

Abstract
Enhanced protein tyrosine phosphorylation is associated with changes in vascular permeability through formation and dissolution of adherens junctions and regulation of stress fiber formation. Inhibition of the protein tyrosine phosphorylase SH2 domain-containing protein tyrosine phosphatase 2 (SHP2) increases tyrosine phosphorylation of vascular endothelial cadherin and β-catenin, resulting in disruption of the endothelial monolayer and edema formation in the pulmonary endothelium. Vascular permeability is a hallmark of acute lung injury (ALI); thus, enhanced SHP2 activity offers potential therapeutic value for the pulmonary vasculature in diseases such as ALI, but this has not been characterized. To assess whether SHP2 activity mediates protection against edema in the endothelium, we assessed the effect of molecular activation of SHP2 on lung endothelial barrier function in response to the edemagenic agents LPS and thrombin. Both LPS and thrombin reduced SHP2 activity, correlated with decreased focal adhesion kinase (FAK) phosphorylation (Y397 and Y925) and diminished SHP2 protein–protein associations with FAK. Overexpression of constitutively active SHP2 (SHP2D61A) enhanced baseline endothelial monolayer resistance and completely blocked LPS- and thrombin-induced permeability in vitro and significantly blunted pulmonary edema formation induced by either endotoxin (LPS) or Pseudomonas aeruginosa exposure in vivo. Chemical inhibition of FAK decreased SHP2 protein–protein interactions with FAK concomitant with increased permeability; however, overexpression of SHP2D61A rescued the endothelium and maintained FAK activity and FAK–SHP2 protein interactions. Our data suggest that SHP2 activation offers the pulmonary endothelium protection against barrier permeability mediators downstream of the FAK signaling pathway. We postulate that further studies into the promotion of SHP2 activation in the pulmonary endothelium may offer a therapeutic approach for patients suffering from ALI.
Keywords: endothelium, SH2 domain-containing protein tyrosine phosphatase 2, pulmonary edema, acute lung injury, focal adhesion kinase
Clinical Relevance
This work studies the mechanism through which SHP2 activation protects the endothelial barrier against injury. In settings of acute lung injury, activation of SHP2 may offer a novel therapeutic approach to treat pulmonary edema.
Acute respiratory distress syndrome (ARDS) is a complex syndrome associated with severe arterial hypoxemia. Onset of ARDS results from several predisposing factors, such as pneumonia, pancreatitis, sepsis, and trauma. At present, patients suffering from ARDS are treated with mechanical ventilation; however, there are insufficient treatment options available, and the mortality rate remains at roughly 40% (1, 2). Vascular barrier dysfunction is one cause of pulmonary edema formation observed in settings of acute lung injury (ALI). The integrity of the vascular barrier is disrupted after the breakdown of endothelial intercellular junctions, which occurs after dissolution of the adherens junction (AJ) and tight junction protein complexes, as well as altered contractile forces and centripetal tensions (3, 4). Thus, understanding signaling mechanisms that regulate the endothelial monolayer may provide therapeutic targets for treatment of ALI.
SH2 domain-containing protein tyrosine phosphatase 2 (SHP2) is a critical mediator in the maintenance of endothelial barrier function. Chemical or molecular inhibition of SHP2 promoted barrier disruption in both cultured microvascular endothelial monolayers and in the intact lung (5). SHP2 associates with vascular endothelial (VE)-cadherin via β-catenin, and regulates VE-cadherin levels at the AJ by maintaining β-catenin in a dephosphorylated state (6). After thrombin-induced loss of SHP2 from the AJ, tyrosine phosphorylation of VE-cadherin, β-catenin, γ-catenin, and p120-catenin is increased, thus altering the protein complex association with α-catenin, leading to AJ disassembly (7). Using substrate trapping studies, Hartman and colleagues (8) identified focal adhesion kinase (FAK) as a substrate of SHP2, with FAK tyrosine 397 (FAKY397) phosphorylation being a key event for the protein–protein interaction. In cardiomyocytes and breast carcinoma cells, SHP2 negatively regulates FAK, maintaining FAK in a dephosphorylated, inactive state (8, 9). The activity of FAK is important in regulation of endothelial barrier function through centripetal tensions, both at focal adhesion complexes (via interactions with the cytoplasmic tail of β-integrin) and through the stabilization of AJ protein complexes (10, 11). The latter is likely mediated through the interactions of key AJ component p120-catenin and/or by modulating the activity of p190RhoGAP through tyrosine phosphorylation (11, 12). Thus, the FAK–SHP2 protein–protein associations may represent a key mechanism regulating endothelial barrier function.
Tyrosine phosphorylation of FAK has been observed at six tyrosine (Y) residues (Y397, Y407, Y576, Y577, Y861, and Y925), with the phosphorylation patterns at these sites differing in response to varying stimuli (13). FAKY397 phosphorylation, an autophosphorylation site of FAK, creates a high-affinity binding site for the SH2 domain of Src, an ubiquitously expressed proto-oncogene—interactions critical to downstream signaling events (14). The formation of a transient FAK–Src dual kinase signaling complex results in the phosphorylation of FAK at Y576, Y577, and Y925 residues, promoting maximal FAK activation (15, 16). FAK autophosphorylation occurs immediately after integrin clustering and associates with different signaling proteins, including Grb2-Sos, p130cas, and paxillin (17, 18), leading to the stabilization of the focal adhesion complexes and enhancing the integrin–cytoskeletal interactions (19). FAK activation has been implicated as both a positive and negative regulator of endothelial barrier function (20). However, the relationship between SHP2 and FAK protein–protein interactions and activities, and the resulting influence on endothelial barrier function, are not well known.
Although SHP2 activity has been shown to influence barrier integrity in vitro and in vivo through regulating tyrosine phosphorylation of key AJ complex proteins (5), the mechanism through which SHP2 regulates FAK to maintain endothelial barrier function has not been described. In the present study, we used the edemagenic agents, LPS and thrombin, and Pseudomonas aeruginosa (strain PA103), and the barrier-enhancing mediator, sphingosine-1-phosphate (S1P), to investigate the link between SHP2, FAK, and barrier function within the pulmonary vasculature. We show that barrier-disrupting agents diminished the activities and protein–protein interactions of FAK and SHP2, whereas barrier-enhancing agents did the opposite. We further show that constitutive activation of SHP2 blocked LPS- or thrombin-induced barrier dysfunction in vitro and in vivo and blunted P. aeruginosa–induced ALI in vivo. Finally, constitutively active SHP2 protected against FAK inactivation or disruption of FAK–SHP2 protein–protein interactions by the chemical inhibitor of FAK, PF-573228. Thus, this work suggests that SHP2 activation protects against pulmonary edema formation through the maintenance of cellular centripetal tensions through interactions with FAK.
Materials and Methods
Cell Lines and Reagents
Rat lung microvascular endothelial cells (LMVECs; Vec Technologies, Rensselaer, NY) as previously described (21).
LPS (endotoxin) from Escherichia coli serotype 055:B5 was obtained from Enzo Life Sciences (Plymouth Meeting, PA); S1P was purchased from Cayman Chemicals (Ann Arbor, MI); 8-Hydroxy-7-(6-sulfonaphthalen-2-yl)diazenyl-quinoline-5-sulfonic acid (NSC-87877) was purchased from Calbiochem (San Diego, CA); and thrombin (from human plasma) and FAK inhibitor PF-573228 were purchased from Sigma (St. Louis, MO). Polyjet was obtained from SignaGen (Gaithersburg, MD).
The vectors encoding activated SHP2 (PJ3-SHP2D61A) and non–kinase C-terminal (0–2186 amino acid region) of FAK (green fluorescent protein [GFP]-FAK-related nonkinase) were purchased from Addgene (Cambridge, MA), and phosphorylated GFP (pGFP-C1) was obtained from Clontech (Mountain View, CA). Antibodies directed against phospho-SHP2 (Y580 and Y542) and phospho-FAK (Y576/577 and Y925) were obtained from Cell Signaling (Beverly, MA). Phospho-FAK (Y397) and FAK (immunogen at amino acids 354–533) antibodies were obtained from Invitrogen (Carlsbad, CA) and BD transduction (San Jose, CA), respectively. Rabbit IgG, VE-cadherin, and SHP2 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). P. aeruginosa strain PA103 bacteria was a kind gift from Dr. Troy Stevens (University of South Alabama, Mobile, AL).
Liposome Preparation
For production of liposomes, dimethyldioctadecyl-ammonium bromide and cholesterol were mixed in chloroform, as described elsewhere (22, 23). Liposomes dissolved in 5% glucose solution were combined with GFP or SHP2D61A complementary DNA (cDNA; 50 μg) and injected into the retrobulbar sinus of the orbit of anesthetized C57 BL/6 mice. Confirmation of cDNA overexpression within the pulmonary vasculature was assessed by endothelial cell isolation and immunoblot analysis of GFP, SHP2, and VE-cadherin, as previously described (24). Transfection efficiency was assessed by densitometry of SHP2.
In Vivo Models of ALI
LPS was administered to adult C57 BL/6 mice via single intraperitoneal injection (5 mg/kg). P. aeruginosa strain PA103 was grown on solid tryptic soy agar and resuspended in sterile PBS to a concentration of 1 × 108 CFU/ml. Under light isofluorane anesthesia, mice were administered 100 μl (containing 107 CFU of P. aeruginosa) intratracheally. At 4 hours (P. aeruginosa) and 24 hours (LPS) after administration, mice were given a lethal dose of anesthesia. Time course studies demonstrated 4-hour P. aeruginosa exposure to cause a significant induction of pulmonary edema. Longer exposure times (> 4 h) resulted in animal discomfort and mortality; thus, this was avoided by using the earlier time points.
Bronchoalveolar lavage (BAL) was performed and BAL cell counts and protein concentrations were determined as previously described (25). In additional experiments, at 24 hours after intraperitoneal injection of LPS or vehicle into mice, lung permeability was determined by ex vivo perfusion of lungs, as previously described (24, 25). Representative plots for lung filtration coefficients experiments are included in the online supplement.
All animal experimental protocols were approved by the Institutional Animal Care and Use Committees of the Providence Veterans Affairs Medical Center and Brown University, and comply with the Health Research Extension Act and U.S. Public Health Service policy.
Endothelial Monolayer Permeability
Changes in endothelial monolayer permeability of transiently transfected LMVECs were assessed using the electrical cell impedance sensor technique (Applied Biophysics, Troy, NY), as previously described (24, 25). Transfection efficiency was assessed by densitometry of phosphorylated SHP2 (Y542 and Y580) and total SHP2 or GFP.
Immunoprecipitations and Immunoblot Analyses
Confluent LMVECs were pretreated with NSC-87877 or vehicle (H2O) for 3 hours followed by treatment with S1P (1 μM, 30 min), thrombin (1 U/ml, 15 min), LPS (1 μg/ml, 2 h), or PF-573228 (0.1 μM, 3 h). Immunoprecipitations and immunoblot analyses were performed as previously described (24), using IgG and nonimmunoprecipitated whole-cell lysate as controls for immunoprecipitations. Immunoprecipitation data are presented as ratios of FAK relative to precipitated SHP2.
Statistical Analysis
Experimental number (n) is presented for each experiment. For three or more groups, variance was assessed using Bartlett’s test, with data sets not reaching significance studied by Kruskal-Wallis test followed by Dunn’s test. For all other data sets, differences among the means were tested for significance by ANOVA with Tukey’s range significance difference test. For two groups, variance in data sets was assessed using the Mann-Whitney test followed by t test. Data are presented as means (± SD). Significance was reached at a P value less than 0.05.
Results
Endothelial Edemagenic Agents Modulate SHP2 Activation
We have previously shown an increase in basal lung endothelial permeability upon either chemical or molecular inhibition of SHP2 (5). In the present study, we further investigated if activation of SHP2 could serve a protective role in the pulmonary circulation. Despite a previously reported median effective concentration (EC50) of 0.32 μM, the chemical inhibitor of SHP2, NSC-87877, was used at 100 μM in the present study due to significant increases in interendothelial cell gapping previously observed by our group (5). We first assayed the activity levels of SHP2 by measuring the phosphorylation state of key tyrosine residues, tyrosine 542 (Y542) and tyrosine 580 (Y580), which reside within the C terminus and have been shown to correlate with PTPase enzymatic activity (26). To investigate the link between SHP2 activation and barrier integrity, we assessed the effect of the permeability agents, LPS and thrombin, and the barrier protective agent, S1P, on SHP2 phosphorylation at Y542 and Y580. We noted a significant decrease in the levels of SHP2 Y542 and Y580 phosphorylation after 15-minute exposure to thrombin or 2-hour exposure to LPS (Figures 1A and 1B). Conversely, S1P promoted an increase in SHP2 Y542 and Y580 phosphorylation, although only Y580 phosphorylation reached significance (Figures 1C and 1D). Interestingly, exposure of the endothelial cells with SHP2 chemical inhibitor, NSC-87877, attenuated the level of SHP2 Y580 phosphorylation; however, S1P treatment ablated this effect (Figure 1D). These data suggest that SHP2 activity may correlate with the state of the barrier function within the pulmonary endothelium.
Figure 1.
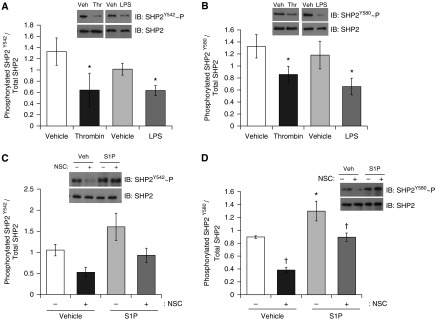
Endothelial edemagenic agents modulate SH2 domain-containing protein tyrosine phosphatase 2 (SHP2) activation. (A and B) Lung microvascular endothelial cells (LMVECs) were treated with LPS (1 μg/ml, 2 h) or thrombin (1 U/ml, 15 min) or vehicle (PBS) for 2 hours (n = 3). (C and D) LMVECs were pretreated with SHP2 inhibitor, 8-hydroxy-7-(6-sulfonaphthalen-2-yl)diazenyl-quinoline-5-sulfonic acid (NSC-87877 [NSC]; 100 μm), or vehicle (H2O) for 3 hours, followed by treatment with sphingosine-1-phosphate (S1P; 1 μM) or vehicle (methanol) for 30 minutes (n = 6). SHP2 activity was assayed by subjecting the lysates to immunoblot analysis and probing with antibodies directed against phosphorylated Y542 (A and C) and Y580 (B and D) residues. Blots were stripped and reprobed for SHP2. Data are presented as a ratio of phosphorylated SHP2 to total SHP2, and as the mean (± SD). *P < 0.05, †P < 0.05, versus vehicle for respective treatments. IB, immunoblot; Thr, thrombin; Veh, vehicle.
Constitutively Active SHP2 Protects Against the Effect of Pulmonary Endothelial Barrier Modulating Agents In Vitro and In Vivo
We next studied the effect of constitutively activated SHP2 on endothelial barrier function in settings of LPS, thrombin, or S1P exposure in vitro by overexpressing constitutively activated SHP2 (SHP2D61A) in LMVECs. In parallel, LMVECs were transfected with cDNA encoding GFP, as a negative control. Overexpression of both SHP2 and GFP proteins was confirmed using Western blotting with antibodies specific to phosphorylated SHP2 (Y542 and Y580), total SHP2, and GFP (Figure 2A, inset). Transfection efficiency, measured as percentage increase in SHP2 protein expression in SHP2D61A overexpressing LMVECs relative to endogenous SHP2 protein expression in cells overexpressing GFP, was 84 (± 11)% (n = 4; P < 0.05; Figure 2A, inset). As expected, SHP2 activity, measured by tyrosine phosphorylation, was significantly increased in LMVECs overexpressing SHP2D61A compared with GFP-overexpressing cells at residues of Y542 (262 ± 87.5%; n = 4; P < 0.05) and Y580 (128.3 ± 7.7%; n = 4; P < 0.05) (Figure 2A, inset). Under baseline conditions, SHP2D61A significantly elevated endothelial monolayer resistance by 137.9 (± 22.7) Ω (P ≤ 0.05; n = 4–7) relative to GFP overexpressing endothelial cells (Figure 2A). In keeping with the noted effect of SHP2 activity on endothelial barrier function, we observed that transient overexpression of SHP2D61A blocked endothelial permeability induced by SHP2 inhibitor, NSC-87877 (Figure 2B). Next, we noted that transient overexpression of SHP2D61A completely blocked LPS-induced increases in endothelial permeability as compared with cells transiently overexpressing cDNA encoding GFP (Figure 2C). Similarly, LMVECs overexpressing SHP2D61A protein displayed no thrombin-induced increase in permeability compared with GFP-overexpressing cells (Figure 2D). In contrast, although S1P enhanced the barrier function of GFP-overexpressing LMVECs relative to vehicle, this was not noted in SHP2D61A-overexpressing cells (Figure 2E). Increased permeability, induced by NSC-87877, was blunted after treatment with S1P (Figure 2F). Thus, SHP2 activity is alternatively modulated in settings of barrier dysfunction versus barrier enhancement.
Figure 2.

Constitutively active SHP2 blocks the effect of pulmonary endothelial barrier–modulating agents in vitro. Equal numbers of LMVECs were transfected with eukaryotic vectors encoding constitutively active SHP2 (SHP2D61A) or green fluorescent protein (GFP) cDNA. (A) The resistance across the monolayers was measured at 48 hours after transfection. SHP2 and GFP overexpression and SHP2 phosphorylation (Y542 and Y580) were confirmed for each experiment by immunoblot analysis of the lysates of transiently transfected LMVECs (inset). Representative images are shown (n = 4). Electric cell impedance system data are presented as means (± SD) (n = 7). *P < 0.05 versus GFP. (B–E) Monolayer resistance was assessed in LMVECs overexpressing SHP2D61A (squares) or GFP (diamonds) in the presence (open symbols) and absence (closed symbols) of NSC-87877 (100 μM; n = 4), LPS (100 ng/ml; n = 11), thrombin (1 U/ml; n = 4), or S1P (1 μM; n = 6). Arrow indicates point of addition of modulating agents. Data are presented as means (± SD). *P < 0.05 versus GFP treated with vehicle. (F) Monolayer resistance was assessed in LMVECs in the presence (open symbols) and absence (closed symbols) of NSC-87877 (100 μM), followed by treatment with S1P (1 μM; square symbols) or vehicle (circle symbols; n = 4). Arrows indicate points of addition of modulating agents. Dashed lines in D and E indicate treated samples (thrombin in D, S1P in E). Data are presented as means (± SD). *P < 0.05 versus vehicle.
We next sought to assess whether SHP2D61A-mediated protection against endothelial barrier disruption in vitro is also observed in the pulmonary circulation of the intact lung. Mice were administered cationic liposomes containing GFP or SHP2D61A cDNA, via retro-orbital injection, and, after 24 hours, LPS was administered. After a further 24 hours, lung permeability in the ex vivo, isolated, perfused lung was assessed, as previously described (24, 25). Transient overexpression of cDNA in the pulmonary vasculature was confirmed via immunological isolation of endothelial cells from the lung, and the homogenates were immunoblotted for SHP2 or GFP; the isolation of endothelial cells was confirmed via immunoblot analysis for VE-cadherin (Figure 3A, insert). Isolated endothelial cells from lungs of mice overexpressing SHP2D61A cDNA exhibit a significant increase (95.5 ± 7%; n = 5; P < 0.05) in SHP2 protein expression compared with those after overexpression of GFP cDNA (Figure 3A, inset). As previously shown (24, 25), LPS exposure increased the filtration coefficient in GFP-overexpressing mice, correlating with lung edema formation. However, mice overexpressing SHP2D61A in the pulmonary vasculature demonstrated no edema formation in response to LPS (Figure 3A). Similar results were noted using the bacteria, P. aeruginosa (PA103), as a model for ALI (27, 28). For these studies, 44 hours after transfection of the SHP2D61A, PA103 was administered intratracheally. At 4 hours after intratracheal injection, lung edema formation was measured. In GFP-overexpressing mice, we noted lung edema formation, as assessed by the increased wet-to-dry lung weight ratio, relative to mice exposed to vehicle. However, SHP2D61A overexpression attenuated PA103-induced increase in lung wet-to-dry weight ratio (Figure 3B). Parallel measurements demonstrated that, in mice overexpressing GFP, the protein content and number of cells infiltrated in the BAL fluid was significantly elevated after PA103 exposure (Figures 3C and 3D). Similar to the lung filtration coefficients measurements, SHP2D61A-overexpressing mice had significantly lower levels of BAL protein in PA103-exposed mice compared with GFP-overexpressing mice exposed to PA103 (Figures 3C). In addition, after PA103 exposure, overexpression of SHP2D61A in mice displayed no increase in cell number in the BAL compared with GFP-overexpressing mice (Figure 3D). Thus, the data strongly suggest that the enzymatic activity of SHP2 is a key component in protection of the pulmonary vasculature against barrier dysfunction and edema formation both in vitro and in vivo.
Figure 3.
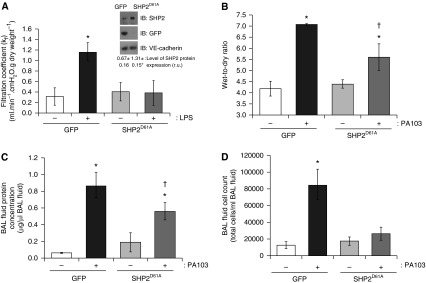
Constitutively active SHP2 blocks bacterial endotoxin (LPS)-induced barrier disruption and attenuates bacteria-induced barrier disruption. Mice were injected with liposomes encapsulating cDNA encoding SHP2D61A or GFP. (A) At 24 hours after liposome injection, mice received an intraperitoneal injection of LPS (5 mg/kg). After an additional 24 hours, lung edema was measured by determining lung permeability in ex vivo isolated, perfused lungs. Inset: overexpression of SHP2 and GFP in the lung endothelium was confirmed by immunological isolation of endothelial cells. The lysates from isolated lung endothelial cells were immunoblotted for SHP2 and GFP. The membranes were stripped and reprobed for vascular endothelial–cadherin expression to confirm that the harvested lung cells were endothelial cells. Level of SHP2 protein expression (in arbitrary units) is indicated. Data are presented as means (± SD). GFP, n = 6; SHP2D61A, n = 7. *P < 0.05 versus GFP, vehicle. (B–D) At 44 hours after liposome injection, mice were administered P. aeruginosa strain PA103 (107 CFU) via intratracheal injection. After an additional 4 hours, lungs were harvested for wet-to-dry weight measurements (B), or bronchoalveolar lavage (BAL) fluid was collected and analyzed for protein concentration (C) and cell infiltration (D). Data are presented as means (± SD). (B) n = 6; (C and D) n = 4. *P < 0.05 PA103-treated versus vehicle-treated mouse lungs; †P < 0.05 versus GFP and PA103. kf, lung filtration coefficient.
SHP2 Activity Increases FAK Phosphorylation and FAK–SHP2 Association
FAK is a tyrosine kinase known to be important in regulating endothelial barrier function through action at both the focal adhesion complex and AJ complexes (29). SHP2 has been implicated in the regulation of FAK phosphorylation, and thus activation (9, 30). Thrombin, LPS, and S1P have all been previously observed to differentially alter FAK phosphorylation status (31, 32). Thus, we next assessed whether FAK plays a role in the mechanism through which SHP2 exerts protection of endothelial barrier function. We assessed the phosphorylation status of FAK at the autophosphorylation residue, Y397, within the activation loop at residue Y576/577, and at Y925, a residue located within the focal adhesion-targeting (FAT) domain and strongly associated with intrinsic FAK activity. In response to both LPS and thrombin exposure, we noted significant decreases in FAK phosphorylation at Y397 (Figure 4A) and Y925 (Figure 4C); however, FAK phosphorylation at residue Y576/577 was unchanged (Figure 4B). Phosphorylation of FAK at Y397 and Y925 was increased by S1P treatment (Figures 4D and 4F), although only increases in FAK Y397 phosphorylation reached significance. Pretreatment with NSC-87877 attenuated S1P-induced phosphorylation at Y397 and Y925 (Figures 4D and 4F). Interestingly, similar to the effects of thrombin and LPS, SHP2 inhibition by NSC-87877 treatment caused significantly decreased phosphorylation of FAK at Y397 and Y925 (Figures 4D and 4F), but exerted no effect on FAK phosphorylation at Y576/577 (Figure 4E).
Figure 4.

SHP2 activity increases focal adhesion kinase (FAK) phosphorylation on select residues. (A–C) LMVECs were treated with LPS (1 μg/ml, 2 h) or thrombin (1 U/ml, 15 min). (D–F) LMVECs were pretreated with SHP2 inhibitor, NSC-87877 (100 μM), or vehicle (H2O) for 3 hours, followed by treatment with S1P (1 μM, 30 min) or methanol vehicle. FAK phosphorylation status was assayed by subjecting the lysates to immunoblot analysis, probing with antibodies directed against phosphorylated Y397 (A and D), Y576 (B and E), and Y925 (C and F) residues. Membranes were then stripped and reprobed for total FAK protein. Data are presented as a ratio of phosphorylated FAK to total FAK, mean (± SD). (A–C) n = 4; (D–F) n = 5. Insets are representative immunoblots for each experiment. *P < 0.05 treatment versus respective vehicle; †P < 0.05 S1P versus vehicle.
FAK phosphorylation at Y397 is essential for FAK–SHP2 association (8). Thus, we next studied whether the alterations in FAK phosphorylation profile, by exposure to LPS, thrombin, and S1P, regulate the direct protein–protein association between SHP2 and FAK. We immunoprecipitated SHP2 from LMVEC lysates treated with LPS or thrombin. We observed a significant decrease in SHP2 association to FAK after treatment with both LPS and thrombin (Figure 5A), similar to the effect of SHP2 chemical inhibition (Figure 5B). In parallel studies, we immunoprecipitated SHP2 from LMVEC lysates treated with S1P in the presence and absence of SHP2 inhibitor, NSC-87877. S1P significantly elevated FAK–SHP2 binding, an effect that was partially reduced (42.5 ± 8.9%) by SHP2 inhibition (Figure 5B). Taken together, these studies imply that FAK dephosphorylation mediated by SHP2 inhibition, and the resulting changes in FAK–SHP2 binding, may be responsible for the alterations in endothelial barrier permeability.
Figure 5.

SHP2 activity increases FAK–SHP2 association. (A) LMVECs were treated with LPS (1 μg/ml, 2 h), thrombin (1 U/ml, 15 min), or vehicle (PBS). (B) LMVECs were pretreated with NSC-87877 (100 μM) or vehicle (H2O) for 3 hours, followed by treatment with S1P (1 μM) or vehicle (methanol) for 30 minutes. Equivalent amounts of lysate were immunoprecipitated for SHP2 using an antibody derived from rabbit or rabbit IgG antibody. In parallel, nonimmunoprecipitated lysate (whole-cell lysate) was resolved by SDS-PAGE and immunoblotted with an FAK antibody derived from mice. Membranes were then stripped and reprobed with a SHP2 antibody, and FAK–SHP2 association was determined as the ratio of FAK bound to total immunoprecipitated SHP2. Inset: representative immunoblot. Data are presented as means (± SD). (A) n = 4; (B) n = 5. *P < 0.05 versus respective vehicle; †P < 0.05 S1P versus vehicle. IP, intraperitoneal.
Effect of FAK Inhibition on Pulmonary Endothelium Is Attenuated by Constitutive Activation of SHP2
Finally, we sought to understand the effect of FAK inhibition on SHP2 activity and endothelial barrier function. Treatment of LMVECs with a FAK chemical inhibitor, PF-573228, showed a diminished level of FAK Y397 phosphorylation, thus decreased FAK activity (Figure 6A). Overexpression of the catalytically deficient FAK-related nonkinase in LMVECs resulted in increased endothelial monolayer permeability under baseline conditions, similar to the level of decreased resistance observed after PF-573228 treatment in GFP-overexpressing cells (Figure 6B). In addition to dephosphorylation of FAK Y397 and increased monolayer permeability, we observed a significant decrease in FAK–SHP2 binding in LMVECs treated with PF-573228 (Figure 6C). Interestingly, overexpression of SHP2D61A abrogated PF-573228-induced inhibition of FAK phosphorylation (Figure 6A) and attenuated endothelial barrier dysfunction (Figure 6E). We further noted that endothelial cells overexpressing SHP2D61A maintained FAK–SHP2 protein–protein interactions in settings of PF-573228 exposure (Figure 6D). Thus, SHP2 activity can override FAK dephosphorylation and inactivation and preserve coassociation, resulting in maintenance of endothelial barrier function.
Figure 6.

FAK regulates endothelial barrier function through SHP2-mediated mechanism. (A, B, D, and E) Equal numbers of LMVECs were transfected with eukaryotic vectors encoding catalytically active SHP2 (SHP2D61A), GFP–FAK-related nonkinase (FRNK), or GFP cDNAs. (A) At 48 hours, LMVECs were treated with FAK inhibitor, PF-573228 (0.1 μM), or vehicle (H2O) for 3 h. FAK phosphorylation status was assayed by subjecting the lysates to immunoblot analysis, probing with antibodies directed against phosphorylated Y397 residue. Membranes were then stripped and reprobed for total FAK protein. Representative blot is shown (n = 4). (B) Representative plot of changes in monolayer resistance were measured in LMVECs overexpressing FRNK (square symbols) or GFP (diamond symbols) cDNA, in the presence (open symbols) and absence (closed symbols) of PF-573228 (0.1 μM). Arrow indicates point of addition (n = 3). FRNK protein overexpression was confirmed by immunoblot analysis of the lysates of transiently transfected LMVECs with an antibody raised against FAK (inset). Representative images are shown (n = 3). (C) LMVECs were treated with FAK inhibitor, PF-573228, or vehicle (H2O) for 3 h. Equivalent amounts of lysate were immunoprecipitated for SHP2 using an antibody derived from rabbits. Immunocomplexes were resolved with total lysate via SDS-PAGE and immunoblotted with a FAK antibody derived from mice. Membranes were then stripped and reprobed with an SHP2 antibody, and FAK–SHP2 association was determined by the ratio of FAK bound to total immunoprecipitated SHP2. Inset: representative immunoblots. Data are presented as means (± SD); n = 3. *P < 0.05 versus vehicle. (D) At 48 hours, LMVECs were treated with FAK inhibitor, PF-573228, or vehicle (H2O) for 3 hours. SHP2 was immunoprecipitated from lysates and subjected to FAK immunoblot analysis as for (C). Inset: representative immunoblots. Data are presented as means (± SD). *P < 0.05 versus vehicle. (E) Changes in monolayer resistance were measured in LMVECs overexpressing SHP2D61A (squares; n = 4) or GFP (diamonds; n = 5) cDNA, in the presence (open symbols) and absence (closed symbols) of PF-573228 (0.1 μM). Arrow indicates point of addition. Data are presented as means (± SD). *P < 0.05 versus GFP vehicle.
Taken together, the data presented here indicate that S1P, LPS, and thrombin modulate the pulmonary vasculature through a FAK–SHP2–dependent pathway. We demonstrate a key role for SHP2 activation on FAK activity, FAK–SHP2 protein associations, and in the regulation of endothelial barrier function. Thus, we propose that the activation of SHP2 represents an interesting therapeutic target in the treatment of ALI.
Discussion
In the current study, we demonstrate that SHP2 activation offers significant protection against barrier disruption through activation of FAK and the maintenance of protein interactions with FAK. We further implicate the effect of the barrier-enhancing agent, S1P, and edemagenic agents, LPS and thrombin, on endothelial barrier function to be mediated via FAK–SHP2–dependent mechanisms. We note that barrier-enhancing agents promoted the tyrosine phosphorylation and activation of FAK and SHP2 and FAK–SHP2 protein–protein interactions. Conversely, LPS and thrombin exposure caused diminished tyrosine phosphorylation of each protein, inhibition of FAK–SHP2 associations, and disruption of endothelial barrier function. We further show that constitutively active SHP2 protein protects against pulmonary endothelial barrier disruption in settings of FAK inhibition by maintaining FAK autophosphorylation and FAK–SHP2 protein interactions. These findings suggest that SHP2 regulates pulmonary endothelial barrier function via maintenance of FAK–SHP2 protein–protein associations.
Activation of SHP2 is dependent on phosphorylation at the C-terminal tyrosine residues, Y542 and Y580. Our findings show that barrier-enhancing S1P increases SHP2 phosphorylation, whereas barrier-disruptive agents, LPS and thrombin, cause dephosphorylation of SHP2 at both these residues. Phosphorylation of Y542 creates an interaction in the N-terminal SH2 domain and appears to relieve basal inhibition of the PTPase, whereas phosphorylation at Y580 causes an interaction at the C terminus that stimulates PTPase activity (26). PTPase activity plays a key role in maintenance of pulmonary endothelial barrier function. In a recent study, we had shown SHP2 inhibition to promote pulmonary barrier dysfunction through the enhanced tyrosine phosphorylation of AJ protein constituents, VE-cadherin, β-catenin, and p190RhoGAP, as well as activation of RhoA (5). Our previous findings suggested SHP2 to act at the level of endothelial intercellular junctions to regulate endothelial monolayer permeability (5). In the present study, we observe SHP2 dephosphorylation at Y542 and Y580 residues concomitant with FAK dephosphorylation at Y397 site upon exposure to the SHP2 chemical inhibitor, NSC-87877. Furthermore, we observe that NSC-87877–induced endothelial cell permeability is blocked by overexpression of SHP2D61A. Thus, we propose that NSC-87877 compound exhibits a dramatic effect on FAK phosphorylation, specifically through inhibition of SHP2.
Although FAK is well defined as a key regulator of focal adhesion complexes, recent data have demonstrated an additional important function in regulating AJ complexes. There remains controversy regarding the effect of FAK activity on endothelial barrier function (33); however, in the present study, we observe that both chemical and molecular inhibition of FAK, via inhibition of FAK Y397 phosphorylation, increased pulmonary endothelial cell permeability. Many other studies have similarly observed FAK activity to play an important role in maintaining barrier integrity. Schmidt and colleagues (34) showed that the knockdown of FAK in the mouse endothelium promoted lung edema and cell infiltration through the impairment of VE-cadherin surface expression and AJ formation. Further studies demonstrated that disruption of FAK signaling caused dysfunction of AJs (12, 29, 35), and have implicated the kinase activity of FAK as vital in maintaining endothelial cell barrier function and localization of VE-cadherin at AJs via select phosphorylation of VE-cadherin Y658 (36). Indeed, Lu and colleagues (37) demonstrated attenuated pulmonary endothelial barrier dysfunction and AJ disruption upon cigarette smoke exposure in cells overexpressing FAK. FAK null cells are not viable (38, 39), and cells overexpressing dominant negative SHP2 form focal adhesions, but do not spread or migrate (40). Thus, it is likely that SHP2 regulates a pool of FAK that functions to establish and/or maintain the intercellular junctions.
Embryonic fibroblasts isolated from SHP2−/− mice exhibit FAK hyperphosphorylation (40). In cardiomyocytes, SHP2 was observed to negatively regulate FAK, maintaining FAK in a tyrosine dephosphorylated, inactive state; events that correlated with increased FAK–SHP2 interactions and cell apoptosis or hypertrophy (9, 30). Similarly, in Sertoli cells, SHP2 activity correlated with reduced tight junction integrity (41). We observed the opposite of these findings, with SHP2 activation correlating with enhanced interendothelial cell junctions. In pulmonary endothelial cells, we showed enhanced FAK Y397 phosphorylation and sustained FAK–SHP2 interactions in response to barrier-enhancing S1P or constitutive SHP2 activation, and the converse was noted with barrier disruption upon thrombin or LPS exposure. It is possible that FAK–SHP2 associations may invoke a conformational change resulting in autophosphorylation of FAK or phosphorylation via another kinase, such as c-Src, leading to the stabilization of AJ. Alternatively, SHP2 association with FAK may act as an anchor or associate with other scaffolding platform proteins, such as receptor of activated C kinase 1, to control FAK phosphorylation (42, 43). Further studies are needed to examine if disruption of FAK–SHP2 interactions affects endothelial barrier function through disruption of intercellular junctions.
Tyrosine phosphorylation of FAK is central to mediating signal transduction in focal adhesions. Data from the present study indicate that SHP2 activity is directly related to FAK phosphorylation status, suggesting that SHP2 mediates FAK activity. Indeed, FAK Y397 phosphorylation promotes a conformational change, revealing sites within the kinase for binding with various signaling mediators, including the tyrosine kinase, Src, phosphatidylinositol-4,5-bisphosphate-3-kinase, and phosphatase and tensin homolog (44, 45). Furthermore, protein interactions with FAK can lead to subsequent phosphorylation, as is the case with FAK–SHP2 binding. SHP2-deficient fibroblasts display increased recruitment of the inhibitory Src kinase, C-terminal Src kinase, to the focal adhesion, resulting in reduced Src activity (46). Likewise, Laird and colleagues (47) showed that chemical inhibition of Src caused FAK dephosphorylation at a putative Src autophosphorylation epitope. Thus, the activation of Src, upon SHP2 activation, may provide a pathway through which SHP2 activity increases FAK phosphorylation and mediates protection of the endothelial barrier function. Several members of the Src family kinase have been observed to increase vascular permeability concomitant with FAK phosphorylation. Indeed, in vitro and in vivo knockdown of Src family kinases, Yamaguchi sarcoma virus oncogene or src-related homolog, Fyn, attenuates vascular permeability associated with dephosphorylation of FAK (15). However, a recent study by Han and colleagues (48) demonstrated the c-Src kinase homolog, Lyn, enhances pulmonary vascular barrier function by promoting tyrosine phosphorylation of FAK at residues Y576 and Y925, but not Y397, and suggested these sites as crucial in stabilizing interendothelial cellular junctions. However, we also observe that SHP2 activation attenuated disruption of the endothelial barrier caused by FAK inhibition, indicating that SHP2 promotes the pulmonary endothelium downstream of FAK signaling. Therefore, the effect of FAK phosphorylation on endothelial barrier function may be dependent on both upstream and downstream tyrosine kinase and phosphatase signaling.
In the present study, we did not assess the localization of phosphorylated FAK after exposure to LPS, thrombin, or S1P or SHP2D61A overexpression. Wang and colleagues (49) demonstrated increased FAK activity in response to the pharmacological S1P analog, FTY720, to enhance barrier function through elevated formation and stabilization of the focal adhesions. G protein βγ–dependent FAK activation localized the kinase to the AJ to promote resealing of the junctions and restore barrier function (22). Likewise, Han and colleagues (48) observed that small interfering RNA knockdown of FAK in human umbilical vein endothelial cells resulted in VE-cadherin breakdown at the endothelial cell border. Thus, the protective effect of FAK activation on the pulmonary vasculature, after SHP2 activation, may be mediated by the localization of FAK–SHP2 protein–protein interactions at the focal adhesion complexes, as well as AJs, in the endothelial cell.
To assess enhanced barrier function after overexpression of SHP2D61A in vitro, thrombin and LPS were used to induce monolayer permeability. Both these agents have been previously observed in vitro to increase endothelial permeability (50–52). Conversely, in vivo studies using these agents causes varying results. Thrombin increases weight gain in isolated perfused lungs as a result of increased capillary pressure (53, 54). LPS increases vascular and airway resistance and leukocyte leak; however, a lack of LPS-induced edema formation in isolated, blood-free, perfused lung preparation suggests a key role for circulating leukocytes in the role of endotoxin-induced vascular permeability (55, 56). However despite the potential differences between in vitro and in vivo mechanisms of vascular permeability, both models increase endothelial permeability and, thus, were used in the current studies to elucidate the protective role of SHP2 activation. In the present study, we induced ALI using both endotoxin (LPS) and bacteria (P. aeruginosa) to study the effect of SHP2 activation on pulmonary edema formation. In addition to permeability of the alveolar epithelium and endothelium, dysregulated inflammation and accumulation of activated leukocytes and platelets are observed in settings of ALI (57). We observed increased SHP2 expression within the pulmonary vasculature after retro-orbital administration of SHP2D61A cDNA, concomitant with attenuation of pulmonary edema formation. Given the ubiquitous distribution of SHP2 within the lung (58–60), we cannot discount that the protective role of SHP2 activation may be mediated by other cell types within the lung, which had taken up the injected cDNA, such as via tightening of epithelial junctions or diminishing the inflammatory response (61, 62). However, given that overexpression of SHP2D61A resulted in enhanced endothelial barrier function in vitro, and that we demonstrate SHP2D61A overexpression in lung endothelial cells in vivo, our studies support the notion that SHP2 is protective against edema formation in the lung vasculature. Thus, we propose that, although in vivo activation of SHP2 may modulate other pathways in settings of ALI, enhanced endothelial barrier function is a key mechanism through which SHP2 activation attenuates pulmonary edema formation.
In conclusion, the present study demonstrates the key role of SHP2 activity in modulating endothelial barrier function in vitro and pulmonary edema formation in vivo through regulation of FAK activity and protein–protein interactions between FAK and SHP2. We further show that constitutive activation of SHP2 in the pulmonary endothelium attenuates the effect of barrier-modulating agents, thereby maintaining a stabilized vasculature. We demonstrate that activation of SHP2 protects FAK against inactivation, thus maintaining activated FAK associated with SHP2, and therefore protecting the pulmonary endothelium from barrier-disruptive agents. Thus, we propose that further study on the FAK-dependent protective mechanisms mediated by SHP2 may allow development of therapeutic agents to restore pulmonary vascular function in ALI.
Acknowledgments
Acknowledgments
The authors thank Katie Grinnell and Milinda Thompson for their contributions to the studies.
Footnotes
This work was supported by Providence Veterans Affairs Medical Center and National Institutes of Health (NIH), National Heart, Lung, and Blood Institute (NHLBI) grant R01 HL-67795, American Heart Association grant 10GRNT4160055, COBRE National Institute of General Medical Sciences award 5P20 GM103652, the University Medicine Foundation of the Rhode Island Hospital (E.O.H.), American Heart Association Fellowship award 13POST16860031 (H.C.), and by the Leadership Alliance and the NIH NHLBI grant R25 HL088992.
Author Contributions: acquisition of data—H.C., J.B., and H.D.; conception and design—H.C. and E.O.H.; analysis and interpretation: H.C. and E.O.H.; drafting the manuscript for important intellectual content—H.C. and E.O.H.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2013-0489OC on October 15, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Johnson ER, Matthay MA. Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv. 2010;23:243–252. doi: 10.1089/jamp.2009.0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 4.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 5.Grinnell KL, Casserly B, Harrington EO. Role of protein tyrosine phosphatase SHP2 in barrier function of pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol. 2010;298:L361–L370. doi: 10.1152/ajplung.00374.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Timmerman I, Hoogenboezem M, Bennett AM, Geerts D, Hordijk PL, van Buul JD. The tyrosine phosphatase SHP2 regulates recovery of endothelial adherens junctions through control of beta-catenin phosphorylation. Mol Biol Cell. 2012;23:4212–4225. doi: 10.1091/mbc.E12-01-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ukropec JA, Hollinger MK, Salva SM, Woolkalis MJ. SHP2 association with VE-cadherin complexes in human endothelial cells is regulated by thrombin. J Biol Chem. 2000;275:5983–5986. doi: 10.1074/jbc.275.8.5983. [DOI] [PubMed] [Google Scholar]
- 8.Hartman ZR, Schaller MD, Agazie YM. The tyrosine phosphatase SHP2 regulates focal adhesion kinase to promote EGF-induced lamellipodia persistence and cell migration. Mol Cancer Res. 2013;11:651–664. doi: 10.1158/1541-7786.MCR-12-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marin TM, Clemente CF, Santos AM, Picardi PK, Pascoal VD, Lopes-Cendes I, Saad MJ, Franchini KG. SHP2 negatively regulates growth in cardiomyocytes by controlling focal adhesion kinase/Src and mTOR pathways. Circ Res. 2008;103:813–824. doi: 10.1161/CIRCRESAHA.108.179754. [DOI] [PubMed] [Google Scholar]
- 10.Sarai K, Shikata K, Shikata Y, Omori K, Watanabe N, Sasaki M, Nishishita S, Wada J, Goda N, Kataoka N, et al. Endothelial barrier protection by FTY720 under hyperglycemic condition: Involvement of focal adhesion kinase, small gtpases, and adherens junction proteins. Am J Physiol Cell Physiol. 2009;297:C945–C954. doi: 10.1152/ajpcell.00606.2008. [DOI] [PubMed] [Google Scholar]
- 11.Sun X, Shikata Y, Wang L, Ohmori K, Watanabe N, Wada J, Shikata K, Birukov KG, Makino H, Jacobson JR, et al. Enhanced interaction between focal adhesion and adherens junction proteins: Involvement in sphingosine 1-phosphate–induced endothelial barrier enhancement. Microvasc Res. 2009;77:304–313. doi: 10.1016/j.mvr.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holinstat M, Knezevic N, Broman M, Samarel AM, Malik AB, Mehta D. Suppression of RhoA activity by focal adhesion kinase–induced activation of p190RhoGAP: role in regulation of endothelial permeability. J Biol Chem. 2006;281:2296–2305. doi: 10.1074/jbc.M511248200. [DOI] [PubMed] [Google Scholar]
- 13.Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435–478. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- 14.Xing Z, Chen HC, Nowlen JK, Taylor SJ, Shalloway D, Guan JL. Direct interaction of v-Src with the focal adhesion kinase mediated by the Src SH2 domain. Mol Biol Cell. 1994;5:413–421. doi: 10.1091/mbc.5.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlaepfer DD, Hunter T. Evidence for in vivo phosphorylation of the GRB2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol. 1996;16:5623–5633. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guan JL. Focal adhesion kinase in integrin signaling. Matrix Biol. 1997;16:195–200. doi: 10.1016/s0945-053x(97)90008-1. [DOI] [PubMed] [Google Scholar]
- 18.Guan JL. Role of focal adhesion kinase in integrin signaling. Int J Biochem Cell Biol. 1997;29:1085–1096. doi: 10.1016/s1357-2725(97)00051-4. [DOI] [PubMed] [Google Scholar]
- 19.Miyamoto S, Teramoto H, Coso OA, Gutkind JS, Burbelo PD, Akiyama SK, Yamada KM. Integrin function: molecular hierarchies of cytoskeletal and signaling molecules. J Cell Biol. 1995;131:791–805. doi: 10.1083/jcb.131.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belvitch P, Dudek SM. Role of FAK in S1P-regulated endothelial permeability. Microvasc Res. 2012;83:22–30. doi: 10.1016/j.mvr.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grinnell K, Duong H, Newton J, Rounds S, Choudhary G, Harrington EO. Heterogeneity in apoptotic responses of microvascular endothelial cells to oxidative stress. J Cell Physiol. 2012;227:1899–1910. doi: 10.1002/jcp.22918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knezevic N, Tauseef M, Thennes T, Mehta D. The G protein betagamma subunit mediates reannealing of adherens junctions to reverse endothelial permeability increase by thrombin. J Exp Med. 2009;206:2761–2777. doi: 10.1084/jem.20090652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou MY, Lo SK, Bergenfeldt M, Tiruppathi C, Jaffe A, Xu N, Malik AB. In vivo expression of neutrophil inhibitory factor via gene transfer prevents lipopolysaccharide-induced lung neutrophil infiltration and injury by a beta2 integrin–dependent mechanism. J Clin Invest. 1998;101:2427–2437. doi: 10.1172/JCI407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grinnell KL, Chichger H, Braza J, Duong H, Harrington EO. Protection against LPS-induced pulmonary edema through the attenuation of protein tyrosine phosphatase-1B oxidation. Am J Respir Cell Mol Biol. 2012;46:623–632. doi: 10.1165/rcmb.2011-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chichger H, Grinnell KL, Casserly B, Chung CS, Braza J, Lomas-Neira J, Ayala A, Rounds S, Klinger JR, Harrington EO. Genetic disruption of protein kinase Cdelta reduces endotoxin–induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2012;303:L880–L888. doi: 10.1152/ajplung.00169.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu W, Gong D, Bar-Sagi D, Cole PA. Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Mol Cell. 2001;8:759–769. doi: 10.1016/s1097-2765(01)00369-0. [DOI] [PubMed] [Google Scholar]
- 27.Ganter MT, Roux J, Su G, Lynch SV, Deutschman CS, Weiss YG, Christiaans SC, Myazawa B, Kipnis E, Wiener-Kronish JP, et al. Role of small GTPases and αvβ5 integrin in Pseudomonas aeruginosa–induced increase in lung endothelial permeability. Am J Respir Cell Mol Biol. 2009;40:108–118. doi: 10.1165/rcmb.2007-0454OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ochoa CD, Alexeyev M, Pastukh V, Balczon R, Stevens T. Pseudomonas aeruginosa exotoxin Y is a promiscuous cyclase that increases endothelial tau phosphorylation and permeability. J Biol Chem. 2012;287:25407–25418. doi: 10.1074/jbc.M111.301440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehta D, Tiruppathi C, Sandoval R, Minshall RD, Holinstat M, Malik AB. Modulatory role of focal adhesion kinase in regulating human pulmonary arterial endothelial barrier function. J Physiol. 2002;539:779–789. doi: 10.1113/jphysiol.2001.013289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rafiq K, Kolpakov MA, Abdelfettah M, Streblow DN, Hassid A, Dell'Italia LJ, Sabri A. Role of protein-tyrosine phosphatase SHP2 in focal adhesion kinase down-regulation during neutrophil cathepsin G–induced cardiomyocytes anoikis. J Biol Chem. 2006;281:19781–19792. doi: 10.1074/jbc.M513040200. [DOI] [PubMed] [Google Scholar]
- 31.Lee HS, Moon C, Lee HW, Park EM, Cho MS, Kang JL. Src tyrosine kinases mediate activations of NF-kappaB and integrin signal during lipopolysaccharide-induced acute lung injury. J Immunol. 2007;179:7001–7011. doi: 10.4049/jimmunol.179.10.7001. [DOI] [PubMed] [Google Scholar]
- 32.Shikata Y, Birukov KG, Garcia JG. S1P induces FA remodeling in human pulmonary endothelial cells: role of Rac, GIT1, FAK, and paxillin. J Appl Physiol. 2003;94:1193–1203. doi: 10.1152/japplphysiol.00690.2002. [DOI] [PubMed] [Google Scholar]
- 33.Arnold KM, Goeckeler ZM, Wysolmerski RB. Loss of focal adhesion kinase enhances endothelial barrier function and increases focal adhesions. Microcirculation. 2013;20:637–649. doi: 10.1111/micc.12063. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt TT, Tauseef M, Yue L, Bonini MG, Gothert J, Shen TL, Guan JL, Predescu S, Sadikot R, Mehta D. Conditional deletion of FAK in mice endothelium disrupts lung vascular barrier function due to destabilization of RhoA and RAC1 activities. Am J Physiol Lung Cell Mol Physiol. 2013;305:L291–L300. doi: 10.1152/ajplung.00094.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quadri SK, Bhattacharjee M, Parthasarathi K, Tanita T, Bhattacharya J. Endothelial barrier strengthening by activation of focal adhesion kinase. J Biol Chem. 2003;278:13342–13349. doi: 10.1074/jbc.M209922200. [DOI] [PubMed] [Google Scholar]
- 36.Zhao X, Peng X, Sun S, Park AY, Guan JL. Role of kinase-independent and -dependent functions of FAK in endothelial cell survival and barrier function during embryonic development. J Cell Biol. 2010;189:955–965. doi: 10.1083/jcb.200912094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu Q, Sakhatskyy P, Grinnell K, Newton J, Ortiz M, Wang Y, Sanchez-Esteban J, Harrington EO, Rounds S. Cigarette smoke causes lung vascular barrier dysfunction via oxidative stress-mediated inhibition of RhoA and focal adhesion kinase. Am J Physiol Lung Cell Mol Physiol. 2011;301:L847–L857. doi: 10.1152/ajplung.00178.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hungerford JE, Compton MT, Matter ML, Hoffstrom BG, Otey CA. Inhibition of pp125FAK in cultured fibroblasts results in apoptosis. J Cell Biol. 1996;135:1383–1390. doi: 10.1083/jcb.135.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu DH, Qu CK, Henegariu O, Lu X, Feng GS. Protein-tyrosine phosphatase SHP-2 regulates cell spreading, migration, and focal adhesion. J Biol Chem. 1998;273:21125–21131. doi: 10.1074/jbc.273.33.21125. [DOI] [PubMed] [Google Scholar]
- 41.Puri P, Walker WH. The tyrosine phosphatase SHP2 regulates Sertoli cell junction complexes. Biol Reprod. 2013;88:59. doi: 10.1095/biolreprod.112.104414. [DOI] [PubMed] [Google Scholar]
- 42.Kiely PA, Sant A, O'Connor R. RACK1 is an insulin-like growth factor 1 (IGF-1) receptor–interacting protein that can regulate IGF-1–mediated Akt activation and protection from cell death. J Biol Chem. 2002;277:22581–22589. doi: 10.1074/jbc.M201758200. [DOI] [PubMed] [Google Scholar]
- 43.Serrels B, Sandilands E, Serrels A, Baillie G, Houslay MD, Brunton VG, Canel M, Machesky LM, Anderson KI, Frame MC. A complex between FAK, RACK1, and PDE4D5 controls spreading initiation and cancer cell polarity. Curr Biol. 2010;20:1086–1092. doi: 10.1016/j.cub.2010.04.042. [DOI] [PubMed] [Google Scholar]
- 44.Chen JS, Huang XH, Wang Q, Huang JQ, Zhang LJ, Chen XL, Lei J, Cheng ZX. Sonic Hedgehog signaling pathway induces cell migration and invasion through focal adhesion kinase/akt signaling-mediated activation of matrix metalloproteinase (MMP)-2 and MMP-9 in liver cancer. Carcinogenesis. 2013;34:10–19. doi: 10.1093/carcin/bgs274. [DOI] [PubMed] [Google Scholar]
- 45.Schaller MD, Parsons JT. Focal adhesion kinase and associated proteins. Curr Opin Cell Biol. 1994;6:705–710. doi: 10.1016/0955-0674(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 46.Zhang SQ, Yang W, Kontaridis MI, Bivona TG, Wen G, Araki T, Luo J, Thompson JA, Schraven BL, Philips MR, et al. SHP2 regulates Src family kinase activity and Ras/Erk activation by controlling csk recruitment. Mol Cell. 2004;13:341–355. doi: 10.1016/s1097-2765(04)00050-4. [DOI] [PubMed] [Google Scholar]
- 47.Laird AD, Li G, Moss KG, Blake RA, Broome MA, Cherrington JM, Mendel DB. Src family kinase activity is required for signal transducer and activator of transcription 3 and focal adhesion kinase phosphorylation and vascular endothelial growth factor signaling in vivo and for anchorage-dependent and -independent growth of human tumor cells. Mol Cancer Ther. 2003;2:461–469. [PubMed] [Google Scholar]
- 48.Han J, Zhang G, Welch EJ, Liang Y, Fu J, Vogel SM, Lowell CA, Du X, Cheresh DA, Malik AB, et al. A critical role for Lyn kinase in strengthening endothelial integrity and barrier function. Blood. 2013;122:4140–4149. doi: 10.1182/blood-2013-03-491423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L, Chiang ET, Simmons JT, Garcia JG, Dudek SM. FTY720-induced human pulmonary endothelial barrier enhancement is mediated by c-Abl. Eur Respir J. 2011;38:78–88. doi: 10.1183/09031936.00047810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adyshev DM, Dudek SM, Moldobaeva N, Kim KM, Ma SF, Kasa A, Garcia JG, Verin AD. Ezrin/radixin/moesin proteins differentially regulate endothelial hyperpermeability after thrombin. Am J Physiol Lung Cell Mol Physiol. 2013;305:L240–L255. doi: 10.1152/ajplung.00355.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Birukova AA, Birukov KG, Smurova K, Adyshev D, Kaibuchi K, Alieva I, Garcia JG, Verin AD. Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J. 2004;18:1879–1890. doi: 10.1096/fj.04-2328com. [DOI] [PubMed] [Google Scholar]
- 52.Joshi AD, Dimitropoulou C, Thangjam G, Snead C, Feldman S, Barabutis N, Fulton D, Hou Y, Kumar S, Patel V, et al. Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am J Respir Cell Mol Biol. 2014;50:170–179. doi: 10.1165/rcmb.2012-0496OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horgan MJ, Fenton JW, II, Malik AB. Alpha-thrombin–induced pulmonary vasoconstriction. J Appl Physiol. 1987;63:1993–2000. doi: 10.1152/jappl.1987.63.5.1993. [DOI] [PubMed] [Google Scholar]
- 54.Waypa GB, Vincent PA, Morton CA, Minnear FL. Thrombin increases fluid flux in isolated rat lungs by a hemodynamic and not a permeability mechanism. J Appl Physiol. 1996;80:1197–1204. doi: 10.1152/jappl.1996.80.4.1197. [DOI] [PubMed] [Google Scholar]
- 55.Chignard M, Balloy V. Neutrophil recruitment and increased permeability during acute lung injury induced by lipopolysaccharide. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1083–L1090. doi: 10.1152/ajplung.2000.279.6.L1083. [DOI] [PubMed] [Google Scholar]
- 56.Uhlig S, Wollin L. An improved setup for the isolated perfused rat lung. J Pharmacol Toxicol Methods. 1994;31:85–94. doi: 10.1016/1056-8719(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 57.Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol. 2005;33:319–327. doi: 10.1165/rcmb.F305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li FF, Shen J, Shen HJ, Zhang X, Cao R, Zhang Y, Qui Q, Lin XX, Xie YC, Zhang LH, et al. SHP2 plays an important role in acute cigarette smoke–mediated lung inflammation. J Immunol. 2012;189:3159–3167. doi: 10.4049/jimmunol.1200197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pazdrak K, Young TW, Stafford S, Olszewska-Pazdrak B, Straub C, Starosta V, Brasier A, Kurosky A. Cross-talk between ICAM-1 and granulocyte–macrophage colony–stimulating factor receptor signaling modulates eosinophil survival and activation. J Immunol. 2008;180:4182–4190. doi: 10.4049/jimmunol.180.6.4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qin XJ, Zhang GS, Zhang X, Qiu ZW, Wang PL, Li YW, Li W, Xie QM, Ke YH, Lee JJ, et al. Protein tyrosine phosphatase SHP2 regulates TGF-beta1 production in airway epithelia and asthmatic airway remodeling in mice. Allergy. 2012;67:1547–1556. doi: 10.1111/all.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coulombe G, Leblanc C, Cagnol S, Maloum F, Lemieux E, Perreault N, Feng GS, Boudreau F, Rivard N. Epithelial tyrosine phosphatase SHP-2 protects against intestinal inflammation in mice. Mol Cell Biol. 2013;33:2275–2284. doi: 10.1128/MCB.00043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang X, Zhang Y, Tao B, Teng L, Li Y, Cao R, Gui Q, Ye M, Mou X, Cheng H, et al. Loss of SHP2 in alveoli epithelia induces deregulated surfactant homeostasis, resulting in spontaneous pulmonary fibrosis. FASEB J. 2012;26:2338–2350. doi: 10.1096/fj.11-200139. [DOI] [PubMed] [Google Scholar]