor proliferation and survival cell signaling, and (3) factors involved in drug metabolism and efflux.
As in our previous studies examining tamoxifen resistance [26], dLEN significantly induced high rates of breast tumor xenograft aerobic glycolysis (Warburg effect) and uptake of LA and its metabolism to its mitogenic end product 13-HODE in the absence or presence of Dox. The mitogenic activity of 13-HODE in breast tumor xenografts may include a unique mechanism involving it’s ability to stimulate the Warburg effect via increasing the phospho-activation of AKT [27]. The presence of melatonin during dLEN markedly suppressed the Warburg effect and uptake of LA and its metabolism to 13-HODE either in the absence or presence of Dox. Activation of the Warburg effect (aerobic glycolysis) has been reported to promote the expression/phospho-activation of ERK1/2, AKT, SRC, and cAMP that feed back to amplify the Warburg effect [33]. Thus, dLEN appears to “push” breast tumor xenografts to generate a mutually reinforcing positive feedback loop, involving metabolic and signaling mechanisms, resulting in a hyper-metabolic state that helps drive intrinsic tumor resistance to Dox.
In addition, dLEN-induced suppression of circadian melatonin stimulates the nighttime expression/activation of key proliferation and survival signaling pathways including the EGFR tyrosine kinase family members HER2, HER3, and downstream pathways and nodes including ERK1/2, AKT, CREB, PKA, PAK-1, PDK1, PKCα, PKCδ, NF-kB, RSK2, STAT3 and PCNA. Nighttime supplementation of melatonin during dLEN suppressed or ablated the expression/phospho-activation of each of these signaling molecules. The importance of each of these signaling molecules in breast cancer proliferation, progression, and drug resistance has been well described [2]. Given that elevated expression/activation of the HER2/3 heterodimer can lead to downstream activation of various signaling nodes and cascades noted above [34, 35], it is possible that the initiating signaling event promoting tumor progression and resistance to Dox is the induction of HER2 and HER3 expression. Since dLEN stimulates rapid tumor uptake of LA and its metabolism to 13-HODE, an activator of EGFR tyrosine kinase [36], it is conceivable that 13-HODE may also promote HER2 or HER3 activation to induce the further activation of downstream signaling pathways.
Recently, Sims et al. [14] reported that STAT3/NF-kB are involved in regulating Dox resistance in human melanoma cells and promote oncogenesis by stimulating the transcription of proliferative, invasive, and anti-apoptotic genes. Our findings strongly suggest that host exposure to dLEN via its induction/activation of STAT3 and NF-kB in breast tumor xenografts promotes intrinsic resistance to Dox. Conversely, melatonin supplementation during dLEN in combination with Dox may induce tumor regression by inhibiting the expression/phospho-activation of various transcription factors including STAT3 and NF-kB.
Dim light exposure at night also resulted in increased expression/phospho-activation of PDK-1 and PAK-1 in breast cancer xenografts. PAK1, a serine-threonine protein kinase regulates the PI3K/AKT, RAS/ERK signaling pathways [37] and is required for full activation of these signaling pathways [38]. Both PDK-1 and PDK-3 via inhibition of pyruvate dehydrogenase, a key regulator of cellular glucose utilization and mitochondrial oxidation, enhance aerobic glycolysis in tumor cells [39] and thus promote chemo-resistance [40, 41]. These findings combined with our current studies provide evidence that dLEN drives tumor xenografts to aerobic glycolysis (Warburg effect) in part by inducing PDK-1 expression and that dLEN-induced hyper-activation of aerobic glycolysis is involved in the development of Dox resistance. Melatonin’s ability to inhibit the expression/phospho-activation of these kinases further supports a new regulatory role for melatonin as a circadian regulated kinase inhibitor [26] that can suppress or inhibit intracellular signaling pathways associated with drug resistance and tumor progression.
At first glance, there seems to be a discrepancy between the dramatic suppressive action of melatonin on kinase and transcription factor expression/activation and cell proliferative activity that exceeds only a 50% decrease in the overall tumor growth rate. This can be explained on the basis of that the endogenous physiological melatonin signal, as well as the melatonin signal simulated via exogenous melatonin supplementation in the present study, is only present during the 12-hr dark phase of the light/dark cycle [26, 27]. Therefore, melatonin is suppressing kinase and transcription factor expression/activation and cell proliferative activity only during dark phase with maximal inhibition occurring during the mid-dark phase (2400 hr) when these parameters were measured. During the light phase when melatonin levels are low to non-detectable, these signaling and proliferative pathways are operating maximally. The net effect is that cell proliferative activity is only maximally inhibited for one-half of the 24-hr day resulting in an overall tumor growth rate that is only 50% slower than when melatonin is suppressed in dLEN [26, 27].
From a mechanistic standpoint, it appears as though the lead events induced by dLEN that drive intrinsic drug resistance include the enhanced expression of HER2 and HER3 that promote RAS expression and the phospho-activation of ERK1/2. Signaling nodes and pathways downstream of ERK1/2 include PDK1, PAK1, PKCα/δ, AKT, NF-kB, CREB, and STAT3. Given the central role of STAT3 in promoting cancer cell survival, cell proliferation, and drug resistance [40], and that cAMP/PKA/CREB, HER2/ERK1/2, PAK1, RSK2, and PKCα all phospho-activate and/or transcriptionally activate STAT3, we postulate that STAT3 is the distal common signal node critical for dLEN/circadian-disruption-induced chemo-resistance in breast cancer. Our data supports that suppression of signaling nodes upstream of STAT3 by melatonin suppress STAT3 phospho-activation to block chemo-resistance. Clearly, additional studies using constitutively-active or dominant-negative forms of these signaling nodes, including STAT3, are needed to define which pathway(s) is most critical in regulating chemo-resistan
Doxorubicin metabolism and clearance are predominantly mediated by the liver by the phase I enzymes CBR1 and AKR1C3 [19, 20, 41, 42]. CBR1 the major hepatic carbonyl reductase in the liver is also expressed in breast tumors [42]. The increased expression of CBR1 and AKR1C3 in breast tumor xenografts in response to dLEN, suggest that Dox is rapidly metabolized to its less active but more cardiotoxic metabolite, doxorubicinol. The suppressed expression of CBR1 and AKR1C3 during dLEN by nocturnal melatonin supplementation, suggests that Dox metabolism is greatly reduced and that intratumoral Dox levels remain high in the presence of melatonin to promote tumor cell killing. The suppressed expression of BCRP, and ABC efflux pump, during dLEN by melatonin supplementation would also suggest that the intratumoral levels of Dox would remain high during the melatonin phase to promoting tumor cell killing. Although we have not yet defined the mechanism(s) involved in melatonin regulation of BCRP specifically in our model system, Martin et al. [43] reported that melatonin induces hypermethylation of the BCRP promoter to suppress BCRP expression. Thus, dLEN, via its disruption of nighttime melatonin may suppress intratumoral levels of Dox by inducing its metabolism and efflux.
Our studies show that melatonin, in addition to enhancing tumor responsiveness to Dox therapy when administered during dLEN, it maintained ERK1/2 and AKT expression in rat cardiomycytes. As caridotoxicity is a significant side effect of Dox therapy [23] and studies report that elevated levels of PDK1, AKT, and ERK1/2 are important for mitigating cardiac toxicity by protecting the heart from ischemia-reperfusion injury [44], the ability to melatonin to maintain the expression of these kinases suggests that it may play a cardio-protective role. Interestingly, melatonin does not suppress ERK1/2 and AKT expression/activity in cardiomyocytes, contrary to its action in breast tumor xenografts, ostensibly protecting cardiomyocytes from doxorubicinnol toxicity. The mechanism(s) involved in this difference between tumor xenograft and cardiomyocytes is currently unknown. However, G-protein switching, as described for other G protein-coupled receptors [45], may be involved such that the activated MT1 receptor may prefer the Gαi2 protein in the tumor xenograft but a Gαq or Gα11 protein in the heart.
The generation of hydroxyl radicals, hydrogen peroxide, and superoxide anions in response to Dox treatment represents another important component of Dox-induced cardiotoxicity [46, 47]. Work by Reiter et al. [48] defined melatonin as a potent scavenger of free radicals and activator of endogenous antioxidant enzymes that protects various organs from oxidative damage. Other groups reported that via its anti-oxidant action melatonin reduces Dox-induced cardiotoxicity [49]. Consequently, our data showing that exogenous melatonin administration during dLEN increases the expression of the antioxidant enzyme SOD2 while maintaining ERK1/2 and AKT signaling in rat cardiomyocytes indicates an additional level of protection against Dox-induced cardiotoxicity.
In conclusion, the present investigation validates the importance of an intact endogenous nocturnal circadian melatonin signal in sensitizing human breast tumor cells to chemotherapy, particularly Dox. As in our previous report, nighttime melatonin significantly represses tumor kinase signaling, and acts broadly to suppress metabolic, -signaling, -proliferative, and –survival activity in breast cancer. As in our tamoxifen-resistance study [26], this study confirms that a comprehensive understanding and maintenance of host/cancer circadian biology and the circadian-regulated nature of cancer metabolism, cell signaling, and drug metabolic and efflux pumping mechanisms is essential to maximize the efficacy of Dox and possibly other chemotherapies. This study also suggests that the actions of melatonin may be cardio-protective through mechanisms that appear to operate differently in heart vs. tumor tissues. The achievement of maximal efficacy and minimal toxicity (e.g., optimal therapeutic index) of such chemotherapies appear to be predicated upon their optimal temporal administration in alignment with a patient’s own intact endogenously regulated circadian timing system (e.g., nocturnal melatonin signal). Given that many, if not all, breast cancer patients are likely subjected to various degrees of LEN as a result of stress, lack of sleep, and/or continued night shift work, and thus are circadian/melatonin disrupted, LEN may represent a unique risk factor that could account for some forms of intrinsic and possibly acquired resistance to Dox and potentially other agents resulting in an increased toxicity and diminished quality of life, shortened survival time and/or possibly a decreased survival rate in breast cancer patients. Therefore, one could envision chemotherapy being administered in a circadian-optimized manner in combination with exogenous melatonin therapy to a breast cancer patient experiencing LEN-induced circadian disruption.
Acknowledgments
Financial Information:
This work was supported by the following grants NIH Grant R21CA129875–04 (to DEB) and an American Association for Laboratory Animal Science GLAS grant (RTD and DEB). Part of the cost of this work was defrayed with the support of the Edmond and Lily Safra Endowed Chair for Breast Cancer Research (SMH) at Tulane Cancer Center.
Footnotes
The Authors disclose no potential conflicts of interest.
References
- 1.AUSTREID E, LONNING PE, EIKESDAL HP. The emergence of targeted drugs in breast cancer to prevent resistance to endocrine treatment and chemotherapy. Expert Opin Pharmacother. 2014;5:681–700. doi: 10.1517/14656566.2014.885952. [DOI] [PubMed] [Google Scholar]
- 2.GARIBOLDI MB, RAVIZZA R, RUGANTI L, et al. Molecular determinants of intrinsic resistance to doxorubicin in human cancer cell lines. Int J Oncol. 2003;22:1057–64. [PubMed] [Google Scholar]
- 3.SINGAL PK, ILISKOVIC N, LI T. Adriamycin cardiomyopathy: pathophysiology and prevention. FASEB J. 1997;11:931–6. doi: 10.1096/fasebj.11.12.9337145. [DOI] [PubMed] [Google Scholar]
- 4.GEWITZ DA. critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57:727–41. doi: 10.1016/s0006-2952(98)00307-4. [DOI] [PubMed] [Google Scholar]
- 5.HEGER Z, CERNEI N, KUDR J, et al. A novel insight into the cardiotoxicity of antineoplastic drug doxorubicin. Int J Mol Sci. 2013;14:21629–46. doi: 10.3390/ijms141121629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.GONZALEZ AM, MORALES-VASQUEZ F, HORTOBAGYI GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol. 2007;608:1–22. doi: 10.1007/978-0-387-74039-3_1. [DOI] [PubMed] [Google Scholar]
- 7.TAVARES-VALENTE D, BALTAZAR F, MOREIRA R, et al. Cancer cell bioenergetics and pH regulation influence breast cancer cell resistance to paclitaxel and doxorubicin. J Bioenerg Biomembr. 2013;5:467–75. doi: 10.1007/s10863-013-9519-7. [DOI] [PubMed] [Google Scholar]
- 8.HUA G, LIU Y, LI X, et al. Targeting glucose metabolism in chondrosarcoma cells enhances the sensitivity to doxorubicin through the inhibition of lactate dehydrogenase-A. Oncol Rep. 2014;6:2727–34. doi: 10.3892/or.2014.3156. [DOI] [PubMed] [Google Scholar]
- 9.BURRIS HA. Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol. 2013;4:829–42. doi: 10.1007/s00280-012-2043-3. [DOI] [PubMed] [Google Scholar]
- 10.ROSKOSKI R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 11.MCCUBREY JA, STEELMAN LS, CHAPPELL WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–84. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LI Q, MULLINS SR, SLOANE BF, et al. P21-activated kinase 1 coordinates aberrant cell survival and pericellular proteolyisis in a three-demensional culture model for premalignant progression of human breast cancer. Neoplasia. 2008;10:314–28. doi: 10.1593/neo.07970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ROMEO Y, ZHANG X, ROUX PP. Regulation and function of the RSK family of protein kinases. Biochem J. 2012;441:553–69. doi: 10.1042/BJ20110289. [DOI] [PubMed] [Google Scholar]
- 14.SIMS JT, GANGULY SS, BENNETT H, et al. Imatinib reverses doxorubicin resistance by affecting activation of STAT3-dependent NF-κB and HSP27/p38/AKT pathways and by inhibiting ABCB1. PLoS One. 2013;8:e55509. doi: 10.1371/journal.pone.0055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LEE SA, KARAZKIEWICZ JW, ANDERSON WB. Elevated level of nuclear protein kinase C in multidrug-resistant MCF-7 human breast carcinoma cells. Cancer Res. 1992;52:3750–9. [PubMed] [Google Scholar]
- 16.DIAZ-BESSONE MI, BERARDI DE, CAMPODONICO PB, et al. Involvement of PKC delta (PKCδ) in the resistance against different doxorubicin analogs. Breast Cancer Res Treat. 2011;126:577–87. doi: 10.1007/s10549-010-0956-2. [DOI] [PubMed] [Google Scholar]
- 17.GOTTESMAN MM, FOJO T, BATES SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 18.ZHANG W, DING W, CHEN Y, et al. Up-regulation of breast cancer resistance protein plays a role in HER2-mediated chemoresistance through PI3K/Akt and nuclear factor-kappa B signaling pathways in MCF7 breast cancer cells. Acta Biochim Biophys Sin. 2011;43:647–53. doi: 10.1093/abbs/gmr050. [DOI] [PubMed] [Google Scholar]
- 19.KASSNER N, HUSE K, MARTIN HJ, et al. Carbonyl reductase 1 is a predominant doxorubicin reductase in the human liver. Drug Metab Dispos. 2008;36:2113–20. doi: 10.1124/dmd.108.022251. [DOI] [PubMed] [Google Scholar]
- 20.NOVOTNA R, WSOL V, XIONG G, et al. Inactivation of the anticancer drugs doxorubicin and oracin by aldo-keto reductase (AKR) 1C3. Toxicol Lett. 2008;181:1–6. doi: 10.1016/j.toxlet.2008.06.858. [DOI] [PubMed] [Google Scholar]
- 21.JOERGER M, HUITEMA AD, MEENHORST PL, et al. Pharmacokinetics of low-dose doxorubicin and metabolites in patients with AIDS-related Kaposi sarcoma. Cancer Chemother Pharmacol. 2005;55:488–96. doi: 10.1007/s00280-004-0900-4. [DOI] [PubMed] [Google Scholar]
- 22.FERRAZZI E, WOYNAROWSKI JN, ARAKALI A, et al. DNA damage and cytotoxicity induced by metabolites of anthracycline antibiotics, doxorubicin and idarubicin. Cancer Commun. 1991;3:173–80. doi: 10.3727/095535491820873308. [DOI] [PubMed] [Google Scholar]
- 23.OLSON RD, MUSHLIN PS, BRENNER DE, et al. Doxoruibicin cardiotoxicity may be due to its metabolite, doxorubicinol. PNAS USA. 1988;85:3585–89. doi: 10.1073/pnas.85.10.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.ARENDT J. Melatonin and human rhythms. Chronobiol Int. 2006;23:21–37. doi: 10.1080/07420520500464361. [DOI] [PubMed] [Google Scholar]
- 25.HILL SM, BLASK DE, XIANG S, et al. Melatonin and associated signaling pathways that control normal breast epithelium and breast cancer. J Mammary Gland Biol Neoplasia. 2011;16:235–45. doi: 10.1007/s10911-011-9222-4. [DOI] [PubMed] [Google Scholar]
- 26.DAUCHY RT, XIANG S, MAO L, et al. Circadian and melatonin disruption by exposure to light at night drives intrinsic resistance to tamoxifen in breast cancer. Cancer Res. 2014;74:4099–119. doi: 10.1158/0008-5472.CAN-13-3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.BLASK DE, DAUCHY RT, DAUCHY EM, et al. Light exposure at night disrupts host/cancer circadian regulatory dynamics: Impact on the Warburg effect, lipid signaling and tumor growth prevention. PLoS One. 2014;9:e102776. doi: 10.1371/journal.pone.0102776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.BLASK DE, BRAINARD GC, DAUCHY RT, et al. Melatonin-depleted blood from premenopausal women exposed to light at night stimulates growth of human breast cancer xenografts in nude rats. Cancer Res. 2005;65:11174–84. doi: 10.1158/0008-5472.CAN-05-1945. [DOI] [PubMed] [Google Scholar]
- 29.LAZENNEC G, THOMAS JA, KATZENELLENBOGEN BS. Involvement of the cAMP response element binding protein (CREB) and estrogen receptor phosphorylation in the synergistic action of the estrogen receptor by estradiol and protein kinase activators. J Steroid Biochem Mol Biol. 2001;77:193–203. doi: 10.1016/s0960-0760(01)00060-7. [DOI] [PubMed] [Google Scholar]
- 30.DZIEGIEL P, JETHON Z, SUDER E, et al. Role of exogenous melatonin in reducing the cardiotoxic effect of daunorubicin and doxorubicin in the rat. Exp Toxicol Pathol. 2002;53:433–9. doi: 10.1078/0940-2993-00217. [DOI] [PubMed] [Google Scholar]
- 31.LIN MC, YIN MC. Preventive effects of ellagic acid against doxorubicin-induced cardio-toxicity in mice. Cardiovasc Toxicol. 2013;13:185–93. doi: 10.1007/s12012-013-9197-z. [DOI] [PubMed] [Google Scholar]
- 32.MARQUETTE C, MABELL L. Chemotherapy-resistant metastatic breast cancer. Curr Treat Options Oncol. 2012;13:263–75. doi: 10.1007/s11864-012-0184-6. [DOI] [PubMed] [Google Scholar]
- 33.HUA G, LIN Y, LI X, et al. Targeting glucose metabolism in chondrosarcoma cells enhances the sensitivity to doxorubicin through the inhibition of lactate dehydrogenase-A. Oncol Rep. 2014;31:2727–34. doi: 10.3892/or.2014.3156. [DOI] [PubMed] [Google Scholar]
- 34.SLAMON DJ, CLARK GM, WONG SG, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 35.HOLBRO T, BEERLI RR, MAURER F, et al. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci USA. 2003;100:8933–38. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.ELING TE, EVERHART AL, ANGERMAN-STEWART J, et al. Modulation of epidermal growth factor signal transduction by linoleic acid metabolites. Adv Exp Med Biol. 1997;407:319–22. doi: 10.1007/978-1-4899-1813-0_47. [DOI] [PubMed] [Google Scholar]
- 37.CHOW HY, JUBB AM, KOCH HN, et al. p21-activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res. 2012;72:5966–75. doi: 10.1158/0008-5472.CAN-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.HIGUCHI M, ONISHI K, KIKUCHI C, et al. Scaffolding function of PAK in the PKD-1-Akt pathway. Nat Cell Biol. 2008;10:1356–64. doi: 10.1038/ncb1795. [DOI] [PubMed] [Google Scholar]
- 39.VELPULA KK, BHASIN A, ASUTHKAR S, et al. Combined targeting of PDK1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013;73:7277–89. doi: 10.1158/0008-5472.CAN-13-1868. [DOI] [PubMed] [Google Scholar]
- 40.FANG B. Genetic interactions of STAT3 and anticancer drug development. Cancers. 2014;6:494–525. doi: 10.3390/cancers6010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.LAL S, SANDANARAJ E, WONG ZW, et al. CBR1 and CBR3 pharmacogenetics and their influence on doxorubicin disposition in Asian breast cancer patients. Cancer Sci. 2008;99:2045–54. doi: 10.1111/j.1349-7006.2008.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.OHARA H, MIYABE Y, DEYASHIKI Y, et al. Reduction of drug ketones by dihydrodiol dehydrogenase, carbonyl reductase and aldehyde reductase of human liver. Biochem Pharmacol. 1995;50:221–227. doi: 10.1016/0006-2952(95)00124-i. [DOI] [PubMed] [Google Scholar]
- 43.MARTIN V, SANCHEZ-SANCHEZ AM, HERRERA F, et al. Melatonin-induced methylation of the ABCG2/BCRP promoter as a novel mechanism to overcome multidrug resistance in brain tumour stem cells. Br J Cancer. 2013;108:2005–12. doi: 10.1038/bjc.2013.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.KLEMENT GL, GOUKASSIAN D, HLATY L, et al. Cancer Therapy Targeting the HER2-PI3K Pathway: Potential Impact on the Heart. Front Pharmacol. 2012;3:113. doi: 10.3389/fphar.2012.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.MAMILLAPALLI R, VANHOUTEN J, ZQWALICH W, et al. Switching of G-protein usage by the calcium-sensing receptor reverses its effect on parathyroid hormone-related protein secretion in normal versus malignant breast cells. J Biol Chem. 2008;283:24435–47. doi: 10.1074/jbc.M801738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LEE V, RANDHAWA AK, SINGAL PK. Adriamycin-induced myocardial dysfunction in vitro is mediated by free radicals. Am J Physiol. 1991;261:H989–95. doi: 10.1152/ajpheart.1991.261.4.H989. [DOI] [PubMed] [Google Scholar]
- 47.SARVAZYAN NA, ASKARI A, HUANG WH. Effects of doxorubicin on cardiomyocytes with reduced level of superoxide dismutase. Life Sci. 1995;57:1003–10. doi: 10.1016/0024-3205(95)02036-i. [DOI] [PubMed] [Google Scholar]
- 48.REITER R, TANG L, GARCIA JJ, et al. Pharmacological actions of melatonin in oxygen radical pathophysiology. Life Sci. 1997;60:2255–71. doi: 10.1016/s0024-3205(97)00030-1. [DOI] [PubMed] [Google Scholar]
- 49.AHMED HH, MANNAA F, ELMEGEED GA, et al. Cardioprotective activity of melatonin and its novel synthesized derivatives on doxorubicin-induced cardiotoxicity. Bioorg Med Chem. 2005;13:1847–57. doi: 10.1016/j.bmc.2004.10.066. [DOI] [PubMed] [Google Scholar]