

Abstract
Acute respiratory distress syndrome (ARDS) is a common and often fatal inflammatory lung condition without effective targeted therapies. Regulatory T cells (Tregs) resolve lung inflammation, but mechanisms that enhance Tregs to promote resolution of established damage remain unknown. DNA demethylation at the forkhead box protein 3 (Foxp3) locus and other key Treg loci typify the Treg lineage. To test how dynamic DNA demethylation affects lung injury resolution, we administered the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (DAC) to wild-type (WT) mice beginning 24 hours after intratracheal LPS-induced lung injury. Mice that received DAC exhibited accelerated resolution of their injury. Lung CD4+CD25hiFoxp3+ Tregs from DAC-treated WT mice increased in number and displayed enhanced Foxp3 expression, activation state, suppressive phenotype, and proliferative capacity. Lymphocyte-deficient recombinase activating gene-1–null mice and Treg-depleted (diphtheria toxin-treated Foxp3DTR) mice did not resolve their injury in response to DAC. Adoptive transfer of 2 × 105 DAC-treated, but not vehicle-treated, exogenous Tregs rescued Treg-deficient mice from ongoing lung inflammation. In addition, in WT mice with influenza-induced lung inflammation, DAC rescue treatment facilitated recovery of their injury and promoted an increase in lung Treg number. Thus, DNA methyltransferase inhibition, at least in part, augments Treg number and function to accelerate repair of experimental lung injury. Epigenetic pathways represent novel manipulable targets for the treatment of ARDS.
Keywords: DNA methylation, epigenetics, Foxp3, 5-aza-2′-deoxycytidine, acute lung injury
Clinical Relevance
No targeted therapies promote resolution of acute lung inflammation as occurs in the acute respiratory distress syndrome. We determined that the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine accelerated resolution of experimental lung injury at least in part via a salutary effect on regulatory T cell phenotype and function. Epigenetic manipulation of the regulatory T cell lineage represents a potential therapeutic strategy for acute respiratory distress syndrome and other acute inflammatory disorders.
Acute respiratory distress syndrome (ARDS), an inflammatory condition caused by direct or indirect lung injury, is a common life-threatening disease (1). Despite extensive investigation into initial events that cause ARDS pathology, no targeted therapies promote repair in the damaged lung. Resolution of inflammation after acute lung injury is an active process (2) that required CD4+CD25+Foxp3+ regulatory T cells (Tregs) in a direct lung injury mouse model: intratracheal LPS administration (3). In that model, surviving wild-type (WT) mice spontaneously resolved their injury 7 to 10 days after receiving LPS. Lymphocytes did not determine initial injury severity; however, injury resolution required lymphocytes as evidenced by unremitting LPS-induced lung inflammation in lymphocyte-deficient recombinase activating gene-1–null (Rag-1−/−) mice. Using adoptive transfer experiments, we identified the Treg subpopulation as the active lymphocyte fraction involved in promoting resolution via pro-repair effects on macrophage inflammatory function, neutrophil efferocytosis, epithelial proliferation (4), and limitation of fibroproliferation (5). Tregs also promote repair in other tissue damage models and in clinical scenarios (6, 7). Although Treg transfer may be a viable management strategy for chronic conditions (7), the briskly evolving nature of ARDS makes cell transfer–based therapy impractical.
Tregs comprise a CD4+ lymphocyte subset that attenuates both innate and adaptive immune responses (8). Treg development and function require epigenetic programming—prominently through DNA demethylation—and forkhead box protein 3 (Foxp3) transcription factor expression (9, 10). Constitutive Foxp3 expression is necessary for Treg suppressive activity (11, 12), and epigenetic marks regulate transcription at the Foxp3 locus and the loci of other key Treg genes (9, 13). In mice and humans, distinct regions within Treg gene loci (including Foxp3) display cytosine-phospho-guanine (CpG) methylation patterns that differ between Tregs and CD4+CD25− conventional T cells, with hypomethylation dominating in Tregs (9, 14–18). Foxp3 expression correlates with Treg suppressive function (10, 19, 20). Epigenetic mechanisms may therefore represent potential targets to rapidly expand Treg numbers and enhance their function (21).
DNA methyltransferases (DNMTs) control de novo and maintenance CpG methylation during cell division and development (22). CpG methylation within promoters and transcription factor binding sites represses gene transcription, and DNMT inhibition or knockdown can lead to DNA demethylation. DNMTs determine Foxp3 expression and Treg identity because DNMT silencing or inhibition via siRNA or 5-aza-2′-deoxycytidine (DAC), respectively, lead to Foxp3 expression and Treg phenotype in naive CD4+ non-Treg cells (16, 23–25).
Because of the vital importance DNA demethylation has in Treg biology, we hypothesized that pharmacologic DNMT inhibition would lead to enhanced repair after lung injury via a beneficial effect on Tregs. Our results demonstrate that Treg DNMT inhibition augmented Treg suppressive phenotype and function and accelerated resolution of direct lung injury. Some of the results of these studies have been previously reported in the form of abstracts (26, 27).
Materials and Methods
Mice
C57BL/6 WT and Rag-1−/− mice were purchased from Jackson Laboratory (Bar Harbor, ME). Foxp3DTR mice were a gift from Alexander Rudensky (Sloan-Kettering Institute, New York, NY). Animals were bred and housed in a pathogen-free facility. All animal protocols were approved by the Johns Hopkins Animal Care and Use Committee. Male mice aged 8 to 10 weeks were used.
Preparation of Mice
After anesthesia and tracheal intubation, Escherichia coli O55:B5 LPS (4 mg/kg) (Sigma-Aldrich, St. Louis, MO) or sterile water was injected into the trachea (3). Beginning 24 hours later, DAC (1 mg/kg) (Sigma-Aldrich) or vehicle was administered via daily intraperitoneal injection. Diphtheria toxin (List Biologicals, Campbell, CA) was administered intraperitoneally as described in the online supplement. For influenza experiments, 600 EID50 units of A/PR/8/34 H1N1 influenza virus (Charles River Laboratories, Wilmington, MA) was administered intratracheally. Vehicle or DAC was given as above on Days 3 through 7 after inoculation. After mice were killed, bronchoalveolar lavage (BAL) fluid analysis and lung histology were performed as previously described (3).
Flow Cytometry
Right lungs were enzymatically digested, and a single cell suspension was created. Cells were prepared for FACS analysis (3) with a live-dead discriminator and fluorochrome-conjugated antibodies described in the online supplement. Our Treg gating strategy is shown in Figure E1 in the online supplement. Mean fluorescence intensity was calculated as the geometric mean of the positive population fluorescence.
Lymphocyte Culture
Splenic CD4+CD25+ cells (> 85% Foxp3+) and CD4+CD25− cells were isolated using magnetic bead separation (Miltenyi Biotec, Auburn, CA). Cells were then plated in media (28) with plate-bound anti-CD3, soluble anti-CD28 (eBioscience, San Diego, CA), and recombinant murine IL-2 (Peprotech, Rocky Hill, NJ). Cells were incubated for 48 hours with vehicle or 10 or 100 nM DAC before downstream application.
DNA Methylation
After DNA isolation with an AllPrep DNA/RNA Micro kit (Qiagen, Valencia, CA), global DNA methylation was measured on 100 ng of DNA using an Imprint Methylated DNA Quantification kit (Sigma-Aldrich) and compared with a methylated DNA control.
Lymphocyte Suppression Assay
Tregs cultured as above were incubated with anti-CD3/CD28–coated latex microbeads and CellTrace Violet-pulsed (Invitrogen, Carlsbad, CA) CD4+CD25− effector T cells purified by magnetic bead separation (28). After 72 hours, effector T cell proliferation was assayed by flow cytometry.
Adoptive Transfer
After 48 hours in culture with vehicle or 100 nM DAC as above, 2 × 105 live Tregs were administered via retro-orbital injection to Treg-depleted Foxp3DTR mice 1 hour after intratracheal LPS.
Statistical Analysis
Groups of five to nine mice were used for all experiments (29). In vitro experiments were performed in triplicate and repeated at least three times. Values are reported as mean ± SEM. Differences between groups were compared using two-tailed Mann-Whitney U tests or Student’s t tests with Holm-Sidak correction for multiple comparisons (mean fluorescence intensity data). Multiple group comparisons were performed using one-way ANOVA or one-way ANOVA on ranks. Mortality differences were analyzed with the Mantel-Cox test. Significance was determined at α values <0.05.
Results
DAC Augments Lung Treg Number
We first determined if the DNMT inhibitor DAC could expand lung Treg number under noninjurious circumstances. As a sham condition for lung injury, WT mice received an intratracheal sterile water dose on Day 0. They then received daily intraperitoneal DAC or vehicle on Days 1 through 4. On Day 5 after water, DAC-treated mice displayed a higher lung Treg number than vehicle-treated mice (Figure E2A). Lung Treg Foxp3 expression increased modestly in the treatment group (Figure E2B).
To ensure that the drug itself did not contribute to lung injury, we performed BAL for total protein concentration—a lung permeability marker—as well as histologic examination. BAL protein was low overall and did not change with DAC treatment compared with vehicle control after intratracheal water (Figure E2C). Lung histology (Figure E2D) was normal after 4 days of DAC or vehicle treatment. Neither group lost weight or experienced mortality (data not shown). Collectively, DAC increased lung Treg number and Foxp3 expression in a dose that did not cause pulmonary or overt systemic toxicity.
DAC Accelerates Resolution of Lung Injury
Because Tregs resolve acute lung inflammation and DAC expanded lung Treg number and frequency in our initial experiments, we examined if DAC could enhance resolution of severe intratracheal LPS-induced lung injury. WT mice received DAC or vehicle starting 24 hours after injury with intratracheal LPS. Mice in the vehicle group had sustained weight loss, whereas DAC-treated mice steadily regained weight after initial injury (Figure 1A). Mortality was 28.5% in the vehicle group and 0% in the DAC group (P = 0.029, Mantel-Cox test). Vehicle- and DAC-treated mice had similar early injury as measured by BAL protein, cell count, neutrophil count, and lung histology 2 days after injury (Figures 1B–1E). At Day 5 after LPS, DAC-treated mice, but not vehicle-treated mice, displayed a resolving phenotype with resolving injury parameters (Figures 1B–1E). In summary, DAC did not affect early injury but accelerated resolution in WT mice.
Figure 1.

5-aza-2′-deoxycytidine (DAC) treatment promotes resolution of lung injury in wild-type (WT) mice. (A) Body weight relative to baseline was plotted after injury. (B–D) Bronchoalveolar lavage (BAL) total protein (B), total cell counts (C), and neutrophil counts (D) were determined in WT mice 2 and 5 days after injury with intratracheal (i.t.) LPS. (E) Lung sections 2 and 5 days after injury were stained with hematoxylin and eosin. Original magnification: ×20; ×200 (insets). *P < 0.05; †P < 0.001 (Mann-Whitney U test; n = 8 per group). Values reported are mean ± SEM. N.S., not significant.
DAC Augments Lung Treg Number and Suppressive Phenotype after LPS Injury
Because Tregs resolve lung injury after LPS injury (3), we characterized the Treg response to systemic DNMT inhibition after injury. Lung Treg number and frequency increased significantly at Day 5 after injury in response to DAC treatment (Figure 2A); at 2 days after injury (after one DAC dose), there was no effect on lung Treg number (Figure 2A) or phenotype (data not shown). Foxp3 expression, the master regulator of Treg function, increased 27% in lung Tregs from DAC-treated mice 5 days after LPS injury (Figure 2B). Figure 2C shows the Treg phenotypic response to DAC 5 days after LPS injury. CD44, a Treg activation marker (30), increased (Figure 2C, top panel), as did CD39, an ecto-enzyme that catalyzes ATP hydrolysis and serves as an important Treg suppressive mediator (Figure 2C, middle panel) (31). CTLA-4, a negative signal to other immune cells (32), was only slightly increased (Figure 2C, bottom panel). Consistent with the overall increase in Treg number, the percentage of proliferating (Ki-67+) Tregs increased in DAC-treated mice (Figure 2D). DAC treatment did not affect lung CD4+Foxp3− cell frequency or phenotype (Figure E3). Splenic Treg frequency paralleled the lung findings (Figure E4). Thus, lung Tregs from DNMT inhibitor–treated mice had an augmentation of their number, activation state, suppressive phenotype, and proliferative capacity.
Figure 2.

Lung regulatory T cell (Treg) number, activation state, suppressive phenotype, and proliferative capacity increase with DAC treatment after injury. (A) Lung CD4+CD25hiFoxp3+ cells are shown as number in the right lung, frequency of lung cells, and frequency of CD4+ cells 2 and 5 days after injury in WT mice. (B–D) Foxp3 (B); CD44, CD39, and CTLA-4 (C) expression; and the percentage of Ki-67+ Tregs (D) were determined by fluorescence in lung Tregs 5 days after injury. Accompanying bar graphs show summary mean fluorescence intensity (MFI) (B and C) and the percentage of Ki-67+ Tregs (D). *P < 0.05, **P < 0.01, and †P < 0.001 (Mann-Whitney U test for cell numbers/frequencies [A and D] or t test with Holm-Sidak correction for multiple comparisons [mean fluorescence intensities, B and C]; n = 8 per group). Values reported are mean ± SEM.
The Pro-resolution Effect of DAC Requires Lymphocytes
DNMT inhibition has profound effects on lymphocyte phenotype and function (13) but can affect virtually any cell type. We administered DAC or vehicle on Days 1 through 4 to LPS-injured, lymphocyte-deficient (Rag-1−/−) mice to test the hypothesis that lymphocytes are involved in the proresolution effect of DNMT inhibition observed in WT mice. Five days after injury, a time point when DAC-treated WT mice exhibited a resolving phenotype, Rag-1−/− mice displayed persistent injury and could not be rescued by DAC. They experienced sustained weight loss, elevated BAL protein, cell count, neutrophil count, and severe histologic lung inflammation 5 days after injury (Figure 3). Mortality was 50% in the vehicle group and 44% in the DAC group (P = 0.7, Mantel-Cox test). The nonresolving phenotype in Rag-1−/− mice demonstrated that lymphocytes are required for DAC to exert its proresolution action.
Figure 3.
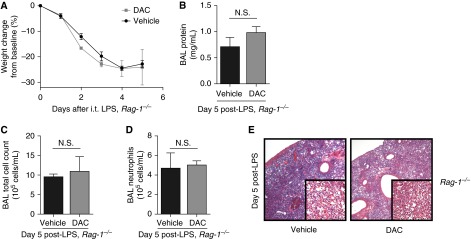
Lymphocyte-deficient (Rag-1−/−) mice do not resolve lung injury in response to DAC treatment. (A) Body weight relative to baseline was plotted after injury. (B–D) BAL total protein (B), total cell counts (C), and neutrophil counts (D) were determined in Rag-1−/− mice 5 days after injury with LPS. (E) Lung sections were stained with hematoxylin and eosin. Original magnification: ×20; ×200 (insets). P > 0.05 (Mann-Whitney U test; n = 5 per group). Values reported are mean ± SEM.
Tregs Are Required for DAC-Enhanced Resolution
We hypothesized that Tregs would be required for DAC to exert its pro-resolution influence. To deplete Tregs, Foxp3DTR mice received diphtheria toxin beginning 2 days before LPS injury and then every other day thereafter. Similar to Rag-1−/− and in contrast to WT mice, DAC treatment in Treg-depleted Foxp3DTR mice did not accelerate lung injury resolution as measured by weight, BAL protein, cell count, neutrophil count, and lung histology 5 days after LPS (Figure 4). Mortality was 33% in both groups. We confirmed lung Treg depletion in diphtheria toxin–treated Foxp3DTR mice compared with diphtheria toxin–treated, LPS-injured WT mice 5 days after injury (Figure E5A). In a pattern similar to WT mice from Figure 1, WT mice treated with diphtheria toxin experienced accelerated lung injury resolution in response to DAC (Figures E5B–E5E). These data confirmed that DAC-enhanced resolution of lung inflammation requires Tregs.
Figure 4.
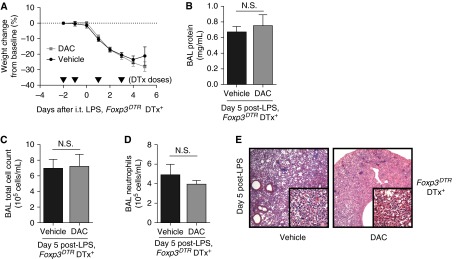
DAC does not promote resolution in Treg-depleted mice. (A) Body weight relative to baseline was plotted beginning with the first diphtheria toxin dose (2 d before injury). Arrowheads represent diphtheria toxin (DTx) doses. (B–D) BAL total protein (B), total cell counts (C), and neutrophil counts (D) were determined in DTx-treated Foxp3DTR mice (Foxp3DTR DTx+) 5 days after injury with LPS. (E) Lung sections were stained with hematoxylin and eosin. Original magnification: ×20; ×200 (insets). P > 0.05 (Mann-Whitney U test; n = 7 per group). Values reported are mean ± SEM.
DAC Augments Foxp3 Expression and In Vitro Suppressive Activity
To determine the effects of DNMT inhibition on CD4+ T cells, we cultured CD4+CD25− cells (conventional T cells) or CD4+CD25+ cells (Tregs, >85% Foxp3+) with anti-CD3/CD28 antibodies and IL-2 with or without DAC. DAC induced Foxp3 expression in conventional T cells and augmented Foxp3 expression in Tregs (Figure 5A). When cultured with DAC, Tregs displayed increased CD44 expression (Figure 5B, top panel). In contrast to the in vivo findings, DAC did not augment Treg CD39 (Figure 5B, middle panel). DAC slightly increased CTLA-4 expression (Figure 5B, bottom panel). A higher percentage of the DAC-treated Tregs expressed Ki-67 compared with vehicle treatment (Figure 5C). After 48 hours in culture, we tested classical Treg suppressive activity in a mixed lymphocyte reaction. DAC-treated Tregs suppressed effector T cell proliferation with greater potency than vehicle-treated Tregs (Figure 5D). Effector T cells did not receive DAC or vehicle treatment. After 48 hours of culture with DAC, global DNA methylation decreased compared with vehicle treatment (Figure 5E). Collectively, DNMT inhibition in culture hypomethylated Tregs and recapitulated some features observed in injured mice (increased Foxp3 expression, activation, and proliferation) but not increased CD39 expression. DNMT inhibitor–treated Tregs exhibited greater suppressive function in a mixed lymphocyte reaction.
Figure 5.

DAC alters CD4+ T cell phenotype and function in vitro. (A) Splenic WT CD4+CD25− conventional T cells (Tconv) or Tregs were cultured with DAC for 48 hours. Foxp3 fluorescence is plotted for shown DAC concentrations (vehicle, 10 and 100 nM). (B and C) CD4+CD25hiFoxp3+ Treg expression of CD44, CD39, and CTLA-4 (B) and the percentage of Ki-67+ Tregs (C) were determined by fluorescence in cultured Tregs treated with vehicle or 100 nM DAC. Accompanying bar graphs show summary MFIs (A and B) and the percentage of Ki-67+ Tregs (C). (D) T effector cells (Teff) were sorted from WT spleens, labeled with CellTrace Violet, and cultured in the presence of anti-CD3/CD28 and varying Treg ratios. Tregs were previously cultured in the presence of vehicle or 100 nM DAC. (E) Global DNA methylation was measured in Tregs cultured with vehicle or 100 nM DAC and compared with methylated control DNA. Experiments were conducted in triplicate and repeated three times. Proliferation data are representative of three independent experiments. *P < 0.05, **P < 0.01, and †P < 0.001 (t test with Holm-Sidak correction for multiple comparisons [MFI, A and B] or Mann-Whitney U test [C–E]). Values reported are mean ± SEM.
Adoptive Transfer of DAC-Treated Tregs Mediates Resolution
Systemic DAC administration likely affects a myriad of cell types, so we sought to determine how ex vivo DAC-treated Tregs specifically influence lung injury resolution. Our previous work suggested that therapeutic adoptive transfer of less than 1 × 106 vehicle-treated Tregs would be an insufficient dose to effect robust resolution (3). Thus, LPS-injured, Treg-depleted Foxp3DTR mice received 2 × 105 WT splenic DAC- or vehicle-treated Tregs via retro-orbital injection 1 hour after intratracheal LPS. Initial injury was similar between the two groups. Seven days after injury, mice that received vehicle-treated Tregs displayed unremitting lung inflammation characterized by sustained weight loss and elevations in BAL cell count, neutrophil count, and protein as well as injured histology. In contrast, lung injury resolution occurred in mice that received DAC-treated Tregs (Figures 6A–6E) with an increase in BAL fluid transforming growth factor (TGF)-β concentration among DAC-treated Treg recipients (Figure 6F). Mortality was 25% in vehicle-treated Treg recipients and 14% in DAC-treated Treg recipients (P = 0.6, Mantel-Cox test). Flow cytometry confirmed successful adoptive transfer and homing to the lung (see Figure E1 for gating). Exogenous lung Tregs (Foxp3-APC+GFP−) increased in mice that received DAC-treated Tregs compared with vehicle-treated Treg recipients (Figure 6G). Among exogenous Tregs, the DAC-treated phenotype resembled the profile observed with systemic DAC administration, with increases in Foxp3, CD44, CD39, and CTLA-4 expression and increases in the percentage of Ki-67+ Tregs (Figures 6H and 6I). In summary, an otherwise subtherapeutic Treg dose—when treated ex vivo with DAC—facilitated lung injury resolution.
Figure 6.

Adoptive transfer (AT) of DAC-treated Tregs rescues the injury phenotype in Treg-depleted mice (Foxp3DTR DTx+). (A) Body weight relative to baseline was plotted after injury. Arrowheads represent DTx doses. (B–D) BAL total protein (B), total cell counts (C), and neutrophil counts (D) were determined in Treg-depleted mice 7 days after injury with LPS. (E) Lung sections 7 days after injury were stained with hematoxylin and eosin. Original magnification: ×20; ×200 (insets). (F) Transforming growth factor (TGF)-β concentrations were measured in BAL fluid. (G) Exogenous (CD4+CD25hiAPC+GFP−) lung Tregs are shown as number in the right lung, frequency of lung cells, and frequency of CD4+ cells 7 days after injury. (H and I) Foxp3 (H); CD44, CD39, and CTLA-4 expression; and the percentage of Ki-67+ Tregs (I) were determined by fluorescence in exogenous lung Tregs 7 days after injury. Accompanying bar graphs show summary MFIs (H and I) and the percentage of Ki-67+ Tregs (I, last panel). *P < 0.05 and †P < 0.001 by Mann-Whitney U test (lung injury parameters, TGF-β levels, and cell numbers/frequencies, A–D, F, G, and I [last panel] or t test with Holm-Sidak correction for multiple comparisons (MFI, H and I [first three panels]) (n = 6 per group). Values reported are mean ± SEM.
DAC Is Beneficial in an Influenza Model
In addition to LPS-induced lung inflammation, we inoculated WT mice intratracheally with a laboratory strain of H1N1 influenza and administered DAC or vehicle on Days 3 through 7 after inoculation. At Day 10 after inoculation, DAC-treated mice had a smaller magnitude of weight loss and decreased BAL protein, cell count, neutrophil count, and histologic injury compared with vehicle-treated mice (Figures 7A–7E). DAC-treated animals displayed an increase in lung Treg number and Foxp3 expression (Figures 7F and 7G). Thus, rescue treatment with DAC had favorable effects on lung injury and Tregs in an infectious model of direct lung inflammation.
Figure 7.

DAC rescue treatment has favorable effects in an influenza (flu) model. (A) Body weight relative to baseline is shown 10 days after inoculation with influenza. (B–D) BAL total protein (B), total cell counts (C), and neutrophil counts (D) were determined in WT mice 10 days after intratracheal inoculation with influenza. (E) Lung sections 10 days after inoculation were stained with hematoxylin and eosin. Original magnification: ×20; ×200 (insets). (F) Lung CD4+CD25hiFoxp3+ cells are shown as number in the right lung, frequency of lung cells, and frequency of CD4+ cells 10 days after inoculation. (G) Foxp3 expression was determined by fluorescence in lung Tregs. The accompanying bar graph shows summary mean fluorescence intensities. *P < 0.05, **P < 0.01, and †P < 0.001 (Mann-Whitney U test; n = 9 per group). Values reported are mean ± SEM.
Discussion
In this report, we establish Foxp3+ regulatory T cell DNMT inhibition as an epigenetic mechanism that accelerates resolution of acute lung injury. The salutary effect of DNMT inhibition required Tregs, and our adoptive transfer experiments confirmed that Tregs, at least in part, mediated the inhibitor’s pro-resolution action. Although systemic DNMT inhibition likely acts on multiple cell types involved in lung injury repair, Tregs exquisitely depend on DNA demethylation and therefore may be predisposed to the effects of a DNMT inhibitor.
ARDS persists as a devastating disease without targeted therapy despite extensive insight into the initial inflammatory injury. Therefore, we focused on events determining resolution of lung inflammation. Resolution of injury requires not only cessation of ongoing pathology but also active repair of damaged tissues (2). In contrast to reports using pretreatment with a histone deacetylase inhibitor (33–35), we show that resolution can be accelerated by a DNMT inhibitor administered after lung injury establishment. DNMT inhibition did not modify early LPS injury in our study, highlighting the conclusion that dynamic changes in DNA methylation patterns likely occur during resolution.
We believe our findings and model have translational relevance to ARDS. Although systemic administration of a DNMT inhibitor represents a potential therapeutic strategy, ex vivo treatment of Tregs with a DNMT inhibitor followed by cell transfer could improve the drug’s therapeutic index. Adoptive transfer of Tregs that have been expanded or modified ex vivo is a validated experimental therapy to promote resolution of acute and chronic inflammatory conditions (7). Diseases with a predictable time course, such as graft-versus-host disease, represent ideal disorders for Treg-based clinical immunotherapy (36–38). However, the timescale required to generate a therapeutic Treg dose is on the order of weeks (39, 40)—too long for a practical ARDS treatment. Epigenetic modification using DNMT inhibition to rapidly augment Treg number and function either in vivo or ex vivo holds translational potential for acute inflammatory diseases, including ARDS. Our model—sterile direct lung injury using the gram-negative bacterial cell wall component LPS—is a well-characterized system to study lung inflammation (41) and recapitulates many ARDS features, including neutrophilic alveolitis, modest mortality, and spontaneous resolution in survivors (3). Our sterile inflammatory model has relevance for many ARDS causes, including aspiration of gastric contents, ventilator-induced lung injury, near drowning, and collateral lung injury associated with treated bacterial infection. Moreover, our data using an influenza model broaden the applicability of epigenetic manipulation as a therapeutic strategy for ARDS.
We chose to use diphtheria toxin–treated WT mice as controls for our experiments using Foxp3DTR mice, which express a normal Foxp3 protein and a diphtheria toxin receptor–green fluorescent protein fusion product. Although mice expressing a Foxp3-green fluorescent protein fusion protein (Foxp3gfp) have facilitated studies of Treg biology (42), these mice exhibit abnormal Treg epigenetic programming due to the altered Foxp3 protein (43). Thus, we selected mice with a normal Foxp3 protein to ensure fidelity of epigenetic responses.
Despite their rarity, Tregs coordinate resolution of direct lung injury via cellular interactions that lead to pro-repair effects on alveolar macrophage responses (3), epithelial regeneration (4), and limitation of fibrocyte-mediated fibrosis (5). However, the specific Treg subset involved in injury resolution remains undefined. Ex vivo treatment of Tregs with DAC followed by adoptive transfer to LPS-injured, Treg-depleted animals increased BAL fluid TGF-β concentration compared with ex vivo vehicle treatment. Tregs require TGF-β to effect repair after LPS injury (3), which indicates that peripherally induced Tregs (pTregs or iTregs) may be the responsible fraction. However, thymus-derived natural Treg (tTreg or nTreg) expansion or recruitment could also contribute to the lymphocyte response after lung injury because adoptive transfer of 1 × 106 splenic nTregs mediates resolution in lymphocyte-deficient mice (3, 5). The CpG methylation signature of Treg gene loci distinguishes committed thymus-derived Tregs and TGF-β–induced Tregs (9). Our study is limited in that we did not directly measure region-specific CpG methylation patterns; however, others have described that Treg induction in the presence of a DNMT inhibitor generates a thymus-derived Treg epigenetic profile (9, 21, 25). Although outside the scope of this report, our group is pursuing lung Treg CpG methylation pattern analysis paired with gene expression profiling to determine the significance of Treg epigenetic signatures after injury and with DNMT inhibitor treatment.
Our flow cytometric molecular phenotyping data suggest several mechanisms involved in DAC-enhanced injury repair. DNMT inhibition modestly augmented Foxp3 expression (18, 24), which leads to increased Treg proliferation and immunoregulatory activity (10, 19, 20). Our in vitro experiments showed that DAC induces Foxp3 expression in non-Tregs. These results lend speculation to the idea that conversion of naive CD4+ T cells into Foxp3+ cells (24) partially underlies the Treg increase observed in DAC-treated mice. However, the functional abilities of these converted cells remain unclear, at least in in vitro systems (44), and our data showed a large difference in Foxp3 expression between DAC-induced Foxp3+ cells and Foxp3+ Tregs cultured with DAC. A recent report demonstrated that deletion of the DNMT adapter protein Uhrf1 within CD4+ cells reduced production of Tregs due to hypomethylation at the locus encoding the cyclin-dependent kinase inhibitor p21 (45). In contrast, our data suggest that demethylation in committed CD4+CD25hiFoxp3+ cells is different than demethylation in undifferentiated CD4+ cells because demethylation in Foxp3+ cells led to proliferation of committed Tregs in vitro, with in vivo data showing nominal effects on CD4+Foxp3− cells. In addition, a study using mice with Dnmt1 deletion restricted to Foxp3+ cells suggested that Tregs require the Dnmt1 isoform to execute their suppressive program (46). Based on our data, we hypothesize that pharmacologic demethylation on an acute timescale leads to different methylation patterns than those accompanying genetic deletion of Dnmt1. We acknowledge that our present study is limited to address this hypothesis in a direct fashion.
Phenotypically, Treg CD39 surface expression increased in response to DAC after lung injury, suggesting extracellular ATP hydrolysis as a mechanism by which Tregs exert their pro-repair program after DNMT inhibition. Damaged cells release ATP into the extracellular milieu, where it exhibits multiple proinflammatory effects; CD39+ Treg-mediated ATP hydrolysis can restore homeostasis to injured tissues (31). DAC did not increase Treg CD39 expression in the absence of inflammation in vitro (Figure 5) or after intratracheal water (data not shown). However, lung Treg CD39 expression increased after adoptive transfer of DAC-treated Tregs to an injured host. These data indicate that environmental conditions in the inflamed lung may combine with the Treg epigenetic state to modulate gene and protein expression profiles. CTLA-4 was only slightly up-regulated with systemic DAC treatment and, although statistically different, does not likely have biological relevance (32). Treg proliferation increased in response to DAC. Indeed, lung Treg number increased significantly in DAC-treated mice compared with vehicle 5 days after LPS injury, and DAC treatment led to robust proliferation after adoptive transfer. Finally, although not directly addressed by our experimental design, more efficient Treg homing to the inflamed lung could also contribute to accelerated repair. Although DAC modestly increases individual Treg markers, the combined effect on Treg function could be synergistic. Collectively, both a qualitative mechanism (augmentation of Treg suppressive function) and a quantitative mechanism (increased Treg proliferation) may underlie the pro-resolution effect of DNMT inhibition.
The present study raises several questions. (1) What other cell types undergo epigenetic changes in response to lung injury? A recent investigation showed that dual therapy with histone acetylation and DNA methylation modifiers can mitigate lung vascular hyperpermeability after systemic LPS injury in mice—an indirect lung injury model—via a beneficial effect on the pulmonary endothelium (47). Dynamic DNA methylation in non-Treg cells after lung injury remains to be explored. (2) What DNA methylation pattern do Tregs that respond to lung injury display? We determined that inhibition of Treg DNA methylation after injury promotes resolution, but it is unclear if induced or thymus-derived Tregs drive lung injury repair. Future experiments with lung Treg CpG methylation pattern analysis will help determine the subset involved in the Treg response to injury. (3) What effect does Treg plasticity have on the lung injury phenotype? Controversy surrounds whether committed Tregs undergo Foxp3 down-regulation or loss and assume T effector function in inflammatory environments (48, 49). Because DNA demethylation stabilizes Tregs, it is possible that pharmacologic DNMT inhibition after injury helps to maintain and stabilize Treg identity and mitigate any effect of Foxp3 loss that might otherwise occur (50). Fate-mapping experiments could help elucidate how Treg plasticity affects lung injury pathology and resolution.
In conclusion, we report a role for regulatory T cell DNMT inhibition in resolution of acute lung injury. DNMT inhibition increases lung Treg frequency and suppressive phenotype and function to promote resolution. Further investigation into the epigenetic marks and mechanisms underlying our findings may lead to therapeutic options for patients with ARDS and other acute inflammatory conditions.
Acknowledgments
Acknowledgments
The authors thank Andre Robinson and James Watkins for their expertise with histologic studies and Raffaello Cimbro and Mark Soloski for assistance in the Johns Hopkins Bayview Flow Cytometry Core.
Footnotes
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health grants R00 HL103973 (F.R.D.'A.) and F32 HL120400 (B.D.S.), by the Baurenschmidt Award of the Eudowood Board (Baltimore, MD) (B.D.S.), and by American Heart Association grant AHA FTF7280014 (N.R.A.).
Author Contributions: B.D.S., J.R.M., N.R.A., B.T.G., V.K.S., K.W.G., S.Y., L.S.K., and F.R.D.'A. participated in the conception, hypotheses delineation, and design of the study. B.D.S., J.R.M., B.T.G., E.C., M.A.F., P.M., and A.T. performed experiments/data acquisition or analysis. B.D.S., J.R.M., N.R.A., B.T.G., S.Y., L.S.K., and F.R.D.'A. wrote the manuscript or provided substantial involvement in its revision.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2014-0327OC on October 8, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 2.Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP. The resolution of inflammation. Nat Rev Immunol. 2013;13:59–66. doi: 10.1038/nri3362. [DOI] [PubMed] [Google Scholar]
- 3.D’Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, Pipeling MR, Brower RG, Tuder RM, McDyer JF, et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest. 2009;119:2898–2913. doi: 10.1172/JCI36498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mock JR, Garibaldi BT, Aggarwal NR, Jenkins J, Limjunyawong N, Singer BD, Chau E, Rabold R, Files DC, Sidhaye V, et al. Foxp3(+) regulatory T cells promote lung epithelial proliferation Mucosal Immunol 2014. May [cited 2014 May 23]. Available from: http://www.nature.com/mi/journal/vaop/ncurrent/full/mi201433a.html [DOI] [PMC free article] [PubMed]
- 5.Garibaldi BT, D’Alessio FR, Mock JR, Files DC, Chau E, Eto Y, Drummond MB, Aggarwal NR, Sidhaye V, King LS. Regulatory T cells reduce acute lung injury fibroproliferation by decreasing fibrocyte recruitment. Am J Respir Cell Mol Biol. 2013;48:35–43. doi: 10.1165/rcmb.2012-0198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, et al. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155:1282–1295. doi: 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singer BD, King LS, D’Alessio FR. Regulatory T cells as immunotherapy. Front Immunol. 2014;5:46. doi: 10.3389/fimmu.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25): breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 9.Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, Osaki M, Tanaka Y, Yamashita R, Nakano N, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity. 2012;37:785–799. doi: 10.1016/j.immuni.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 10.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 11.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 12.Josefowicz SZ, Rudensky A. Control of regulatory T cell lineage commitment and maintenance. Immunity. 2009;30:616–625. doi: 10.1016/j.immuni.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. 2009;9:91–105. doi: 10.1038/nri2487. [DOI] [PubMed] [Google Scholar]
- 14.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang H-D, Bopp T, Schmitt E, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janson PCJ, Winerdal ME, Marits P, Thörn M, Ohlsson R, Winqvist O. FOXP3 promoter demethylation reveals the committed Treg population in humans. PLoS One. 2008;3:e1612. doi: 10.1371/journal.pone.0001612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim H-P, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med. 2007;204:1543–1551. doi: 10.1084/jem.20070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mantel P-Y, Ouaked N, Rückert B, Karagiannidis C, Welz R, Blaser K, Schmidt-Weber CB. Molecular mechanisms underlying FOXP3 induction in human T cells. J Immunol. 2006;176:3593–3602. doi: 10.4049/jimmunol.176.6.3593. [DOI] [PubMed] [Google Scholar]
- 18.Toker A, Engelbert D, Garg G, Polansky JK, Floess S, Miyao T, Baron U, Düber S, Geffers R, Giehr P, et al. Active demethylation of the Foxp3 locus leads to the generation of stable regulatory T cells within the thymus. J Immunol. 2013;190:3180–3188. doi: 10.4049/jimmunol.1203473. [DOI] [PubMed] [Google Scholar]
- 19.Chauhan SK, Saban DR, Lee HK, Dana R. Levels of Foxp3 in regulatory T cells reflect their functional status in transplantation. J Immunol. 2009;182:148–153. doi: 10.4049/jimmunol.182.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CPJ, Pals CEGM, Meerding J, Berkers CR, Barbi J, Gröne A, et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity. 2013;39:259–271. doi: 10.1016/j.immuni.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lal G, Bromberg JS. Epigenetic mechanisms of regulation of Foxp3 expression. Blood. 2009;114:3727–3735. doi: 10.1182/blood-2009-05-219584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leonhardt H, Page AW, Weier HU, Bestor TH. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell. 1992;71:865–873. doi: 10.1016/0092-8674(92)90561-p. [DOI] [PubMed] [Google Scholar]
- 23.Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, Reid SP, Levy DE, Bromberg JS. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. 2009;182:259–273. doi: 10.4049/jimmunol.182.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moon C, Kim SH, Park KS, Choi BK, Lee HS, Park JB, Choi GS, Kwan JH, Joh JW, Kim SJ. Use of epigenetic modification to induce FOXP3 expression in naïve T cells. Transplant Proc. 2009;41:1848–1854. doi: 10.1016/j.transproceed.2009.02.101. [DOI] [PubMed] [Google Scholar]
- 25.Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, Olek S, Hamann A, von Boehmer H, Huehn J. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38:1654–1663. doi: 10.1002/eji.200838105. [DOI] [PubMed] [Google Scholar]
- 26.Singer BD, Mock JR, Garibaldi BT, Chau E, Aggarwal NR, Sidhaye VK, King LS, D’Alessio FR. Pharmacologic epigenetic manipulation rescues experimental lung injury and promotes lung regulatory T cell number and suppressive phenotype [abstract] Am J Respir Crit Care Med. 2013;187:A1213. [Google Scholar]
- 27.Singer BD, Mock JR, Giibs KW, Garibaldi BT, Aggarwal NR, Sidhaye VK, King LS, D’Alessio FR. DNA demethylation promotes regulatory T cell-mediated resolution of acute lung injury [abstract] Am J Respir Crit Care Med. 2014;189:A5575. [Google Scholar]
- 28.Collison LW, Vignali DAA. In vitro Treg suppression assays. Methods Mol Biol. 2011;707:21–37. doi: 10.1007/978-1-61737-979-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Festing MFW, Altman DG. Guidelines for the design and statistical analysis of experiments using laboratory animals. ILAR J. 2002;43:244–258. doi: 10.1093/ilar.43.4.244. [DOI] [PubMed] [Google Scholar]
- 30.Bollyky PL, Falk BA, Long SA, Preisinger A, Braun KR, Wu RP, Evanko SP, Buckner JH, Wight TN, Nepom GT. CD44 costimulation promotes FoxP3+ regulatory T cell persistence and function via production of IL-2, IL-10, and TGF-beta. J Immunol. 2009;183:2232–2241. doi: 10.4049/jimmunol.0900191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, Höpner S, Centonze D, Bernardi G, Dell’Acqua ML, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 32.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 33.Ni Y-F, Wang J, Yan X-L, Tian F, Zhao J-B, Wang Y-J, Jiang T. Histone deacetylase inhibitor, butyrate, attenuates lipopolysaccharide-induced acute lung injury in mice. Respir Res. 2010;11:33. doi: 10.1186/1465-9921-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kochanek AR, Fukudome EY, Li Y, Smith EJ, Liu B, Velmahos GC, deMoya M, King D, Alam HB. Histone deacetylase inhibitor treatment attenuates MAP kinase pathway activation and pulmonary inflammation following hemorrhagic shock in a rodent model. J Surg Res. 2012;176:185–194. doi: 10.1016/j.jss.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Liu B, Zhao H, Sailhamer EA, Fukudome EY, Zhang X, Kheirbek T, Finkelstein RA, Velmahos GC, deMoya M, et al. Protective effect of suberoylanilide hydroxamic acid against LPS-induced septic shock in rodents. Shock. 2009;32:517–523. doi: 10.1097/SHK.0b013e3181a44c79. [DOI] [PubMed] [Google Scholar]
- 36.Trzonkowski P, Bieniaszewska M, Juścińska J, Dobyszuk A, Krzystyniak A, Marek N, Myśliwska J, Hellmann A. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol. 2009;133:22–26. doi: 10.1016/j.clim.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 37.Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, Defor T, Levine BL, June CH, Rubinstein P, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117:1061–1070. doi: 10.1182/blood-2010-07-293795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, Del Papa B, Zei T, Ostini RI, Cecchini D, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117:3921–3928. doi: 10.1182/blood-2010-10-311894. [DOI] [PubMed] [Google Scholar]
- 39.Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, McKenna DH, Bromberg JS, Levine BL, Riley JL, et al. Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Sci Transl Med. 2011;3:83ra41. doi: 10.1126/scitranslmed.3001809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoffmann P, Eder R, Kunz-Schughart LA, Andreesen R, Edinger M. Large-scale in vitro expansion of polyclonal human CD4(+)CD25high regulatory T cells. Blood. 2004;104:895–903. doi: 10.1182/blood-2004-01-0086. [DOI] [PubMed] [Google Scholar]
- 41.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L379–L399. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 43.Bettini ML, Pan F, Bettini M, Finkelstein D, Rehg JE, Floess S, Bell BD, Ziegler SF, Huehn J, Pardoll DM, et al. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity. 2012;36:717–730. doi: 10.1016/j.immuni.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kehrmann J, Tatura R, Zeschnigk M, Probst-Kepper M, Geffers R, Steinmann J, Buer J. Impact of 5-aza-2′-deoxycytidine and epigallocatechin-3-gallate for induction of human regulatory T cells. Immunology. 2014;142:384–395. doi: 10.1111/imm.12261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Obata Y, Furusawa Y, Endo TA, Sharif J, Takahashi D, Atarashi K, Nakayama M, Onawa S, Fujimura Y, Takahashi M, et al. The epigenetic regulator Uhrf1 facilitates the proliferation and maturation of colonic regulatory T cells. Nat Immunol. 2014;15:571–579. doi: 10.1038/ni.2886. [DOI] [PubMed] [Google Scholar]
- 46.Wang L, Liu Y, Beier UH, Han R, Bhatti TR, Akimova T, Hancock WW. Foxp3+ T-regulatory cells require DNA methyltransferase 1 expression to prevent development of lethal autoimmunity. Blood. 2013;121:3631–3639. doi: 10.1182/blood-2012-08-451765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thangavel J, Malik AB, Elias HK, Rajasingh S, Simpson AD, Sundivakkam PK, Vogel SM, Xuan Y-T, Dawn B, Rajasingh J. Combinatorial therapy with acetylation and methylation modifiers attenuates lung vascular hyperpermeability in endotoxemia-induced mouse inflammatory lung injury. Am J Pathol. 2014;184:2237–2249. doi: 10.1016/j.ajpath.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, Huehn J, Hori S. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. 2012;36:262–275. doi: 10.1016/j.immuni.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 49.Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. 2014;20:62–68. doi: 10.1038/nm.3432. [DOI] [PubMed] [Google Scholar]
- 50.Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, Fehling HJ, Bluestone JA. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity. 2013;39:949–962. doi: 10.1016/j.immuni.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]