

Abstract
Autopsy specimens from human victims or experimental animals that die due to acute chlorine gas exposure present features of cardiovascular pathology. We demonstrate acute chlorine inhalation–induced reduction in heart rate and oxygen saturation in rats. Chlorine inhalation elevated chlorine reactants, such as chlorotyrosine and chloramine, in blood plasma. Using heart tissue and primary cardiomyocytes, we demonstrated that acute high-concentration chlorine exposure in vivo (500 ppm for 30 min) caused decreased total ATP content and loss of sarcoendoplasmic reticulum calcium ATPase (SERCA) activity. Loss of SERCA activity was attributed to chlorination of tyrosine residues and oxidation of an important cysteine residue, cysteine-674, in SERCA, as demonstrated by immunoblots and mass spectrometry. Using cardiomyocytes, we found that chlorine-induced cell death and damage to SERCA could be decreased by thiocyanate, an important biological antioxidant, and by genetic SERCA2 overexpression. We also investigated a U.S. Food and Drug Administration–approved drug, ranolazine, used in treatment of cardiac diseases, and previously shown to stabilize SERCA in animal models of ischemia–reperfusion. Pretreatment with ranolazine or istaroxime, another SERCA activator, prevented chlorine-induced cardiomyocyte death. Further investigation of responsible mechanisms showed that ranolazine- and istaroxime-treated cells preserved mitochondrial membrane potential and ATP after chlorine exposure. Thus, these studies demonstrate a novel critical target for chlorine in the heart and identify potentially useful therapies to mitigate toxicity of acute chlorine exposure.
Keywords: chlorine, inhalation, cardiac, sarcoendoplasmic reticulum Ca2+ATPase, ranolazine
Clinical Relevance
This study defines impact of inhalation of a toxic gas, chlorine, on a critical cardiac calcium pump, sarcoendoplasmic reticulum Ca2+ATPase (SERCA). It also demonstrates that therapeutic strategies that protect or modify SERCA function could be useful approaches for emergent resuscitation of severe chlorine inhalation victims.
Chlorine is a commonly used chemical in industry and society. Acute chlorine inhalation toxicity can occur due to accidents at swimming pools and/or involving water purification systems, after transportation accidents, upon industrial exposure, with misuse of domestic cleaners, during military operations, and, more recently, through chemical terrorism. In the United States, over 14 million tons of chlorine are produced and transported annually, enhancing the potential for accidents.
Chlorine has twice the density of air, and settles in areas where released, causing very high local concentrations in that vicinity. Inhalation of chlorine concentrations of 400 ppm or greater is generally fatal over 30 minutes. Asphyxia, respiratory failure, acute burns of the upper and lower airways, pulmonary edema, acute pulmonary hypertension, cardiomegaly, pulmonary vascular congestion, and death are some clinical features noted in victims after high-level chlorine exposures (1). Cardiomegaly was observed in autopsy of victims of acute chlorine poisoning in a major accidental cargo train derailment (2). Cardiomegaly has been observed in a number of other cases. However, it remains unclear whether cardiac enlargement results from primary cardiovascular dysfunction or a combined cardiopulmonary process. In animal models, some studies noted elevated pulmonary vascular resistance and diminished cardiac output after a decline in lung compliance (3). Chlorine inhalation may cause injury to both pulmonary and systemic vasculature (4, 5).
Altered cardiomyocyte Ca2+ homeostasis is a central pathophysiological mechanism of various cardiac diseases, including cardiac hypertrophy and heart failure (6). Sarcoendoplasmic reticulum Ca2+ ATPase (SERCA) critically regulates Ca2+ homeostasis. By sequestering cytosolic Ca2+ in the endoplasmic reticulum, SERCA mediates smooth, cardiac, and skeletal muscle relaxation, and helps regulate cell growth, proliferation, and apoptosis (7). Of 10 known isoforms, SERCA2 is most highly expressed in the heart. SERCA2 is highly susceptible to oxidant injury, and especially to HOCl, a major reaction product of chlorine gas with water (8). SERCA knockout mice do not survive to birth, and defects in SERCA occur in aging tissues and heart disease (9).
Here, we investigated the effect of chlorine exposure on SERCA in hearts of animals exposed to high concentrations of chlorine. We evaluated strategies to mitigate the loss of SERCA function and explored potential mechanisms. We demonstrate that damage to cardiac SERCA by chlorine exposure could be prevented or reversed by antioxidants, such as sodium thiocyanate (NaSCN), and ranolazine, a U.S. Food and Drug Administration (FDA)–approved drug for heart disease.
Materials and Methods
Additional details are included in the online supplement.
Animals
Male Sprague-Dawley rats (250–300 g) were obtained from Harlan Laboratories (Indianapolis, IN). Animals were acclimated to Denver altitude for 1 week with ad libitum food and water. All procedures were performed according to approved Institutional Animal Care and Use Committee protocols of the University of Colorado Denver (Denver, CO).
Chlorine Exposure
Animals were exposed in sealed polysulfone biocontainment cages (Allentown Cage Equipment, Allentown, NJ). Dry air was supplied at 15 L/min and chlorine was provided to attain the desired final chlorine concentration (500 ppm). Rats were exposed for 30 minutes. After exposure, chlorine gas flow was discontinued, and cages were vented with compressed air until no residual chlorine content was detected in effluent gas. Rats were removed and returned to cages, given room air for 3 hours, and then killed, necropsied, and hearts were removed for microsome preparation or cardiomyocyte isolation. In vitro chlorine exposures were performed as previously described (10).
Isolation of Rat Cardiomyocytes and Cell Culture
Primary rat cardiomycoytes were isolated as described previously (10). Detailed descriptions of procedures are provided in the online supplement. H9c2, an embroyonic rat heart cell line, was obtained from ATCC (Manassas, VA) and cultured in Dulbecco’s modified Eagles medium containing 10% FBS, according to manufacturer’s instructions. HL-1 cells were a gift of Dr. W. C. Claycomb (Louisiana State University Health Sciences Center, New Orleans, LA), and cultured using protocols provided by that lab. Live cell (trypan blue–excluding) counts were performed using a Bio-Rad cell counter (Bio-Rad, Hercules, CA).
Statistical Analysis
Statistical analysis was performed using JMP software (SAS Institute, Cary, NC). Means were compared by two-tailed t test for comparison between two groups, or one-way ANOVA followed by Tukey-Kramer test for multiple comparisons. P values less than 0.05 were considered significant.
Results
Cardiac Dysfunction and SERCA Modulation due to Chlorine Inhalation
Rats were exposed to chlorine (500 ppm) for 30 minutes. Heart rate and oxygen saturation were measured in control and chlorine-exposed rats at different time intervals. An acute decline in heart rate in the first 1 hour after exposure was observed. However, at 2 hours there was some recovery, but heart rate was still lower than in nonexposed controls (Figure 1A, left panel). Similarly, oxygen saturations were reduced in chlorine-exposed animals (Figure 1A, right panel). We used pimonidazole-HCL staining to further investigate the extent of hypoxia in hearts of chlorine-exposed animals (11). This compound forms adducts at tissue O2 below 10 mm Hg. The results demonstrate that heart tissue was as hypoxic as detected in hearts in a hypoxic pulmonary hypertension model, used as a positive control (see Figure E1A in the online supplement). Hematoxylin and eosin–stained sections of chlorine-exposed rat hearts (Figure E1B, lower left panel) also demonstrated cell damage and swelling. We also found elevation of cardiac injury markers in the plasma, viz. N-terminal pro-brain natriurectic peptide and troponin I, 3 hours after chlorine exposure (data not shown). The toxicity of Cl2 is often attributed primarily to hypochlorite (HOCl + OCl−) (12). HOCl can oxidize many important biomolecules, chlorinate critical targets, such as protein, DNA, and cholesterol, and cause cell death. Tyrosine chlorination in airways of chlorine-exposed rats has been observed previously (13). To investigate sources of chlorine toxicity in the heart, we examined the plasma of chlorine-exposed rats for chlorotyrosine and chloramines, two important reaction products of chlorine (13). There was an enhanced intensity of chlorotyrosine in a band at approximately 100 kD in immunoblots of plasma proteins from chlorine-exposed animals. A higher molecular weight band of approximately 200 kD was also observed in some animals, but was very labile (Figure 1B, top left panel, lane 4). We also performed a time course to evaluate the duration of chlorotyrosine formation. Enhanced chlorotyrosine reactivity was observed immediately after chlorine exposure, and persisted until 1 hour (Figure 1B, top right panel). Chlorotyrosination of albumin in blood plasma was suggested, due to the proximity of the band to that of BSA treated with HOCl, which was used as a positive control (Figure 1B, top right panel, lane 12). Chloramine content was also increased in plasma of chlorine-exposed rats (Figure 1C), further demonstrating off-site generation of chlorine reaction products upon chlorine inhalation beyond the lung and airways.
Figure 1.

Effect of chlorine (Cl2) inhalation on rat heart. Rats were exposed to Cl2 (500 ppm, 30 min) as described in Materials and Methods. Heart rate (A, left panel) and O2 saturation (Oxy Sat) (A, right panel) was measured using pulse oximetry at various time intervals as indicated. (B) Immunoblots demonstrate chlorotyrosine (Chlorotyr) formation in the plasma of rats inhaling Cl2. The left panel shows chlorotyrosine reactivity of plasma of control or Cl2-exposed rats 1 hour after exposure. The right panel shows time course of chlorotyrosine formation. The lane labeled “BSA” contains bovine serum albumin (1 mg/ml) treated with HOCl (1 mM) to generate chlorotyrosine for positive control. The lower panels of (B) are loading controls developed using Ponceu dye, and lane 1 contained molecular weight markers. (C) Time-dependent chloramine formation in the plasma of Cl2-exposed rats. (D) ATP contents of hearts of rats exposed to Cl2 harvested at various times after exposure. Sarcoendoplasmic reticulum Ca2+ATPase (SERCA) activity was also measured, and is shown in (E). (F) Apoptotic death in cardiomyocytes from hearts of Cl2-exposed animals isolated and fixed at various time intervals after exposure, as described in Materials and Methods. (G) The fixed cardiomyoctes were also stained with chlorotyrosine antibody and quantitated using flow cytometry, as described in Materials and Methods. Values shown are mean ± SEM (n = 4). *Significant difference (P < 0.05) from control (0 ppm Cl2). Ctrl, control; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
To further evaluate cardiac effects of chlorine inhalation, we measured total ATP content. Total ATP content of chlorine-exposed rat hearts decreased in a time-dependent fashion (Figure 1D). SERCA plays an important role in chemomechanical energy transduction of the heart (14). Chlorine inhalation markedly (> 50%) decreased SERCA activity (Figure 1E). Because chlorine-exposed hearts were hypoxic, we measured SERCA activity after hypoxia (5 or 10% O2) in cardiomyocytes. Tissue hypoxia appears not to be a factor in loss of SERCA activity after chlorine exposure, as hypoxic exposure of cardiomyocytes did not affect SERCA activity (Figure E1C). To assess if loss of SERCA activity was due to tissue/cell death, we measured apoptosis in cells isolated from control and chlorine-exposed rat hearts. Only 5–10% of cells were terminal deoxynucleotidyl transferase dUTP nick end labeling positive after chlorine exposure (Figure 1F). This would not account for a greater than 50% decline in SERCA activity. Interestingly, increased chlorotyrosine formation was detected on the cardiomyocyte cell surface (Figure 1G) as well. Thus, there was decreased total ATP content and loss of SERCA activity in hearts of chlorine-exposed rats that did not appear to be accounted for by cell death or tissue hypoxia.
To further study SERCA activity, rat heart microsomes were prepared. Figure E2A illustrates expression of SERCA and sarcomeric actin protein in crude fractions and in purified microsomes. Sarcomeric actin expression was observed in crude fractions of cardiac muscle. SERCA was enriched several fold in purified microsomal membranes (Figure E2A). Microsomes were also isolated from hearts of rats exposed to chlorine (500 ppm for 30 min) and observed for SERCA expression (Figure E2B), using microsome-specific calnexin and calreticulin as loading controls. As shown, no significant change in SERCA expression was observed. The presence of a functional SERCA in microsomal fractions was demonstrated by thapsigargin-inhibitable Ca2+ATPase assay and 45Ca2+ uptake assays (Figures E2C and E2D). Chlorine exposure caused greater than 50% reduction in Ca2+ATPase activity (Figure 1C). We sought to determine if chlorine exposure also affected Ca2+ transport activity. Notably, chlorine exposure also caused a greater than 75% decrease in the Ca2+ uptake activity of the heart microsomal membranes (Figure E2D). Thus, short-term, high-level chlorine exposure markedly decreased cardiac SERCA activity without changing SERCA protein expression.
To investigate potential SERCA modification, we first studied chlorotyrosine formation in cardiac tissue of control and chlorine-exposed rats. Immunoblots of immunoprecipitated proteins obtained using chlorotyrosine antibody (see the online supplement for details) demonstrated increased chlorotyrosine formation in hearts of chlorine-exposed animals in a time-dependent manner. Chlorotyrosine formation was more pronounced in hearts immediately after 30-minute chlorine exposure, and proteins of roughly 100 kD were targeted (Figure 2A). There was also a band (∼ 50 kD) of constitutively expressed chlorinated proteins. We further confirmed the presence of SERCA in the chlorotyrosine-bearing protein band using Western blots of immunoprecipitated proteins with chlorotyrosine and SERCA2 antibodies (Figure 2B). Oxidation of SERCA cysteine-674 residues also occurred upon chlorine inhalation, as demonstrated by immunoblot of immunoprecipitated proteins using SERCA antibody (Figure 2C).
Figure 2.

Chlorine inhalation–induced chemical modification of rat heart proteins and SERCA2. Rats were exposed to Cl2 (500 ppm, 30 min), killed at various time intervals after exposure, and hearts were snap frozen in liquid nitrogen. Tissue lysates were prepared and immunoprecipitation was performed using pooled tissue lysates of all animals in a group (n = 4). (A) An immunoblot of chlorotyrosine antibody–immunprecipitated (IP) proteins that were also detected by the same antibody. Here, time 0 (lane 3) rat hearts had already seen chlorine exposure for 30 minutes, whereas those in lane 4 (time 0.5) had seen 30 minutes of exposure plus 30 minutes of recovery. The lower two panels labeled “s-actin” (sarcomeric actin) and “β-actin” indicate the protein content of the input lysates used for the IP. (B) A double IP for detection of SERCA2 in the chlorotyrosinated proteins. (C) Oxidation of cysteine (Cys)-674 of SERCA2 using the specific antibody in the immunoprecipitates of SERCA2 from the tissue lysates of control and Cl2-exposed animals after 3 hours of exposure. (D) Deconvoluted fragmentation spectrum of the quadruply charged tryptic peptide sequence [836–876] with m/z 1,140.5295. The b and y fragment ions are labeled in blue and red, respectively. The b ions and y ions combined with a loss of CO and NH3, respectively, are labeled in green. Tyrosine (Tyr)-836, Tyr-842, and Tyr-867 are chlorinated. Cyst-841 is oxidized to sulfonic acid. Cyst-875 is unmodified. Gln-868 is deamidated. “M+H” represents the protonated molecule. (E) Comparison of the isotopic distribution of the ion with m/z 1,140.5295 (z = 4; black) with the theoretical isotopic distribution (red) of the chlorinated model peptide as described in (D). (G) The six panels represent the zoom-ins of the mass spectrum shown in (D). (F) Demonstration of the modifications in the SERCA peptide sequence [836–876]. WB, Western blot.
To further identify the proteins, mass spectrometry was performed for the 100-kD band (band 1) obtained in rat hearts 0.5 hour after chlorine exposure, where proteins were immunoprecipitated with anti-chlorotyrosine antibody. Interestingly, the results demonstrate that SERCA2 tryptic peptides were abundant (24 peptides) in this band (Figure E3). The fragmentation pattern of the tentatively assigned SERCA2A tryptic peptide [-836 to -876] was consistent with the presence of chlorinated tyrosine (Tyr) residues (Figure 2D). Indeed, the b and y ions revealed that three Tyr residues, viz. Tyr-836, Tyr-842, and Tyr-867, were chlorinated. The y38 and b5 ions allowed for identification of Tyr-836-Cl. The y9 and y10 ions permitted identification of Tyr-867-Cl. The b6 and b7 ions allowed for identification of Tyr-842-Cl (Figures 2D–2G). A comparison between the measured isotopic distribution of the experimental tryptic peptide ion (m/z 1,140.5295, z = 4) and the calculated isotopic distribution of such a chlorinated model peptide was consistent with the presence of three chlorine atoms within the tryptic sequence [-836 to -876] of SERCA2A (Figure 2E). These results demonstrated chlorination of proteins in hearts of animals exposed to chlorine. These results also indicated that SERCA2 is a critical target of chlorine inhalation.
Loss of SERCA Activity and Calcium-Signaling Ability in Primary Rat Cardiomyocytes upon Chlorine Exposure
Cardiomyocytes were prepared from hearts of air- or chlorine-exposed animals. Cardiomyocytes were released readily from chlorine-exposed heart tissue, and, when observed by light microscopy, the cells appeared swollen (Figure 3A, inset) relative to air-exposed controls. Homogenates were prepared from freshly isolated cardiomyocytes, and Ca2+ATPase activity was measured. There was complete loss of Ca2+ATPase activity in cardiomyocytes isolated from chlorine-exposed rat hearts (Figure 3A). This was further confirmed by ligand (ATP)–induced intracellular calcium mobilization assay in freshly-isolated cardiomyocytes from air- or chlorine-exposed rat hearts (Figures 3B and 3C). ATP stimulation of cardiomyocytes from air-exposed rat hearts caused enhanced cytosolic calcium mobilization (Figures 3B and 3C, filled column in 3C) as compared with buffer controls. Cytosolic calcium was already increased in cardiomyocytes from hearts of chlorine-exposed rats, and it was not further enhanced upon ATP stimulation (Figures 3B and 3C). Thus, short-term, high-level chlorine exposure caused elimination of SERCA activity in isolated cardiomyocytes that were chlorine exposed in vivo. Furthermore, intracellular calcium signaling was markedly impaired in freshly isolated cardiomyocytes from chlorine-exposed versus air-exposed rats. Cell death contributed only partially to loss of SERCA activity, because fractionated live cells (4′,6-diamidino-2-phenylindole–excluding) demonstrated similar declines in SERCA activity (Figures E4A and E4B). Presence of chlorotyrosine was also demonstrated in these cells, both by flow cytometry and by Western blot (Figures E4C and E4D).
Figure 3.

Effect of chlorine exposure on SERCA2 activity and calcium signaling capacity in primary rat cardiomyocytes. Cardiomyocytes were isolated from rat hearts (0 or 500 ppm) (inset of [A] depicts the morphology of freshly isolated cells), as described in the Materials and Methods. Cells were homogenized in SERCA activity buffer (see details in Materials and Methods) and Ca2+ATPase activity was measured as described in Materials and Methods. Values shown are mean ± SEM (n = 4). *Significant difference (P < 0.05) from control (0 ppm chlorine) (A). (B) Freshly isolated cardiomyocytes were plated on laminin-coated 96-well plates for 4 hours, and diluent- (Dil; assay buffer) or ATP (50 μM) -induced increases in intracellular calcium were measured as described in Materials and Methods. (C) The peak values obtained in (B). Values shown are mean ± SEM (n = 4). *Significant difference (P < 0.05) from diluent control (0 ppm chlorine). [Ca2+]i, intracellular calcium concentration; Dil., diluent; Fluo, fluorescence; Pi, inorganic phosphate.
NaSCN and Hypothiocyanate Prevent SERCA Inactivation and Cell Death due to Chlorine
Thiocyanate (SCN−) in biological fluids rapidly scavenges HOCl generated by myeloperoxidase action (15). This antioxidant reaction limits HOCl-mediated cytotoxicity, and a similar reaction of SCN− with protein chloramines has been reported (16). We used NaSCN and freshly prepared hypothiocyanate (HOSCN) to preserve SERCA activity of cardiomyocytes exposed to chlorine (Figure 4) (16). Exposure of primary rat cardiomyocytes in vitro to chlorine (50 ppm, 15 min) caused complete loss of SERCA Ca2+ATPase activity (Figure 4A). Pretreatment of cells with NaSCN (500 μM) or HOSCN (100 μM) preserved a fraction of SERCA activity (∼ 20–30% with NaSCN and ∼ 50% with HOSCN) in chlorine-exposed cardiomyocytes (Figure 4A). Cell exposure to chlorine in vitro also caused enhanced cell death (Figures 4B and 4C). Treatment (before or 10 min after chlorine exposure) with NaSCN or HOSCN also protected against chlorine-mediated cell death (Figure 4C and inset). Thus, potential chemical scavengers of chloramines and HOCl/OCl− can modulate cardiomyocyte SERCA activity and cell survival after chlorine exposure in vitro.
Figure 4.
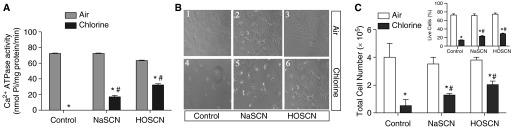
Effects of sodium thiocyanate (NaSCN) and hypothiocyanate (HOSCN) on SERCA inactivation and cell death due to chlorine. Cardiomyocytes were isolated and cultured from naïve rat hearts, as described in Materials and Methods. Approximately 50 × 104 cells/well were plated on laminin-coated 12-well plates and then exposed to 0 or 50 ppm chlorine in an in vitro exposure chamber, as described in Materials and Methods. Cell were treated with NaSCN (500 μM) or HOSCN (100 μM; freshly prepared) 10 minutes before chlorine exposures, as described in Materials and Methods. (A) Ca2+ATPase activity was determined in whole-cell homogenates prepared in SERCA activity buffer (detailed in Materials and Methods). Values shown are mean ± SEM (n = 4). *Significant difference (P < 0.05) from control (0 ppm chlorine); #significant difference from chlorine-exposed cells without pretreatment. (B) A representative pictogram illustrating cardiomyocyte morphology in these experiments. (C) Total cell counts (live cells only) obtained in cardiomyocyte cultures after exposure to 0 ppm or 50 ppm chlorine, both in the presence and absence of NaSCN and HOSCN. The inset in (C) shows SCN-induced rescue of chlorine-exposed cardiomyocytes where NaSCN or HOSCN were supplemented 10 minutes after chlorine exposure. Percent live cells that excluded trypan blue were counted in a Bio-Rad cell counter. Values shown are mean ± SEM (n = 4). *Significant difference (P < 0.05) from control (0 ppm Cl2); #significant difference from chlorine-exposed cells without pretreatment.
SERCA2 Overexpression Protects against Primary Rat Cardiomyocyte Death due to Chlorine
Because NaSCN and HOSCN preserved SERCA activity and protected against cell death, we were interested in determining if SERCA2 overexpression would affect cellular responses to chlorine. We used our previously published system of adenoviral SERCA2 overexpression to enhance SERCA2 protein of cardiomyocytes (17). Adenoviral green fluorescent protein was used to monitor transduction efficiency (Figure 5A, inset to panel 2). Fluorescence microscopy revealed strong green fluorescence in the majority of cells, signifying a high efficiency of adenoviral transduction and infection. Here, we used the same system that we have previously shown to increase SERCA2 mRNA by roughly 30-fold and to increase protein and activity by two- to threefold. The increase in SERCA protein in this expression system was demonstrated by Western blot (Figure 5B, inset). Overexpression of SERCA2 enhanced survival of cardiomyocytes in chlorine by more than twofold (Figure 5B). Thus, increasing the expressed protein of potential biochemical targets of chlorine toxicity increased cell survival after chlorine exposure.
Figure 5.

Effect of SERCA2 overexpression on primary rat cardiomyocyte death due to chlorine exposure. Cardiomyocytes were cultured as described in the legend for Figure 3, and transduced with adenoviral (Ad) green fluorescent protein (GFP) (Ad.GFP) or Ad.SERCA2 (described in Materials and Methods). At 48 hours after transduction, cells were exposed to 0 ppm (air) or 50 ppm chlorine. (A) Cell morphology after exposure, and the inset (A2) shows use of GFP fluorescence as an indicator of transduction efficiency. Inset of (B) also demonstrates SERCA2 protein expression in the cell lysates of control and Ad.SERCA-expressing cells. Live cells were counted and are shown in (B). Values shown are mean ± SEM (n = 4). *Significant difference (P < 0.05) from nontransduced control (0 ppm chlorine); #significant difference from chlorine-exposed cells.
Ranolazine, an Ionotropic Agent, Is a SERCA Stabilizer and Modifier of Chlorine Toxicity
Ranolazine reverses left ventricular dysfunction by stabilizing SERCA (18), and is an FDA-approved drug to treat various cardiovascular disorders (19). We used ranolazine to determine effects on chlorine-induced loss of SERCA activity and cardiomyocyte death. Ranolazine pretreatment protected against cell death due to chlorine (25–100 ppm for 15 min; Figure 6A and inset). This protection was attributed, at least in part, to preservation of SERCA Ca2+ATPase activity (Figure 6B). Besides using primary rat cardiomyocytes, we used rat cardiomyocyte cell line H9c2 and a mouse cardiac cell line with a beating phenotype, HL-1, to explore the effects of ranolazine and to further study relevant mechanism(s). We used even lower chlorine concentrations (12.5 ppm for 15 min) to preserve some live cells, as higher-level chlorine exposure was deleterious to cardiomyocytes. Chlorine exposure at 12.5 ppm also caused death (∼50%) of the three types of cardiomyocytes, and ranolazine helped prevent death due to lower chlorine concentrations (Figure 6C). Thus, a pharmacologic agent with direct effects on SERCA activity could protect SERCA activity and cell survival after chlorine exposure.
Figure 6.
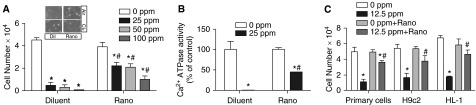
Effect of ranolazine, an ionotropic agent that stabilizes SERCA, on chlorine toxicity. Primary rat cardiomyocytes were cultured as described in the legend for Figure 3. At 30 minutes before exposure, cells were treated with saline (diluent) or ranolazine (Rano; 1.0 μM). (A) Live cell counts in 0, 25, 50, or 100 ppm chlorine–exposed cells in the absence or presence of ranolazine. Inset of (A) shows a representative pictogram depicting the morphology of the cells. Values shown are mean ± SEM (n = 4). *Significant difference (P < 0.05) from control (0 ppm chlorine). #Significant difference from chlorine-exposed diluent controls. (B) Ca2+ATPase activity was determined in whole-cell homogenates prepared in SERCA activity buffer (details in Materials and Methods). Values shown are mean ± SEM (n = 4). *Significant difference (P < 0.05) from control (0 ppm chlorine). #Significant difference from chlorine-exposed diluent controls. (C) The effect of ranolazine on viability of chlorine-exposed primary rat cardiomyocytes, or in cardiomyocyte cell lines H9c2 and HL-1 (cultures prepared as described in Materials and Methods). Values shown are means ± SEM (n = 4). *Significant difference (P < 0.05) from control (0 ppm chlorine). #Significant difference from chlorine-exposed diluent controls.
Ranolazine and Istaroxime Protect Mitochondrial Function
Loss of SERCA activity and the accompanying enhancement of cytosolic calcium (calcium overload) can cause damage to mitochondria, loss of mitochondrial function (ATP production), and subsequent apoptotic cell death. We have previously shown chlorine-induced apoptotic cell death in primary rat cardiomyocytes (10). Ranolazine can protect against calcium overload and preserve mitochondrial integrity in an ischemia–reperfusion model of isolated guinea pig heart (20). Because ranolazine preserves SERCA function, we determined its effect on mitochondrial function. Exposure to chlorine (25 or 50 ppm for 15 min) of H9c2 cells decreased intracellular ATP (Figure 7A). Ranolazine preserved ATP content in chlorine-exposed cells (Figure 7A). H9c2 and HL-1 cells were exposed to 12.5 ppm chlorine with and without ranolazine, and mitochondrial membrane potential was determined using 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanine iodide (JC-1) dye. Notably, the ranolazine-treated surviving cells had improved mitochondrial membrane potential (MMP), relative to untreated cells, in both H9c2 (Figure 7B) and HL-1 (data not shown). These data confirm that SERCA stabilization by ranolazine protects mitochondrial function after lethal chlorine exposure. Because ranolazine can have other effects, we also used another specific SERCA activator, istaroxime (21). Istaroxime pretreatment also prevented cell death (data not shown) and loss of MMP (Figures 7B and E5).
Figure 7.

Effect of ranolazine on mitochondrial function. (A) H9c2 cells were cultured on six-well plates and exposed to 0, 25, or 50 ppm chlorine in the absence or presence of ranolazine. Cells were pelleted and lysed, and ATP assay was performed as described in Materials and Methods. Values shown are mean ± SEM (n = 4). *Significant difference (P < 0.05) from control (0 ppm chlorine); #significant difference from chlorine-exposed cells without ranolazine. (B) Mitochondrial membrane potential of H9c2 cells measured using 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanine iodide (JC-1) as described in Materials and Methods. Values shown are mean ± SEM (n = 4). *Significant difference (P < 0.05) from control (0 ppm chlorine). Istar, istaroxime; MFIFL1, mean fuorescence intensity in FL1 channel.
Discussion
Besides pulmonary edema and respiratory failure, more than 90% of fatalities due to acute high-level chlorine exposure may be due to cardiovascular complications (1, 22–24). Our earlier studies with environmental oxidant gas exposures revealed enhanced airway epithelial cell death and subsequent loss of SERCA activity (17, 25). We also demonstrated that diseased cystic fibrosis airway epithelium with already decreased SERCA function was highly susceptible to oxidant gas exposures (17, 25). SERCA dysfunction in the heart causes severe cardiac dysfunction and death in animal models (26).
In the current study, we found that rats exposed to acute high chlorine concentrations had decreased heart rate and diminished blood O2 saturations. The hearts of chlorine-exposed animals were also hypoxic. Further investigations revealed presence of chlorotyrosine and chloramines in the plasma, suggesting circulation of chlorine reaction products. Markers of cardiac injury were elevated in the plasma, and a decreased total cardiac ATP content was observed. There was a time-dependent loss of SERCA activity in the heart. Loss of SERCA activity was not due to tissue hypoxia, as exposure of cardiomyocytes to hypoxia (for the same duration as in chlorine exposure) did not cause loss of SERCA activity. There was a time-dependent enhancement of apoptotic cell death, but only 10% of cells died. This likely does not account for more than 50% loss of SERCA. Our studies revealed that chlorine exposure caused chlorotyrosination of cardiac proteins. This was observed within the first 30 minutes of chlorine exposure. Such reversibility of chlorotyrosine formation has been observed previously (27). SERCA was identified as a major component of chlorotyrosinated proteins. There was oxidative modification of SERCA as well. Similar results were observed in in vitro experiments using primary rat cardiomyocytes. Importantly, treatment of cardiomyocytes with NaSCN or HOSCN reversed losses of SERCA due to chlorine exposure and prevented chlorine-induced cell death. SERCA overexpression by adenoviral SERCA2 transduction of cardiomyocytes, or treatment with SERCA modulators, such as ranolazine and istaroxime, also were beneficial. Further studies to define mechanisms of protection revealed that SERCA stabilization reversed chlorine exposure–induced loss of total cellular ATP, potentially by restoring mitochondrial function.
Inhalation of oxidant gases and other pollutants can compromise cardiac function (28). Inhalation of environmentally persistent free radicals, potentially derived from combustion of chlorine-containing hydrocarbons in rats, also caused cardiac inflammation and loss of function (29). These studies proposed a role for particulate matter–induced cardiovascular inflammation and oxidative stress (29). Cardiac effects of chlorine, especially cardiomegaly, were reported in autopsies of human victims of acute high-concentration chlorine gas exposure (1, 22–24). Similarly, experimental dogs that died of acute chlorine exposure had dilated right ventricles (30). Studies using chlorine inhalation in anesthetized, intubated pigs described loss of cardiac output and excessive pulmonary vasoconstriction (3, 31). Chlorine inhalation–induced injury to pulmonary and systemic vascular endothelial cells also can occur (4, 5). However, comprehensive reports of cardiac effects of chlorine inhalation in animal models are lacking, and additional mechanistic studies of cardiac dysfunction upon chlorine exposure are needed.
Upon reacting with water in moist airway epithelial surfaces, Cl2 forms hypochlorite (HOCl + OCl−) (12, 32). The bronchial and/or pulmonary circulations might then transport HOCl, or its secondary reaction products, to the heart. Granted, chlorine gas and HOCl are highly reactive, and therefore likely very short lived after chlorine inhalation. Thus, HOCl and other chlorinating compounds, such as chloramines, could react with Tyr residues to form 3-chlorotyrosines (33), which are more stable reaction products of chlorine. Sochaski and colleagues (13) demonstrated formation of 3-chlorotyrosine and 3,5-dichlorotyrosine upon chlorine inhalation in rat airways. The presence of chlorinating compounds in circulating blood after chlorine inhalation has not been shown before. We demonstrated persistence of chlorine-reactive products upon chlorine inhalation in the plasma and cardiac tissue of rats. This is a unique finding, and may identify and highlight offsite targets of toxic gas inhalation.
Decreased total ATP content and diminished mitochondrial function were observed in murine lungs after phosgene inhalation (34). However, such studies in hearts of chlorine-exposed animals are unknown. SERCA plays an important role in chemomechanical energy transduction of the heart (14). By sequestering cytosolic calcium in the endoplasmic reticulum, SERCA further regulates important signaling mechanisms critical for normal cell function, growth, and survival. Under conditions of heart failure, decreased Ca2+ uptake due to loss of SERCA activity is observed in humans, as well as in animal models of heart failure (35). Irreversible oxidation of SERCA and subsequent loss of activity occurs in the presence of higher levels of reactive oxygen species (36). Although cardiomyocyte SERCA has not been studied in chorine inhalation models, evidence of extensive protein oxidation due to high-level chlorine inhalation in organs, such as lungs, has been observed (37). Our findings of swollen morphology of cardiomyocytes isolated from chlorine-exposed animals are consistent with those of other studies of oxidative stress due to superoxides, where cardiomyocyte swelling was detected in heart tissues (38). Altered calcium homeostasis and loss of membrane integrity also were observed in oxidant-exposed rat cardiomyocytes (39). Furthermore, complete loss of stimulus-induced cytosolic calcium mobilization can occur upon brief oxidant exposure of rat cardiomyocytes (40). Thus, our observations of effects of chlorine exposure on myocardium or isolated cardiomyocytes are at least consistent with those of previous studies of oxidative cardiac injury.
HOCl exposure decreases SERCA activity, and this can be reversed by antioxidants (41). In turn, SCN− in biological fluids serves as a scavenger of HOCl. HOCl can be generated by myeloperoxidase activity (15). SCN− rapidly reacts with HOCl to form HOSCN, which is a less reactive and more selective oxidant than HOCl, which may mitigate oxidative damage to SERCA by HOCl. Moreover, HOSCN can react with chloramines, longer-lived reaction products, and restore them to amines (42). It is also conceivable that Cl2 produces chloramines in SERCA that are then repaired by HOSCN (43). However, our findings contrast with those in which SCN-induced decreases in SERCA activity were described (8). Cook and colleagues (8) used isolated sarcoplasmic reticulum (SR) vesicles in many of the studies directly assessing SERCA activity. This is not a comparable exposure method to those with intact cells, which can regulate both accessibility of different subcellular compartments to chemicals as well as dynamic responses after exposure. Furthermore, HOSCN is well “sensed” by cell signaling pathways (44, 45), and only slowly permeates whole cells (46). This could greatly influence resulting SERCA activity in intact cells compared with isolated SR vesicles. Although the duration of pretreatment was too short to result in changes in protein expression, it is possible that cell signals mobilized existing antioxidants in ways that were protective, or that additional signaling took place during Cl2 exposure. Experiments by Cook and colleagues (8) in whole cells used HOCl applied in a 500–5,000× excess of SCN− assuming that HOSCN would be generated. However, studies have shown that large excesses of SCN− in the presence of HOSCN can lead to rapid HOSCN breakdown, generating cyanate (OCN−), cyanide (CN−), and H2SO4 (47). These experiments could have favored rapid generation of HOSCN breakdown products, which is why HOSCN is usually generated enzymatically, or, if it is chemically generated, it is done at high pH to stabilize it (as OSCN−) (48). Furthermore, culture conditions can greatly impact effects of HOSCN on cells. Cook and colleagues (8) used serum-free conditions, whereas cells in this study were exposed to SCN− or HOSCN with 10% FBS, before Cl2 gas exposure, in addition to different exposure times. Dilute serum is a useful surrogate for the complex milieu of extracellular fluid, and significantly enhances cell-signaling responses after HOSCN exposure (44), and some or all protective effects of HOSCN on cardiomyocytes may have been indirectly mediated by inclusion of FBS in cell culture medium. In our study, SERCA activity buffers contained DTT. However, the results were not affected, as DTT may reverse sulfenic acid moieties, but not terminal sulfur oxidations (sulfi/onic). The importance of SERCA in chlorine-induced cardiotoxicity was further revealed in our studies using SERCA overexpression as a protective strategy against chlorine-induced cardiomyocyte death. SERCA overexpression selectively increased cytosolic calcium uptake in sarcoplasmic reticulum (49). SERCA overexpression using adenoviral vectors was protective against apoptotic cardiomyocyte death (50). Thus, there are precedents for protection by SERCA overexpression such as we have observed here.
Ranolazine is an FDA-approved drug for treatment of acute myocardial infarction and stable angina (19). It may have short-term efficacy in protecting against antitumor chemotherapy–induced cardiotoxicity (51). Therapeutic effects of ranolazine occur via two mechanisms: (1) inhibition of late inward sodium current and, hence, prevention of Ca2+ overload in ischemic hearts; and (2) inhibition of fatty acid oxidation and shift of metabolism toward glucose oxidation (52). Therefore, ranolazine may preserve mitochondrial supercomplexes, in part by restoring cardiolipin and, hence, electron flux in mitochondrial Fe-S clusters (53). Ranolazine also has been shown to indirectly stabilize SERCA in hearts of dogs with mild cardiac failure (18). SERCA plays a dynamic role in controlling cytosolic calcium content (54). Forster resident energy transfer studies of SR luminal calcium with specifically labeled SERCA2a showed that SERCA2a conformational changes directly coincided with calcium pump activity and appearance of luminal SR calcium. In addition, in dual-imaging studies of cytosolic calcium and SERCA2a, Forster resident energy transfer showed that SERCA2a activity coincided with cytosolic calcium oscillations triggered by extracellular ATP. Cytosolic calcium overload can cause mitochondrial damage, cellular energy depletion, and apoptotic cardiomyocyte death. Thus, SERCA2a has a dual role, acting as a “rapid switch” to refill SR calcium stores used in signaling, while protecting mitochondrial function.
In addition to changes in cell viability, we also demonstrated loss of mitochondrial membrane potential and intracellular ATP content concurrent with chlorine exposure. Gene microarray studies by Leikauf and colleagues (55) indicated that energy metabolism may be targeted in chlorine toxicity. To our knowledge, disruption of mitochondrial membrane potential and cellular ATP homeostasis due to chlorine gas exposure has not been shown previously. Furthermore, we are not aware of a published demonstration of downstream damage to the heart after chlorine gas inhalation. In tissues such as the heart, where energy demands are high, mitochondria, along with SR, are highly critical targets in disease pathogenesis. Under normal steady-state conditions, SERCA activity consumes roughly 40% of cell ATP. Once SERCA is damaged, maintenance of SR function likely consumes much more ATP. By preserving SERCA activity, one could minimize excessive ATP consumption by the SR, which could preserve ATP and MMP. However, whether ATP preservation is a primary or secondary effect of SERCA modulation is unknown. Others previously found preservation of intracellular ATP content and mitochondrial function by ranolazine during protection against ischemia–reperfusion (20). Because ranolazine may impact other pathways, we also used istaroxime, a more specific SERCA activator (56). Thus, ranolazine and istaroxime may protect against chlorine-induced cardiotoxicity by protecting SERCA, thereby preserving mitochondrial and cellular functions. We continue to investigate the efficacy of these two agents in rescuing chlorine-induced cardiotoxicity.
In conclusion, our studies demonstrated critical effects of in vivo chlorine exposure on the heart, and demonstrate a novel molecular target, cardiac SERCA2, for protection. Importantly, this investigation reports the presence of circulating chloramines in blood plasma after acute chlorine inhalation, and chlorotyrosine formation on specific Tyr residues in SERCA2 in the heart. The functional importance of these modifications in SERCA was suggested by our in vitro studies. Because in vitro chlorine gas exposure studies are rare, and our studies included both in vivo and in vitro exposures, we have used a novel approach. Early treatment with SCN− or HOSCN to modify HOCl and/or chloramines, or treatments to change SERCA expression or stability, were sufficient to modify SERCA activity and cell survival. Most deaths related to chlorine inhalation occur before the patient can be removed from the chlorine exposure, and autopsies of these victims have often shown cardiac pathology. Those surviving have gone on to show varying degrees and duration of lung dysfunction. Therapeutic strategies that protect or modify SERCA function could be useful approaches for emergent resuscitation of victims of severe chlorine inhalation.
Acknowledgments
Acknowledgments
The authors thank Rhonda B. Garlick and Jacqueline S. Rioux for their excellent technical support. All FACS analyses were performed at the Flow Cytometry Shared Resource (University of Colorado Cancer Center, Denver, Colorado).
Footnotes
This work was supported by the CounterACT Program, National Institutes of Health, Office of the Director, and the National Institute of Environmental Health Sciences grant U54 ES015678 (C.W.W.), and by Children’s Hospital of Colorado/Colorado School of Mines Pilot Award G0100394 and a Children’s Hospital of Colorado Research Institute’s Pilot Award (S.A.).
Author Contributions: conception and design—S.A., A.A., L.A.V., and C.W.W.; analysis and interpretation—T.B.H.-H., J.E.L., W.C.C., O.M., C.S., J.D.C., B.J.D., N.R., and R.L.P.; drafting the manuscript for important intellectual content—S.A., A.A., O.M., C.S., J.D.C., and C.W.W.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2014-0005OC on September 4, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.White CW, Martin JG. Chlorine gas inhalation: human clinical evidence of toxicity and experience in animal models. Proc Am Thorac Soc. 2010;7:257–263. doi: 10.1513/pats.201001-008SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Sickle D, Wenck MA, Belflower A, Drociuk D, Ferdinands J, Holguin F, Svendsen E, Bretous L, Jankelevich S, Gibson JJ, et al. Acute health effects after exposure to chlorine gas released after a train derailment. Am J Emerg Med. 2009;27:1–7. doi: 10.1016/j.ajem.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gunnarsson M, Walther SM, Seidal T, Bloom GD, Lennquist S. Exposure to chlorine gas: effects on pulmonary function and morphology in anaesthetised and mechanically ventilated pigs. J Appl Toxicol. 1998;18:249–255. doi: 10.1002/(sici)1099-1263(199807/08)18:4<249::aid-jat507>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 4.Honavar J, Bradley E, Bradley K, Oh JY, Vallejo MO, Kelley EE, Cantu-Medellin N, Doran S, Dell’italia LJ, Matalon S, et al. Chlorine gas exposure disrupts nitric oxide homeostasis in the pulmonary vasculature. Toxicology. 2014;321:96–102. doi: 10.1016/j.tox.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Honavar J, Samal AA, Bradley KM, Brandon A, Balanay J, Squadrito GL, MohanKumar K, Maheshwari A, Postlethwait EM, Matalon S, et al. Chlorine gas exposure causes systemic endothelial dysfunction by inhibiting endothelial nitric oxide synthase–dependent signaling. Am J Respir Cell Mol Biol. 2011;45:419–425. doi: 10.1165/rcmb.2010-0151OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo M, Anderson ME. Mechanisms of altered Ca²⁺ handling in heart failure. Circ Res. 2013;113:690–708. doi: 10.1161/CIRCRESAHA.113.301651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 8.Cook NL, Viola HM, Sharov VS, Hool LC, Schöneich C, Davies MJ. Myeloperoxidase-derived oxidants inhibit sarco/endoplasmic reticulum Ca2+-ATPase activity and perturb Ca2+ homeostasis in human coronary artery endothelial cells. Free Radic Biol Med. 2012;52:951–961. doi: 10.1016/j.freeradbiomed.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Periasamy M, Kalyanasundaram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve. 2007;35:430–442. doi: 10.1002/mus.20745. [DOI] [PubMed] [Google Scholar]
- 10.Ahmad S, Ahmad A, Neeves KB, Hendry-Hofer T, Loader JE, White CW, Veress L. In vitro cell culture model for toxic inhaled chemical testing. J Vis Exp. 2014. ;(87) [DOI] [PMC free article] [PubMed]
- 11.Arteel GE, Thurman RG, Raleigh JA. Reductive metabolism of the hypoxia marker pimonidazole is regulated by oxygen tension independent of the pyridine nucleotide redox state. Eur J Biochem. 1998;253:743–750. doi: 10.1046/j.1432-1327.1998.2530743.x. [DOI] [PubMed] [Google Scholar]
- 12.Squadrito GL, Postlethwait EM, Matalon S. Elucidating mechanisms of chlorine toxicity: reaction kinetics, thermodynamics, and physiological implications. Am J Physiol Lung Cell Mol Physiol. 2010;299:L289–L300. doi: 10.1152/ajplung.00077.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sochaski MA, Jarabek AM, Murphy J, Andersen ME. 3-chlorotyrosine and 3,5-dichlorotyrosine as biomarkers of respiratory tract exposure to chlorine gas. J Anal Toxicol. 2008;32:99–105. doi: 10.1093/jat/32.1.99. [DOI] [PubMed] [Google Scholar]
- 14.Boardman NT, Aronsen JM, Louch WE, Sjaastad I, Willoch F, Christensen G, Sejersted O, Aasum E. Impaired left ventricular mechanical and energetic function in mice after cardiomyocyte-specific excision of Serca2. Am J Physiol Heart Circ Physiol. 2014;306:H1018–H1024. doi: 10.1152/ajpheart.00741.2013. [DOI] [PubMed] [Google Scholar]
- 15.Ashby MT, Carlson AC, Scott MJ. Redox buffering of hypochlorous acid by thiocyanate in physiologic fluids. J Am Chem Soc. 2004;126:15976–15977. doi: 10.1021/ja0438361. [DOI] [PubMed] [Google Scholar]
- 16.Chandler JD, Nichols DP, Nick JA, Hondal RJ, Day BJ. Selective metabolism of hypothiocyanous acid by mammalian thioredoxin reductase promotes lung innate immunity and antioxidant defense. J Biol Chem. 2013;288:18421–18428. doi: 10.1074/jbc.M113.468090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmad S, Nichols DP, Strand M, Rancourt RC, Randell SH, White CW, Ahmad A. SERCA2 regulates non-CF and CF airway epithelial cell response to ozone. PLoS One. 2011;6:e27451. doi: 10.1371/journal.pone.0027451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rastogi S, Sharov VG, Mishra S, Gupta RC, Blackburn B, Belardinelli L, Stanley WC, Sabbah HN. Ranolazine combined with enalapril or metoprolol prevents progressive LV dysfunction and remodeling in dogs with moderate heart failure. Am J Physiol Heart Circ Physiol. 2008;295:H2149–H2155. doi: 10.1152/ajpheart.00728.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kloner RA. Current state of clinical translation of cardioprotective agents for acute myocardial infarction. Circ Res. 2013;113:451–463. doi: 10.1161/CIRCRESAHA.112.300627. [DOI] [PubMed] [Google Scholar]
- 20.Aldakkak M, Camara AK, Heisner JS, Yang M, Stowe DF. Ranolazine reduces Ca2+ overload and oxidative stress and improves mitochondrial integrity to protect against ischemia reperfusion injury in isolated hearts. Pharmacol Res. 2011;64:381–392. doi: 10.1016/j.phrs.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrandi M, Barassi P, Tadini-Buoninsegni F, Bartolommei G, Molinari I, Tripodi MG, Reina C, Moncelli MR, Bianchi G, Ferrari P. Istaroxime stimulates SERCA2a and accelerates calcium cycling in heart failure by relieving phospholamban inhibition. Br J Pharmacol. 2013;169:1849–1861. doi: 10.1111/bph.12278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kose A, Kose B, Acikalin A, Gunay N, Yildirim C. Myocardial infarction, acute ischemic stroke, and hyperglycemia triggered by acute chlorine gas inhalation. Am J Emerg Med. 2009;27:1022.e1–4. doi: 10.1016/j.ajem.2008.12.029. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki S, Sakamoto S, Maniwa K, Saitoh A, Hirayama Y, Kobayashi H, Matsuo T. Fatal pulmonary arterial thrombosis associated with chlorine gas poisoning. Clin Appl Thromb Hemost. 2001;7:356–358. doi: 10.1177/107602960100700420. [DOI] [PubMed] [Google Scholar]
- 24.Wenck MA, Van Sickle D, Drociuk D, Belflower A, Youngblood C, Whisnant MD, Taylor R, Rudnick V, Gibson JJ. Rapid assessment of exposure to chlorine released from a train derailment and resulting health impact. Public Health Rep. 2007;122:784–792. doi: 10.1177/003335490712200610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmad S, Ahmad A, Dremina ES, Sharov VS, Guo X, Jones TN, Loader JE, Tatreau JR, Perraud AL, Schöneich C, et al. Bcl-2 suppresses sarcoplasmic/endoplasmic reticulum Ca2+-ATPase expression in cystic fibrosis airways: role in oxidant-mediated cell death. Am J Respir Crit Care Med. 2009;179:816–826. doi: 10.1164/rccm.200807-1104OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinis FI, Andersson KB, Christensen G, Metzger JM. Prominent heart organ–level performance deficits in a genetic model of targeted severe and progressive SERCA2 deficiency. PLoS ONE. 2013;8:e79609. doi: 10.1371/journal.pone.0079609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curtis MP, Hicks AJ, Neidigh JW. Kinetics of 3-chlorotyrosine formation and loss due to hypochlorous acid and chloramines. Chem Res Toxicol. 2011;24:418–428. doi: 10.1021/tx100380d. [DOI] [PubMed] [Google Scholar]
- 28.Devlin RB, Duncan KE, Jardim M, Schmitt MT, Rappold AG, Diaz-Sanchez D. Controlled exposure of healthy young volunteers to ozone causes cardiovascular effects. Circulation. 2012;126:104–111. doi: 10.1161/CIRCULATIONAHA.112.094359. [DOI] [PubMed] [Google Scholar]
- 29.Lord K, Moll D, Lindsey JK, Mahne S, Raman G, Dugas T, Cormier S, Troxlair D, Lomnicki S, Dellinger B, et al. Environmentally persistent free radicals decrease cardiac function before and after ischemia/reperfusion injury in vivo. J Recept Signal Transduct Res. 2011;31:157–167. doi: 10.3109/10799893.2011.555767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winternitz MC, Lambert RA, Jackson L, Smith GH. New Haven, CT: Yale University Press; 1920. The pathophysiology of chlorine poisoning. [Google Scholar]
- 31.Wang J, Abu-Zidan FM, Walther SM. Effects of prone and supine posture on cardiopulmonary function after experimental chlorine gas lung injury. Acta Anaesthesiol Scand. 2002;46:1094–1102. doi: 10.1034/j.1399-6576.2002.460907.x. [DOI] [PubMed] [Google Scholar]
- 32.Evans RB. Chlorine: state of the art. Lung. 2005;183:151–167. doi: 10.1007/s00408-004-2530-3. [DOI] [PubMed] [Google Scholar]
- 33.Hazen SL, Crowley JR, Mueller DM, Heinecke JW. Mass spectrometric quantification of 3-chlorotyrosine in human tissues with attomole sensitivity: a sensitive and specific marker for myeloperoxidase-catalyzed chlorination at sites of inflammation. Free Radic Biol Med. 1997;23:909–916. doi: 10.1016/s0891-5849(97)00084-1. [DOI] [PubMed] [Google Scholar]
- 34.Qin XJ, Li YN, Liang X, Wang P, Hai CX. The dysfunction of ATPases due to impaired mitochondrial respiration in phosgene-induced pulmonary edema. Biochem Biophys Res Commun. 2008;367:150–155. doi: 10.1016/j.bbrc.2007.12.111. [DOI] [PubMed] [Google Scholar]
- 35.Shareef MA, Anwer LA, Poizat C. Cardiac SERCA2A/B: therapeutic targets for heart failure. Eur J Pharmacol. 2014;724:1–8. doi: 10.1016/j.ejphar.2013.12.018. [DOI] [PubMed] [Google Scholar]
- 36.Lancel S, Qin F, Lennon SL, Zhang J, Tong X, Mazzini MJ, Kang YJ, Siwik DA, Cohen RA, Colucci WS. Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq-overexpressing mice. Circ Res. 2010;107:228–232. doi: 10.1161/CIRCRESAHA.110.217570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin JG, Campbell HR, Iijima H, Gautrin D, Malo JL, Eidelman DH, Hamid Q, Maghni K. Chlorine-induced injury to the airways in mice. Am J Respir Crit Care Med. 2003;168:568–574. doi: 10.1164/rccm.200201-021OC. [DOI] [PubMed] [Google Scholar]
- 38.Burton KP, McCord JM, Ghai G. Myocardial alterations due to free-radical generation. Am J Physiol. 1984;246:H776–H783. doi: 10.1152/ajpheart.1984.246.6.H776. [DOI] [PubMed] [Google Scholar]
- 39.Buja LM, Burton KP, Chien KR, Willerson JT. Altered calcium homeostasis and membrane integrity in myocardial cell injury. Adv Exp Med Biol. 1988;232:115–124. doi: 10.1007/978-1-4757-0007-7_13. [DOI] [PubMed] [Google Scholar]
- 40.Burton KP, Hagler HK, Nazeran H. Exposure to free radicals alters ionic calcium transients in isolated adult rat cardiac myocytes. Am J Cardiovasc Pathol. 1992;4:235–244. [PubMed] [Google Scholar]
- 41.Strosová M, Skuciová M, Horáková L. Oxidative damage to Ca2+-ATPase sarcoplasmic reticulum by HOCl and protective effect of some antioxidants. Biofactors. 2005;24:111–116. doi: 10.1002/biof.5520240113. [DOI] [PubMed] [Google Scholar]
- 42.Pattison DI, Hawkins CL, Davies MJ. Hypochlorous acid–mediated protein oxidation: how important are chloramine transfer reactions and protein tertiary structure? Biochemistry. 2007;46:9853–9864. doi: 10.1021/bi7008294. [DOI] [PubMed] [Google Scholar]
- 43.Xulu BA, Ashby MT. Small molecular, macromolecular, and cellular chloramines react with thiocyanate to give the human defense factor hypothiocyanite. Biochemistry. 2010;49:2068–2074. doi: 10.1021/bi902089w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang JG, Mahmud SA, Nguyen J, Slungaard A. Thiocyanate-dependent induction of endothelial cell adhesion molecule expression by phagocyte peroxidases: a novel HOSCN-specific oxidant mechanism to amplify inflammation. J Immunol. 2006;177:8714–8722. doi: 10.4049/jimmunol.177.12.8714. [DOI] [PubMed] [Google Scholar]
- 45.Wang JG, Mahmud SA, Thompson JA, Geng JG, Key NS, Slungaard A. The principal eosinophil peroxidase product, HOSCN, is a uniquely potent phagocyte oxidant inducer of endothelial cell tissue factor activity: a potential mechanism for thrombosis in eosinophilic inflammatory states. Blood. 2006;107:558–565. doi: 10.1182/blood-2005-05-2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lloyd MM, Grima MA, Rayner BS, Hadfield KA, Davies MJ, Hawkins CL. Comparative reactivity of the myeloperoxidase-derived oxidants hypochlorous acid and hypothiocyanous acid with human coronary artery endothelial cells. Free Radic Biol Med. 2013;65:1352–1362. doi: 10.1016/j.freeradbiomed.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 47.Thomas EL, Bates KP, Jefferson MM. Peroxidase antimicrobial system of human saliva: requirements for accumulation of hypothiocyanite. J Dent Res. 1981;60:785–796. doi: 10.1177/00220345810600040401. [DOI] [PubMed] [Google Scholar]
- 48.Ashby MT. Hypothiocyanite. Adv Inorg Chem. 2012;64:263–303. [Google Scholar]
- 49.Morimoto S, Hongo K, Kusakari Y, Komukai K, Kawai M, J OU, Nakayama H, Asahi M, Otsu K, Yoshimura M, Kurihara S. Genetic modulation of the serca activity does not affect the ca leak from the cardiac sarcoplasmic reticulum. Cell Calcium. 2014;55:17–23. doi: 10.1016/j.ceca.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 50.Ma TS, Mann DL, Lee JH, Gallinghouse GJ. SR compartment calcium and cell apoptosis in SERCA overexpression. Cell Calcium. 1999;26:25–36. doi: 10.1054/ceca.1999.0049. [DOI] [PubMed] [Google Scholar]
- 51.Minotti G. Pharmacology at work for cardio-oncology: ranolazine to treat early cardiotoxicity induced by antitumor drugs. J Pharmacol Exp Ther. 2013;346:343–349. doi: 10.1124/jpet.113.204057. [DOI] [PubMed] [Google Scholar]
- 52.Stone PH. Ranolazine: new paradigm for management of myocardial ischemia, myocardial dysfunction, and arrhythmias. Cardiol Clin. 2008;26:603–614. doi: 10.1016/j.ccl.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 53.Gadicherla AK, Stowe DF, Antholine WE, Yang M, Camara AK. Damage to mitochondrial complex I during cardiac ischemia reperfusion injury is reduced indirectly by anti-anginal drug ranolazine. Biochim Biophys Acta. 2012;1817:419–429. doi: 10.1016/j.bbabio.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Satoh K, Matsu-Ura T, Enomoto M, Nakamura H, Michikawa T, Mikoshiba K. Highly cooperative dependence of sarco/endoplasmic reticulum calcium ATPase SERCA2a pump activity on cytosolic calcium in living cells. J Biol Chem. 2011;286:20591–20599. doi: 10.1074/jbc.M110.204685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leikauf GD, Pope-Varsalona H, Concel VJ, Liu P, Bein K, Berndt A, Martin TM, Ganguly K, Jang AS, Brant KA, et al. Integrative assessment of chlorine-induced acute lung injury in mice. Am J Respir Cell Mol Biol. 2012;47:234–244. doi: 10.1165/rcmb.2012-0026OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Micheletti R, Palazzo F, Barassi P, Giacalone G, Ferrandi M, Schiavone A, Moro B, Parodi O, Ferrari P, Bianchi G. Istaroxime, a stimulator of sarcoplasmic reticulum calcium adenosine triphosphatase isoform 2a activity, as a novel therapeutic approach to heart failure. Am J Cardiol. 2007;99:24A–32A. doi: 10.1016/j.amjcard.2006.09.003. [DOI] [PubMed] [Google Scholar]