

Abstract
A large number of DNA copy number alterations (CNAs) exist in human breast cancers, and thus characterizing the most frequent CNAs is key to advancing therapeutics because it is likely that these regions contain breast tumor ‘drivers’ (i.e., cancer causal genes). This study aims to characterize the genomic landscape of breast cancer CNAs and identify potential subtype-specific drivers using a large set of human breast tumors and genetically engineered mouse (GEM) mammary tumors. Using a novel method called SWITCHplus, we identified subtype-specific DNA CNAs occurring at a 15 % or greater frequency, which excluded many well-known breast cancer-related drivers such as amplification of ERBB2, and deletions of TP53 and RB1. A comparison of CNAs between mouse and human breast tumors identified regions with shared subtype-specific CNAs. Additional criteria that included gene expression-to-copy number correlation, a DawnRank network analysis, and RNA interference functional studies highlighted candidate driver genes that fulfilled these multiple criteria. Numerous regions of shared CNAs were observed between human breast tumors and GEM mammary tumor models that shared similar gene expression features. Specifically, we identified chromosome 1q21-23 as a Basal-like subtype-enriched region with multiple potential driver genes including PI4KB, SHC1, and NCSTN. This step-wise computational approach based on a cross-species comparison is applicable to any tumor type for which sufficient human and model system DNA copy number data exist, and in this instance, highlights that a single region of amplification may in fact harbor multiple driver genes.
Electronic supplementary material
The online version of this article (doi:10.1007/s10549-015-3476-2) contains supplementary material, which is available to authorized users.
Keywords: Copy number alterations, Intrinsic subtypes, Driver genes, Gene expression, Genetically engineered mouse, Network analysis
Introduction
Breast cancer is a heterogeneous disease that is characterized by distinct histological forms, genetic alterations, and patient outcomes [1–6]. Consistent with these observations, differential gene expression can distinguish molecular subtypes that separate breast cancer into distinct groups including Basal-like, Claudin-low, HER2-enriched, Luminal A, and Luminal B subtypes [2–4, 7–9]. These so called “intrinsic subtypes” are predictive of relapse-free survival, overall survival, and responsiveness to treatment [7–11]. Previous work highlighted numerous somatic mutations [12] and DNA copy number alterations (CNAs) [13] that are linked to specific intrinsic subtypes, suggesting that these genetic events may be causative of these subtypes. Beyond a few well-known drivers, the identification of genetic drivers present in many of these recurrent regions of DNA copy number change remains to be determined. Specifically, numerous CNAs are located on chromosome 1 and occur at high frequency among various cancer types including breast and liver [12, 14]. In breast cancer, copy number loss frequently occurs at 1p while copy number gains are frequent at 1q [13]. Furthermore, copy number gains at 1q often encompass the majority of the 1q arm, which include hundreds of genes.
To identify additional genetic drivers of breast cancer in common regions of amplification, we have taken a cross-species conservation approach based on the hypothesis that important etiological events in breast tumors will occur both in human breast cancers and mouse mammary tumor models. Through combined DNA copy number analyses of human breast tumors and multiple genetically engineered mouse (GEM) mammary tumor models, we identified 662 CNA regions conserved between these two species. Our ultimate selection strategy also incorporated gene expression data, an RNAi screen, and a network analysis to focus the list on the most likely driver genes within CNAs. Furthermore, using published functional studies, we provide new insights on the potential implications of Basal-like tumor-specific chromosome 1 drivers, some of which are therapeutically targetable.
Methods
Breast cancer tumor datasets
For these comparative studies, two human datasets and one mouse dataset were used that contained both gene expression and DNA copy number data (Table 1). The two human datasets were: (1) tumors collected at the University of North Carolina at Chapel Hill and the Oslo University Hospital, Radiumhospitalet, Norway (“UNC”, n = 159, GSE52173), and (2) The Cancer Genome Atlas (TCGA) Project dataset [12] (“TCGA”, n = 485). The third dataset contained tumors from numerous mouse mammary tumor models including GEM mammary models with inactivation of TP53, BRCA1, BRG1, and over-expression of cMYC, HER2/ERBB2/Neu, PyMT, and WNT1 (“mouse”, n = 73, GSE52173) (Supplemental Table 1). The publically available level 3 segmented copy number data for the TCGA dataset was downloaded through the TCGA data portal and the published PAM50 subtype calls were used [12]. Demographic and clinical characteristics of the UNC tumors are provided in Supplemental Table 2.
Table 1.
Copy number array sample information of (a) human and (b) mouse tumors
| (a) Subtypes | Number of samples | Total |
|---|---|---|
| Basal-like | UNC: 54, TCGA: 89 | 143 |
| Claudin-low | UNC: 20, TCGA: 8 | 28 |
| HER2-enriched | UNC: 16, TCGA: 55 | 71 |
| Luminal A | UNC: 35, TCGA: 213 | 248 |
| Luminal B | UNC: 34, TCGA: 120 | 154 |
| (b) Expression SigClust group | Number of samples |
|---|---|
| Wap Myc | 10 |
| Neu/PyMT | 11 |
| Wnt1 | 16 |
| C3Tag | 8 |
| Mixed | 6 |
| p53null-Basal | 9 |
| p53null-Luminal | 13 |
Cross-species assessment of subtype-specific changes in genomic DNA copy number
To identify subtype-specific CNAs from segmentation data generated by the various copy number array platforms (see Supplemental File 1 for details), we produced an add-on script to the SWITCHdna method of DNA copy number change point detection [13]. We created an R suite of functions called SWITCHplus, which can identify segments of the genome with copy number changes specific for a user-determined set of tumors, thus providing a supervised method for analyzing copy number data. SWITCHplus is provided as a source script in R and available for download at: https://genome.unc.edu/SWITCHplus/. Note, that we did not perform multiple hypothesis testing corrections as we chose alternative biologically based filtering criteria (Fig. 1) based upon cross-species conservation.
Fig. 1.

Data analysis pipeline to identify candidate driver genes within subtype-specific CNAs
Computational analysis of candidate driver genes within conserved CNAs
In order to identify putative driver alterations within regions of copy number gains or losses, we began with all the conserved CNAs with a subtype segment frequency of 15 % or greater. To distinguish putative drivers from passengers, three further criteria were used. We first identified genes within a CNA that demonstrate concordance between the DNA and RNA expression. The second criterion filtered for conserved CNAs that contained genes with a breast cell line RNAi-associated phenotype as published in the Solimini et al. 2012 RNAi screen on human mammary epithelial cells [15]. The third criterion was to identify top ranking genes when scored using DawnRank [16]. By combining all these features together, we further decrease the false positive genes by filtering out genes without functional implications (Supplemental Table 3). A more extensive and detailed “Methods” section can be found as Supplemental File 1.
Results
Subtype-specific breast cancer copy number landscapes
In order to identify both known and novel genetic drivers of breast cancer on the DNA copy number level, we developed a multi-step and multi-platform computational strategy (Fig. 1). This strategy is predicated on using a “cross-species” comparative genomics approach where we searched for spontaneous copy number events across two different species (human and mouse). For this study, we created a new murine genomic resource of 73 mammary tumors profiled by both gene expression and DNA copy number microarray data (GSE52173); this new resource complements our human data set that contains 644 human breast tumors that have both gene expression and DNA copy number data (GSE52173 and http://tcga-data.nci.nih.gov/tcga).
We began using gene expression data to identify subtypes, separately for human tumor samples and GEM mammary models. For clarity, we refer to the classification of mouse tumors as “groups” to distinguish them from human classes that are termed “subtypes”. Using the PAM50 [8] algorithm and the Claudin-low predictor [9] we assigned each of the human tumor samples within the dataset to a specific intrinsic breast cancer subtype (Table 1). However, since there is no established expression-based classifier for mouse mammary tumors, we performed a supervised hierarchical cluster analysis of the murine mRNA expression data using the Herschkowitz et al. 2007 intrinsic mouse list of 866 genes. SigClust [17] analysis was used to identify 7 significant mouse groups (Supplemental Fig. 1), which were given a unique group name based on the majority mouse model contributor in that group (i.e., Myc, Neu/PyMT, Wnt1, C3Tag, Mixed, p53null-Basal, and p53null-Luminal). The “Mixed” mouse group lacked a single dominant mouse model contributor, however, this group comprised mouse tumors that all demonstrate the previously described Claudin-low gene expression features [18, 19], and hence forth this mouse group is referred to as “ClaudinLow”.
To identify subtype-specific, and mouse group-specific regions of DNA copy number gains and/or losses we developed a new bioinformatics visualization tool called SWITCHplus. Applying this tool to the mouse dataset identified group-specific DNA copy number changes for each of the seven expression-defined groups (Fig. 2). These results suggest that most mouse groups are characterized by numerous DNA copy number changes, many of which are specific to a given model/group (Supplemental Table 4). However, by comparing the copy number landscape between mouse groups, we also identified CNAs that were present in multiple models (Fig. 2; Supplemental Table 4), which can be considered as common CNAs of murine mammary oncogenesis. Therefore, these data support the notion that common spontaneous events may occur within different GEM mammary models irrespective of the initiating genetic event (i.e., transgene). Consistent with previous work, we identified multiple GEM mammary p53null groups based on gene expression patterns [18, 19]. Interestingly, these p53null groups demonstrated not only differences in mRNA expression patterns, but also exhibited differences in the DNA copy number landscapes (Fig. 2c, d). Additionally, we noticed that the p53null-Luminal, p53null-Basal, and C3Tag groups contained more group-specific CNAs than any of the other mouse groups (Supplemental Table 4); this observation is likely due to the loss of TP53 in these three groups. On average, each mouse group exhibited nearly twice the number of group-specific copy number gains versus losses.
Fig. 2.

Copy number frequency landscape plots from SWITCHplus showing mouse group-specific CNAs. Segments of group-specific copy number gains are plotted above the x-axis in red and segments of copy number loss are plotted below the x-axis in green. Regions shaded gray indicate segments that are not group-specific or highly frequent (greater than or equal to 15 %). The frequency of alterations in each mouse group is indicated on the y-axis from 0 to 100 %. a C3Tag, b Neu/PyMT, c p53null-Basal, d p53null-Luminal, e Myc, f Wnt1, and g Claudin-Low copy number landscapes
We next analyzed the human DNA copy number landscape in the combined UNC/TCGA breast cancer dataset (Supplemental Fig. 2; Supplemental Table 5). Our results, not surprisingly, were consistent with previous publications [6, 12, 13]. For example, our analyses confirmed previously identified breast cancer copy number gains of 8q that is common and present irrespective of breast cancer subtype, as well as a number of subtype-specific CNAs. For instance, we again identified Basal-like-specific DNA copy number losses at 4q, 5q, and gains of 10p; Luminal A-specific copy number gains at 16p; Luminal B-specific copy number gains at 17q; and a Luminal-associated (encompassing both Luminal A and Luminal B) copy number loss at 16q (Supplemental Fig. 2; Supplemental Table 5) [6, 12, 13, 20, 21]. The HER2-enriched subtype contained few subtype-specific CNAs, noting that the HER2/ERBB2 amplicon was not a HER2-enriched subtype-specific copy number gain event as it also occurred in many Luminal tumors. Additionally, the Basal-like subtype contained the highest number of subtype-specific CNAs (Supplemental Table 5). In contrast to what was observed in the mouse groups, human tumors on average demonstrated more frequent subtype-specific regions of copy number loss compared to copy number gains (Supplemental Table 5).
Comparisons of copy number landscapes of mouse and human breast tumors
The extent to which mouse models of breast cancer recapitulate human phenotypes has been examined at the gene expression level [18–20], as well as on the copy number level, albeit only in a much smaller subset of these data [20]. We examined sub-chromosomal events and compared human subtype-specific copy number landscape plots to mouse group-specific landscape plots and identified shared cross-species CNA events [after re-ordering the mouse chromosomal landscape into human chromosome order (see “Methods” section)]. We first selected for “conserved regions”, which were DNA segments/regions that were altered at high frequency (≥15 %) and in the same direction (i.e., amplified or lost) in both human and mouse copy number landscapes. Applying this selection criterion reduced the search space for potential subtype-specific drivers more than 2-fold, leaving a total of 662 conserved regions when all mouse groups and human subtypes were considered (Supplemental Fig. 3; Supplemental Table 7).
In comparison among subtypes, the Claudin-low subtype had the fewest number of conserved regions (and the fewest CNAs overall) (Supplemental Table 7). Conversely, the Basal-like subtype contained the most conserved CNAs; however, this may be due to the fact that the Basal-like subtype also contained the most subtype-specific CNAs (Supplemental Table 7). Consistent with a previous publication [20], shared Basal-like-specific and murine p53null-Basal-specific regions of DNA copy number loss was observed spanning human 4q31-q35.2 and encompassing INPP4B, and also spanning 14q22.1-23.1 (Supplemental Table 7). By comparing shared sub-chromosomal CNAs between the human Basal-like subtype and all mouse groups, we noted that the C3Tag mouse group contained the most human Basal-like-specific copy number amplified regions, while the p53null-Basal mouse group contained the most human Basal-like-specific copy number loss regions (Supplemental Table 7). Both of these mouse models were previously shown to have the Basal-like tumor gene expression phenotypes [18, 20], therefore, for this study, we largely focused on copy number commonalities between human Basal-like tumors and these two mouse groups.
Identification of Basal-like tumor chromosome 1 amplification driver genes
Across all breast tumors, amplification of human chromosome 1q was the most frequent copy number altered event (not depicted). However, as can be seen in Supplemental Figs. 2 and 3, the “shape” of the chromosome 1 amplification varies by subtype, with the subtype-enriched amplification regions being identified within this largest of human chromosome arms. Among the 662 conserved regions identified across the genome, chromosome 1 harbored 18 % of all conserved CNAs (Supplemental Table 7). Focusing on chromosome 1, we determined that chromosome 1q harbored more than twice the number of conserved segments when compared to the 1p arm (Supplemental Table 7). Of particular note, a number of 1q amplified regions that were identified as human Basal-like-specific were also altered in the mouse C3Tag and/or p53null groups (Fig. 3b; Supplemental Table 7); thus our results indicate that this region of human chromosome 1q21-23 is being repeatedly selected for both mouse and human Basal-like breast cancers.
Fig. 3.
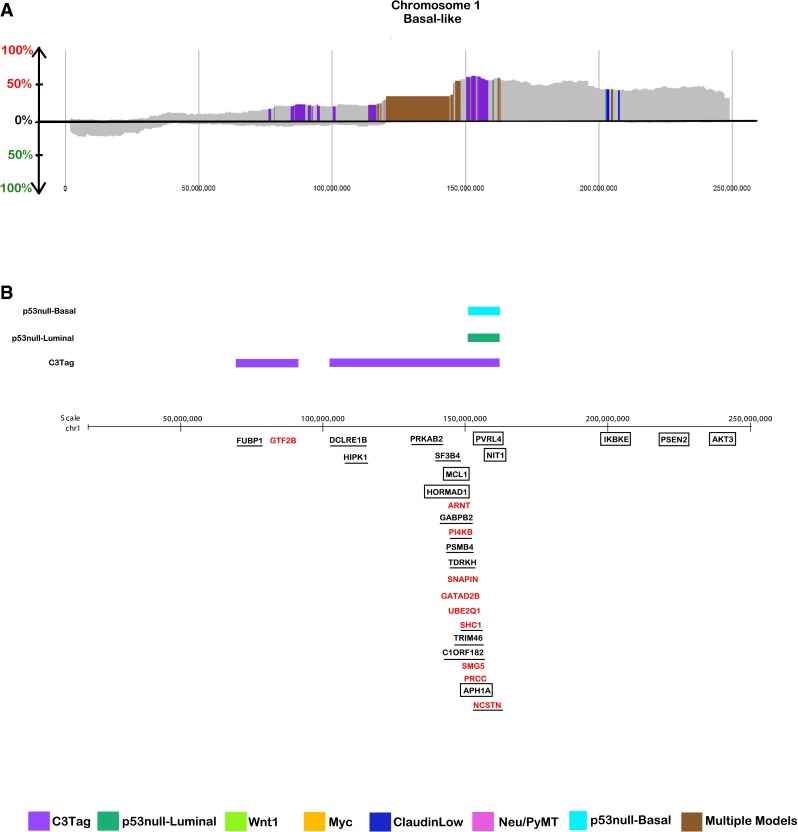
Expanded view of a chromosome 1 Basal-like conserved copy number frequency landscape plots from SWITCHplus. Segments of copy number gains are plotted above the x-axis and segments of copy number loss are plotted below the x-axis. The conserved segments are colored according to the mouse model(s) in which they appear. The frequency of alterations is indicated on the y-axis. Regions shaded gray indicate segments that are any combination of either not subtype-specific, not mouse group-specific, or not high frequent (greater than or equal to 15 %). b View of the genomic location of candidate chromosome 1 driver genes. Genes colored red are Basal-like-specific or subtype-associated, demonstrate DNA and RNA concordance in human tumors and had a top DawnRank score; genes underlined are Basal-like-specific or Basal-like-associated, demonstrate DNA and RNA concordance in human tumors and labeled as a growth enhancer and oncogene (“GO gene”) in the Solimini et al. [15] RNAi screen on human mammary epithelial cells; the remaining genes surrounded by a box are additional potential drivers in this region. A color bar is placed above the genes conserved for a particular mouse group
In order to identify the driver(s) present on chromosome 1, we next applied our filtering criteria outlined in Fig. 1. Of the 120 chromosome 1 conserved CNAs, 79 contained at least one gene that showed DNA–RNA concordance (Supplemental Table 8); 25 CNAs contained at least one RNAi-identified essential gene (Supplemental Table 9), and 20 CNAs contained genes showing DNA–RNA concordance and a RNAi-identified essential gene (Supplemental Table 10). Interestingly, all 20 CNAs were copy number gained segments, even among the 1p CNAs (Supplemental Table 10).
To further study the biology of the conserved chromosome 1 genes, we performed a cohort-based DawnRank [16] analysis using genes from human chromosome 1. DawnRank uses gene–gene interaction networks to measure the impact of genomic alterations on the differential gene expression of downstream genes in the network. Then, DawnRank scores (as previously described [16]) the level of perturbation on the gene interaction network caused by the alteration (either amplification or deletion) of the gene of interest. We selected human chromosome 1 gene blocks with shared synteny with the mouse genome for the DawnRank analysis. There were 7 such gene blocks, totaling 1509 genes (Supplemental Table 11). Using the chromosome 1 syntenic regions, we identified 44 chromosome 1 genes that represented the top 5 % DawnRank scores (Supplemental Table 12) using DNA copy number changes as the input “mutation” features along with the gene expression for each human tumor sample. The 44 DawnRank genes mapped to 9 copy number gained segments, which also harbored genes with DNA–RNA concordance, or an RNAi-identified essential gene (Supplemental Table 10). Within the 9 CNAs, encompassing a total of 182 potential genes, only 3 genes met all four filtering criteria of (1) subtype-specific CNA, (2) DNA–RNA concordance, (3) a RNAi “GO” gene, and (4) a DawnRank hit: these genes were phosphatidylinositol 4-kinase (PI4KB), src homology 2 domain containing (SHC1), and nicastrin (NCSTN) (Fig. 3; Supplemental Table 10).
The three chromosome 1 potential driving genes span 1q21-q23 and are altered with an average segment subtype frequency of 47 % (Supplemental Table 10). Interestingly, PI4KB and SHC1 span 1q21, falling less than the average Basal-like subtype segment length apart (Fig. 3), thus suggesting that on chromosome 1q21-23 multiple target genes lie within a single amplicon. Furthermore, SHC1 is in a subtype-specific high frequency altered segment among Basal-like tumors only (Fig. 3; Supplemental Table 5), while NCSTN and PI4KB CNAs appeared across multiple subtypes, passing the significance threshold in the Basal-like and Luminal A subtypes (Supplemental Table 5). However, NCSTN and PI4KB also passed the significance threshold for the p53null-Luminal, p53null-Basal, and C3Tag mouse groups (Supplemental Table 7), the last two of which are models linked to human Basal-like disease as determined in previous gene expression comparative studies [18, 19].
Notch pathway features in 1q21-23 amplified Basal-like breast cancers
Numerous studies have implicated the Notch signaling pathway in Basal-like breast and/or triple-negative breast cancers [22, 23]. Importantly, numerous studies on the functional role of NCSTN have already been performed [24–26]. To evaluate the effect of 1q21-23/NCSTN amplification, we first examined the DawnRank network space around NCSTN and noted that when NCSTN was amplified NOTCH1–3 were also more highly expressed (Fig. 4). In addition, NCSTN is one of the three components of the gamma-secretase complex (GSC), a protein complex that cleaves and activates Notch receptors. Two other GSC members, namely APH1A and PSEN2, were also both altered within the network (Fig. 4), and were also higher in NCSTN amplified samples versus not amplified (Supplemental Fig. 4a). Also, APH1A and PSEN2 are physically located on human chromosome 1q21.2 and 1q42, and are often co-amplified along with NCSTN (although PSEN2 is not within a Basal-like-specific CNA). Thus, three components of the GSC are often co-amplified together, and are more highly expressed, and the NCSTN/Notch network is perturbed in these NCSTN amplified tumors. Following up on these network findings, NCSTN amplification was also correlated with higher NOTCH1 and NOTCH3 mRNA levels (Supplemental Fig. 4b), with this feature showing an even greater difference when examined just among Basal-like breast cancers (Supplemental Fig. 4c). As expected from the previous work, Basal-like tumors as a whole exhibited significantly lower LFNG expression (i.e., a negative regulator of Notch signaling) along with significantly higher expression of NOTCH1, NCSTN, APH1A, MYC, and HEY2 mRNAs (Supplemental Fig. 5), the latter two of which are thought to be targets of activated Notch pathway.
Fig. 4.

DawnRank identified NCSTN gene expression network. The plot represents the local neighborhood of NCSTN and all direct genes downstream. The green and red colors represent whether the gene is up-regulated or down-regulated, respectively. The intensity of the color represents the magnitude of gene expression change. The size of each node indicates the DawnRank score of that gene, representing the gene’s impact on downstream expression changes in the network
Conclusion
In breast cancer, there are many copy number gains and losses, a few of which like amplification of ERBB2, are of known clinical and biological significance. Over the years, many of these CNAs have been studied and candidate genes identified [12, 13, 27–30], but there are still many regions for which the genetic drivers remain unknown. The simultaneous analysis of DNA copy number change in both human and mouse tumors, and their corresponding gene expression patterns, provides for a biologically meaningful way to identify important regions of CNAs. The basic hypothesis being that a CNA found to spontaneously occur in two different mammalian species breast cancers is being repeatedly selected and must therefore contain an important tumor causing gene(s).
Although many studies have identified frequent CNAs within groups of human breast tumors [13, 21], most do not functionally narrow down the candidate genes within a specific segment. In addition to the mere presence of a highly frequent CNAs being identified across species, we took a biologically based approach to refine the list of genes within a given segment into a subset of candidate driver genes. These analyses prompted the development of a new bioinformatics tool (SWITCHplus) to identify and highlight subtype-specific DNA copy number events using a visual display in a user-friendly format. Using this tool and a systematic data-mining schema that includes identifying regions that show: (1) shared DNA CNAs cross-species, (2) concordance between mRNA expression and relative DNA copy number value, (3) functional effects in a genome-wide RNAi screen, and (4) functional effects in a network analysis (i.e., DawnRank), we identified a limited number of CNAs that harbored potential breast cancer driver genes. From these analyses, we identified human chromosome 1q21-23 as a region of amplification consistently present in human and mouse Basal-like tumors, and which contains at least three potential driver genes (Fig. 3).
The first of these three genes, PI4KB encodes for a lipid kinase member of the phosphoinositide signaling pathway. The phosphoinositide signaling system regulates cell migration [31–33]and proliferation [31–33], and activation of this signaling pathway is observed in many aggressive tumors [33–35]. Specifically, phosphatidylinositol 4-phosphate is utilized by phosphoinositide kinases, such as PI3KCA, to signal to downstream protein kinase targets including AKT and PDK1 [33, 35, 36]. In the 2012 TCGA publication on breast cancer, it was noted that Basal-like cancers showed high activity of the PIK3CA/AKT pathway, and that these tumors tended to show few PIK3CA mutations, but frequent loss of PTEN and/or INPP4B (negative regulators of the pathway) and amplification of PIK3CA and AKT3 (positive regulators of the pathway) [12]. Here we show that yet another positive regulator of the pathway is amplified in Basal-like cancers.
SHC1 encodes for a member of the Shc family of adapter proteins. SHC1 is composed of multiple protein domains that can bind to multiple transmembrane receptors including phosphorylated insulin-like growth factor 1 receptor, and the platelet-derived growth factor receptor (PDGFR), thus potentially activating multiple pathways involved in cell proliferation and differentiation [37, 38]. Specifically, SHC1 is a key signaling mediator, and can act as a scaffold between an activated receptor and downstream signaling proteins [39]. In addition, growth factor signaling through PDGFR is known to occur in many TNBC [40], and thus SHC1 amplification may be contributing to these key signaling processes.
NCSTN encodes for a component of the GSC, which is a multi-protein complex that cleaves a number of transmembrane proteins to typically activate their functions [41, 42]; the GSC targets include Notch 1–4, ErBB4, CD44, and E-cadherin [24, 41, 42]. Importantly, Hu et al. 2002 demonstrated, in Drosophila, that NCSTN provides structural support and is required for GSC cleavage of Notch receptor [43]. In our data, when Basal-like tumors were examined, those with copy number gains at NCSTN showed (1) perturbation/activation of the Notch pathway via the DawnRank network analysis (Fig. 4), (2) significantly higher expression of NOTCH1 and NOTCH3 (Supplemental Fig. 4c), and (3) high expression of other markers of the Notch pathway (Supplemental Fig. 4d). Further support for Notch pathway importance comes from previous mouse model experiments where genetic inactivation of a negative regulator of Notch signaling (i.e., lunatic fringe) resulted in Basal-like mammary tumors [22]. Interestingly, Notch activity is also higher in Basal-like breast cancer cell lines compared with Luminal breast cancer cell lines [44]. In vitro, by RNAi-mediated silencing of NCSTN in the TNBC cell line MDA-MB-231, Filipović et al. 2011 showed reduced transcription of Notch pathway targets, and a reduction in cell motility and invasion [41]. In total, these results strongly suggest that activation of Notch pathway signaling is occurring within Basal-like/TNBC tumors, and we now provide additional evidence for a mechanistic explanation for this in vivo.
Other investigators using different computational approaches have also identified this region, but identified other genes (i.e., NIT1 and PVRL4) as potential drivers [45]. The observed differences in potential driver genes are mostly likely due to the “filtering criteria”, where we focused on species conservation, and they focused on somatic mutation targets. It is clear that a multitude of targets and drivers are present, and that 1q21-23 is a region that is the target of selection as opposed to any single gene being the target of selection. In conclusion, our work here provides an objective analysis path for identifying potential driver genes using a cross-species computational approach, which can be applied to any tumor type for which sufficient mouse and human tumor data exist.
Electronic supplementary material
Fig. S1 Supervised cluster of mouse gene expression data using an 866 intrinsic gene list. The cluster analysis identified seven murine tumor groups, which were further used to supervise subsequent DNA copy number analyses. Each group is labeled according to the majority representing GEM mammary model within that group
Fig. S2 Copy number frequency plots from SWITCHplus showing human subtype-specific CNAs. Segments of subtype-specific copy number gains are plotted above the x-axis in red and segments of copy number loss are plotted below the x-axis in green. Regions shaded gray indicate segments that are not subtype-specific or high frequent (greater than or equal to 15 %). The frequency of alterations in each subtype is indicated on the y-axis from 0 to 100 %. a Basal-like, b Her2-enriched, c Claudin-low, d Luminal A, and e Luminal B copy number landscapes
Fig. S3 Copy number frequency plots from SWITCHplus showing conserved CNAs. Segments of copy number gains are plotted above the x-axis and segments of copy number loss are plotted below the x-axis. Regions shaded gray indicate segments that are any combination of either not subtype-specific, not mouse group-specific or not high frequent (greater than or equal to 15 %). The conserved segments are colored according to the mouse group(s) in which they appear. The frequency of alterations in each subtype is indicated on the y-axis from 0 to 100 %. a Basal-like, b Her2-enriched, c Claudin-low, d Luminal A, and e Luminal B copy number landscapes
Fig. S4Box-and-whisker plots for expression of Notch signaling pathway targets. aAPH1A and PSEN2 mRNA expressions across all tumors with NCSTN amplification versus all other tumors without NCSTN amplification. bNOTCH1 and NOTCH3 mRNA expressions across all tumors with NCSTN amplification versus all tumors without NCSTN amplification. cNOTCH1 and NOTCH3 mRNA expressions across all Basal-like tumors with NCSTN amplification versus Basal-like tumors without NCSTN amplification. dAPH1A and PSEN2 mRNA expressions across all Basal-like tumors with NCSTN amplification versus Basal-like tumors without NCSTN amplification
Fig. S5Box-and-whisker plots of the mRNA expression of LFNG, NOTCH1, NCSTN, APH1A, MYC, and HEY2 across breast cancer subtypes
Acknowledgments
This study was supported by Funds from the Initiative for Maximizing Student Diversity Grant 5R25GM055336, the UNC Bioinformatics and Computational Biology Diversity Fellowship, the NCI Breast SPORE Program Grant P50-CA58223-09A1, RO1-CA148761, and the Breast Cancer Research Foundation. We would like to thank the Translational Breast Cancer Research Consortium for providing some of the tumor specimens used for gene and copy number profiling, which came from TBCRC001.
Conflict of interests
C.M.P. is an equity stock holder, and Board of Director Member of BioClassifier LLC., C.M.P., and J.S.P. are also listed as inventors on a patent application on the PAM50 assay.
Contributor Information
Grace O. Silva, silvag@email.unc.edu
Xiaping He, Email: xiaping_he@med.unc.edu.
Joel S. Parker, parkerjs@email.unc.edu
Michael L. Gatza, mgatza@email.unc.edu
Lisa A. Carey, lisa_carey@email.unc.edu
Jack P. Hou, Email: jackhou2@illinois.edu
Stacy L. Moulder, Email: smoulder@mdanderson.org
Paul K. Marcom, Email: marco001@mc.duke.edu
Jian Ma, Email: jianma@illinois.edu.
Jeffrey M. Rosen, Email: jrosen@bcm.edu
Charles M. Perou, Phone: 919-843-5740, Email: cperou@med.unc.edu
References
- 1.Kravchenko J, Akushevich I, Seewaldt VL, et al. Breast cancer as heterogeneous disease: contributing factors and carcinogenesis mechanisms. Breast Cancer Res Treat. 2011;128:483–493. doi: 10.1007/s10549-011-1347-z. [DOI] [PubMed] [Google Scholar]
- 2.Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 2011;5:5–23. doi: 10.1016/j.molonc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sotiriou C, Neo S-Y, McShane LM, et al. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci USA. 2003;100:10393–10398. doi: 10.1073/pnas.1732912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nordgard SH, Johansen FE, Alnaes GIG, et al. Genes harbouring susceptibility SNPs are differentially expressed in the breast cancer subtypes. Breast Cancer Res. 2007;9:113. doi: 10.1186/bcr1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergamaschi A, Kim Y. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosom Cancer. 2006;1040:1033–1040. doi: 10.1002/gcc.20366. [DOI] [PubMed] [Google Scholar]
- 7.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parker JS, Mullins M, Cheang MCU, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prat A, Parker JS, Karginova O, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu Z, Fan C, Oh DS, et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics. 2006;7:96. doi: 10.1186/1471-2164-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheang MCU, Chia SK, Voduc D, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst. 2009;101:736–750. doi: 10.1093/jnci/djp082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer T, Atlas G. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weigman VJ, Chao H-H, Shabalin AA, et al. Basal-like Breast cancer DNA copy number losses identify genes involved in genomic instability, response to therapy, and patient survival. Breast Cancer Res Treat. 2012;113:865–880. doi: 10.1007/s10549-011-1846-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishida N, Nishimura T, Ito T, Komeda T. Chromosomal instability and human hepatocarcinogenesis. Histol Histopathol. 2003;18:897–909. doi: 10.14670/HH-18.897. [DOI] [PubMed] [Google Scholar]
- 15.Solimini NL, Xu Q, Mermel CH, et al. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science. 2012;337:104–109. doi: 10.1126/science.1219580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hou JP, Ma J. DawnRank: discovering personalized driver genes in cancer. Genome Med. 2014;6:56. doi: 10.1186/s13073-014-0056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Hayes DN, Nobel A, Marron JS. Statistical significance of clustering for high-dimension, low-sample size data. J Am Stat Assoc. 2008;103:1281–1293. doi: 10.1198/016214508000000454. [DOI] [Google Scholar]
- 18.Herschkowitz JI, Simin K, Weigman VJ, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfefferle AD, Herschkowitz JI, Usary J, et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol. 2013;14:R125. doi: 10.1186/gb-2013-14-11-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herschkowitz JI, Zhao W, Zhang M, et al. Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Acad Sci USA. 2012;109:2778–2783. doi: 10.1073/pnas.1018862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curtis C, Shah SP, Chin S-F, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu K, Usary J, Kousis PC, et al. Lunatic fringe deficiency cooperates with the Met/Caveolin gene amplicon to induce basal-like breast cancer. Cancer Cell. 2012;21:626–641. doi: 10.1016/j.ccr.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stoeck A, Lejnine S, Truong A, et al. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov. 2014;4:1154–1167. doi: 10.1158/2159-8290.CD-13-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lombardo Y, Filipović A, Molyneux G et al (2012) Nicastrin regulates breast cancer stem cell properties and tumor growth in vitro and in vivo. Proc Natl Acad Sci USA 109:16558–16563. doi:10.1073/pnas.1206268109 [DOI] [PMC free article] [PubMed]
- 25.Murphy MP, Das P, Nyborg AC, et al. Overexpression of nicastrin increases Aβ production. FASEB J. 2003;17:1138–1140. doi: 10.1096/fj.02-1050fje. [DOI] [PubMed] [Google Scholar]
- 26.Sarajlić A, Filipović A, Janjić V et al (2014) The role of genes co-amplified with nicastrin in breast invasive carcinoma. Breast Cancer Res Treat 143:393–401. doi:10.1007/s10549-013-2805-6 [DOI] [PubMed]
- 27.Shadeo A, Lam WL. Comprehensive copy number profiles of breast cancer cell model genomes. Breast Cancer Res. 2006;8:R9. doi: 10.1186/bcr1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taylor BS, Barretina J, Socci ND, et al. Functional copy-number alterations in cancer. PLoS ONE. 2008;3:e3179. doi: 10.1371/journal.pone.0003179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kristensen VN, Lingjærde OC, Russnes HG, et al. Principles and methods of integrative genomic analyses in cancer. Nat Rev Cancer. 2014;14:299–313. doi: 10.1038/nrc3721. [DOI] [PubMed] [Google Scholar]
- 30.Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katso R, Okkenhaug K. Cellular function of phosphoinositide 3-kinases: implications for development, immunity, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- 32.Cantley L. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1658. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 33.Bunney TD, Katan M. Phosphoinositide signalling in cancer: beyond PI3K and PTEN. Nat Rev Cancer. 2010;10:342–352. doi: 10.1038/nrc2842. [DOI] [PubMed] [Google Scholar]
- 34.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 35.Chu KME, Minogue S, Hsuan JJ, Waugh MG. Differential effects of the phosphatidylinositol 4-kinases, PI4KIIα and PI4KIIIβ, on Akt activation and apoptosis. Cell Death Dis. 2010;1:e106. doi: 10.1038/cddis.2010.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balla A, Balla T. Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol. 2006;16:351–361. doi: 10.1016/j.tcb.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 37.Wagner K, Hemminki K, Grzybowska E, et al. The insulin-like growth factor-1 pathway mediator genes: SHC1 Met300Val shows a protective effect in breast cancer. Carcinogenesis. 2004;25:2473–2478. doi: 10.1093/carcin/bgh263. [DOI] [PubMed] [Google Scholar]
- 38.Yu H, Rohan T. Role of the insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst. 2000;92:1472–1489. doi: 10.1093/jnci/92.18.1472. [DOI] [PubMed] [Google Scholar]
- 39.Zheng Y, Zhang C, Croucher DR, et al. Temporal regulation of EGF signalling networks by the scaffold protein Shc1. Nature. 2013;499:166–171. doi: 10.1038/nature12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duncan JS, Whittle MC, Nakamura K, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Filipović A, Gronau JH, Green AR, et al. Biological and clinical implications of nicastrin expression in invasive breast cancer. Breast Cancer Res Treat. 2011;125:43–53. doi: 10.1007/s10549-010-0823-1. [DOI] [PubMed] [Google Scholar]
- 42.Kopan R, Ilagan M. γ-Secretase: proteasome of the membrane? Nat Rev Mol Cell Biol. 2004;5:499–504. doi: 10.1038/nrm1406. [DOI] [PubMed] [Google Scholar]
- 43.Hu Y, Ye Y, Fortini ME. Nicastrin is required for γ-secretase cleavage of the Drosophila Notch receptor. Dev Cell. 2002;2:69–78. doi: 10.1016/S1534-5807(01)00105-8. [DOI] [PubMed] [Google Scholar]
- 44.D’Angelo RC, Ouzounova M, Davis A, et al. Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol Cancer Ther. 2015;14:779–787. doi: 10.1158/1535-7163.MCT-14-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanchez-Garcia F, Villagrasa P, Matsui J, et al. Integration of genomic data enables selective discovery of breast cancer drivers. Cell. 2014;159:1461–1475. doi: 10.1016/j.cell.2014.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]