

Abstract
Angiogenesis in ischemic organs is modulated by immune cells. Systemic neovascularization of the ischemic lung requires macrophages, with chemokines playing a central role in new vessel growth. Because regulatory T (Treg) cells modulate tumor-induced neovascularization, we questioned whether this CD4+ lymphocyte subset impacts blood vessel growth during ischemia. In a model of left lung ischemia, an increase in CD4+ CD25+ forkhead homeobox protein-3 (Foxp3)+ cells was observed 3–5 days after the onset of ischemia in wild-type C57Bl/6 mice. Using transgenic mice where Foxp3+ Treg cells can be depleted with diphtheria toxin (DT; Foxp3DTR), we unexpectedly found that Foxp3+ Treg depletion led to markedly reduced lung angiogenesis (90% reduction from Foxp3gfp controls). Adoptive transfer studies using CD4+ CD25+ splenocytes from congenic CD45.1 mice into Foxp3+ Treg–depleted mice showed an almost complete recovery of the angiogenic phenotype (80% of Foxp3gfp controls). A survey of lung gene expression of angiogenic (lipopolysaccharide-induced CXC chemokine [LIX], IL-6, IL-17) and angiostatic (IFN-γ, transforming growth factor-β, IL-10) cytokines showed Treg-dependent differences only in LIX (CXCL5) and IL-6. Protein confirmation demonstrated a significant reduction in LIX in Treg-deficient mice compared with controls 5 days after the onset of ischemia. Phenotyping other inflammatory cells in the lung by multicolor flow cytometry demonstrated a significantly reduced number of macrophages (major histocombatibility complex class II [MHCII]int, CD11C+) in Treg-deficient lungs compared with Treg-sufficient lungs. Treg cells are essential for maximal systemic angiogenesis after pulmonary ischemia. One likely mechanism responsible for the decrease in angiogenesis in Treg-depleted mice was the decline in the essential CXC chemokine, LIX.
Keywords: angiogenesis, ischemia, regulatory T cells, CXC chemokines
Clinical Relevance
Angiogenesis in the lung is understudied, yet it is a common feature of a variety of lung pathologies. Control of the growth of new blood vessels by regulatory T cells offers a potential therapy in situations where new vessels are required.
Angiogenesis is pivotal for diverse physiological processes, including embryonic development and growth, wound healing, and endometrial and gestational maintenance (1, 2). Modulation of angiogenesis in the lung is necessary for tissue repair, but is susceptible to dysregulation, and can become prominent and contribute to pathology in chronic inflammatory conditions, such as cystic fibrosis, asthma, chronic obstructive pulmonary disease, interstitial lung diseases, chronic thromboembolic disease, and lung cancer (3–8). The systemic vasculature in and surrounding the lung is proangiogenic, whereas the pulmonary vasculature does not proliferate under most conditions (9–11). Understanding the balance of proangiogenic and angiostatic factors is of paramount importance to understanding the regulation of the process of neovascularization. However, the underlying cellular mechanisms involved in lung angiogenesis remain largely unknown. Using a murine model of total left pulmonary artery ischemia, our laboratory has shown that subsequent neovascularization arises exclusively from the bronchial artery (12) and intercostal arteries (10), is initiated by reactive oxygen species (ROS) (13), and is dependent on early up-regulation of CXC chemokine growth factors (14). Furthermore, our laboratory has demonstrated the importance of mature lung macrophages in promoting angiogenesis (15), whereas lymphocytes limited the inflammation and ischemia-induced lung angiogenic responses (16). Several reports suggest that lymphocytes are involved in both homeostatic and pathological angiogenesis in peripheral tissue (17, 18). However, the role of lymphocytes or their specific subpopulations in lung angiogenesis has not been fully explored.
Forkhead homeobox protein-3 (Foxp3)+ regulatory T (Treg) cells are a distinct subpopulation of CD4+ lymphocytes that also express CD25 (IL-2 receptor) and the transcription factor, Foxp3, which is the master regulator of Treg development and suppressive function. Given the important regulatory role of Foxp3+ Treg cells in resolving lung inflammation and promoting repair (19), we questioned whether this lymphocyte population similarly served a regulatory role after ischemic injury. Furthermore, we recently demonstrated that Foxp3+ Treg cells modulated activated lung macrophages in response to endotoxin-induced lung injury (19). Given the importance of macrophages in promoting angiogenesis after ischemic insult (15), we hypothesized that Foxp3+ Treg cells moderate lung angiogenesis through modulation of macrophage function.
In our experimental model of left lung ischemia, we found that wild-type (WT) mice had increased Foxp3+ Treg cells over time in their left ischemic lung and draining lymph nodes. Using transgenic mice where Foxp3+ Treg cells can be depleted with diphtheria toxin (DT; Foxp3DTR), we found that Foxp3+ Treg depletion led to markedly reduced lung angiogenesis. Contrary to expectations, our findings demonstrate that Foxp3+ Treg cells are required for lung angiogenesis. These observations may have important clinical implications, as Foxp3+ Treg cells could be used therapeutically to enhance angiogenesis in ischemic organs or to limit pathological blood vessel formation.
Materials and Methods
Mice
C57BL/6 WT (Jackson Laboratories, Bar Harbor, ME) and Foxp3gfp and Foxp3DTR mice (gifts of Dr. A. Y. Rudensky, Sloan-Kettering Institute [New York, NY]) were housed in a pathogen-free facility. Procedures were approved by the Johns Hopkins Animal Care and Use Committee (protocol no. MO13M239). The lung ischemic injury model was used as previously described (10, 15). Left pulmonary artery ligation (LPAL) was performed in mice and the left lung was harvested at specified time points.
Preparation of Cell Suspensions
Left lungs were collected in dissociator tubes (2 mg/ml Dulbecco’s modified Eagles medium, collagenase D; 40 U/ml DNase I; and HEPES). Tissues were homogenized, incubated (37°C, 30 min), strained (70 μm), red blood cells removed (ammonium chloride potassium buffer), and cells were washed (cold PBS). Endothelial cell analysis followed the digestion protocol previously described (20).
Antibodies and Flow Cytometry
Fluorescence-conjugated anti-mouse antibodies were used to identify lung T cells, macrophages, and endothelial cells. A complete list of antibodies, concentrations, vendors, and detailed staining methods with gating strategy are provided in the Materials and Methods section in the online supplement. Cells were acquired using BD LSRII (Becton Dickinson Life Science Research II, Franklin Lakes, NJ) and analyzed with FlowJo (Tree Star, Ashland, OR).
Foxp3DTR Mice and DT Administration
To eliminate Foxp3+ Treg cells, we used transgenic Foxp3DTR mice (21) and eliminated Foxp3+ Treg cells in vivo through DT (Sigma, St. Louis, MO) administration (20 ng/g or 10 ng/g, intraperitoneal) (21, 22). Foxp3gfp reporter mice that express an N-terminal green fluorescent protein–Foxp3 fusion protein served as controls (23).
Angiogenic Index
Functional angiogenic perfusion of the left lung was determined by infusing fluorescent microspheres (10 μm; Invitrogen, Grand Island, NY) into the aorta as described previously (15).
Isolation of CD4+ CD25+ T Cells and Adoptive Transfer
Splenocytes were collected from naive congenic CD45.1, C57BL/6 mice. CD4+CD25+ T cells were purified by magnetic cell sorting (Treg isolation kit; Miltenyi, San Diego, CA). Isolated cells (1 × 106) or PBS (100 μl) were given to recipient mice intravenously 24 hours before LPAL.
Real-Time PCR
RNA was isolated from left lung using standard techniques followed by quantitative PCR (SYBR Green gene-specific primers; Table 1). Results were normalized (β2-microglobulin) and fold change reported (versus 0 h left lung). We selected genes with expected proangiogenic (IL-6, lipopolysaccharide-induced CXC chemokine [LIX], IL-17) and antiangiogenic (IFN-γ, transforming growth factor [TGF]-β, IL-10) effects.
Table 1.
Cytokine Primer Sequences
| Gene | Forward/Reverse | Sequence 5′ to 3′ |
|---|---|---|
| Mouse IL-10 | Forward | TCG GCC AGA GCC ACA TG |
| Reverse | TTA AGG AGT CGG TTA GCA AGT ATG TTG | |
| Mouse IL-17A | Forward | GGA CTC TCC ACC GCA ATG AA |
| Reverse | GCA CTG AGC TTC CCA GAT CAC | |
| Mouse IFN-γ | Forward | TTG CCA AGT TTG AGG TCA ACA A |
| Reverse | TGG TGG ACC ACT CGG ATG A | |
| Mouse IL-6 | Forward | TCG GAG GCT TAA TTA CAC ATG TTC |
| Reverse | TGC CAT TGC ACA ACT CTT TTC T | |
| Mouse LIX | Forward | GCCGCTGGCATTTCTGTT |
| Reverse | GGGCAGCTTCAGCTAGATGCT | |
| Mouse TGF-β | Forward | CGGAGAGCCCTGGATACCA |
| Reverse | GCCGCACACAGCAGTTCTT | |
| Mouse β2 microglobulin | Forward | AAA TGC TGA AGA ACG GGA AAA |
| Reverse | ATA GAA AGA CCA GTC CTT GCT GAA G |
Definition of abbreviations: LIX, lipopolysaccharide-induced CXC chemokine; TGF-β, transforming growth factor-β.
Protein Analysis
Left lung was homogenized, centrifuged, and supernatants collected to determine LIX (CXCL5) and IL-6 by ELISA (R&D Systems, Minneapolis, MN).
Immunohistochemistry
Sections of left lungs of mice were stained with fluorescent anti-F4/80 for lung macrophages, anti-CXCL5 (LIX), and 4′,6-diamidino-2-phenylindole to identify cell nuclei (see the Materials and Methods section in online supplement). Fluorescent images were obtained with a microscope (Olympus IX-51; Olympus, Center Valley, PA) using a Sensicam High Performance camera (Cooke, Auburn Hills, MI).
In Vitro Macrophage Cytokine Secretion
Single-cell suspensions of left lung (1 × 106 cells/ml) were dispensed (six-well plates, 10% fenugreek seed extract in Dulbecco’s modified Eagles medium) and incubated (2 h at 37°C). Nonadherent cells were discarded, and fresh media with/without 100 μmol H2O2 was added to adherent fraction and incubated. Supernatants at 4 hours were analyzed for LIX and IL-6 (ELISA; R&D Systems).
Statistical Analysis
Data are presented as the mean (± SE); t tests were used to compare single times between strains. Time courses were analyzed with repeated measures ANOVA and Newman-Keuls multiple comparisons. A P value less than 0.05 was accepted as significant.
Results
Foxp3+ Treg Cells Increase after Lung Ischemia
We sought to determine whether lung lymphocyte subpopulations were dynamically regulated in the ischemic lung. The percentage of CD4+Foxp3+ Treg cells increased significantly 3 days after LPAL and peaked 7 days after lung ischemia (Figures 1A and 1B; n = 5–7 mice/time point, P < 0.0001). There was no change in Foxp3+ Treg cell proliferation in the lung by intracellular ki67 staining (Figure 1C).
Figure 1.
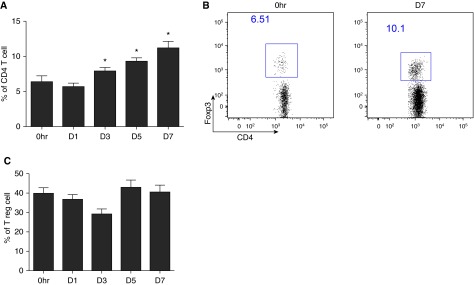
Regulatory T (Treg) profiles after the induction of pulmonary ischemia in wild-type mice (n = 5–7 mice/time point). *P < 0.05, different from 0 h control. (A) Average changes in lung Treg cells (% of CD4+ T cells) at the onset of left lung ischemia (0 h) through the first 7 days (D7) after ligating the left pulmonary artery. A progressive increase in the percent of lung Treg cells is seen over the course of the first 7 days of ischemia. (B) Representative dot plot showing changes in CD4+ forkhead homeobox protein-3 (Foxp3)+ cells immediately (0 h) and 7 days (D7) after the onset of ischemia. (C) Treg cells (CD4+, Foxp3+ cells) showed no changes in the intracellular proliferation marker, ki67, over the time course of 7 days.
Lung Angiogenesis Is Markedly Reduced in the Absence of Foxp3+ Treg Cells
To determine whether Foxp3+ Treg cells participate in lung angiogenesis after ischemia, we used DT-treated transgenic Foxp3DTR mice and Foxp3gfp mice (controls). Foxp3gfp and WT mice had similar increases in the percent of Foxp3+ Treg cells (9% versus 6% of CD4+ T cells at 5 d [D5] after LPAL, respectively). We optimized the DT treatment scheme to deplete Foxp3+ Treg cells over a 2-week period and achieved over 85% depletion of Foxp3+ Treg cells (Figures 2A and 2B). Systemic angiogenesis of the left lung was assessed 14 days after LPAL (Figure 2C). Foxp3+ Treg–depleted animals had a 90% reduction in angiogenic response in the left ischemic lung compared with Foxp3gfp controls (Figure 2B; n = 6–8 mice/group; P < 0.0001). Consistent with the functional angiogenic index, flow cytometry to detect CD31+CD45− endothelial cells as early as 5 days after LPAL showed an average 25% reduction in the Foxp3+ Treg–depleted mice (Figure 2D; n = 2–3/group; P = 0.003). Gating strategy for endothelial cells is shown in Figure E2 in the online supplement. These results confirmed that, in the absence of Treg cells, systemic angiogenesis and lung endothelial cell numbers are significantly reduced during ischemia.
Figure 2.

Treg cell depletion prevents angiogenesis. (A) Treatment regimen to deplete Treg cells in the Foxp3DTR mouse strain before and after left pulmonary artery ligation (LPAL) with diphtheria toxin (DT), and time of determination of angiogenesis (blood flow at Day 14). (B) Representative dot plot showing effectiveness of depletion treatment. CD4+ Foxp3+ cells in Treg-sufficient (Foxp3gfp mouse strain) spleens are much greater than in Treg-deficient (Foxp3DTR) spleens 14 days after LPAL. (C) Functional angiogenesis index assessed by perfusion to the left lung 14 days after LPAL. A significant reduction in angiogenesis was observed in mice without Treg cells (n = 6–8 mice/group). *P < 0.0001. (D) CD31+ CD45− endothelial cells (% live cells) in the left lung 5 days after LPAL. A significant reduction in endothelial cells was seen in left lungs from Foxp3DTR mice compared with control Foxp3gfp lungs by 5 days after the induction of ischemia (n = 2–3/group). *P = 0.003.
Transfer of Foxp3+ Treg Cells Restores Lung Angiogenesis
To further confirm that Foxp3+ Treg cells are required to promote lung angiogenesis after ischemia, we used the Treg depletion scheme shown in Figure 2A. In addition, 1 day before LPAL, mice were given a retro-orbital injection of PBS or Treg cells isolated from congenic CD45.1 spleens and sorted using sterile technique. In contrast to endogenous Treg cells, the adoptively transferred cells were not susceptible to exogenous DT depletion. After 14 days of lung ischemia, the Foxp3+ Treg–depleted group, that received PBS, showed a marked reduction in angiogenesis (Figure 3A; n = 5–6 mice/treatment) similar to that seen in Figure 2B. In contrast, the Foxp3+ Treg–depleted group that was adoptively transferred with congenic WT Treg cells 1 day before LPAL showed a significant restoration of the normal angiogenic response observed after LPAL (P = 0.001). Adoptively transferred CD45.1 Treg cells were confirmed in spleens 14 days after lung ischemia (Figure 3B), and were approximately the same as seen in Foxp3gfp controls (Figure 2B). These experiments reinforced the observation demonstrating a role for Foxp3+ Treg cells in promoting lung neovascularization after ischemia.
Figure 3.

Transfer of normal Foxp3+ Treg cells restores lung angiogenesis. (A) Foxp3DTR mice treated with DT and vehicle showed reduced angiogenesis (each point represents one mouse and the horizontal line is group mean). However, a significant increase in angiogenesis occurred in Treg-depleted mice after adoptive transfer of CD45.1+CD4+CD25+ splenocytes before LPAL (n = 5–6 mice/group). *P = 0.001. (B) Representative dot plot of cells from spleens showing effectiveness of depletion treatment (PBS) and adoptive transfer (CD4+CD25+). CD4+Foxp3+ cells in the in Treg-sufficient (Foxp3gfp) spleen are much greater than in Treg-deficient (Foxp3DTR) spleens 14 days after LPAL.
Lung Inflammatory Milieu Modulated by Foxp3+ Treg Cells
To begin to understand mechanisms underlying Foxp3+ Treg–mediated lung angiogenesis after ischemia, we measured at intervals, several prominent proangiogenic and antiangiogenic cytokines known to be involved in ischemia-induced vascular responses. Ischemic lung homogenate mRNA levels for IL-6 and LIX (CXCL5) were significantly higher in mice with intact Foxp3+ Treg cells compared with Treg-depleted animals (Figure 4A; n = 3–4 mice/time point/group; P < 0.05). Other mediators studied (IFN-γ, TGF-β, IL-17 and IL-10) were not different between Foxp3+ Treg–sufficient or –depleted mice. To validate the mRNA results, we performed ELISA on ischemic lung homogenates to confirm significantly higher levels of LIX, but not IL-6 in Foxp3+ Treg–sufficient mice (Figure 4B; n = 5–6 mice/time point/group; P < 0.001). Of note, the differences in LIX were measured at D5, whereas differences in mRNA levels were detected earlier at D3 after lung ischemia. Our studies support the notion that Treg cells allow a more favorable proangiogenic milieu in the ischemic lung characterized by higher lung LIX levels.
Figure 4.

Lung inflammatory milieu is modulated by Foxp3+ Treg cells. (A) Time course of mRNA expression (fold induction from 0 h) of proangiogenic (IL-6, lipopolysaccharide-induced CXC chemokine [LIX], IL-17) and angiostatic (IFN-γ, transforming growth factor [TGF]-β, IL-10) cytokines in the left lungs of Treg-sufficient (Foxp3gfp) and Treg-deficient (Foxp3DTR) mice. A significant difference in IL-6 and LIX expression was seen between the two strains 3 days after the onset of ischemia (n = 3–4 mice/time point/group). *P < 0.05. (B) LIX and IL-6 protein of the upper left lung was confirmed at Day 3 (D3) and D5 after LPAL in Foxp3gfp and Foxp3DTR mice (n = 5–6 mice/time point/group). *P < 0.001. Although significant increases in both LIX and IL-6 were seen in both groups between D3 and D5, the change in LIX protein was significantly attenuated in Foxp3DTR mice. No difference in IL-6 between Foxp3gfp and Foxp3DTR mice was apparent at either time point.
Foxp3+ Treg Cells Promote Increased Macrophages during Lung Ischemia
To explore lung macrophage modulation by Foxp3+ Treg cells, we further immunophenotyped macrophages in dissociated left lung by multicolor flow cytometry at the onset of ischemia (0 h) and at intervals after LPAL. We found similar percent live macrophages in the lungs (major histocombatibility complex class II [MHCII]int, CD11C+) initially and during the first 3 days after LPAL in Treg-depleted Foxp3DTR and Treg-sufficient Foxp3gfp mice, with ever-increasing numbers. However, by D5 after LPAL, Foxp3gfp ischemic left lung had nearly threefold more macrophages than their Foxp3DTR counterparts (Figure 5A; n = 6–9 mice/time point/group; P < 0.01). Evaluation of activation markers CD86 (M1 activation), and mannose receptor (M2 activation) showed no difference between Foxp3gfp and Foxp3DTR mice as assessed by mean fluorescence intensity. However, overall mannose receptor staining was much greater than CD86 in both groups (data not shown). To begin to determine the source of LIX in left lungs, immunohistochemistry was performed using frozen sections from both groups of mice (n = 1 mouse/group at 0 h, 2 mice/group at 5D). Figure 5B shows a representative example of a 5D Foxp3gfp left lung showing F4/80+ cells colocalized with LIX protein. In all sections evaluated, macrophage–LIX colocalization was confirmed.
Figure 5.

Foxp3+ Treg cells promote increased macrophage numbers during lung ischemia. (A) The time course of macrophages (major histocombatibility complex class IIint, CD11C+) in left lungs showed a progressive increase in Treg-sufficient Foxp3gfp mice. However, by D5 after LPAL, Foxp3gfp ischemic left lungs had nearly threefold more macrophages than their Foxp3DTR counterparts (n = 6–9 mice/time point/group). *P < 0.01, Foxp3gfp versus Foxp3DTR. (B) Representative immunostaining of macrophage F4/80 (green), LIX protein (red), and the merge showing colocalization (yellow-green) of a left lung section of Foxp3gfp lung 5 days after LPAL. 4′,6-diamidino-2-phenylindole (blue) labels all cell nuclei. Scale bar, 10 μm; original magnification, ×400.
To determine whether there were inherent differences in lung macrophages between the two groups of mice, we studied in vitro secretion of proangiogenic cytokines, LIX and IL-6. Macrophages isolated on D5 from Treg-depleted Foxp3DTR and Treg-sufficient Foxp3gfp lungs showed similar patterns of cytokine secretion. Figure 6 demonstrates that, whether under basal conditions or after stimulation with an ROS mimic (H2O2), macrophages from the two strains responded in a similar manner. ROS stimulation caused a significant increase in cytokine release in macrophages from both strains (P = 0.01).
Figure 6.

Macrophages from Treg-sufficient Foxp3gfp mice and Treg-deficient Foxp3DTR show similar levels of cytokine expression in vitro. Macrophages isolated from the lungs of Treg-sufficient Foxp3gfp mice and Treg-deficient Foxp3DTR mice 5 days after the onset of ischemia showed similar basal secretion of LIX and IL-6. Stimulation with a reactive oxidant (H2O2) showed a significant increase in cytokine secretion (n = 3–4 experiments/group; P = 0.01), but no difference between strains.
Discussion
Organ ischemia is among the leading pathophysiological causes of morbidity and mortality. New blood vessel formation in response to ischemia is critical for the restoration of organ homeostasis. The goal of the present study was to determine the modulating influence of Treg cells (CD4+CD25+Foxp3+) on the process of lung neovascularization during ischemia. Our previous work demonstrated that angiogenesis in the lung is dependent on macrophage-derived chemokine release early after the induction of pulmonary vascular ischemia. Consequently, we predicted that limiting Treg control of chemokine-secreting macrophages would enhance angiogenesis. Contrary to these expectations, results demonstrated that Treg depletion eliminated angiogenesis completely. The prominent CXC chemokine, LIX, was significantly reduced in lung homogenate in Treg-depleted mice, as were the number of macrophages. We conclude that Treg cells are essential for maximal neovascularization after pulmonary ischemia. One mechanism by which Treg cells control the angiogenic process is by limiting the number of chemokine-producing macrophages in the lung.
Our work describes a novel role for Treg cells in modulating angiogenesis after lung ischemia. In general, the role of lymphocytes in angiogenesis has become more apparent. Recently, Du and colleagues (24) demonstrated that tumor-infiltrating lymphocytes produce IL-17 and promote tumor growth by enhancing angiogenesis. Moreover, Tayade (18) reported that lymphocytes promoted endometrial angiogenesis, and thereby contributed to fetal health. An earlier study of graft versus host disease showed that injection of lymphocytes into histoincompatible hosts resulted in formation of networks of new blood vessels (25). These reports suggest that lymphocytes are involved in both homeostatic and pathological angiogenesis in peripheral tissues. However, to our knowledge, the role of lymphocytes or their specific subtypes in lung angiogenesis has not been described.
Treg cells have been regarded as master regulators of exuberant immune responses (23). Work from the Rudensky laboratory described profound autoimmunity when using transgenic mice where specific depletion of Treg cells can be achieved (e.g., Foxp3DTR). We expected that depletion of Treg cells would have led to an enhanced lung proinflammatory milieu and, subsequently, a greater angiogenic response in the ischemic lung. The robust and consistent differences in phenotype between Treg-sufficient and Treg-deficient mice with regard to measurements of functional angiogenesis were somewhat surprising, given the complex process of angiogenesis after ischemia. Furthermore, the gain of function when Treg cells from CD45.1 splenocytes were adoptively transferred into mice without Treg cells was highly reproducible with little variability (Figure 3). These results highlight the capacity of Treg cells to ostensibly control the process of neovascularization, and reinforce the critical and novel nature of Treg cells as potent proangiogenic cells.
To begin to determine the mechanism for the differences in angiogenesis, we surveyed cytokines known to be involved in ischemia-induced neovascularization. The survey of proangiogenic and suppressive cytokines in the ischemic left lung revealed differences between the Treg-sufficient and Treg-deficient mice in two cytokines that we have shown to be critical growth factors for neovascularization (Figure 4A). Previously, we showed that the CXC chemokines (LIX, MIP-2α, and KC) and IL-6 were up-regulated early after the onset of ischemia and when blocked with neutralizing antibody for the CXCR1/2 receptor, which binds these chemokines, and, in IL-6 null mice, typical neovascularization was prevented (26, 27). With regard to the CXC chemokines, our work was supportive of the work of Strieter and colleagues (28), who showed, in a number of proangiogenic pathologies, that these proteins bind CXCR1/2 on endothelial cells and promote proliferation and chemotaxis. In the current study, we focused on LIX as representative of the chemokines and IL-6. LIX mRNA and protein were significantly decreased in the Treg-deficient mice, suggesting a possible mechanism for decreased angiogenesis in this experimental group (Figure 4B). Although LIX is produced by several lung cell types, immunohistochemistry showed clear colocalization of LIX protein and F4/80+ lung macrophages (Figure 5B). The observation that the number of macrophages was significantly reduced coincident with the decline in LIX protein was suggestive of a role for macrophages in the observed difference in LIX protein. However, when studied in vitro, macrophages isolated from the lungs of the two mouse strains did not show differences in their in vitro capability to secrete cytokines, either under basal conditions or upon oxidant stimulation. Thus, we suggest that one explanation for the decrease in LIX protein in the lung of Treg-deficient mice is the decrease in chemokine-producing macrophages, and not in the inherent ability of these cells to secrete chemokine. At this time, we cannot explain the observation that IL-6 protein appeared similar in both strains. We speculate that the kinetics of this protein may differ from LIX, thereby contributing to the lack of significant changes at the time points of measurement.
It should be noted that, because we saw no differences in gene expression of known suppressive cytokines, such as IL-10, TGF-β, or IFN-γ, between Treg-sufficient and Treg-deficient lungs, we pursued the proangiogenic cytokines that showed differences. We acknowledge that, at later time points, the suppressive cytokines may play an additional role in modulating the extent of angiogenesis. Because of past work implicating macrophages as critical to the process of ischemia-induced angiogenesis in the lung and the strain-dependent changes in lung macrophages at the time when there were measureable differences in an important proangiogenic chemokine, we focused on the macrophage as a source of growth factors. However, there are likely to be other cells interacting with Treg cells that may contribute as much, if not more, to the profound differences in angiogenesis observed between strains. Our laboratory has recently described that Treg cells can modulate alveolar epithelial proliferation and repair (20); thus, a Treg cell–epithelial cell cross-talk might also exist that could further promote the angiogenic response. Similarly, a Treg cell–endothelial cell interaction might directly promote angiogenesis. ROS-induced Treg activation has been described (29), and, thus, the ROS release during pulmonary ischemia confirmed by our laboratory and others might promote a compensatory angiogenic response requiring Treg cells (13, 30). For example, Treg cells can express high levels of neuropillin that can bind to vascular endothelial growth factor receptor on endothelial cells to promote their sprouting and, ultimately, new vessel formation (31, 32). Although, in this nonhypoxic model, neither vascular endothelial growth factor nor other hypoxia-inducible factor-1-alpha (HIF-1α)–regulated growth factors were shown to be up-regulated (14), the direct effect of Treg cell activation of pulmonary endothelium remains to be elucidated and is ongoing in our laboratory.
Also of interest is the observation that differences between Treg-sufficient and Treg-deficient mice become most apparent at 5 days, the point in time at which the new systemic vasculature becomes functional in WT mice (10). Whether the lack of Treg cells prevents macrophage proliferation within the ischemic left lung beyond 3 days, or whether the lack of a fully functional vasculature prevents the influx of macrophages (and chemokine availability) in the recovering ischemic lung, cannot be determined from these studies. Future studies evaluating lung morphometry, including microvessel density, with macrophage and Treg localization, may provide additional insight into the process of lung neovascularization.
In summary, it is clear that Treg cells are vitally important to the process of lung neovascularization. Although our efforts to understand the mechanism by which Treg cells control ischemia-induced angiogenesis focused on changes in macrophage-derived growth factor release, there are likely other processes, such as direct Treg–endothelial cell proliferative interactions, as well as suppression of effector T cells. Enhancing and understanding Treg-mediated proangiogenic effects could become a novel strategy for therapeutic angiogenesis required for ischemic organs.
Acknowledgments
Acknowledgments
The authors thank Dr. A. Y. Rudensky for use of the Foxp3gfp and Foxp3DTR mice strains.
Footnotes
This work was supported by National Institutes of Health grants HL10342 (Project 3) (E.M.W.) and R00HL103973 (F.R.D.’A.).
Author Contributions: conception and design—F.R.D.’A., Q.Z., and E.M.W.; experimental work—Q.Z., J.J., and A.M.; analysis and interpretation—F.R.D.’A., Q.Z., J.J., A.M., and E.M.W.; manuscript preparation—F.R.D.’A., Q.Z., and E.M.W.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2014-0278OC on October 2, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8:464–478. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 3.Wilson JW, Robertson CF. Angiogenesis in paediatric airway disease. Paediatr Respir Rev. 2002;3:219–229. doi: 10.1016/s1526-0542(02)00200-2. [DOI] [PubMed] [Google Scholar]
- 4.Li X, Wilson JW. Increased vascularity of the bronchial mucosa in mild asthma. Am J Respir Crit Care Med. 1997;156:229–233. doi: 10.1164/ajrccm.156.1.9607066. [DOI] [PubMed] [Google Scholar]
- 5.Kranenburg AR, De Boer WI, Van Krieken JH, Mooi WJ, Walters JE, Saxena PR, Sterk PJ, Sharma HS. Enhanced expression of fibroblast growth factors and receptor FGFR-1 during vascular remodeling in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2002;27:517–525. doi: 10.1165/rcmb.4474. [DOI] [PubMed] [Google Scholar]
- 6.Antoniou KM, Tzouvelekis A, Alexandrakis MG, Sfiridaki K, Tsiligianni I, Rachiotis G, Tzanakis N, Bouros D, Milic-Emili J, Siafakas NM. Different angiogenic activity in pulmonary sarcoidosis and idiopathic pulmonary fibrosis. Chest. 2006;130:982–988. doi: 10.1378/chest.130.4.982. [DOI] [PubMed] [Google Scholar]
- 7.Endrys J, Hayat N, Cherian G. Comparison of bronchopulmonary collaterals and collateral blood flow in patients with chronic thromboembolic and primary pulmonary hypertension. Heart. 1997;78:171–176. doi: 10.1136/hrt.78.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strieter RM. Out of the shadows: CXC chemokines in promoting aberrant lung cancer angiogenesis. Cancer Prev Res (Phila) 2008;1:305–307. doi: 10.1158/1940-6207.CAPR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu L, Hales CA. Hypoxia does neither stimulate pulmonary artery endothelial cell proliferation in mice and rats with pulmonary hypertension and vascular remodeling nor in human pulmonary artery endothelial cells. J Vasc Res. 2011;48:465–475. doi: 10.1159/000327005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mitzner W, Lee W, Georgakopoulos D, Wagner E. Angiogenesis in the mouse lung. Am J Pathol. 2000;157:93–101. doi: 10.1016/S0002-9440(10)64521-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner EM, Petrache I, Schofield B, Mitzner W. Pulmonary ischemia induces lung remodeling and angiogenesis. J Appl Physiol. 2006;100:587–593. doi: 10.1152/japplphysiol.00029.2005. [DOI] [PubMed] [Google Scholar]
- 12.Sukkar A, Jenkins J, Sánchez J, Wagner E. Inhibition of CXCR2 attenuates bronchial angiogenesis in the ischemic rat lung. J Appl Physiol. 2008;104:1470–1475. doi: 10.1152/japplphysiol.00974.2007. [DOI] [PubMed] [Google Scholar]
- 13.Nijmeh J, Moldobaeva A, Wagner EM. Role of ROS in ischemia-induced lung angiogenesis. Am J Physiol Lung Cell Mol Physiol. 2010;299:L535–L541. doi: 10.1152/ajplung.00002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srisuma S, Biswal SS, Mitzner WA, Gallagher SJ, Mai KH, Wagner EM. Identification of genes promoting angiogenesis in mouse lung by transcriptional profiling. Am J Respir Cell Mol Biol. 2003;29:172–179. doi: 10.1165/rcmb.2002-0276OC. [DOI] [PubMed] [Google Scholar]
- 15.Moldobaeva A, van Rooijen N, Wagner E. Effects of ischemia on lung macrophages. PLoS One. 2011;6:e26716. doi: 10.1371/journal.pone.0026716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagner EM, Sanchez J, McClintock JY, Jenkins J, Moldobaeva A. Inflammation and ischemia-induced lung angiogenesis. Am J Physiol Lung Cell Mol Physiol. 2008;294:L351–L357. doi: 10.1152/ajplung.00369.2007. [DOI] [PubMed] [Google Scholar]
- 17.Sidky YA, Auerbach R. Lymphocyte-induced angiogenesis in tumor-bearing mice. Science. 1976;192:1237–1238. doi: 10.1126/science.5775. [DOI] [PubMed] [Google Scholar]
- 18.Tayade C, Fang Y, Croy BA. A review of gene expression in porcine endometrial lymphocytes, endothelium and trophoblast during pregnancy success and failure. J Reprod Dev. 2007;53:455–463. doi: 10.1262/jrd.18170. [DOI] [PubMed] [Google Scholar]
- 19.D’Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, Pipeling MR, Brower RG, Tudor RM, McDyer JF, et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest. 2009;119:2898–2913. doi: 10.1172/JCI36498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mock JR, Garibaldi BT, Aggarwal NR, Jenkins J, Limjunyawong N, Singer BD, Chau E, Rabold R, Files DC, Sidhaye V, et al. Foxp3 regulatory T cells promote lung epithelial proliferation. Mucosal Immunol. doi: 10.1038/mi.2014.33. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 22.Boehm F, Martin M, Kesselring R, Schiechl G, Geissler EK, Schlitt HJ, Fichtner-Feigl S. Deletion of Foxp3+ regulatory T cells in genetically targeted mice supports development of intestinal inflammation. BMC Gastroenterol. 2012;12:97. doi: 10.1186/1471-230X-12-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 24.Du J, Luan J, Liu H, Daniel TO, Peiper S, Chen TS, Yu Y, Horton LW, Nanney LB, Strieter RM, et al. Potential role for Duffy antigen chemokine–binding protein in angiogenesis and maintenance of homeostasis in response to stress. J Leukoc Biol. 2002;71:141–153. [PMC free article] [PubMed] [Google Scholar]
- 25.Sidky YA, Auerbach R. Lymphocyte-induced angiogenesis: a quantitative and sensitive assay of the graft-vs.-host reaction. J Exp Med. 1975;141:1084–1100. doi: 10.1084/jem.141.5.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McClintock JY, Wagner EM. Role of IL-6 in systemic angiogenesis of the lung. J Appl Physiol. 2005;99:861–866. doi: 10.1152/japplphysiol.00006.2005. [DOI] [PubMed] [Google Scholar]
- 27.Sánchez J, Moldobaeva A, McClintock J, Jenkins J, Wagner E. The role of CXCR2 in systemic neovascularization of the mouse lung. J Appl Physiol. 2007;103:594–599. doi: 10.1152/japplphysiol.00037.2007. [DOI] [PubMed] [Google Scholar]
- 28.Keeley EC, Mehrad B, Strieter RM. Chemokines as mediators of neovascularization. Arterioscler Thromb Vasc Biol. 2008;28:1928–1936. doi: 10.1161/ATVBAHA.108.162925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraaij MD, Savage ND, van der Kooij SW, Koekkoek K, Wang J, van den Berg JM, Ottenhoff TH, Kuijpers TW, Holmdahl R, van Kooten C, et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc Natl Acad Sci USA. 2010;107:17686–17691. doi: 10.1073/pnas.1012016107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chatterjee S, Chapman KE, Fisher AB. Lung ischemia: a model for endothelial mechanotransduction. Cell Biochem Biophys. 2008;52:125–138. doi: 10.1007/s12013-008-9030-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pan Q, Chathery Y, Wu Y, Rathore N, Tong RK, Peale F, Bagri A, Tessier-Lavigne M, Koch AW, Watts RJ. Neuropilin-1 binds to VEGF121 and regulates endothelial cell migration and sprouting. J Biol Chem. 2007;282:24049–24056. doi: 10.1074/jbc.M703554200. [DOI] [PubMed] [Google Scholar]
- 32.Delgoffe GM, Woo SR, Turnis ME, Gravano DM, Guy C, Overacre AE, Bettini ML, Vogel P, Finkelstein D, Bonnevier J, et al. Stability and function of regulatory T cells is maintained by a neuropilin-1–semaphorin-4a axis. Nature. 2013;501:252–256. doi: 10.1038/nature12428. [DOI] [PMC free article] [PubMed] [Google Scholar]