

Summary
Turnover and exchange of nucleosomal histones and their variants, a process long believed to be static in post-replicative cells, remains largely unexplored in brain. Here, we describe a novel mechanistic role for HIRA (histone cell cycle regulator) and proteasomal degradation associated histone dynamics in the regulation of activity-dependent transcription, synaptic connectivity and behavior. We uncover a dramatic developmental profile of nucleosome occupancy across the lifespan of both rodents and humans, with the histone variant H3.3 accumulating to near saturating levels throughout the neuronal genome by mid-adolescence. Despite such accumulation, H3.3 containing nucleosomes remain highly dynamic–in a modification independent manner–to control neuronal- and glial-specific gene expression patterns throughout life. Manipulating H3.3 dynamics in both embryonic and adult neurons confirmed its essential role in neuronal plasticity and cognition. Our findings establish histone turnover as a critical, and previously undocumented, regulator of cell-type specific transcription and plasticity in mammalian brain.
Introduction
Mammalian transcription is a highly complex process, which controls fundamental aspects of cell diversity and organismal adaptation. Neurons, in particular, exhibit remarkable specialization and plasticity, which is mediated, in part, by activity-dependent changes in gene expression (Greer and Greenberg, 2008). One method to control activity-dependent gene expression is by modulating the accessibility of genes to the transcriptional machinery via alterations in chromatin structure, the mechanisms of which are poorly understood (Borrelli et al., 2008; Maze et al., 2013). Nucleosomal histones have long been considered highly stable proteins that exhibit slow turnover kinetics with predicted half lives of months to years in post-mitotic cells (Commerford et al., 1982). Previous studies in the central nervous system (CNS) have therefore emphasized histone post-translational modifications (PTMs) and chromatin remodeling as the primary mechanisms of chromatin structural variation regulating transcriptional and behavioral plasticity. However, recent analyses of histone dynamics in lower eukaryotes have challenged the notion of nucleosomal stability by demonstrating rapid incorporation of newly synthesized histone variants within active regions of the genome (Deal et al., 2010; Dion et al., 2007). These findings raise the possibility that rapid turnover of subpopulations of nucleosomes may directly influence patterns of gene expression in mammalian brain.
Both historical (Commerford et al., 1982) and recent (Savas et al., 2012; Toyama et al., 2013) analyses of protein turnover in rodent brain indicate that canonical histones (e.g., H3.1, H3.2), whose incorporation into chromatin is replication-dependent (RD), are remarkably stable throughout the lifetime of an animal. Variant histones, such as H3.3, can be incorporated into chromatin in a replication-independent (RI) manner, and might therefore exhibit more rapid turnover rates relative to their canonical counterparts. Indeed, recent studies have suggested potential roles for histone variant ‘exchange’ during periods of activity-dependent gene expression in neurons (Michod et al., 2012; Santoro and Dulac, 2012; Zovkic et al., 2014). For example, in neuronal cultures, the H3.3-specific chaperone Daxx was found to be dephosphorylated in a calcium-dependent manner and is required for activity-dependent H3.3 deposition at a subset of immediate early gene (IEG) promoters (Michod et al., 2012). This exciting finding implicates histone variant exchange as a potential mechanism of gene regulation in response to neural activity. However, the degree to which histone turnover itself contributes to lifelong transcriptional plasticity, as well as the physiological impact of such events in vivo, has yet to be described.
Here, we demonstrate that H3.3 dramatically accumulates in neuronal and glial chromatin with age and remains highly dynamic throughout life to control cell-type specific gene expression programs and physiological plasticity. We also find that the majority of activity-dependent H3.3 deposition is mediated by the histone cell cycle regulator Hira, rather than Daxx/Atrx, and that active proteasomal degradation is required for efficient nucleosomal exchange. Moreover, manipulations of H3.3 in neurons, which stall its incorporation and eviction from chromatin, reveal histone turnover as a critical mediator of neuronal activity-dependent gene expression, synaptic connectivity and cognition.
Results
H3.3 accumulates with age but remains dynamic in neuronal chromatin throughout life
To assess the prevailing dogma that histones are highly stable and non-dynamic proteins in post-replicative cells, we began with a focused analysis of histone variant expression and incorporation into neuronal chromatin. Based on preliminary evidence suggesting that H3.3 accumulates with age in rodent brain (Pina and Suau, 1987), we began by testing the relative abundance of chromatin associated H3.1, H3.2 and H3.3 proteins in mouse neurons (NeuN+) isolated by fluorescence activated cell sorting (FACS) (validations, Figures S1A–D). Consistent with data from proliferating cells, H3.3 was found to constitute only a small percentage of the total H3 pool in embryonic (E16.5) neuronal chromatin, with canonical H3 proteins (H3.1 and H3.2) representing the dominant isoforms. However, by old age (2 years in mice), H3.3 levels approached >94% of the total H3 pool (Figure 1A). These results indicate that H3.3 accumulates with age to become the dominant, if not exclusively expressed, H3 isoform occupying the adult neuronal genome. These findings contrast with work in dividing cells, where H3.3 has been proposed to ‘barcode’ regions of active chromatin to promote locus-specific gene expression patterns (Hake and Allis, 2006).
Figure 1. H3.3 accumulates with age but remains dynamic in the CNS.

(A) LC-MS/MS quantified H3.1 and H3.2 vs. H3.3 protein expression in NeuN+ mouse chromatin with age. (frontal cortex, n = 3 biological replicates/age). (B) SILAC LC-MS/MS analysis of H3.1/2 vs. H3.3 in NeuN+ mouse chromatin from multiple brain structures after two or 4 wks of feeding on a heavy lysine (6 Da) diet. (n = 3–6 biological replicates/brain region). (C) LC-MS/MS quantified H3.1/2 vs. H3.3 protein expression in fetal (n = 5) vs. adult (n = 30) human postmortem brain. (D) The rate of change of C14 levels in Histone H3.3 pools from brain tissue depends on the rates of histone H3.3 production and degradation. We model production rates as proportional to the current atmospheric 14C/12C levels, and degradation rates as proportional to current cellular 14C/12C levels. (E) F14C (14C/12C) bomb pulse accelerator mass spectrometry of HPLC purified H3.3 protein from postmortem human brain (n = 18). Human H3.3 F14C levels (red squares) are plotted against the atmospheric record for 14C (black line) as a function of time (each n is plotted along the x-axis by respective dates of birth). The range of death dates is indicated (blue line). ‘Error’ bars represent one σ deviation above the average for each sample. Modern F14C correction assessments were made using H3.3 protein purified from fetal human brain collected in 2012 (mid-gestation). The atmospheric record of F14C was constructed from published tree ring and CO2 data of Northern Hemisphere growing season averages (see Supplemental Information for references). (F and G) Mathematical descriptions of 14C/12C dynamics in cerebellar H3.3 pools were used to predict 14C/12C ratios, given various assumptions with regards to the nature of H3.3 turnover in healthy post-mitotic cells, and then compared to observed data. Data represented as means ± SEM, unless otherwise noted. See also Figure S1 and Supplemental Spreadsheet 1.
We next sought to assess whether accumulated histones continue to exhibit active turnover in neuronal chromatin during early postnatal development and in adulthood. To do so, we employed SILAC (Stable Isotope Labeling of Amino acids in Cell culture) in mice, where animals were fed a special diet containing heavy labeled lysines beginning at P21 (post-weaning) for a period of 2–4 weeks—a period during which H3.3 levels remain stable in neuronal chromatin (Figure 1A). Neurons from frontal cortex, hippocampus and cerebellum were FACS-purified and analyzed for histone incorporation. Consistent with its role as a RI histone, newly made H3.3, but not H3.1 or H3.2, displayed active incorporation in adult neuronal chromatin with >30% of the total H3.3 pool being replaced in all brain regions examined throughout the course of four weeks (Figures 1B and S1E). Since the loss of ‘old’ and replacement by ‘new’ H3 proteins indicates partial to complete nucleosomal exchange, our data indicate that histones are constitutively turned over in neuronal chromatin throughout life.
We next examined the dynamics and relative abundance of H3 proteins in chromatin from postmortem human brain. Consistent with data from mouse neurons, H3.3 was found to constitute ~31% of the total H3 pool in fetal brain; however, H3.3 gradually accumulates over the first approximate decade of life, with its levels then remaining stable at >93% of the total H3 pool in individuals ranging from 14 to 72 years old (Figure 1C). To measure histone turnover in the human CNS, we employed 14C/12C bomb pulse dating of high-performance liquid chromatography (HPLC) purified H3.3 from postmortem human brain tissue (Figure S1F) via accelerator mass spectrometry. This technique works on the principle that high levels of radioactive carbon (14C) released into the atmosphere during periods of open-air nuclear bomb testing permit direct assessments of organic synthesis and decay in human tissues. To assess histone dynamics in human brain, multiple distinct variables, all of which may contribute to observed H3.3 14C/12C ratios at the time of death, can be mathematically defined (Figure 1D). Following bomb pulse dating of purified H3.3 proteins from human brain, 14C/12C ratios were normalized against modern 14C/12C levels determined by analysis of fetal brain samples and plotted by date of birth against the 14C/12C atmospheric record (Figure 1E). Deviations from the atmospheric record at the subjects’ time of death indicate H3.3 turnover events throughout life.
To first determine if cell turnover alone could account for the H3.3 14C/12C levels observed in pre-bomb subjects, we plotted actual vs. predicted 14C/12C ratios based on constraints defined solely by DNA 14C/12C measurements (i.e., cell turnover). Accounting for DNA synthesis/decay (i.e., neurogenesis/neurodegeneration) alone severely underestimated 14C/12C levels (Figure 1F, black X’s) indicating that additional turnover processes must contribute to observed ratios. Indeed, considering both cell turnover and H3.3 dynamics was able to fully account for all 14C/12C levels observed in pre-bomb subjects (Figure 1F, red X’s). These data demonstrate that slow constitutive turnover of H3.3 occurs in human brain throughout life, even following periods during which H3.3 has accumulated to levels >93% of the total H3 pool.
Next, to investigate H3.3 turnover in post-bomb subjects, all of whom were exposed to elevated levels of 14C during early periods of neurodevelopment, we again assessed the contribution of cell turnover alone to the 14C/12C ratios observed. Similar to analyses of pre-bomb subjects, post-bomb 14C/12C levels could not be fully explained using constraints of cell turnover alone (Figure 1G, black X’s). Given that H3.3 protein levels in neuronal chromatin approximately triple in abundance during the first ~14 years of life, we examined whether accounting for slow rates of histone turnover derived from pre-bomb subjects, as well as histone accumulation (described in Figure 1C), could better describe the data. Doing so reduced estimates for 14C/12C ratios constrained to cell turnover alone, however, a single, time-invariant rate model remained inadequate to fully explain the data (Figure 1G, green X’s). We then allowed for histone turnover to be fast in the short-term (i.e., rapid dynamics, khd ~1 yr−1; during the histone accumulation phase), and slow in the long-term (i.e., constitutive dynamics, khd ~0.001 yr−1) following a calculated ‘switching’ age. This enabled our data from post-bomb subjects to be fully explained (Figure 1G, red X’s). Consistent with histone accumulation patterns during early brain development, 14C/12C measurements estimated the fast-to-slow switching age to be ~13.9 ± 7.5 years. In total, these data provide the first evidence for constitutive histone dynamics in human brain throughout life and indicate that such turnover rates in brain are differentially regulated during early periods of neurodevelopment vs. those observed in adulthood. Such differences may indicate varying needs for histone turnover throughout different stages of brain development.
Modeling embryonic H3.3 nucleosomal dynamics in neuronal culture
We next examined whether embryonic nucleosomal exchange could be effectively modeled in cultured embryonic neurons, a system extensively employed to examine various aspects of neurodevelopment and activity-dependent transcription. To ensure that the primary culture system accurately reflects the embryonic state in vivo, we compared gene expression profiles of cultured neurons [eight days in vitro (DIV)] to freshly dissected embryonic cortical tissues [embryonic day 16.5 (E16.5)]. RNA-seq analysis confirmed highly significant, positive correlations between gene expression profiles from these samples (Figure 2A). We next sought to verify that H3.3 genomic enrichment patterns in cultured neurons mimic those observed in freshly dissected tissues. To this end, chromatin immunoprecipitation coupled to massively parallel sequencing (ChIP-seq) was performed comparing H3.3 genomic enrichment in cultured neurons (DIV 8) vs. FACS purified E16.5 NeuN+ cells using an extensively validated in-house H3.3 antibody (Figure S2A–E). Basal H3.3 peak enrichment profiles between cultured and embryonic neurons were found to strongly correlate with high significance (Figure 2B) indicating the validity of this system in modeling embryonic histone dynamics.
Figure 2. ‘De-coupling’ of histone turnover from PTMs in neuronal chromatin.
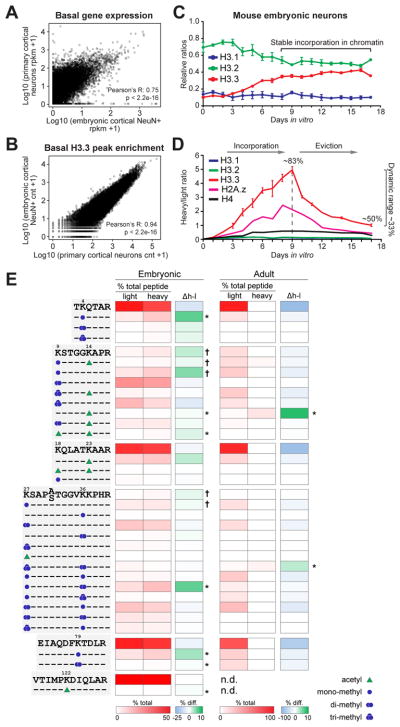
(A) Correlation of basal gene expression profiles from primary cortical neurons (DIV 8) vs. E16.5 cortical tissues. (B) Comparison of basal H3.3 peak enrichment profiles via ChIP-seq from primary neurons (DIV 8) vs. NeuN+ E16.5 cortical neurons. (C) LC-MS/MS quantified H3.1/2 vs. H3.3 protein expression in mouse embryonic neuronal chromatin from DIV 0–17 (n = 4 biological replicates). (D) SILAC LC-MS/MS analysis of H3.1, H3.2, H3.3, H4 vs. H2A.Z embryonic chromatin incorporation and eviction in primary cortical neurons. Percentages reflect H3.3 peptide labeling by SILAC indicating constitutive H3.3 turnover in embryonic neurons (n = 4 biological replicates). (E) Heatmap depicting NanoLC-MS/MS data for relative enrichment values of (un)modified histone ‘H3’ N-terminal tail peptides (non-SILAC/light vs. SILAC/heavy) in embryonic vs. adult neurons. Absolute values (% total peptide) for each (un)labeled peptide (unmodified vs. modified) are provided. Δh–l indicates % enrichment differences between SILAC vs. non-SILAC labeled peptides (n = 3 biological replicates, embryonic and adult). Heatmap data represented as means, *P ≤ 0.05 (2-tail), † P ≤ 0.05 (1-tail).
n values for sequencing experiments are provided in the Extended Experimental Procedures. See also Figure S2.
We next examined the relative abundance of chromatin associated H3 proteins in primary neurons over the course of 17 DIV, during which time primary neurons undergo rapid and sustained changes in morphology and synapse formation. Directly following dissociation and plating, H3.3 was expressed as only a small fraction of the total H3 pool (<20%), however, over the course of the first eight DIV, H3.3 accumulated to levels similar to those observed at E16.5 (Figure 1A). H3.3 was found to reach sustained levels in neuronal chromatin over the remaining nine DIV (Figure 2C) providing a validated time window in which to perform subsequent analyses related to H3.3 dynamics in ‘embryonic’ neurons. Next, to model H3.3 turnover in cultured neurons, SILAC was employed in a manner similar to that described in vivo. SILAC labeling revealed high levels of chromatin incorporation for newly made H3.3 (~83% of the total H3.3 pool was labeled during the first nine DIV), but not H3.1 or H3.2. Equally rapid rates of H3.3 eviction were observed (Figures 2D and S2F). Consistent with these findings, newly synthesized H4, which exists as a single protein species, displayed lower levels of incorporation/eviction (H4 eviction t1/2 = ~7.7 days) in comparison to H3.3 (H3.3 eviction t1/2 = ~3.5 days). H4 is present in all H3 containing nucleosomes and therefore turns over at a rate equal to that of the total H3 pool. Although high levels of incorporation of newly made H3.3 during the first eight DIV may be explained, in part, by H3.3 accumulation, high rates of H3.3 eviction from DIV 9–17 indicate that H3.3 turnover in nucleosomes remains dynamic even during periods in which total H3.3 levels in chromatin remain stable. Indeed, labeling of H3.3 plateaued with ~50% of the H3.3 pool labeled by DIV 17, indicating that ~33% of the total H3.3 pool remains constitutively dynamic in embryonic neurons, a turnover rate that is significantly more rapid than that observed in adult mouse cortical neurons (i.e., ~13.4 days). These findings are consistent with analyses of H3.3 dynamics in human brain, which indicate varying rates of H3.3 exchange between early periods of neurodevelopment and adulthood. Of note, the histone variant H2A.Z was found to turn over at a similar rate to that of H3.3 in primary neurons (t1/2 = ~3.5 days), however, significantly lower labeling of H2A.Z was observed indicating that global pools of dynamic H2A.Z are less abundant than those of H3.3.
To understand the relationship between H3.3 genomic occupancy and gene expression in brain, we performed ChIP-seq for H3.1/2 and H3.3 in embryonic vs. adult neurons, followed by correlational analyses between H3 variant protein enrichment and transcript abundance (RNA-seq). Consistent with literature in dividing cells (Goldberg et al., 2010), embryonic H3.1/2 was found to enrich primarily at intergenic regions of the genome, with minimal localization observed at genic loci (Spearman’s rank correlation, H3.1/2 vs. gene expression: promoter = −0.40, gene body = −0.51, p < 2.2e-16). In contrast, embryonic H3.3 was found to enrich most significantly at gene bodies and promoters of active genes (Spearman’s rank correlation, H3.3 vs. gene expression: promoter = 0.62, gene body = 0.50, p < 2.2e-16). Interestingly, as H3.3 accumulates to become the dominant H3 isoform in adult neurons, associations between H3.3 enrichment and active gene expression become significantly reduced (Spearman’s rank correlation, H3.3 vs. gene expression: promoter = 0.46, gene body = 0.18, p < 2.2e-16). These data indicate that H3.3’s purported function as an active histone ‘mark’ requires revision with respect to adult neurons.
Life-long H3.3 turnover in neurons occurs independently from ‘active’ PTMs
We next evaluated PTMs associated with dynamic H3.3 using NanoLC-MS/MS of SILAC labeled histones purified from either ‘embryonic’ or NeuN+ adult neurons. Specifically, we identified combinatorial modifications enriched on dynamic (heavy, H3.3) vs. non-dynamic (light, H3.1/2 or ‘static’ H3.3) chromatin associated H3 proteins. In agreement with H3.3’s localized enrichment at active genes in proliferating cells, dynamic embryonic H3.3 significantly enriched for active PTMs, such as H3K4me1, H3K9acK14ac and H3K79me2 (Figure 2E). In adult neurons, however, associations between active PTMs and dynamic H3.3 were all but lost. These data indicate that H3.3 turnover in the adult CNS may be ‘de-coupled’ from active histone PTM enrichment.
Neuronal activity promotes histone turnover
To better understand the functional significance of histone turnover in the CNS, we examined H3.3 gene (H3f3a and H3f3b) expression in response to neuronal stimulation. We found that H3f3b, but not H3f3a, was transcriptionally induced by numerous activating stimuli (Figure 3A). To assess whether membrane depolarization in adult neurons in vivo could similarly promote increased expression of H3.3 transcripts, we performed optogenetic stimulations of medial prefrontal cortex neurons. qPCR analysis of H3f3a and H3f3b from stimulated tissues yielded similar increases in expression to those observed in culture (Figure S3), while no expression changes in H3.1 or H3.2 transcripts were observed following stimulation in either cultured or adult neurons.
Figure 3. Neuronal activity promotes histone turnover.

(A) qPCR analysis of H3f3a and H3f3b mRNA expression in mouse embryonic neurons following 2 hrs of stimulation (n = 3–4 biological replicates/group). (B) Western blot analysis of H3f3b (HA), total H3, H3.1/2, H3.3 and H3S10p (control for stimulation) levels in H3.3-HA tagged mouse embryonic neuronal chromatin −/+ KCl stimulation (5 hrs) (n = 3–4 biological replicates/group). (C) LC-MS/MS SILAC analysis of H3.1/2 vs. H3.3 chromatin incorporation (top) and eviction (bottom) −/+ KCl stimulation (5 hrs) in mouse embryonic neurons (n = 4–5 biological replicates/group). (D) (G) qPCR analysis of H3f3a and H3f3b mRNA expression in frontal cortex (FC), hippocampus (Hipp.), striatum (Str.) and cerebellum (Cer.) of normally housed vs. EE mice (4 wks) (n = 9–10 animals/group). (E) SILAC LC-MS/MS analysis of H3.1/2 vs. H3.3 in NeuN+ (hippocampal) mouse chromatin from normally housed vs. EE mice (n = 4–5 biological replicates/group). Data represented as means ± SEM. *P ≤ 0.05, ***P ≤ 0.001. See also Figure S3.
Since H3f3b (the least abundant of the two transcripts) expression was observed to be specifically increased in response to KCl-mediated depolarization, we next examined whether H3f3b protein incorporation into chromatin was similarly affected. H3f3b protein was selectively examined in neuronal chromatin isolated from H3.3-HA tagged mice, in which the H3f3b, but not H3f3a, locus is genetically tagged. H3.3-HA, but not total H3.1/2 or H3.3, levels were increased in expression by KCl in neuronal chromatin (Figure 3B), consistent with mRNA analyses. These results suggest that increased H3f3b expression reflects an increased rate of H3.3 turnover. To test this directly, we employed SILAC coupled to KCl depolarization. Consistent with our hypothesis, H3.3, but not H3.1 or H3.2, turnover was accelerated by cellular stimulation (Figure 3C). Analysis of KCl-induced nucleosomal eviction rates similarly revealed a considerable reduction in H3.3 half-life in response to stimulation [i.e., 0.7 days (KCl) vs. 3.5 days (controls)].
To extend our in vitro findings to a physiologically relevant model of behavioral plasticity, we subjected mice to chronic periods of environmental enrichment (EE), a paradigm previously demonstrated to enhance synaptic plasticity and learning/memory (Fischer et al., 2007). After four weeks of EE vs. normal housing, tissue was collected from multiple brain regions and subjected to mRNA analysis. Similar to findings in stimulated embryonic neurons, H3f3b, but not H3f3a, was significantly upregulated in the hippocampus of EE mice, with no effects seen in other brain regions (Figure 3D). We next examined whether incorporation of newly made H3.3 protein was similarly increased in adult neuronal chromatin following EE. To do so, mice were fed on a SILAC diet and were exposed to either EE or normal housing for 4-weeks. Following EE, FACS purified hippocampal neurons were examined via LC-MS/MS. Consistent with mRNA findings, a significant increase in H3.3 incorporation was observed (Figure 3E).
H3.3 turnover in neurons is Hira-dependent and requires active proteasomal degradation
To better understand the molecular mechanisms contributing to activity-dependent H3.3 turnover in neurons, we next examined whether H3.3 interactions with known chaperone proteins are altered by neuronal depolarization. Following cellular depolarization, neurons from H3.3-HA mice were subjected to nuclear isolation and ammonium sulfate precipitation. H3.3-HA was immunoprecipitated and probed for chaperone binding. Consistent with the literature (Goldberg et al., 2010), H3.3 was found to interact with Daxx, Atrx and Hira. Although we observed no differences in protein expression among the chaperones following membrane depolarization, a selective increase in binding of H3.3 to Hira was observed (Figure 4A). To assess the contribution of these interactions to activity-dependent histone turnover in neuronal chromatin, we utilized lentiviral delivery of shRNAs to efficiently knockdown Daxx or Hira in neurons cultured in SILAC media (Figure 4B, insert), and applied KCl depolarization. Following stimulation, cells were examined via LC-MS/MS for incorporation of newly made H3.3 into neuronal chromatin. Consistent with previous results, scrambled controls responded to depolarization with an enhanced rate of H3.3 incorporation. Although Daxx knockdown had no effect on ‘bulk’ H3.3 turnover, either at basal state or following KCl stimulation, knockdown of Hira did result in significant deficits in H3.3 incorporation, with the most dramatic effects observed following cellular depolarization (Figure 4B).
Figure 4. Neuronal histone dynamics are Hira-dependent and require active proteasomal degradation.

(A) Immunoprecipitations of free H3.3-HA from embryonic neuronal soluble nuclear fractions following depolarization. IgG controls indicate selective immunoprecipitations of H3.3 interacting proteins. (B) LC-MS/MS SILAC analysis of H3.1/2 vs. H3.3 chromatin incorporation −/+ KCl stimulation (5 hrs) in mouse embryonic neurons transduced with one of three lentiviruses (scramble, shRNA-Daxx or shRNA-Hira) (n = 3–4 biological replicates/group). Insert: western blot analysis indicating knockdown of targeted proteins vs. a scrambled control. (C) H3.3 polyubiquitylation in response to KCl depolarization in embryonic neuronal chromatin. Primary neurons were transduced with lenti-Flag-Ub, lenti-H3.3-HA or both, and Flag-tagged ubiquitin was immunoprecipitated from fractionated neuronal chromatin followed by Western blot analysis for ubiquitin (Flag) and ubiquitylated H3.3 (HA). (D) LC-MS/MS SILAC analysis of H3.3 chromatin incorporation (beginning at DIV 5 of SILAC labeling) in mouse embryonic neurons following treatment with DMSO (control) or MG132 (proteasome inhibitor). Cells were analyzed at DIV 0, 1, 2 and 3 post-treatment (n = 3 biological replicates/group). (E) LC-MS/MS SILAC analysis of H3.3 chromatin eviction (beginning at DIV 9 of SILAC labeling) in mouse embryonic neurons following treatment with DMSO or MG132 (n = 3–5 biological replicates/group). Data represented as means ± SEM. *P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001. See also Figure S4.
Although our data indicate that Hira is responsible for H3.3 incorporation in neuronal chromatin during periods of heightened cellular activity, regulation of neuronal nucleosomal eviction remained unclear. Given that H3.3 transcripts and protein are upregulated in response to cellular activity, we sought to investigate whether active degradation of evicted histones is required to allow for H3.3 turnover in neuronal chromatin. First, to assess if activity-dependent histone turnover may be associated with proteasomal degradation, neurons were virally transduced with tagged forms of ubiquitin and/or H3.3 in the absence or presence of KCl. Following immunoprecipitation of tagged ubiquitin from neuronal extracts, a process that successfully purifies ubiquitylated proteins from chromatin, H3.3 was found to display increased levels of polyubiquitylation in response to depolarization (Figure 4C). Such H3.3-specific polyubiquitylation can similarly be observed in chromatin extracts from adult brain (Figure S4A).
Next, to more directly assess the role of proteasomal degradation in the regulation of histone turnover in neurons, chromatin-associated H3.3 dynamics were monitored using SILAC in the absence or presence of the proteasome inhibitor MG132. Following proteasome inhibition, H3.3 incorporation (Figure 4D) and eviction (Figure 4E) from chromatin were found to be significantly reduced in comparison to vehicle treated controls, further suggesting that histone incorporation and eviction represent coupled processes. In summary, these data demonstrate a novel mechanism controlling histone turnover in the CNS, whereby the euchromatic chaperone Hira, in concert with chromatin associated proteasomal activity, are required to facilitate both constitutive and activity-dependent nucleosomal exchange (Figure S4B).
Histone dynamics mediate late-responsive activity-dependent gene expression in embryonic neurons
To address the role of histone turnover in activity-dependent transcription, we next performed transcriptional profiling of embryonic neurons in response to depolarization across numerous time points (30 min, 2 hrs or 5 hrs vs. respective non-stimulated controls). Clustering analysis resulted in 27 categories of transcriptional responses (three stimulation time points by three transcriptional outcomes: neutral, up or down), with the largest cluster predictably representing transcriptionally unresponsive genes (‘N’, neutral 40.2% of all genes detected). Closer examination of individual clusters revealed unique characteristics across groups, in which various response patterns could be extracted for further analysis. For example, ‘U1’ and ‘D1’ clusters represent classical IEGs that are characteristically up- or downregulated at 30 min following stimulation and then are returned to neutral expression at 2 and 5hrs. ‘U2’ and ‘D2’ clusters, on the other hand, represent another classification of IEGs that remain transcriptionally altered at extended time points. Numerous late response genes were also identified (e.g., early-late: ‘U3’ and ‘D3,’ and late-late: ‘U4’ and ‘D4’). These nine clusters, which include >80% of all transcripts identified, are displayed as heatmaps in Figure 5A (left column).
Figure 5. Neuronal histone dynamics correlate with late responsive activity-dependent enhancements in gene expression.

(A) Clustering analysis of primary cortical neuron transcriptional responses to KCl depolarization across time (30 min, 2 hrs and 5 hrs vs. respective non-stimulated controls). Data are displayed as heatmaps of nine representative clusters encompassing >83% of all detected genes. Up- (‘U’) and down- (‘D’) regulated genes were clustered into immediate early response genes (‘U1/D1’), immediate early-maintained genes (‘U2/D2’), early-late response genes (‘U3/D3’) and late-late response genes (‘U4/D4’), as well as unresponsive transcripts (‘N’). Histone dynamics (H3.3: 30 min, 2 hrs and 5 hrs; H3.1/2: 5 hrs) are depicted for each cluster of transcriptional response after depolarization. Basal chromatin peak enrichment profiles (H3.1/2, H3.3, H3K4me1, H3K4me3, H3K27me3 and RNA PolII), as well as basal state transcript expression, are similarly depicted. (B) Odds ratio analyses of differential enrichment events for H3.3 vs. activity-dependent gene expression across time for the nine clusters depicted in Figure 4A. Insert numbers (white) represent P values. (C) Genomic distribution of H3.3 differential enrichment events in response to KCl depolarization (5 hrs – late responsive genes) in mouse embryonic neurons. (D) Ingenuity Pathway Analysis (IPA) of late responsive genes displaying H3.3 differential enrichment following KCl stimulation (5 hrs) in mouse embryonic neurons. n values for sequencing experiments are provided in the Extended Experimental Procedures. See also Figure S5 and Supplemental Spreadsheet 2.
To examine associations between activity-dependent gene expression and histone dynamics, we performed H3.3 ChIP-seq in parallel to RNA-seq, such that primary neurons were collected following the absence or presence of KCl at 30 min, 2 hrs or 5 hrs. Following differential analysis of H3.3 enrichment, odds ratios were calculated to correlate changes in H3.3 deposition/eviction with global alterations in gene expression for each time point examined. Strong correlations between H3.3 dynamics and activity-dependent transcription were observed almost exclusively for upregulated genes at extended time points (i.e., 2 and 5 hrs) (Figure S5A). Further analyses overlapping H3.3 dynamics with individual clusters of transcriptionally active genes indicated highly significant correlations between H3.3 enrichment and upregulated gene expression, specifically within clusters representing late response genes (e.g., ‘U3’ and ‘U4’), as well as non-classical IEGs, which remain upregulated over extended periods of time (e.g., ‘U2’) (Figures 5A and 5B). Interestingly, classical IEGs did not correlate with H3.3 dynamics demonstrating a more specified role for histone turnover in the regulation of activity-dependent gene expression. Neutral genes and downregulated gene clusters did not display significant correlations between H3.3 dynamics and activity-dependent gene expression. Furthermore, significant dynamics for RD histones H3.1/2 were not observed in response to stimulation (Figures 5A and 5B).
To validate our interpretation that H3.3 enrichment corresponds to nucleosomal incorporation/eviction in response to neuronal activity, we performed CATCH-IT-Seq (Covalent Attachment of Tags to Capture Histones and Identify Turnover), which allows for direct genome-wide identification of sites of newly incorporated histones (H3/H4) in neuronal chromatin (Figure S5B) (Deal et al., 2010). Differential analysis of CATCH-IT-Seq, comparing control vs. KCl stimulated neurons, revealed highly significant overlaps with H3.3 differential enrichment, events that were associated with identical classes of upregulated late response genes (Figure S5C). Furthermore, differential analysis of genes displaying altered CATCH-IT signals revealed highly significant increases in enrichment for newly made histones in response to stimulation (Figure S5E), data that are in agreement with our SILAC LC-MS/MS findings.
Further analysis of genomic distributions of activity-dependent H3.3 dynamics revealed that the largest percentage of events occurred within the gene bodies of highly expressed genes (Figure 5C). Subsequent pathway analyses conducted on late responsive genes displaying altered H3.3 enrichment identified pathways essential to synaptic development (e.g., CREB signaling, axon guidance), function (e.g., glutamate receptor signaling, G protein receptor signaling) and plasticity (e.g., LTP, LTD) (Figure 5D). These results demonstrate high levels of H3.3 turnover within gene bodies of late response activity-dependent genes suggesting that these events may be functionally important in the regulation of embryonic neuronal plasticity.
To determine if histone dynamics are indeed necessary for activity-dependent transcription in neurons, we sought to directly disrupt turnover using a knockdown (KD) approach. Specifically, we employed lentivirus to infect cultured neurons with chained microRNAs (miRs) targeting both H3.3 genes [miR (H3f3a/b)]. Relative to the negative control [miR(−)], miR(H3f3a/b) significantly down-regulated both H3f3a and H3f3b mRNAs (Figure 6A). However, due to the abundance of H3.3 already present in chromatin, we were unable to detect a difference in H3.3 protein levels by western blot following KD (Figure S6A and 6C). Nevertheless, we hypothesized that H3f3b protein levels may be sensitive to viral KD, as its transcripts are responsive to depolarization. We therefore infected H3f3b-HA neurons with either miR (H3f3a/b) or miR(−), subjected the neurons to depolarization, and fractionated soluble nuclear vs. chromatin pools. Notably, KCl depolarization increased the expression of H3f3b-HA protein in both chromatin and soluble nuclear fractions (Figure 6C) for miR(−)-infected but not miR(H3f3a/b)-infected neurons, without loss of existing H3.3 in chromatin. To further investigate the effect of KD on H3.3 turnover, we employed SILAC labeling and found a significantly decreased rate of H3.3 turnover in cells infected with miR (H3f3a/b) (t1/2 = ~197.5 days) relative to miR(−) controls (t1/2 = ~4.8 days) (Figure 5B). Importantly, blocking histone turnover via H3.3 KD did not affect general aspects of neuronal health, as measured in numerous assays (e.g., dendritic arborization/Scholl Analysis, apoptosis/TUNEL staining, etc. – data not shown). Moreover, deficits in turnover induced by H3.3 KD were efficiently rescued via exogenous replacement of a miR (H3f3a/b) resistant H3.3 isoform (Figure S6A). Thus, infection with miR (H3f3a/b) provides a novel method to directly manipulate RI nucleosome turnover in the absence of global nucleosomal depletion.
Figure 6. RI histone turnover is necessary for activity-dependent transcription and synaptic connectivity in embryonic neurons.

(A) qPCR analysis of H3f3a and H3f3b mRNA expression after viral transduction with lenti-miR(−) (control) or lenti-miR (H3f3a/b) (H3.3 KD) in mouse embryonic neurons (n = 3–4 biological replicates/group). (B) LC-MS/MS SILAC analysis of H3.1/2 vs. H3.3 eviction from chromatin in mouse embryonic neurons following viral KD of H3f3a/b. (n = 3 biological replicates/group). (C) Western blot analysis of soluble nuclear vs. chromatin fractions from virally infected H3.3-HA tagged embryonic neurons [miR (H3f3a/b) vs. miR(−)] −/+ KCl (5 hrs). (D) RNA-seq analysis of transcriptional response cluster genes from Figure 4A −/+ KCl (5 hrs) after viral transduction with lenti-miR(−) or lenti-miR (H3f3a/b). (E) Dendritic spine analysis (spines/10 μm) of mouse embryonic cortical neurons virally infected with lenti-miR(−), lenti-miR (H3f3a/b), lenti-miR (H3f3a/b) + H3.1 or lenti-miR (H3f3a/b) + H3.3 add backs (n = 20–29 dendrites). Representative ICC images are shown below. (F) Left: quantification of synaptic co-puncta labeling for (top) excitatory (PSD-95/synapsin) and (bottom) inhibitory (GABAAγ2/GAD67) synapses on mouse embryonic neurons virally transduced with lenti-miR(−), lenti-miR (H3f3a/b), lenti-miR (H3f3a/b) + H3.1 or lenti-miR (H3f3a/b) + H3.3 (n = 40–50 dendrites/group). Right: representative ICC images of synaptic co-puncta labeling for excitatory and inhibitory synapses. Data represented as means ± SEM. */#P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. n values for sequencing experiments are provided in the Extended Experimental Procedures. See also Figure S6 and Supplemental Spreadsheet 3.
Next, to investigate causal relationships between H3.3 turnover and activity-dependent gene expression in neurons, we infected primary neurons with lenti-miR (H3f3a/b) or miR(−) and performed RNA-seq. In the absence of cellular stimulation, disrupting H3.3 dynamics altered the expression of numerous synaptic development/plasticity associated genes (e.g., Fos, Bdnf, Grm4, etc.), most of which could be successfully rescued by simultaneous add back of miR-resistant H3.3, but not H3.1 (Figures S6B). Subsequent qPCR analysis of virally manipulated neurons from independent cohorts of animals validated these KD/add back effects (Figure S6C) indicating a critical role for basal H3.3 turnover in the maintenance of neuronal specific gene expression.
To assess the impact of disrupting histone turnover on activity-dependent gene expression in neurons, we subjected embryonic neurons infected with lenti-miR (H3f3a/b) or miR(−) to depolarization, followed by RNA-seq. Transcriptional responses were clustered as described in Figure 5A and stimulus-dependent transcription was analyzed to assess whether H3.3 dynamics are necessary for mounting appropriate activity-dependent transcriptional responses. miR(−) expressing neurons responded similarly to uninfected cells in response to stimulation (Figure 6C), and, consistent with our earlier RNA-seq time course, genes clustering into non-responsive categories (e.g., ‘U1,’ ‘N,’ and ‘D1’) remained unchanged in response to depolarization. Stalling H3.3 turnover with viral H3.3 KD, however, significantly attenuated activity-dependent responses in ‘U3’ and ‘U4’ clusters specifically (Figure 5D) providing direct causal evidence of a role for histone turnover in the mediation of late responsive activity-dependent neuronal transcription.
Histone turnover is necessary for controlling synaptic connectivity
Given our finding that H3.3 turnover is required for activity-dependent neuronal transcription, we next examined the physiological impact of histone turnover in cultured neurons. We observed a significant reduction in the density of dendritic spines, classical sites of glutamatergic synapses, in neurons infected with miR(H3f3a/b) vs. miR(−) control. This effect could be rescued by simultaneous add back of miR-resistant H3.3, but not H3.1 (Figure 6E). We next employed a pre-/postsynaptic co-puncta labeling approach to examine excitatory vs. inhibitory synapse development. In response to H3.3 KD, both types of synapses were significantly reduced relative to miR(−) controls (Figure 6F). We similarly validated this finding by infection with an independent miR(H3f3a/b) targeting construct (Figure S6D–E). Deficits in both dendritic spine number and synaptic copuncta labeling resulting from H3.3 KD could be fully rescued by simultaneous add back of miR-insensitive H3.3, but not H3.1. These data indicate that histone dynamics are critically important to synaptic connectivity and are not specific to synapse type.
Neuronal histone turnover controls life-long transcriptional and behavioral plasticity
To examine the significance of histone turnover in adult brain, we next assessed H3.3 dynamics in adult hippocampal neurons purified from EE vs. normally housed mice via ChIP-seq. Comparable to results in embryonic neurons, H3.3 displayed high levels of differential enrichment throughout active coding regions, specifically within gene bodies (Figure 7A). These data indicate that while H3.3 accumulates over time, plasticity-associated histone dynamics persist within active genic regions of adult neurons. We next examined associations between EE-induced H3.3 dynamics and alterations in gene expression. Gene body associated H3.3 turnover was found to correlate strongly with EE-induced gene expression in adult brain, with similar classes of plasticity-associated genes experiencing these dynamics (Figure 7B).
Figure 7. Histone turnover is required for life-long transcriptional and behavioral plasticity.

(A) Genomic distribution of H3.3 differential enrichment sites in response to EE (4 wks) in adult mouse hippocampal NeuN+ neurons. (B) Top: IPA of genes displaying H3.3 differential enrichment after EE (4 wks) in adult mouse hippocampal NeuN+ neurons. Bottom: Odds ratio analysis correlating differential enrichment events for H3.3 vs. gene expression in response to EE. Insert numbers (white) represent P values. (C) Representative immunohistochemistry image of intra-hippocampal (CA1) infections with AAVs expressing either miR(−)-IRES-GFP or miR (H3f3a/b)-IRES-GFP 4 wks following surgery. (D) qPCR analysis of H3f3a and H3f3b, as well as H3.3 dynamics associated candidate genes after intra-CA1 viral infections with AAV-miR(−)-IRES-GFP or AAV-miR (H3f3a/b)-IRES-GFP in adult mouse hippocampus (n = 3–4 animals/group). (E) Dendritic spine analysis (spines/10 μm) of adult hippocampal CA1 pyramidal neurons infected with AAV-miR(−)-IRES-GFP or AAV-miR (H3f3a/b)-IRES-GFP (n = 28–37 dendrites/group in 3–4 mice/group). (F) Representative traces and quantification of mEPSCs from adult hippocampal CA1 pyramidal cells infected with AAV-miR(−)-IRES-GFP or AAV-miR (H3f3a/b)-IRES-GFP. Adjacent uninfected cells from experimental animals were recorded as additional controls for viral infection (n = 3–4 animals/group, 5–7 cells/animal). (G) Locomotor activity of mice infected intra-CA1 with AAV-miR(−)IRES-GFP or AAV-miR (H3f3a/b)-IRES-GFP. Data reported as total distance travelled (cm) (n = 10 animals/group). (H) Novel object recognition in mice infected intra-CA1 with AAV-miR(−)-IRES-GFP or AAV-miR (H3f3a/b)-IRES-GFP. Data reported as difference scores (time spent exploring the novel vs. old object in seconds) (n = 10 animals/group). (I) Contextual fear conditioning in mice infected intra-CA1 with AAV-miR(−)-IRES-GFP or AAV-miR (H3f3a/b)-IRES-GFP. Data reported as % time freezing (n = 9 animals/group). Data represented as means ± SEM. *P ≤ 0.05, **P ≤ 0.01. n values for sequencing experiments are provided in the Extended Experimental Procedures. See also Figure S7 and Supplemental Spreadsheet 4.
To further address the physiological impact of H3.3 turnover in adult brain, mice were stereotaxically injected intra-hippocampus (CA1) with adenoassociated viral (AAV) vectors expressing either miR (H3f3a/b) or miR(−). Four weeks later, infected tissues were collected, validated for placement/expression/health (Figure 7C) and analyzed by qPCR to examine the impact of H3.3 KD on the expression of candidate genes known to be basally occupied by H3.3 vs. H3.1/2 based on previous ChIP-seq analyses. Consistent with results from embryonic neurons, H3f3a/H3f3b KD reduced the expression of these genes in adult hippocampus (Figure 7D) indicating that inhibiting histone turnover in adult neurons similarly leads to deficits in neuronal specific gene expression. To examine the contribution of H3.3 dynamics to synaptic maintenance in the adult CNS, CA1 pyramidal cell dendritic spines were analyzed. Similar to embryonic neurons, H3.3 KD resulted in decreased numbers of dendritic spines on adult pyramidal cells (Figure 7E). Further electrophysiological analyses of miniature excitatory postsynaptic currents (mEPSCs) on CA1 pyramidal neurons confirmed these effects, with the frequency of mEPSCs (Figure 7F) being reduced by KD compared to miR(−) expressing and uninfected controls. In contrast, mEPSC amplitudes remained unaffected by KD indicating that the strength of remaining synapses remained normal. Global reductions in synapse number were further confirmed via transmission electron microscopy (TEM), where both asymmetric (excitatory) and symmetric (inhibitory) synapses were reduced after H3.3 KD (Figure S7A). In total, these data demonstrate that H3.3 turnover remains functionally significant throughout the lifetime of an animal.
Given the critical role of H3.3 turnover in mediating synaptic connectivity in adult hippocampal neurons, we next chose to behaviorally characterize the impact of H3.3 dynamics in the adult CNS. H3.3 KD in CA1 neurons was found to have no effect on basal locomotion or exploratory behavior in an open field (Figures 7F and S7B, respectively), behaviors that are traditionally disassociated from dorsal hippocampal function. However, disrupting H3.3 turnover significantly impaired animals’ abilities to distinguish between novel vs. previously encountered objects (Figure 7H), a classic hippocampal-dependent task. Similarly, KD animals displayed significantly decreased freezing behavior in a long-term memory task of contextual fear conditioning (Figure 7I). To ensure that reduced freezing responses were not the result of anxiolytic behaviors precipitated by H3.3 KD, animals were tested in an elevated plus maze (EPM), where viral KD of H3.3 was found to be anxiogenic (Figure S7B), thus strengthening our interpretation that disrupting H3.3 turnover significantly impairs learning and memory.
Histone dynamics are essential for cell-type specific gene expression in mammals
Given the vital role of nucleosomal dynamics in regulating neuronal gene expression and function, we next sought to determine if histone turnover might play an equally significant role in other mammalian cell types. Using SILAC, we measured the incorporation and eviction rates of H3 variants in cellular chromatin from rapidly proliferating mESCs and slowly dividing astrocytes (cultured from postnatal cortex), both of which express H3 isoforms in similar ratios to those observed in embryonic neurons (Figures S8A and S7B). Beginning with asynchronous mESCs, all three H3 proteins were found to incorporate equally into chromatin (t1/2 = ~0.1 days for H3.1, H3.2 and H3.3) (Figure 8A), consistent with historical predictions. Following thymidine blockade, which disrupts DNA synthesis and halts the cell cycle at the G1/S boundary (Figure 8A inset), we observed reduced incorporation rates of canonical H3.1 (t1/2 = ~2.2 days) and H3.2 (t1/2 = ~1.8 days), but not H3.3 (t1/2 = ~1.3 days). Next, we examined the incorporation/eviction profiles of fully differentiated mouse astrocytes. While all three H3 proteins were found to actively incorporate into chromatin, the rate of H3.3 turnover was considerably greater than that of its canonical counterparts [eviction t1/2 = ~4.5 days (H3.1), ~4.9 days (H3.2) vs. ~1.7 days (H3.3)], likely owing to slower rates of cell cycling (Figure 8B).
Figure 8. RI nucleosomal dynamics are essential for cell-type specific mammalian gene expression.

(A) LC-MS/MS SILAC analysis of H3.1/2 vs. H3.3 chromatin incorporation −/+ thymidine (DNA synthesis inhibitor) in rapidly proliferating mESCs. Inset depicts DAPI FACS analysis of mESCs −/+ thymidine indicating a shift toward the G1/S boundary due to inhibition of replication. (B) LC-MS/MS SILAC analysis of H3.1/2 vs. H3.3 chromatin incorporation and eviction in mouse primary astrocytes from postnatal cortex. (C) Venn diagrams of genes dysregulated by H3f3a/b KD in mESCs, postnatal astrocytes and embryonic neurons. (D) IPA of dysregulated genes following KD of H3f3a/b in mESCs, postnatal astrocytes and embryonic neurons. (E) LC-MS/MS quantified H3.1/2 vs. H3.3 relative protein expression in NeuN− mouse chromatin with age. (n = 3 biological replicates/age). (F) SILAC LC-MS/MS analysis of H3.1/2 vs. H3.3 in mouse NeuN− glial chromatin from multiple brain structures after 2 or 4 wks of feeding on a heavy lysine (6 Da) diet. (n = 3–6 biological replicates/brain region). (G) Overlap and number of genes displaying differential H3.3 enrichment in adult mouse hippocampal NeuN− vs. NeuN+ cells after 4 wks of EE. (H) IPA of genes displaying H3.3 differential enrichment following EE (4 wks) in adult mouse hippocampal NeuN− cells indicating enrichment for glial specific/enriched transcriptional programs. (I) Odds ratio analysis of differential enrichment events for H3.3 in NeuN− glia vs. gene expression in response to EE (4 wks). Numbers (white) represent P values. Data represented as means ± SEM. n values for sequencing experiments are provided in the Extended Experimental Procedures. See also Figure S8 and Supplemental Spreadsheets S5–S6.
We next sought to determine if H3.3 turnover is required for gene expression in mESCs and astrocytes. For mESCs, RNA-seq data were extracted from a previous study (Banaszynski et al., 2013), in which H3.3 KD resulted in a global reductions in nucleosomal turnover. Using our bioinformatics pipeline, we uncovered 80 genes whose expression was significantly altered by H3.3 KD; most genes were upregulated (Figure S8C). Although it is unclear whether H3.1 and/or H3.2 are capable of compensating for H3.3 loss in mESCs, such rescue is plausible given that equal rates of RI vs. RD nucleosomal exchange were observed. In contrast, viral KD of H3.3 in primary astrocytes revealed robust alterations in gene expression (449 genes displaying significant changes vs. 272 in neurons) indicating a critical role for H3.3 turnover in the regulation of astrocytic transcription. Unlike in neurons, however, where only H3.3 add back was sufficient to rescue KD mediated alterations in gene expression, both H3.3 and H3.1 were able to efficiently rescue transcriptional deficits in astrocytes (Figure S8D). These data suggest that deficits in RI nucleosomal exchange can be partially rescued by canonical H3 proteins in dividing cells and are consistent with a more dominant role for RI turnover in post-mitotic vs. rapidly proliferating cells.
Genes impacted by H3.3 KD in mESCs vs. astrocytes vs. neurons minimally overlapped (Figure 8C) revealing cell type-specific gene regulation patterns by H3.3 dynamics. Pathway analyses on these subsets of genes highlighted the cellular specificity of transcriptional programs mediated by RI nucleosomal turnover (Figure 8D). Consistent with H3.3’s role in the recruitment of PRC2 complexes in mESCs (Banaszynski et al., 2013), H3.3 KD was found to primarily affect expression of weakly expressed/silenced genes in mESCs. In contrast, altering nucleosomal dynamics had a more profound impact on active gene expression in astrocytes and neurons (Figure S8E) indicating distinct functions for histone turnover in mESCs vs. slowly dividing/post-replicative cells.
Finally, to validate these findings in vivo, we examined H3.3 accumulation in aging mouse glial (NeuN−) cells obtained via FACS, the vast majority of which represent astrocytes and oligodendrocytes. Consistent with data obtained from cultured astrocytes, H3.3 was found to represent only ~30% of the total H3 chromatin pool during embryonic development (E16.5). However, H3.3 was observed to accumulate with glial age, reaching sustained levels of >80% of the total H3 pool in two year old mice (Figure 8E). Although a small portion of glial cells in adult brain continue to divide throughout life, recent evidence suggests that bulk NeuN− cell population becomes post-mitotic within the first decade or so of postnatal existence in humans (Yeung et al., 2014) indicating that observed similarities between NeuN− and NeuN+ cells with age may reflect transitions to the post-replicative state. Of interest, however, are developmental differences observed between neurons and non-neuronal cells with respect to the timing of H3.3 accumulation, whereby glial cells display slightly delayed accumulation patterns of H3.3 during early postnatal development in comparison to neurons. This delay is not surprising, as both oligodendrocytes and astrocytes require signals from mature axons to fully differentiate and transition to the post-mitotic state.
Given similar patterns of H3.3 accumulation between glial cells and neurons, we next examined whether glia also display active histone turnover throughout life in adult brain. Using the SILAC mouse model, we determined that glial cells display continued patterns of RI histone turnover in the absence of accumulation (e.g., H3.3 t1/2 = ~17.0 days, hippocampus), (Figure 8F) suggesting a role for histone dynamics in the regulation of adult glial transcription. To assess this possibility, adult glial cells were similarly isolated from adult hippocampus of animals exposed to EE vs. normal housing, and H3.3 dynamics were examined via ChIP-seq. Similar to that observed in neurons, chronic EE dramatically altered H3.3 enrichment patterns throughout the glial genome (538 genic differential enrichment events vs. 843 identified in NeuN+ hippocampal neurons), however, little overlap was identified between genes regulated in these distinct cell types from brain (Figure 8G). These findings are consistent with data obtained from cultured cells, whereby distinct patterns of gene regulation were observed following H3.3 KD in mESCs vs. astrocytes vs. neurons. Pathway analysis further confirmed such cell type distinctions indicating that transcriptional programs specific to glia (e.g., VEGF signaling, GDNF family ligand receptor interactions, etc.) were highly affected by altered nucleosomal dynamics in NeuN− cells (Figure 8H). Moreover, altered H3.3 enrichment in NeuN− cells were found to strongly correlate with increased levels of gene expression resulting from EE (Figure 8I), with the majority of these events occurring throughout gene bodies, similar to patterns observed in embryonic and adult neurons.
Discussion
Previous interpretations of histone dynamics in proliferating cells have suggested that RI histone variant exchange may simply represent a general mechanism of nucleosomal replacement following nucleoprotein engagement and transcriptional elongation (Ray-Gallet et al., 2011). Such mechanisms have been proposed to explain how cells prevent exposure of ‘naked’ DNA to potentially deleterious nuclease activities. With the exception of H3.3 “gap-filling” in the maintenance of genomic integrity, additional roles for histone turnover in the regulation of cellular plasticity in vivo have not yet been described. Results of the present study reveal causal evidence supporting a more interactive role for H3.3 turnover in the regulation of neuronal activity-dependent gene expression, and indicate histone dynamics as being essential for maintenance of basal patterns of cell-type specific gene expression in brain required to support synaptic connectivity and behavioral plasticity. Although critical roles for histone modifications and chromatin remodeling are well documented in the regulation of neural plasticity, this work provides the first direct evidence that neuronal histone turnover itself may represent an equally powerful mechanism governing transcriptional plasticity in brain.
Our results indicate that the majority of H3.3 turnover in brain occurs within gene bodies of active genes. In agreement with these results, our data show that binding interactions between free/soluble nuclear H3.3 and Hira are potentiated in response to cellular activity, with subsequent knockdown studies confirming that Hira is responsible for the majority of H3.3 turnover in neurons. (Goldberg et al., 2010; Tagami et al., 2004). This does not suggest that Daxx, which has previously been shown to promote H3.3 deposition at a subset of IEG promoters (Michod et al., 2012), does not play an important role in euchromatic H3.3 turnover, but rather that its role in such processes is likely more specialized and does not account for high levels of turnover observed in activated neuronal chromatin. Our data further indicate a novel role for proteasome-dependent degradation in the regulation of histone dynamics suggesting that the proteasome itself, which harbors ATPase activity and has previously been shown to associate with chromatin (Geng and Tansey, 2012), may directly contribute to activity-dependent histone turnover. These data are in agreement with previous studies suggesting a fundamental role for proteasome-dependent degradation during activity-dependent synaptic development (Hamilton et al., 2012). Our data suggest that during periods of heightened neuronal activity, active engagement of the transcriptional machinery results in nucleosomal H3.3 being ‘marked’ for chromatin eviction (i.e., polyubiquitylated), before being rapidly degraded in a proteasome-dependent manner. Meanwhile, H3.3 associated with Hira in the soluble nuclear pool acts to quickly replace nucleosome-depleted regions, likely preventing cryptic transcription. Finally, newly-synthesized histones replace depleted pools of free histones in the nucleus and form new associations with Hira to allow for subsequent turnover events. This process, which is essential for maintenance of neuronal and glial gene expression in brain, is likely coordinated by a multitude of additional factors, and future studies are needed to fully delineate the entirety of mechanisms required to support histone turnover in the mammalian CNS.
Previous studies in actively dividing cells have demonstrated that mammalian H3 variants, although strikingly similar in amino acid sequence, exhibit distinct PTM “signatures” (Hake et al., 2006). These findings led to the “H3 barcode hypothesis,” which posits that histone variants intrinsically enrich for specific histone PTMs, are localized to distinct genomic loci and are functionally distinct from their canonical counterparts due to the formation of unique chromosomal domains (Hake and Allis, 2006). Although our data suggest that such ‘barcoding’ may similarly be important in embryonic neurons, our findings from adult neurons indicate that, with age, H3.3 and its associated turnover, are, at least partially, ‘de-coupled’ from ‘active’ PTMs indicating life-long histone turnover as an independent and critical histone modulatory process controlling neuronal transcription. Furthermore, as H3.3 effectively saturates the neuronal genome with age, correlations between its enrichment and basal transcript abundance become significantly reduced. These data indicate that as H3.3 becomes more evenly distributed across the neuronal genome, including throughout regions typically underrepresented by active histone PTMs, its ascribed role as a active histone ‘mark’ becomes less representative of its overall function.
We propose that such patterns of H3.3 accumulation and turnover may be similar in other fully differentiated, non-proliferating cells, which raises the intriguing possibility that such mechanisms may play critical roles in the regulation of developmental potentials and adulthood plasticity across all post-mitotic tissues. Our findings that H3.3 KD in mESCs similarly regulates cell-type specific gene expression, albeit to a lesser extent than that observed in fully differentiated/post-replicative cells, indicates a more global role for histone turnover in the regulation of mammalian transcription. Although the precise role of nucleosomal turnover in the regulation of gene expression may vary across cell types, it is clear that this mechanism is critically important for cell-type specific transcription and physiological plasticity in response to environmental perturbations. Therefore, future studies aimed at investigating the role of histone turnover in distinct neuronal subtypes, as well as in other supporting cells of the CNS, during periods of altered activity are needed to fully delineate the contribution of this mechanism to mammalian behavioral plasticity.
Finally, our data challenge the notion that H3.3 itself is functionally distinct from its canonical counterparts. Rather, it appears that it is the dynamic nature of H3.3, and not specific intrinsic functions, that mediates critical aspects of neurodevelopment and lifelong synaptic and behavioral plasticity. Our observation that complete histone turnover–a mechanism distinct from so-called histone variant ‘exchange’–within gene bodies of active genes is directly linked to cell-type specific transcriptional plasticity suggests a novel molecular mechanism controlling mammalian cellular diversity and brain specific functions. Moreover, because the two genes encoding H3.3 can be readily knocked down in rodents to promote stalling of H3.3 turnover in the absence of complete nucleosomal depletion, our discovery of a dominant role for life-long histone turnover establishes a novel paradigm by which the contribution of highly dynamic nucleosomal turnover to neurodevelopmental and neuropsychiatric phenomena can now be studied in diverse animal models. Future studies aimed at investigating the interplay between histone turnover and other modes of epigenetic regulation will greatly aid in our understanding of the mechanisms underlying human brain development and disease.
Experimental Procedures
Detailed procedures are described in the Extended Experimental Procedures.
Mass Spectrometry
Histone H3.x protein samples, separated by reversed-phase chromatography or by 1D-gel electrophoresis, were reduced and alkylated prior to trypsinization. In-solution digests were desalted and concentrated using a double Empore C18 membrane inserted into a P200 pipette tip (Ishihama et al., 2006). Peptides were measured by nano LC-MS/MS using Q-Exactive or Orbitrap XL mass spectrometers coupled to a Dionex NCP3200RS HPLC setup. MS/MS spectra were extracted using ProteomeDiscoverer v. 1.4 and queried against Uniprots complete Human or Mouse proteome databases concatenated with common known contaminants. Ion traces (10 ppm) were generated using the second carbon 13 isotope, since this isotope is most abundant for H3.x peptides (see Supplementary Figure S1B). All ion traces were generated using QuanBrowser (Xcalibur 2.2) and were manually validated. For consistency, this strategy was used for both label free quantitation, as well as SILAC (13C6-Lys in-vivo mouse and 13C615N2-Lys in-vitro) experiments.
ChIP-seq Peak Calling and Differential Analysis
For histones H3.1/2 and H3.3, MACS was used for peak calling with biological triplicates for each control group merged with fragment size set as 250bp. Differential analysis of H3.1/2 and H3.3 was performed using diffReps with a negative binomial test in nucleosome mode and a fragment size of 250bp. A default p-value cutoff of 1E-4 was used with a fold change cutoff of 3.0. For CATCH-IT data, DANPOS was used for peak calling and differential analysis. The KCl or EE treatment groups were compared to control groups with corresponding inputs used as background samples. Peaks and differential sites were further annotated to nearby genes or intergenic regions using the diffReps package region analysis tool.
RNA-seq Analysis and Correlations with ChIP-seq
RNA-seq reads were aligned against Ensembl gene annotations using Tophat. Cufflinks was used for differential analysis adjusted for sequencing bias and corrected for multiple alignments in order to obtain more accurate estimates of transcript abundance. For in vitro RNA-seq experiments, an FDR of <5% was used as a cutoff. Since in vivo RNA-seq data were more variable, relaxed cutoffs (FDR <15% and fold change >1.3) were used. RNA-seq differential lists were compared to ChIP-seq differential lists based on gene names, and heat maps were drawn using the GeneOverlap (https://github.com/shenlab-sinai/geneoverlap/) package.
Supplementary Material
Acknowledgments
We would like to thank members of the Allis laboratory for critical readings of the manuscript. We would also like to thank Dr. Kunihiro Uryu (The Rockefeller University Electron Microscopy Resource Center) for assistance with TEM experiments, Ms. Kelly Gleason for help with human postmortem brain dissections, and Mr. Charlie Li for assistance with mESC FACS. This work was supported by grants from the National Institute of Mental Health (NIMH): 5R01 MH094698 and P50 MH096890. Benjamin A. Garcia gratefully acknowledges funding from the following NIH grants: R21MH102679 and RO1GM110174. Support for accelerator mass spectrometry analyses was provided by NIH 8P41GM103483. This work was performed, in part, under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under contract DE-AC52-07NA27344. LLNL-JRNL-653296.
Footnotes
Competing Financial Interests
The authors declare no competing financial interests.
Accession Numbers
The National Center for Biotechnology Information Gene Expression Omnibus accession number for all sequencing data described in this paper is TBD.
Author Contributions
I.M. and C.D.A conceived of the project. I.M. designed the experiments. I.M., W.W., K.M.N, R.C.B., N.T., S.J.E, Y.G., L.F., M.R.B, B.A.B., and H.M. collected and analyzed the data. I.P. and L.S. performed the bioinformatics analyses. Z.F.Y and B.A.G. performed the combinatorial histone PTM mass spectrometry analyses. Y.L.H, C.A.T., and T.H. provided human tissue. C.I., S.A., R.D.B., and E.J.N. provided essential resources and reagents. A.A.S. helped with illustrations. D.W., and S.R. helped with breeding and maintenance of the H3.3-HA mouse line. I.M. and C.D.A. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Banaszynski LA, et al. Hira-Dependent Histone H3.3 Deposition Facilitates PRC2 Recruitment at Developmental Loci in ES Cells. Cell. 2013;155:107–120. doi: 10.1016/j.cell.2013.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrelli E, et al. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commerford SL, et al. Histone turnover within nonproliferating cells. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:1163–1165. doi: 10.1073/pnas.79.4.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal RB, et al. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science. 2010;328:1161–1164. doi: 10.1126/science.1186777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion MF, et al. Dynamics of replication-independent histone turnover in budding yeast. Science. 2007;315:1405–1408. doi: 10.1126/science.1134053. [DOI] [PubMed] [Google Scholar]
- Fischer A, et al. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Geng F, Tansey WP. Similar temporal and spatial recruitment of native 19S and 20S proteasome subunits to transcriptionally active chromatin. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6060–6065. doi: 10.1073/pnas.1200854109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AD, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PL, Greenberg ME. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2008;59:846–860. doi: 10.1016/j.neuron.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Hake SB, Allis CD. Histone H3 variants and their potential role in indexing mammalian genomes: the “H3 barcode hypothesis”. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:6428–6435. doi: 10.1073/pnas.0600803103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hake SB, et al. Expression patterns and post-translational modifications associated with mammalian histone H3 variants. The Journal of biological chemistry. 2006;281:559–568. doi: 10.1074/jbc.M509266200. [DOI] [PubMed] [Google Scholar]
- Hamilton AM, et al. Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron. 2012;74:1023–1030. doi: 10.1016/j.neuron.2012.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihama Y, et al. Modular stop and go extraction tips with stacked disks for parallel and multidimensional Peptide fractionation in proteomics. J Proteome Res. 2006;5:988–994. doi: 10.1021/pr050385q. [DOI] [PubMed] [Google Scholar]
- Maze I, et al. Histone regulation in the CNS: basic principles of epigenetic plasticity. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2013;38:3–22. doi: 10.1038/npp.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michod D, et al. Calcium-dependent dephosphorylation of the histone chaperone DAXX regulates H3.3 loading and transcription upon neuronal activation. Neuron. 2012;74:122–135. doi: 10.1016/j.neuron.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pina B, Suau P. Changes in histones H2A and H3 variant composition in differentiating and mature rat brain cortical neurons. Developmental Biology. 1987;123:51–58. doi: 10.1016/0012-1606(87)90426-x. [DOI] [PubMed] [Google Scholar]
- Ray-Gallet D, et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Molecular Cell. 2011;44:928–941. doi: 10.1016/j.molcel.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Santoro SW, Dulac C. The activity-dependent histone variant H2BE modulates the life span of olfactory neurons. Elife. 2012;1:e00070. doi: 10.7554/eLife.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savas JN, et al. Extremely Long–Lived Nuclear Pore Proteins in the Rat Brain. Science. 2012 doi: 10.1126/science.1217421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami H, et al. Histone H3.1 and H3.3 Complexes Mediate Nucleosome Assembly Pathways Dependent or Independent of DNA Synthesis. Cell. 2004;116:51–61. doi: 10.1016/s0092-8674(03)01064-x. [DOI] [PubMed] [Google Scholar]
- Toyama BH, et al. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell. 2013;154:971–982. doi: 10.1016/j.cell.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung MS, et al. Dynamics of oligodendrocyte generation and myelination in the human brain. Cell. 2014;159:766–774. doi: 10.1016/j.cell.2014.10.011. [DOI] [PubMed] [Google Scholar]
- Zovkic IB, et al. Histone H2A.Z subunit exchange controls consolidation of recent and remote memory. Nature. 2014;515:582–586. doi: 10.1038/nature13707. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.