

Abstract
Laminins are heterotrimeric proteins that are secreted by the alveolar epithelium into the basement membrane, and their expression is altered in extracellular matrices from patients with pulmonary fibrosis. In a small number of patients with pulmonary fibrosis, we found that the normal basement membrane distribution of the α3 laminin subunit was lost in fibrotic regions of the lung. To determine if these changes play a causal role in the development of fibrosis, we generated mice lacking the α3 laminin subunit specifically in the lung epithelium by crossing mice expressing Cre recombinase driven by the surfactant protein C promoter (SPC-Cre) with mice expressing floxed alleles encoding the α3 laminin gene (Lama3fl/fl). These mice exhibited no developmental abnormalities in the lungs up to 6 months of age, but, compared with control mice, had worsened mortality, increased inflammation, and increased fibrosis after the intratracheal administration of bleomycin. Similarly, the severity of fibrosis induced by an adenovirus encoding an active form of transforming growth factor-β was worse in mice deficient in α3 laminin in the lung. Taken together, our results suggest that the loss of α3 laminin in the lung epithelium does not affect lung development, but plays a causal role in the development of fibrosis in response to bleomycin or adenovirally delivered transforming growth factor-β. Thus, we speculate that the loss of the normal basement membrane organization of α3 laminin that we observe in fibrotic regions from the lungs of patients with pulmonary fibrosis contributes to their disease progression.
Keywords: laminin, matrix, pulmonary fibrosis, development
Clinical Relevance
Pulmonary fibrosis is defined by changes in the amount and composition of extracellular matrix proteins in the lung. In this article, we use a lung-specific knockout strategy in mice to show that the loss of a protein found only in the extracellular matrix contributes to the development of fibrosis. These data complement findings from other groups suggesting that the altered composition of the matrix plays a causal role in the progression of lung fibrosis.
The flattened conformation of the alveolar type I cells and alveolar capillary endothelial cells, and their separation by only a thin basement membrane, facilitates the exchange of oxygen and carbon dioxide in the healthy lung (1). In patients with pulmonary fibrosis, an excess deposition of extracellular matrix proteins results in thickening and distortion of the alveolar basement membrane, eventually obliterating the alveolar architecture, with an accompanying increase in the elastic recoil of the lung and impairment in gas exchange (2). Although the diagnosis of pulmonary fibrosis is made by radiographic or histologic detection of these excess matrix proteins (3), abnormalities of the matrix have traditionally been considered as a consequence of the cellular injury or dysfunction that underlies the pathogenesis of the disorder, rather than causal factors in its development (3). Recent studies, in which normal human lung fibroblasts have been seeded onto decellularized scaffolds from the lungs of patients with idiopathic pulmonary fibrosis (IPF) or individuals without lung disease, show that the fibrotic matrices can induce a profibrotic expression program in the overlying fibroblasts, suggesting a vicious circle of increasing fibrosis with the matrix playing a causal role (4).
Laminins are heterotrimeric matrix proteins comprised of an α, a β, and a γ subunit, and the 16 identified laminin heterotrimer combinations resulting from the five α, three β, and three γ chains show tissue-specific patterns of expression (5–8). In the adult lung, the α3 laminin is a major component of the alveolar basement membrane (5, 6); however, its function is not known, as mice and humans with disruptions in the gene expressing α3 laminin die of a blistering skin disease (juvenile epidermolysis bullosa in humans) early in life (9, 10). When Booth and colleagues (4) conducted a proteomic analysis of lung matrix scaffolds from patients with IPF, they found a plethora of matrix proteins that were increased in fibrotic compared with control lungs. Intriguingly, on the other hand, the expression of several laminins, including laminin α-3, β-3, and γ-2 chains, was reduced (4). In this study, we show that α3 laminin is homogenously distributed in the basement membrane in the normal human lung and in unaffected areas of the lungs of patients with pulmonary fibrosis. However, this pattern is disrupted in fibrotic regions of lungs from patients with pulmonary fibrosis, where the expression of α3 laminin subunit proteins is reduced. To determine whether loss of α3 laminin plays a causal role in the development of fibrosis, we examined the susceptibility of mice with a lung epithelial–specific deletion of α3 laminin to bleomycin and transforming growth factor (TGF)-β–induced lung fibrosis.
Materials and Methods
Human Subjects
Studies using human subjects were approved by the Northwestern University Institutional Review Board (Chicago, IL) and the Bioethics Committee at the Instituto Nacional de Enfermedades Respiratorias (Mexico City, Mexico). Deidentified human lung tissue obtained from donors whose lungs were unsuitable for transplantation was provided by the Regional Organ Bank of Illinois (Chicago, IL). Stored lung tissues and frozen lung sections from patients with IPF were obtained from the National Institute of Respiratory Diseases (Mexico City, Mexico). The diagnosis of IPF was based on published consensus guidelines and determined on the basis of high-resolution computed tomography or surgical lung biopsy showing a definite usual interstitial pneumonia pattern (11).
Mice
All experiments using mice were approved by the Animal Care and Use Committee at Northwestern University (Chicago, IL). Unless indicated otherwise, we used mice that were 8–12 weeks of age (20–25 g). Mice harboring floxed alleles in the gene encoding α3 laminin (Lama3fl/fl) were created by generating a vector that targets exon 42 of the mouse α3 laminin allele flanked by loxP (locus of x-ing over bacteriophage P) sequences and introduced this into embryonic stem cells, as we previously described (12). To generate a line of mice deficient in α3 laminin in the lung epithelium, we crossed Lama3fl/fl animals with transgenic mice expressing Cre recombinase driven by the surfactant protein C promoter (SPC-Cre) (a kind gift of Dr. B. Hogan, Duke University, Durham, NC) (13).
The Administration of Bleomycin and Infection with Adenoviruses Encoding Active TGF-β to Induce Lung Fibrosis
Mice were anesthetized with isoflurane and their lungs were intubated orally with a 20-gauge Angiocath (Becton-Dickinson, Franklin Lakes, NJ) for the intratracheal administration of bleomycin, saline, or adenoviruses, as previously described (14). For all substances, a total volume of 50 μl was instilled in two equal aliquots through the endotracheal catheter. After each aliquot, the mice were placed first on their right side and then on their left side for 10–15 seconds. Bleomycin was purchased from APP Pharmaceuticals LLC (Schaumburg, IL), and sterile saline for injection was from Baxter (Deerfield, IL). An adenovirus encoding an active form of TGF-β (Ad–TGF-β) was a kind gift from Dr. R. Derynck (University of California San Francisco, San Francisco, CA) (15). This adenovirus and an adenovirus encoding no transgene were commercially amplified, tittered, and confirmed to be replication deficient (Viraquest, Liberty City, Iowa). For experiments using Ad–TGF-β or adenovirus encoding no transgene, mice were intratracheally infected with 1 × 108 plaque-forming units/mouse in Tris-EDTA buffer, as previously described (16). Mice were killed 21 and 28 days after the instillation of bleomycin and Ad–TGF-β, respectively.
Tissue Analysis and Staining
A 20-gauge Angiocath was sutured into the trachea, the lungs and heart were removed en bloc, and the lungs were inflated to 15 cm H2O with 4% paraformaldehyde. The heart and lungs were fixed in paraffin, and 5-μm sections were stained with hematoxylin and eosin and Masson’s trichrome stains (17). Images of murine lungs were obtained using TissueGnostics system and software (Vienna, Austria).
For immunofluorescence analysis of human lung tissue, the tissue was rapidly frozen in Tissue-Tek OCT compound (Electron Microscopy Sciences, Hatfield, PA) and 5-μm sections were prepared on a cryomicrotome. Frozen sections were mounted onto microscope slides, fixed for 2 minutes in −20°C acetone, air dried, and then incubated for 2 hours at room temperature with antibody against the mouse α3 laminin subunit, diluted in PBS. The sections were then washed extensively and subsequently incubated for 1 hour at 37°C in rhodamine-conjugated secondary antibody. After further washing, the sections were covered with mounting medium and a coverslip. Tissue sections were viewed on a Leica SP8X TCS confocal imaging microscope (Leica Microsystems Inc., Buffalo Grove, IL). Microscope images were exported as .tif files and figures generated using Adobe Photoshop software (Adobe Systems, San Jose, CA).
Immunoblotting
Protein immunoblotting was performed as previously described (17). Cell lysates were mixed with sample loading buffer (125 mM Tris base [pH 6.8], 4% [wt/vol] SDS, 20% [vol/vol] glycerol, 200 mM dithiothreitol, 0.02% [wt/vol] bromphenol blue). After heating, the protein was resolved on a SDS–15% polyacrylamide gel and transferred to a Hybond-ECL nitrocellulose membrane (Amersham, Piscataway, NJ). After transfer, the gel was stained with Ponceau S to verify uniform transfer. Membranes were blocked with 5% (wt/vol) nonfat milk in TBS-T (100 mM Tris base [pH 7.5], 0.9% [wt/vol] NaCl, 0.1% [vol/vol] Tween 20) for 1 hour at room temperature and subsequently incubated with the appropriate primary antibody overnight at 4°C. The membrane was washed with TBS-T three times and incubated for 1 hour at room temperature with horseradish peroxidase–conjugated secondary antibody. The membrane was washed three times with TBS-T and analyzed by enhanced chemiluminescence (Amersham). Densitometry was assessed using Image J software (Bethesda, MD) (18).
Measurement of Total Lung Collagen
Lung collagen was measured using a modification of a previously described method for the precipitation of lung collagen using picrosirius red (19). Mouse lungs were harvested and suspended in 1 ml of 0.5 M acetic acid and then homogenized, first with a tissue homogenizer (60 s on ice) and then using 15 strokes in a Dounce homogenizer (on ice). The resulting homogenate was spun (12,000 × g) for 10 minutes, and the supernatant was used for subsequent analyses. Collagen standards were prepared in 0.5 M acetic acid using rat tail collagen (Sigma-Aldrich, St. Louis, MO). Picrosirius red dye was prepared by mixing 0.2 g of Sirius red F3B (Sigma-Aldrich) with 200 ml of water; 1 ml of the Picrosirius red dye was added to 100 μl of the collagen standard or the lung homogenates and then mixed continuously at room temperature on an orbital shaker for 30 minutes. The precipitated collagen was then pelleted and washed once with 0.5 M acetic acid (12,000 × g for 15 min each). The resulting pellet was resuspended in 1 ml of 0.5 M NaOH and Sirius red staining was quantified spectrophotometrically (540 nm) using a colorimetric plate reader (Bio-Rad, Hercules, CA).
Measurement of Lung Mechanics
Measurements of lung mechanics were performed using a FlexiVent mouse ventilator (Scireq, Montreal, PQ, Canada) according to the protocols established by Scireq (20). A standard ventilation history for each mouse was obtained with three total lung capacity maneuvers before the forced oscillation and quasistatic pressure–volume curve protocols that were used to calculate airway resistance, dynamic and quasistatic tissue compliance, and elastance.
Generation of Lung Scaffolds for Electron Microscopy
The lungs were decellularized using a previously described procedure (21). Briefly, the lungs were rinsed five times by instilling 3 ml of distilled H2O through the trachea and right ventricle. The lungs were then instilled with 0.1% Triton X-100 and incubated at 4°C for 24 hours, followed by 2% deoxycholate at 4°C for 24 hours. Between each incubation period, the lungs were rinsed five times with 3 ml of distilled H2O. Finally, lungs were incubated at room temperature for 1 hour in 1 M NaCl, followed by DNase (30μg/ml) in 1.3 mM MgSO4 and 2 mM CaCl2. Tissue materials were prepared for electron microscopy as detailed elsewhere (22). Thin sections of tissues were viewed on an FEI Tecnai Spirit G2 transmission electron microscope (FEI Co., Hillsboro, OR). Microscope images were exported as .tif files and figures generated using Adobe Photoshop software.
Quantitative Real-Time RT-PCR Measurement of RNA
We isolated total RNA using a commercially available system (TRIzol; Invitrogen, Carlsbad, CA) from mouse lung homogenates and performed quantitative real-time RT-PCR (qRT-PCR) reactions using IQ SYBR Green superscript with the primers listed in the online supplement, analyzed on a Bio-Rad CFX384 real-time PCR detection system. We employed the ΔΔCt method to analyze the normalized data (23).
Quantification of Inflammatory Cell Populations Using Flow Cytometry
Inflammatory cell populations in whole-lung homogenates were measured as previously described (24). Briefly, the lungs were perfused, removed en bloc, cut into 3- to 4-mm pieces, and processed using a Lung Dissociation Kit (Miltenyi, Auburn, CA) and GentleMACS tissue dissociator (Miltenyi), according to the manufacturer’s instructions and passed through a 40-μm nylon mesh to obtain a single cell suspension. Remaining red blood cells were lysed using BD Pharm Lyse (BD Biosciences, San Jose, CA). Immune cells were isolated using anti-CD45 microbeads (Miltenyi) and positive selection on AutoMACS separator. CD45-positive cells were counted using Countess automated cell counter (Invitrogen); dead cells were excluded using trypan blue. After live/dead staining with eFluor506 viability dye (eBioscience, San Diego, CA), cells were incubated with FcBlock (BD Biosciences) and stained with a mixture of fluorochrome-conjugated antibodies (see Table E1 in the online supplement for the list of antibodies, clones, fluorochromes, and manufacturers). Data were acquired on a BD LSR II flow cytometer (BD Biosciences; see Table E2 for instrument configuration). Compensation and data analysis were performed using FlowJo software (TreeStar, Ashland, OR). After gating out cell aggregates, debris, and dead cells, immune cells were identified using the panhematopoietic marker, CD45. Specific cell types were identified as follows: eosinophils as CD11bintermediateCD11c−Siglec FintermediateSSChigh; neutrophils as CD11b+Ly6G+; alveolar macrophages as highly autofluorescent and CD11chighCD11bintermediateSiglec Fhigh CD64high; plasmacytoid dendritic cells as mPDCA-1+CD11cintermediateLy6C+; CD103+ dendritic cells as CD11chighCD11bintermediateMHC II+CD103+; CD11b+ dendritic cells as CD11chighCD11b+MHC II+CD103−; interstitial macrophages as CD11b+CD11cintermediateCD64+Siglec F−; and monocytes as CD11b+CD11c−CD64lowMHC II−. Data are presented as mean number of cells in both lung (±SEM).
Statistical Analysis
Differences between groups were explored using ANOVA. When the ANOVA indicated a significant difference, individual differences were explored using Student’s t tests with a Bonferonni correction for multiple comparisons. Mortality differences were determined using a Kaplan–Meier analysis. All analyses were performed using GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego, CA). Data are shown as means (± SEM).
Results
The α3 Subunit of Laminin Is Localized to the Alveolar Basement Membrane in Normal Human Lung Tissue, but Not in Fibrotic Regions from Patients with Pulmonary Fibrosis
We obtained lung tissue and frozen lung sections from four deidentified subjects who had undergone transplant for IPF and individuals without a history of lung disease from normal-appearing regions of lungs that were unsuitable for transplantation. We found increased levels of mRNA encoding Lama3 and collagen type IV (ColIVa) in lung specimens from patients with pulmonary fibrosis compared with normal individuals (Figures 1A and 1B). The levels of Lama3 expression in control subjects were near the lower limits of detection. Staining for α3 laminin in frozen lung sections from normal individuals and in normal regions of lung in patients with lung fibrosis revealed a linear pattern morphologically consistent with the basement membrane and colocalizing with the basement membrane protein collagen type IV (Figure 1B). However, in fibrotic areas, the basement membrane pattern of staining and colocalization with collagen type IV was lost (Figure 1C). Instead, aggregates positive for collagen type IV, but not α3 laminin, were scattered throughout the fibrotic regions of the lung (Figure 1D). To provide a means of comparison, images from normal and fibrotic regions of the same lung are shown.
Figure 1.
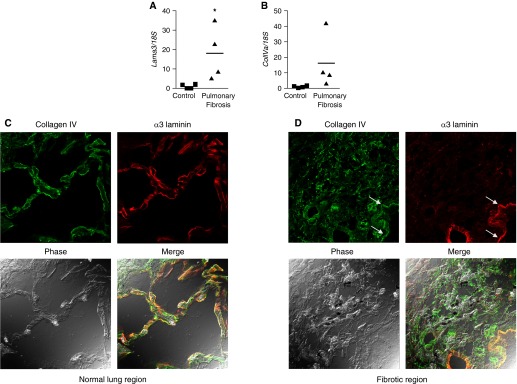
The α3 laminin subunit is aberrantly expressed in fibrotic regions of human lungs from patients with pulmonary fibrosis. (A) Distal lung tissue (∼ 1 cm2) from four organ donors whose lungs were not suitable for transplantation and four patients with pulmonary fibrosis was homogenized for RNA extraction and the expression of the mRNA encoding α3 laminin subunit (Lama3) (A) and collagen type IV (ColIVa) (B) was measured using quantitative RT-PCR. Values are normalized to the levels of 18S ribosomal RNA. *P < 0.05. (C and D) Frozen sections of distal lung tissue were generated from the normal subjects and those with pulmonary fibrosis and stained using antibodies that recognize collagen type IV (green) and α3 laminin (red). Shown are normal and fibrotic regions from the same lung of a patient with pulmonary fibrosis. The bottom panels show the phase (left) and merged (right) images. (C) In normal regions of the lung, linear staining for α3 laminin was observed in the alveoli and colocalized with the basement membrane protein collagen type IV (merged panel) (D). In fibrotic regions of the lung, collagen type IV was found in disordered clumps throughout the fibrotic regions, whereas staining for α3 laminin was lost. A relatively spared region adjacent to the fibrotic region continues to demonstrate linear staining for both collagen type IV and α3 laminin (white arrows). Representative images are shown; all images were obtained at 400× using identical microscope and imaging settings.
Loss of the α3 Laminin Subunit Does Not Result in a Detectable Phenotype in Adult Mice
Mutations in the laminin α3 gene in humans are associated with the blistering skin disease, junctional epidermolysis bullosa (9). Global deletion of the laminin α3 gene in mice results in a similar blistering skin disease, which is the cause of death of the animals shortly after birth, before lung development is complete (25). Thus, we developed a mouse line lacking the α3 laminin subunit specifically in the lung epithelium by crossing mice expressing Cre recombinase driven by the SPC promoter (SPC-Cre) with mice expressing floxed alleles encoding the α3 laminin gene (Lama3fl/fl), which we have previously described (12). Expression of the SPC gene in early progenitors of the lung epithelium during development results in Cre-mediated gene recombination in the entire airway epithelium (13, 26). The resulting SPC-Cre/Lama3fl/fl mice exhibited a complete absence of α3 laminin mRNA (Figure 2A) and protein in total lung homogenates (Figure 2B). A gene–dose relationship was also seen in the heterozygous animals (Figure 2B), without a compensatory increase in α5 laminin (Figure 2C). We were not able to detect differences upon careful histologic evaluation of the airways, pulmonary vessels, or lung parenchyma in hematoxylin and eosin–stained lung sections from SPC-Cre, Lama3fl/fl, and SPC-Cre/Lama3fl/fl mice, and trichrome staining revealed no apparent differences in lung collagen (Figure 2D). Similar results were obtained in mice 6 months of age (data not shown). Similarly, the static compliance of the lung and airway resistance were similar in untreated Lama3fl/fl and SPC-Cre/Lama3fl/fl (Figures 2E and 2F). To more closely examine the morphology of the basement membrane, we generated decellularized lung scaffolds from Lama3fl/fl and SPC-Cre/Lama3fl/fl mice, and examined them using electron microscopy. We were unable to detect significant differences in basement membrane morphology in the two strains of mice (Figures 2G and 2H).
Figure 2.

Lung epithelial–specific deletion of α3 laminin is not associated with detectable morphologic or mechanical changes in the lungs of adult mice. (A) Transgenic mice expressing Cre recombinase driven by the surfactant protein C promoter (SPC-Cre) were crossed with mice harboring floxed alleles in the gene encoding α3 laminin (Lama3fl/fl) to generate SPC-Cre/Lama3fl/fl mice. (A) mRNA was isolated from whole-lung homogenates generated from untreated mice with the indicated genotypes, and expression of α3 laminin was measured using quantitative RT-PCR. (B) Lung homogenates (solubilized in 8 M urea) from mice with the indicated genotypes were immunoblotted and stained using an antibody against (B) α3 laminin or (C) α5 laminin; nuclear lamin and tubulin were used as loading controls, respectively. Representative blots from three independent experiments are shown. (D) Lung sections were obtained from mice with the indicated genotypes and stained using hematoxylin and eosin or trichrome and examined using light microscopy. Significant differences between the genotypes were not observed. Representative images from more than six animals in each group are shown. The panels on the left are montage images (100×). The panels on the right are higher-magnification views of the areas indicated by squares. (E) Quasistatic lung compliance (Cst) and (F) total airway resistance (R) was measured in Lama3fl/fl and SPC-Cre/Lama3fl/fl mice (five animals each group) using a FlexiVent system. (G and H) Decellularized lung scaffolds were generated from (G) Lama3fl/fl and (H) SPC-Cre/Lama3fl/fl mice and examined using electron microscopy. No significant differences were seen in the basement membrane morphology. Representative images from three animals in each group are shown. Values are means ± SEM.
Lung-Specific Loss of the α3 Laminin Subunit Increases the Susceptibility to Bleomycin-Induced Injury and Fibrosis
We treated SPC-Cre, Lama3fl/fl, and SPC-Cre/Lama3fl/fl mice with a dose of intratracheal bleomycin that induces fibrosis, but seldom results in a sufficiently severe illness to require killing in wild-type mice (0.025 IU/animal). We observed an unexpectedly high mortality in SPC-Cre/Lama3fl/fl mice, which was significantly higher than that observed in the Lama3fl/fl mice (lethal dose, 50% [LD50] undefined in Lama3fl/fl and 15 d in the SPC-Cre/Lama3fl/fl mice; Figure 3A). No mortality was observed in SPC-Cre mice treated with the same dose of bleomycin (data not shown). The deaths in these animals all occurred more than 10 days after the instillation of bleomycin, a time when acute lung injury has usually resolved and lung fibrosis has begun (17). Excess mortality was also observed at a dose of 0.020 IU bleomycin/animal (see Figure E1), and even at a dose of 0.01 IU bleomycin/animal, significant excess mortality was still observed in the SPC-Cre/Lama3fl/fl compared with the Lama3fl/fl animals (Figure 3B). We measured the numbers and activation state of inflammatory cells in the lungs of Lama3fl/fl and SPC-Cre/Lama3fl/fl mice 5 days after the administration of bleomycin, before lung injury had resolved. The total number of alveolar macrophages, but not interstitial macrophages or neutrophils, was increased in the SPC-Cre/Lama3fl/fl mice (Figures 3C–3E). The numbers of classical (lymphocyte antigen 6C high [Ly6Chi]) and nonclassical (lymphocyte antigen 6C low [Ly6Clo]; monocytes), CD103+ and CD11b+ dendritic cells, and eosinophils were similar in the two strains (Figures E1A–E1E). Markers of M1 macrophage activation (CD40, CD80, and CD86) were lower in alveolar macrophages from SPC-Cre/Lama3fl/fl compared with Lama3fl/fl mice, whereas CD206—a marker of M2 polarization in alveolar macrophages (AMs)—was similar (Figures E2A–E2E). Interstitial macrophages from SPC-Cre/Lama3fl/fl mice exhibited higher levels of M2 markers when compared with those from Lama3fl/fl mice (Figures E2F–E2I). We measured the severity of lung fibrosis in the surviving animals 21 days after treatment with 0.01 IU of bleomycin. Ashcroft scores generated from the histologic examination of trichrome-stained lung sections showed increased lung collagen in SPC-Cre/Lama3fl/fl compared with Lama3fl/fl animals (Figures 3F and 3G). Consistent with these findings, picrosirius red collagen precipitation from lung homogenates was higher in SPC-Cre/Lama3fl/fl compared with Lama3fl/fl mice (Figure 3H). To confirm the physiologic effects of the increased fibrosis, we measured lung compliance in the three strains of mice using the FlexiVent system. Whereas low-dose bleomycin (0.01 IU/animal) resulted in only modest reductions in lung compliance in the Lama3fl/fl mice, significant reductions in lung compliance were observed in the SPC-Cre/Lama3fl/fl animals (Figure 3I). Total lung collagen was similar in SPC-Cre and Lama3fl/fl mice (Figure E3), and the administration of bleomycin to mice in which gene recombination was accomplished through delivery of adenoviral Cre resulted in enhanced bleomycin-induced fibrosis (Figure E4).
Figure 3.

Lung epithelial–specific deletion of α3 laminin worsens pulmonary fibrosis in response to bleomycin. (A) Mice with the indicated genotype were treated with bleomycin (0.025 IU/mouse), and unanticipated mortalities were recorded (lethal dose, 50% [LD50] Lama3fl/fl, undefined, n = 16; LD50 SP-C-Cre/Lama3fl/fl, 15 d, n = 11; P < 0.001). (B) Mice with the indicated genotype were treated with bleomycin (0.01 IU/animal), and unanticipated mortalities were recorded (LD50 undefined in both groups; n = 22 for both groups; P = 0.01). (C–E) Mice were treated with bleomycin 0.01 IU/mouse and 5 days later the lungs were homogenized and the number of CD45-positive cells were counted. (C) neutrophils, (D) alveolar macrophages, and (E) interstitial macrophages were identified from their pattern of cell surface marker expression using flow cytometry. (F–I) Mice with the indicated genotype were treated with bleomycin (bleo) 0.01 IU/animal or saline, and the lungs of the surviving mice were harvested 21 days later. (F) Trichrome-stained lung sections of the bleomycin-treated mice were imaged at 100× using a TissueGnostics system. Left panels are montage images; right panels are higher-magnification views of the areas indicated by the squares. (G) Fibrosis severity was quantified using Ashcroft scores (n = 4 animals per group). (H) Total lung collagen was measured in whole-lung homogenates by picrosirius red collagen precipitation (n = 8–10 mice in both saline control groups and n = 22 and 23 mice in the bleomycin-treated groups). (I) Lung compliance was measured using the FlexiVent system before harvest (n = 4 animals per group). All animals were 8–12 weeks of age at the time of bleomycin administration. When present, significant differences between groups are indicated in the figures. Error bars indicate SEM. ns, not significant.
Lung-Specific Loss of the α3 Laminin Subunit Worsens TGF-β–Mediated Lung Fibrosis
The development of fibrosis in response to bleomycin requires the activation of TGF-β (27). To determine whether the loss of α3 laminin altered the activation of TGF-β in response to bleomycin, we treated Lama3fl/fl and SPC-Cre/Lama3fl/fl mice with intratracheal bleomycin (0.01 IU/animal), and measured the expression of TGF-β target genes 5 days later. After treatment with bleomycin, the expression of the TGF-β target genes, connective tissue growth factor (CTGF), plasminogen activator inhibitor-1 (Pai-1), and TGF-β, were all increased in SPC-Cre/Lama3fl/fl mice when compared with Lama3fl/fl mice (Figures 4A–4C). We then examined whether α3 laminin loss worsened lung fibrosis downstream of the activation of TGF-β by administering an adenovirus encoding an active form of TGF-β (1 × 108 plaque-forming units/animal) to Lama3fl/fl and SPC-Cre/Lama3fl/fl mice and measuring lung fibrosis 28 days later. Lung fibrosis, as assessed by Ashcroft scores generated by examination of trichrome-stained lung sections, was worse in SPC-Cre/Lama3fl/fl compared with Lama3fl/fl mice (Figures 4D and 4E). Whereas total soluble collagen measured using picrosirius red collagen precipitation was higher in the SPC-Cre/Lama3fl/fl compared with Lama3fl/fl mice, these differences did not reach statistical significance, even with the relatively large numbers of animals examined (Figures 4E). Lung compliance was significantly lower in the SPC-Cre/Lama3fl/fl compared with Lama3fl/fl mice (Figures 4F).
Figure 4.

Lung epithelial–specific deletion of α3 laminin worsens pulmonary fibrosis induced by active transforming growth factor-β (TGF-β) (A–C) Mice with the indicated genotype were treated with bleomycin (0.01 IU/mouse) and, 5 days later, the levels of mRNA encoding the TGF-β target gene, connective tissue growth factor (CTGF), plasminogen activator inhibitor-1 (Pai-1), and TGF-β (which induces its own transcription) were measured in lung homogenates (n = 5 animals per group). (D) Mice with the indicated genotype were treated intratracheally with an adenovirus encoding no transgene (Ad-Null) or an adenovirus encoding an active form of TGF-β (Ad–TGF-β) both at 1 × 108 plaque-forming units [pfu]/animal, and the lungs of the mice were harvested 28 days later. Trichrome-stained lung sections of the TGF-β–treated mice were imaged at 100× using a TissueGnostics system. Left panels are montage images; right panels are higher-magnification views of the areas indicated by the squares. (E) Fibrosis severity was quantified using Ashcroft scores (n = 4 animals per group). (F) Total lung collagen was measured in whole-lung homogenates by picrosirius red collagen precipitation (n = 5–10 mice in the (Ad-Null) control groups and n = 12 and 10 mice in the TGF-β–treated groups). (G) Lung compliance was measured using the FlexiVent system before harvest (n = 4 animals per group). All animals were 8–12 weeks of age at the time of bleomycin administration or adenoviral infection. When present, significant differences between groups are indicated in the figures. Error bars indicate SEM.
Discussion
The laminins are essential components of the basement membrane, where they serve as important points of adhesion to the overlying cells and can serve as ligands for signaling receptors on the cellular surface (10). The α3 laminin subunit is expressed throughout the basement membranes of the lung in mice and rats (28–30). Here, we show that α3 laminin is expressed uniformly throughout the alveolar basement membrane in normal human lungs. Despite the widespread expression of α3 laminin protein in the normal lung, α3 laminin mRNA was nearly undetectable in lung homogenates, suggesting that α3 laminin may have a very long half-life in the human lung, as we have previously observed in mice (3 mo) (12). In patients with lung fibrosis, a normal basement membrane pattern of laminin staining was observed in areas of the lung without fibrosis. However, in the fibrotic regions, this normal basement membrane organization of laminin was lost, and staining overall for α3 laminin was reduced, despite a significant increase in the expression of α3 laminin mRNA. These findings are consistent with those of Booth and colleagues (4), who used a proteomic approach to show that the expression of α3 laminin was reduced in decellularized matrices from patients with IPF. We and others have suggested that α3-containing laminins play an important signaling role in the lung (12, 28, 31–33). Therefore, we used a lung epithelial–specific knockout approach in mice to show that the loss of α3 laminin plays a causal role in the development and/or progression of lung fibrosis.
Because humans and mice deficient in the α3 laminin gene die prematurely as a result of blistering skin disease (junctional epidermolysis bullosa in humans), a lung epithelial–specific promoter was used to delete the α3 laminin gene specifically in the adult lung (9, 12, 25). Specifically, we used mice generated by Hogan and colleagues, which express Cre recombinase driven by the SPC promoter (13). SPC is active in the lung epithelium in some of the earliest stages of lung development; therefore, Cre-mediated recombination of the floxed α3 laminin allele removes the gene from the developing lung epithelium and all of its daughter cells (13, 26). In lung homogenates from the SPC-Cre/Lama3fl/fl mice, we observed very little if any expression of α3 laminin mRNA or protein, suggesting that a large majority of laminins comprised of α3 subunits in the lung are secreted by the alveolar epithelium. We did not detect evidence of a compensatory increase in the expression of α5 laminin in the lungs of the SPC-Cre/Lama3fl/fl mice, and we have previously reported that Cre-mediated recombination in cells from these mice does not result in the expression of a truncated protein (12). In untreated mice, we found no difference in lung histology, lung mechanics, or basement membrane organization in adult mice deficient in α3 laminin when compared with wild-type controls.
The lack of an unstressed phenotype in the lungs of mice deficient in α3 laminin subunit protein is consistent with previous findings in humans and mice. In the lung, α3 laminin binds with either a β1 and γ1 chain to form laminin 311, or a β2 and γ2 chain to form laminin 322 (28). Loss-of-function mutations in either the laminin α3 or laminin γ2 gene are associated with junctional epidermolysis bullosa, and, although these individuals typically succumb to their illness by early adulthood, no reported phenotype in the lung has been reported (5, 10, 34). In mice, global loss of either the laminin α3 or laminin γ2 gene is associated with a similar blistering skin disease that results in death of the animal within the first several days of life (5, 10, 34). Although death in these animals occurs before lung development is complete, no abnormalities have been reported in their lungs. Together, these findings provide strong support for the conclusion that, unlike the α1, α2, and α5 laminin subunits, expression of the α3 laminin subunit is not required for normal lung development in the mouse (29).
We found that the loss of α3 laminin worsened lung fibrosis in mice treated with intratracheal bleomycin. The administration of intratracheal bleomycin results in an acute lung injury, which is maximal 3–5 days after the administration of bleomycin, and then resolves within 10 days after infection (35). In response to the injury, there is an increase in TGF-β activity in the lung, which is both necessary and sufficient for the development of lung fibrosis (16, 36). We found that mice lacking α3 laminin in the lung exhibited unexpected morality in response to bleomycin, with all of the deaths occurring 10 days or more after the intratracheal administration of bleomycin, a time when lung injury is resolving and fibrosis is developing (35). In mice deficient in α3 laminin, we found that lung inflammation, as measured by the number and activation state of alveolar macrophages in the lung, was enhanced, and the expression of several TGF-β target genes was increased, 5 days after the administration of bleomycin. These results suggest that the loss of α3 laminin might increase or prevent the resolution of bleomycin-induced acute lung injury. Although the effects were smaller, the loss of α3 laminin also worsened lung fibrosis induced by an adenovirus encoding an active form of TGF-β, suggesting that the loss of α3 laminin also contributes to fibrosis downstream of the activation of TGF-β. Our results complement those of Booth and colleagues (4), who found reduced levels of laminin in decellularized matrices from patients with IPF. Both Booth and colleagues (4) and Parker and colleagues (37) showed that matrices from patients with pulmonary fibrosis can activate normal human lung fibroblasts.
The mechanism by which α3 laminin contributes to the development of fibrosis is not clear from our studies. However, we and others have reported that α3 laminin in the basement membrane of the skin serves as a ligand for two key integrins expressed on the surface of epithelial cells—α3β1 and α6β4—which are involved in epithelial repair (32). In humans, mutations in the α3β1 integrin result in abnormal epithelial barrier function in the lung, skin, and kidney (38). In mice, global deletion of the α3 integrin is developmentally lethal; however, Chapman and colleagues (39) reported that specific deletion of the α3 integrin from alveolar type II cells resulted in reduced fibrosis following exposure to bleomycin. In vitro, they showed that binding between α3 integrin and laminin promoted TGF-β signaling through Smad phosphorylation. Our finding that TGF-β induced fibrosis was worse in α3 laminin deficient mice compared with wild-type controls, suggest that this is not the dominant mechanism by which α3 laminin influences fibrosis in vivo. However, Chapman and colleagues (33) also reported a rare cell population expressing the α6β4 integrin in the lungs of mice, which are increased in number after bleomycin treatment, and show many features of a progenitor cell population. In the skin, we have shown that binding of α6β4 to α3 laminin facilitates the migration of progenitor cells in response to wounding (31). It is therefore tempting to speculate that the loss of α3 laminin impairs the migration of α6β4 integrin–positive cells in the lung, resulting in prolonged inflammation, increased TGF-β target gene expression, and enhanced fibrosis. Consistent with this hypothesis, we observed increased mortality in α3 laminin–deficient mice treated with bleomycin at a time when the animals were recovering from lung injury and developing fibrosis. This hypothesis may also explain our previous report in which we deleted α3 laminin from the lung epithelium using a high-titer adenovirus expressing Cre recombinase. Although the acute inflammation induced by infection with an adenovirus encoding Cre recombinase resolves without fibrosis in wild-type mice, the use of this adenovirus to delete α3 laminin in the lung epithelium resulted in an increase in lung collagen that persisted for as long as 3 months after the infection (12).
In summary, we show that the α3 laminin subunit is distributed throughout the basement membrane in the normal lung. In patients with pulmonary fibrosis, the transcription of α3 laminin is increased; however, the protein is not present in the fibrotic regions of the lung. Moreover, in mice, although not being required for lung development, the loss of α3 subunit–containing laminins results in enhanced fibrosis in response to intratracheal bleomycin and an adenovirus encoding an active form of TGF-β. These results lend support to the hypothesis that changes in the composition of the extracellular matrix play a causal role in the development of lung fibrosis. Future investigations will focus on the role of laminins that include the α3 laminin subunit in the resolution of acute lung injury and the development of TGF-β–induced fibrosis.
Acknowledgments
Acknowledgments
The authors thank Dr. Brigid Hogan for providing the transgenic mice expressing Cre recombinase driven by the surfactant protein C promoter (SPC-Cre).
Footnotes
This work was supported by National Institute of Health grants HL092963, ES015024, ES013995, HL071643, Northwestern University Clinical and Translational Sciences Institute Center for Translational Innovation Pilot Award NCCR UL1 RR025741, and the Veterans Administration.
Author Contributions: L.I.M.-N. conducted the bulk of the experiments and assisted in preparing the manuscript; M.R.R., J.L.E., K.J.H., S.S., R.N., M.C., T.C., K.A.R., K.M.R., A.V.M., A.W., S.H., and H.P. assisted with the performance of the experiments; A.P. and M.S. assisted with the performance of the studies involving human lung tissue; G.M.M., J.C.R.J., and G.R.S.B. oversaw the experimental design and the presentation of the data; the manuscript was written in draft form by L.I.M.-N. and edited by G.R.S.B. and J.C.R.J. before receiving input from all of the other authors.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2014-0057OC on September 4, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Budinger GR, Sznajder JI. The alveolar–epithelial barrier: a target for potential therapy. Clin Chest Med. 2006;27:655–669. (abstract ix.). doi: 10.1016/j.ccm.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Selman M, King TE, Jr, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 3.King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 4.Booth AJ, Hadley R, Cornett AM, Dreffs AA, Matthes SA, Tsui JL, Weiss K, Horowitz JC, Fiore VF, Barker TH, et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med. 2012;186:866–876. doi: 10.1164/rccm.201204-0754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miner JH. Laminins and their roles in mammals. Microsc Res Tech. 2008;71:349–356. doi: 10.1002/jemt.20563. [DOI] [PubMed] [Google Scholar]
- 6.Pierce RA, Griffin GL, Susan Mudd M, Moxley MA, Longmore WJ, Sanes JR, Miner JH, Senior RM. Expression of laminin alpha 3, alpha 4, and alpha 5 chains by alveolar epithelial cells and fibroblasts. Am J Respir Cell Mol Biol. 1998;19:237–244. doi: 10.1165/ajrcmb.19.2.3087. [DOI] [PubMed] [Google Scholar]
- 7.Schuger L, O’Shea S, Rheinheimer J, Varani J. Laminin in lung development: effects of anti-laminin antibody in murine lung morphogenesis. Dev Biol. 1990;137:26–32. doi: 10.1016/0012-1606(90)90004-3. [DOI] [PubMed] [Google Scholar]
- 8.Willem M, Miosge N, Halfter W, Smyth N, Jannetti I, Burghart E, Timpl R, Mayer U. Specific ablation of the nidogen-binding site in the laminin γ1 chain interferes with kidney and lung development. Development. 2002;129:2711–2722. doi: 10.1242/dev.129.11.2711. [DOI] [PubMed] [Google Scholar]
- 9.Laimer M, Lanschuetzer CM, Diem A, Bauer JW. Herlitz junctional epidermolysis bullosa. Dermatol Clin. 2010;28:55–60. doi: 10.1016/j.det.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Jones JC, Dehart GW, Gonzales M, Goldfinger LE. Laminins: an overview. Microsc Res Tech. 2000;51:211–213. doi: 10.1002/1097-0029(20001101)51:3<211::AID-JEMT1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 11.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urich D, Eisenberg JL, Hamill KJ, Takawira D, Chiarella SE, Soberanes S, Gonzalez A, Koentgen F, Manghi T, Hopkinson SB, et al. Lung-specific loss of the laminin α3 subunit confers resistance to mechanical injury. J Cell Sci. 2011;124:2927–2937. doi: 10.1242/jcs.080911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okubo T, Knoepfler PS, Eisenman RN, Hogan BLM. Nmyc plays an essential role during lung development as a dosage-sensitive regulator of progenitor cell proliferation and differentiation. Development. 2005;132:1363–1374. doi: 10.1242/dev.01678. [DOI] [PubMed] [Google Scholar]
- 14.Urich D, Soberanes S, Burgess Z, Chiarella SE, Ghio AJ, Ridge KM, Kamp DW, Chandel NS, Mutlu GM, Budinger GR. Proapoptotic noxa is required for particulate matter–induced cell death and lung inflammation. FASEB J. 2009;23:2055–2064. doi: 10.1096/fj.08-114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng X-H, Filvaroff EH, Derynck R. Transforming growth factor-β (TGF-β)–induced down-regulation of cyclin A expression requires a functional TGF-β receptor complex. J Biol Chem. 1995;270:24237–24245. doi: 10.1074/jbc.270.41.24237. [DOI] [PubMed] [Google Scholar]
- 16.Bonniaud P, Kolb M, Galt T, Robertson J, Robbins C, Stampfli M, Lavery C, Margetts PJ, Roberts AB, Gauldie J. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta–mediated pulmonary fibrosis. J Immunol. 2004;173:2099–2108. doi: 10.4049/jimmunol.173.3.2099. [DOI] [PubMed] [Google Scholar]
- 17.Budinger GR, Mutlu GM, Eisenbart J, Fuller AC, Bellmeyer AA, Baker CM, Wilson M, Ridge K, Barrett TA, Lee VY, et al. Proapoptotic bid is required for pulmonary fibrosis. Proc Natl Acad Sci USA. 2006;103:4604–4609. doi: 10.1073/pnas.0507604103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Budinger GR, Tso M, McClintock DS, Dean DA, Sznajder JI, Chandel NS. Hyperoxia induced apoptosis does not require mitochondrial reactive oxygen species and is regulated by bcl-2 proteins. J Biol Chem. 2002;277:15654–15660. doi: 10.1074/jbc.M109317200. [DOI] [PubMed] [Google Scholar]
- 19.Mutlu GM, Budinger GRS, Wu M, Lam AP, Zirk A, Rivera S, Urich D, Chiarella SE, Go LHT, Ghosh AK, et al. Proteasomal inhibition after injury prevents fibrosis by modulating TGF-β1 signalling. Thorax. 2012;67:139–146. doi: 10.1136/thoraxjnl-2011-200717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anonymous. Scireq science: measurements. 2010[cited 2010 September 1, 2010]. Available from: http://www.scireq.com/science/measurements/
- 21.Price AP, England KA, Matson AM, Blazar BR, Panoskaltsis-Mortari A. Development of a decellularized lung bioreactor system for bioengineering the lung: the matrix reloaded. Tissue Eng Part A. 2010;16:2581–2591. doi: 10.1089/ten.tea.2009.0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klatte DH, Kurpakus MA, Grelling KA, Jones JC. Immunochemical characterization of three components of the hemidesmosome and their expression in cultured epithelial cells. J Cell Biol. 1989;109:3377–3390. doi: 10.1083/jcb.109.6.3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmittgen TD, Livak KJ. Analyzing real-time pcr data by the comparative C(t) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 24.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GRS, Perlman H. Flow cytometric analysis of the macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. 2013;49:503–510. doi: 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryan MC, Lee K, Miyashita Y, Carter WG. Targeted disruption of the lama3 gene in mice reveals abnormalities in survival and late stage differentiation of epithelial cells. J Cell Biol. 1999;145:1309–1324. doi: 10.1083/jcb.145.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou L, Lim L, Costa RH, Whitsett JA. Thyroid transcription factor-1, hepatocyte nuclear factor-3beta, surfactant protein B, C, and Clara cell secretory protein in developing mouse lung. J Histochem Cytochem. 1996;44:1183–1193. doi: 10.1177/44.10.8813084. [DOI] [PubMed] [Google Scholar]
- 27.Sheppard D. Transforming growth factor {beta}: a central modulator of pulmonary and airway inflammation and fibrosis. Proc Am Thorac Soc. 2006;3:413–417. doi: 10.1513/pats.200601-008AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones JC, Lane K, Hopkinson SB, Lecuona E, Geiger RC, Dean DA, Correa-Meyer E, Gonzales M, Campbell K, Sznajder JI, et al. Laminin-6 assembles into multimolecular fibrillar complexes with perlecan and participates in mechanical-signal transduction via a dystroglycan-dependent, integrin-independent mechanism. J Cell Sci. 2005;118:2557–2566. doi: 10.1242/jcs.02395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen NM, Senior RM. Laminin isoforms and lung development: all isoforms are not equal. Dev Biol. 2006;294:271–279. doi: 10.1016/j.ydbio.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 30.Hamill KJ, Kligys K, Hopkinson SB, Jones JC. Laminin deposition in the extracellular matrix: a complex picture emerges. J Cell Sci. 2009;122:4409–4417. doi: 10.1242/jcs.041095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sehgal BU, DeBiase PJ, Matzno S, Chew TL, Claiborne JN, Hopkinson SB, Russell A, Marinkovich MP, Jones JC. Integrin beta4 regulates migratory behavior of keratinocytes by determining laminin-332 organization. J Biol Chem. 2006;281:35487–35498. doi: 10.1074/jbc.M606317200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldfinger LE, Hopkinson SB, deHart GW, Collawn S, Couchman JR, Jones JC. The alpha3 laminin subunit, alpha6beta4 and alpha3beta1 integrin coordinately regulate wound healing in cultured epithelial cells and in the skin. J Cell Sci. 1999;112:2615–2629. doi: 10.1242/jcs.112.16.2615. [DOI] [PubMed] [Google Scholar]
- 33.Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, Sonnenberg A, Wei Y, Vu TH. Integrin α6β4 identifies an adult distal lung epithelial population with regenerative potential in mice. J Clin Invest. 2011;121:2855–2862. doi: 10.1172/JCI57673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin GR, Timpl R. Laminin and other basement membrane components. Annu Rev Cell Biol. 1987;3:57–85. doi: 10.1146/annurev.cb.03.110187.000421. [DOI] [PubMed] [Google Scholar]
- 35.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L379–L399. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 37.Parker MW, Rossi D, Peterson M, Smith K, Sikström K, White ES, Connett JE, Henke CA, Larsson O, Bitterman PB. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest. 2014;124:1622–1635. doi: 10.1172/JCI71386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Has C, Spartà G, Kiritsi D, Weibel L, Moeller A, Vega-Warner V, Waters A, He Y, Anikster Y, Esser P, et al. Integrin α3 mutations with kidney, lung, and skin disease. N Engl J Med. 2012;366:1508–1514. doi: 10.1056/NEJMoa1110813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim Y, Kugler MC, Wei Y, Kim KK, Li X, Brumwell AN, Chapman HA. Integrin alpha3beta1–dependent beta-catenin phosphorylation links epithelial Smad signaling to cell contacts. J Cell Biol. 2009;184:309–322. doi: 10.1083/jcb.200806067. [DOI] [PMC free article] [PubMed] [Google Scholar]