

Abstract
Introduction
Sclerostin (SOST), a soluble antagonist of Wnt signaling, is expressed in chondrocytes and contributes to chondrocytes’ hypertrophic differentiation; however its role in osteoarthritis (OA) pathogenesis is not well known. Based on our previous findings on the interaction between Wnt/β-catenin pathway and BMP-2 in OA, we aimed to investigate the role of DNA methylation and BMP-2 on SOST’s expression in OA chondrocytes.
Methods
SOST mRNA and protein expression levels were investigated using real-time polymerase chain reaction (PCR) and Western blot, respectively. The methylation status of SOST promoter was analysed using methylation-specific PCR (MSP), quantitative methylation-specific PCR (qMSP) and bisulfite sequencing analysis. The effect of BMP-2 and 5’-Aza-2-deoxycytidine (5-AzadC) on SOST’s expression levels were investigated and Smad1/5/8 binding to SOST promoter was assessed by Chromatin Immunoprecipitation (ChΙP).
Results
We observed that SOST’s expression was upregulated in OA chondrocytes compared to normal. Moreover, we found that the CpG region of SOST promoter was hypomethylated in OA chondrocytes and 5-AzadC treatment in normal chondrocytes resulted in decreased SOST methylation, whereas its expression was upregulated. BMP-2 treatment in 5-AzadC-treated normal chondrocytes resulted in SOST upregulation, which was mediated through Smad 1/5/8 binding on the CpG region of SOST promoter.
Conclusions
We report novel findings that DNA methylation regulates SOST’s expression in OA, by changing Smad 1/5/8 binding affinity to SOST promoter, providing evidence that changes in DNA methylation pattern could underlie changes in genes’ expression observed in OA.
Introduction
Osteoarthritis (OA), a chronic degenerative disease of the joints, is a major health burden linked to high morbidity in the aging population [1, 2]. The central pathological features of OA are the progressive degradation of articular cartilage, new bone formation at joint margins (osteophytes) and changes in subchondral bone structure (sclerosis) [3]. OA is considered a multifactorial disease and several risk factors contribute to its pathogenesis, including genetic predisposition, aging, obesity and joint malignment [2, 4].
Articular chondrocytes may be the most important cells that are involved in OA pathogenesis [5, 6]. The disruption of matrix equilibrium between synthesis and degradation of extracellular matrix (ECM) components and progressive loss of cartilage tissue are associated with changes in their anabolic and catabolic activities following exposure to multiple signals [7, 8]. Recently, it was demonstrated that one of the genes that are deregulated in OA chondrocytes is SOST [9]. Sclerostin (SOST), encoded by the SOST gene, is specifically expressed by osteocytes and is involved in bone homeostasis [10, 11]. SOST is a soluble antagonist of Wnt signaling [12] and it has been demonstrated that SOST loss-of-function mutations cause abnormal skeletal phenotypes in humans, characterized by high bone mineral density [13, 14], whereas transgenic mice that overexpress SOST are osteopenic due to reduced bone formation [15]. In OA, which is characterized by new bone formation, it has been reported that SOST is implicated in OA disease processes in both bone and cartilage with opposing effects, by promoting subchondral bone sclerosis while inhibiting cartilage degradation [9].
Besides the well-known role of SOST as a Wnt signaling inhibitor, it has been recently suggested that SOST interacts with other signaling pathways, such as bone morphogenic proteins (BMPs) and affects the biology of the skeleton [16–18]. The canonical BMP-Smad pathway induces human mesenchymal stem cells to differentiate into chondrocytes and osteoblasts and BMP-2 is a crucial local factor responsible for chondrocyte proliferation and maturation during endochondral ossification [19, 20]. Although the interaction between SOST and BMPs is not yet clear, it has been shown that in osteoblasts, SOST binds to BMPs and modulates the activity of osteoblastic cells by reducing the expression of alkaline phosphatase (ALP), synthesis of type I collagen, and mineralization [15]. Despite the role of SOST as a Wnt and BMP signaling inhibitor, little is known about its gene regulation. Previous studies have reported that different molecular mechanisms are able to modulate SOST expression, among which BMPs and parathyroid hormone (PTH) [21–24]. Moreover, recent studies point towards the involvement of DNA methylation in the regulation of SOST expression in human osteocytes and bone cells [18, 25, 26]. In the present study, we sought to investigate first whether DNA methylation regulates SOST expression in OA chondrocytes, and the role of BMP-2 on changes in SOST expression in OA.
Materials and methods
Bioinformatic analysis
The 1,500 bp upstream of the SOST transcript start site (TSS) were obtained from Ensembl genome browser and putative CpG islands were identified using Metlyl Primer Express software v1.0 (available from Applied Biosystems). A CpG island was defined as a region of at least 200 bp, with GC content greater than 50 %, and observed-to-expected (O/E) CpG ratio >0.6 [27]. CpG islands were tested for Smad binding sites (SBEs, 5’-GCCGnGCG-3’) using ChIP bioinformatics tools.
Patients and cartilage samples
Articular cartilage samples were obtained from femoral condyles and tibial plateaus of 14 patients (11 female/3 male; mean age 67.8 ± 9.6 years) with primary hypertrophic OA, undergoing knee replacement surgery at the Orthopaedics Department of the University Hospital of Larissa. Radiographs were obtained before surgery and graded using the Kellgren-Lawrence system according to the following criteria: grade 1 (doubtful narrowing of joint space and possible osteophytes), grade 2 (definite osteophytes and possible narrowing of joint space), grade 3 (moderate multiple osteophytes, definite narrowing of joint space and some sclerosis and possible deformity of bone ends) and grade 4 (large osteophytes, marked narrowing of joint space, severe sclerosis and definite deformity of bone ends). All patients had a Kellgren-Lawrence grade ≥3. The assessment of the radiographs by two independent expert observers was blinded. Normal articular cartilage was obtained from 10 individuals (7 female/3 male; mean age 56.9 ± 10.8 years), undergoing knee fracture repair surgery, with no history of joint disease and who did not show clinical manifestations compatible with OA when specifically explored by radiography. Written informed consent was obtained from all individuals in the study. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the local ethical committee of the University Hospital of Larissa.
Primary cultures of normal and OA human articular chondrocytes
Articular cartilage was dissected and subjected to sequential digestion with 1 mg/ml pronase and 1 mg/ml collagenase P (Roche Applied Science, Mannheim, Germany). Chondrocytes were counted and checked for viability using trypan blue staining. More than 95 % of the cells were viable after isolation. Isolated chondrocytes from individual specimens were separately cultured with DMEM/F-12 (GIBCO, Life Technologies, Paisley, UK) plus 5 % FBS (Invitrogen, Life Technologies, Paisley, UK) at 37 °C under a humidified 5 % CO2 atmosphere until reaching confluence for 4–6 days. Half of cultured chondrocytes were then harvested by trypinization and were used for DNA, RNA and protein extraction. The other half was cultured again without treatment until confluence for 1 week (passage-1 chondrocytes).
Passage-1 normal and OA chondrocytes were seeded on six-well plates at 3 × 105 cells/well and 3 days post-seeding cells were treated with 50 ng/ml of BMP-2 (Sigma-Aldrich, MO, USA) for 24 and 48 h or with 5 μM 5-AzadC (Sigma-Aldrich) in dimethyl sulfoxide (DMSO). Media containing DMSO or DMSO+5-AzadC was exchanged daily and lasted for 5 days. Moreover, for BMP-2 experiments, chondrocytes were treated with or without 5-AzadC for 3 days, then media was removed and 50 ng/ml of BMP-2 was added for 48 h.
RNA extraction and quantification of mRNA expression
Total cellular RNA was extracted from cultured chondrocytes using Trizol reagent (Invitrogen, Life Technologies, Paisley, UK). Preservation of 28S and 18S ribosomal RNA (rRNA) species was used to assess RNA integrity. All the samples included the study were with prominent 28S and 18S rRNA components. The yield was quantified spectrophotometrically. Transcription of 1 μg RNA to cDNA was performed using SuperScript III reverse transcriptase (Invitrogen, Life Technologies, Paisley, UK) and random primers (Invitrogen, Life Technologies, Paisley, UK). Quantification of SOST mRNA expression was performed by real-time PCR (ABI 7300, Applied Biosystems, Foster, CA, USA). The oligonucleotide primers used for SOST amplification are shown in Table 1. Reactions were done in triplicate using 2 μl of cDNA per reaction. Real-time PCR validation was carried out using the 2-ΔΔCT method. Normalized gene expression values for each gene based on cycle threshold (CT) values for each of the genes and the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were generated.
Table 1.
Primer sequences for PCR, real-time PCR, methylation-specific PCR (MSP), quantitative MSP (Qmsp) and bisulfite (Bis.) sequencing analysis
| Name | Sequence (5′-3) | Experiment |
|---|---|---|
| M-SOST-forward | GAATAGGTCGGGTTTAGTTTC | MSP, qMSP |
| M-SOST-reverse | ACCTCCCACGTACTAACGA | MSP, qMSP |
| U-SOST-forward | GGAATAGGTTGGGTTTAGTTTT | MSP, qMSP |
| U-SOST-reverse | CACCTCCCACATACTAACAA | MSP, qMSP |
| SOST-forward | CCGGAGCTGGAGAACAACAAG | RT-PCR |
| SOST-reverse | GGTGTGCTCCGGCCAGTGC | RT-PCR |
| Promoter SOST-forward | GGGACCAATGGGATTTCTTT | PCR |
| Promoter SOST-reverse | TGAGCTCCGGCTTTTAATTG | PCR |
| BSP SOST-forward | TTATTTGTTGGTGGGGTGATAA | Bis. sequencing |
| BSP SOST-reverse | ACAAAACCCAAACCTACTCTCC | Bis. sequencing |
Protein extraction and western blot analysis
Chondrocytes were lysed using radioimmunoprecipitation assay (RIPA) buffer containing 10 mM Tris (pH 7.5), 150 mM NaCl, 1 % Triton X-100, 1 % sodium deoxycholate, 0.1 % SDS, 1 mM EDTA, and a cocktail of protease inhibitors. Protein concentration was quantified using the Bio-Rad Bradford protein assay (Bio-Rad Protein Assay, BioRad, Hercules, CA, USA) with bovine serum albumen as standard. Cell lysates from chondrocytes were electrophoresed and separated on 12 % acrylamide gels and transferred to PVDF membranes (Millipore, Billerica, MA, USA). The membrane was probed with anti- SOST (1:100 dilution) (Novus biologicals, CO, USA) and signal was detected using anti-rabbit immunoglobulin IgG conjugated with horseradish peroxidase (1:10.000 dilution) (Invitrogen, Life Technologies, Paisley, UK). The results were normalized using anti-β-actin polyclonal antibody (1:3.000 dilution) (Sigma-Aldrich, MO, USA). PVDF membranes were then exposed to photographic film and western blot bands from several different blots were quantified using the NIH Scion Image according to the software guidelines.
DNA methylation analysis by MSP
Genomic DNA was extracted from normal and OA cultured chondrocytes using the Genomic DNA Isolation Kit (Qiagen, Valencia, CA, USA) and was treated with bisulfite conversion reagents using the MethylCode™ Bisulfite Conversion Kit (Invitrogen, Life Technologies, Paisley, UK) according to the manufacturer’s instruction. The region of interest in the SOST promoter was amplified by PCR using primers for MS-PCR derived from the Methlyl Primer Express (software v1.0) (Table 1). PCR reaction was confirmed by electrophoresis in a 3 % agarose gel and was stained with ethidium bromide. Quantification analysis of bands was performed using the NIH Scion Image according to the software guidelines.
DNA methylation analysis by qMSP
Quantitative methylation-specific PCR (qMSP) for the CpG island of the SOST promoter was performed using a real-time PCR instrument (ABI 7300, Applied Biosystems, Foster, CA, USA). In the qMSP reaction, 2 μl of bisulfite-treated genomic DNA were amplified with 2 × EpiTect Master Mix (Qiagen, Valencia, CA, USA) and 0,75 μΜ primers (Table 1) in a total volume of 25 μl. Amplification conditions were: 95 °C for 5 minutes, followed by 40 cycles of 95 °C for 10 s, 55 °C for 30 s, and 72 °C for 27 s, with a final extension of 72 °C for 10 minutes. DNA methylation values were calculated by interpolating the cycle threshold gap (CtU-CtM) in a standard curve, conducted using mixtures of methylated and unmethylated human control samples with 0 %, 10 %, 25 %, 50 %, 75 %, 90 % and 100 % methylated DNA (Qiagen, Valencia, CA, USA).
Bisulfite DNA sequencing analysis
Bisulfite-treated DNA was amplified by PCR using primers for BSP-PCR derived from the Methlyl Primer Express (software v1.0) (Table 1).). In the PCR reaction, 2 μl of bisulfite-treated genomic DNA were amplified with 10 × PCR buffer, 400 μΜ dNTPs, 1 U of AmpliTaq Gold DNA polymerase (Applied Biosystems, Foster, CA, USA) and 0,5 μΜ of each primer in a total volume of 25 μl. Amplification conditions were: 95 °C for 10 minutes, followed by 40 cycles of 95 °C for 10 s, 54 °C for 30 s, and 72 °C for 1 minute, with a final extension of 72 °C for 5 minutes. PCR products were cleaned using QIAquick PCR Purification kit (Qiagen, Valencia, CA, USA) and then were sequenced using a Bigdye terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster, CA, USA) and analyzed on the ABI 3130 Genetic Analyzer (Applied Biosystems). Sequencing was performed using the forward primer and the methylation percentage for each CpG site in the CpG island was quantified by measuring the ratio between peak height values of cytosine (C) and thymine (T), yielding the basic equation for the methylation percentage to be (C/(C + T) *100) [28].
Chromatin immunoprecipitation (ChIP) assay
ChIP was performed using a ChIP assay kit (Upstate USA, Inc., Charlottesville, VA, USA) on normal and OA chondrocytes. Cell lysates were pre-cleared by incubation with G-Sepharose beads and were incubated with monoclonal antibody Smad-1/5/8 (Cell signalling Technology, Boston, MA, USA) overnight at 4 °C. Antibody human purified IgG was used as control (R&D Systems, McKinley Place, MN, USA). The immunoprecipitated DNAs were used for PCR amplification. The primers were designed according to the nucleotide sequence of SOST promoter and the PCR fragment covered 250–400 bp of the promoter. Table 1 shows the primer sets that amplify the promoter region containing putative sites as observed after bioinformatic analysis. The PCR products were fractionated on 3 % agarose gels and were stained with ethidium bromide. Quantification analysis of bands was performed using the NIH Scion Image according to the software guidelines.
Statistical analysis
Data were analyzed using the SPPS software 20. Statistical significance was determined using Student’s t test and a confidence level of 95 % (p <0.05).
Results
SOST mRNA and protein expression levels are increased in OA chondrocytes
Using real-time PCR, we found higher SOST mRNA expression levels in OA compared to normal chondrocytes (p = 0.005) (Fig. 1a). SOST protein levels confirmed our real-time PCR findings. Western blot analysis revealed that SOST protein expression is elevated in OA chondrocytes compared to normal (p = 0.038) (Fig. 1b).
Fig. 1.
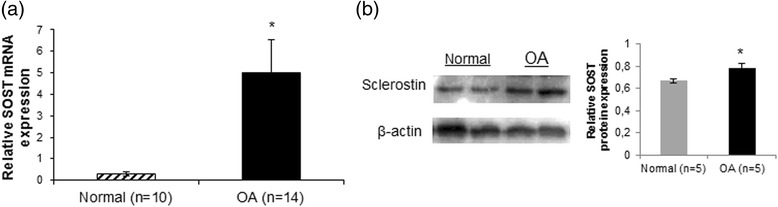
SOST mRNA and protein expression levels in normal and osteoarthritis (OA) chondrocytes. a Quantitative SOST mRNA expression in cultured normal (n = 10) and OA chondrocytes (n = 14). GAPDH was used for normalization of the real-time PCR data (error bars = standard error, *p = 0.005). b Representative western blot of SOST protein expression in cultured normal and OA chondrocytes and a bar graph showing relative SOST protein expression normalized to β-actin in normal (n = 5) and OA chondrocytes (n = 5) (error bars = standard error, *p = 0.038)
DNA methylation status of SOST promoter is different between normal and OA chondrocytes
Taking into consideration recent reports that demonstrated involvement of epigenetics in the regulation of SOSTs expression, we tested whether the DNA methylation status of the SOST promoter is different between normal and OA chondrocytes. Bioinformatic analysis revealed the presence of two CpG islands surrounding the TSS of the SOST gene. One CpG island was located upstream of the TSS and the other within exon 1, downstream of the transcription start codon. We then tested the CpG islands for the presence of transcription factors binding sites and especially Smad binding sites, as we wanted to investigate the role of BMP-2, mediated through Smad proteins, on SOST expression. As we found Smad binding sites only in the first CpG island located upstream of the TSS in the region of the SOST promoter, we investigated the methylation status of this CpG island. This CpG island was identified between −516 bp and −256 bp upstream of the TSS, as a region of DNA spanning over 200 bp with a GC content over 50 %. Using MSP technology, we evaluated the methylation status of this region in genomic DNA isolated from normal and OA chondrocytes. Our results showed a significant difference in the methylation status between normal and OA chondrocytes in this CpG island located at the SOST promoter (Fig. 2a). Moreover, analysis by qMSP demonstrated that this CpG-rich region at the SOST promoter was highly methylated in normal chondrocytes (76,18 % ± 0,842) compared to OA chondrocytes (72,68 % ± 0,654) (p = 0.004) (Fig. 2b). To identify the particular CpG sites in this region whose methylation status is associated with SOST expression, we analyzed six CpG dinucleotides in the CpG island located at the SOST promoter using bisulfite DNA sequencing. We found that CpG sites 1, 4 and 6 were highly methylated (p = 0.05, p = 0.05 and p = 0.046, respectively) in normal compared to OA chondrocytes (Fig. 2c and d).
Fig. 2.
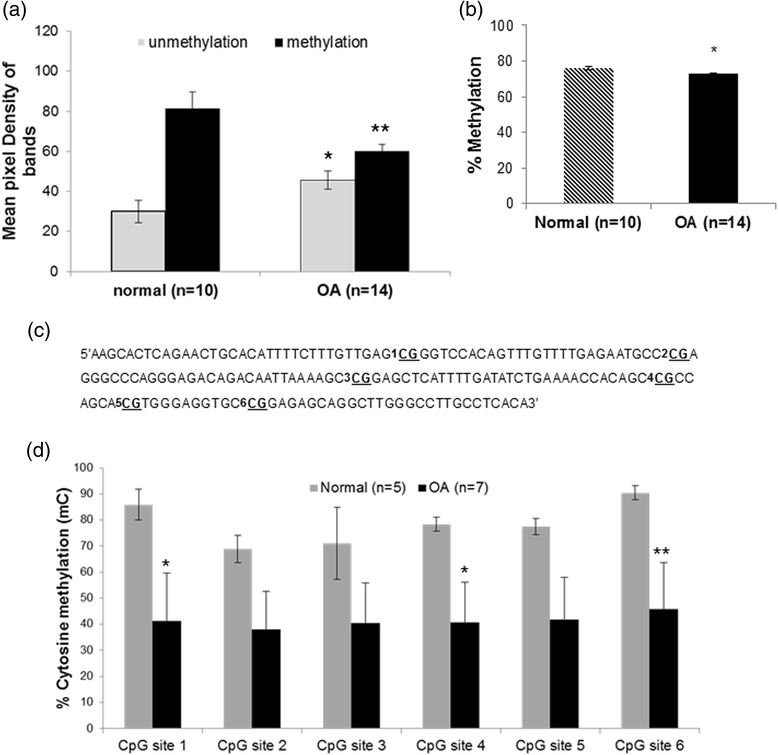
DNA methylation status of the SOST promoter in normal and osteoarthritis (OA) chondrocytes. a DNA methylation status of CpG island located upstream of the transcript start site (TSS) in the region of the SOST promoter in cultured normal (n = 10) and OA chondrocytes (n = 14) (error bars = standard error, *p = 0.05 versus normal unmethylation status, **p = 0.043 versus normal methylation status). b DNA methylation values in the CpG-rich region of the SOST promoter in cultured normal (n = 10) and OA chondrocytes (n = 10) by qMSP (error bars = standard error, *p = 0.004). c Schematic representation of the CpG-rich region of the SOST promoter showing the six CpG sites that were analyzed using the bisulfite DNA sequencing method. d The percentage of cytosine methylation of each CpG site located at the CpG region of the SOST promoter in cultured normal (n = 5) and OA chondrocytes (n = 7), after bisulfite DNA sequencing analysis (error bars = standard error, *p = 0.05 and **p = 0.046)
SOST expression is induced by 5-AzadC through modification of DNA methylation status
To confirm the causal inverse association between SOST promoter methylation and gene expression, we evaluated the ability of the demethylating agent 5-AzadC to promote SOST expression in chondrocytes. Normal chondrocytes, for which the promoter was found to be methylated, were treated with 5-AzadC and subsequently we evaluated SOST mRNA and protein levels by real-time PCR and western blot analysis, respectively. We found that SOST mRNA and protein levels (p = 0.041, p = 0.009, respectively) were markedly increased in 5-AzadC-treated cells compared to untreated (Fig. 3a, b) and this 5-AzadC-induced change in gene expression was associated with a decrease in DNA methylation in the CpG-rich region of the SOST promoter (p = 0.032) (Fig. 3c, d).
Fig. 3.
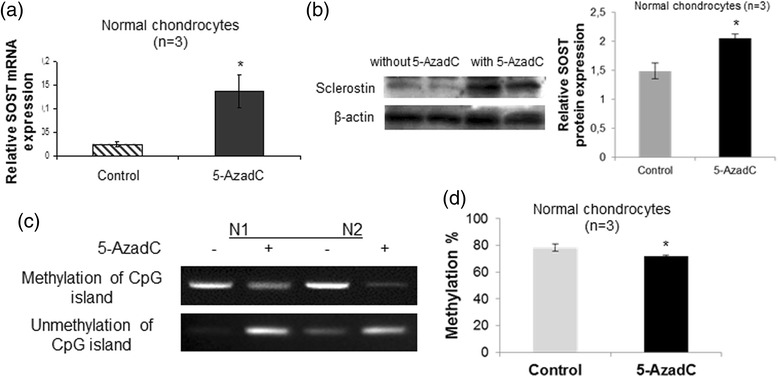
Effect of 5-AzadC treatment on SOST expression and DNA methylation status in the CpG-rich region of the SOST promoter. a Quantitative SOST mRNA expression in cultured normal chondrocytes (n = 3) after treatment with 5 μM 5-AzadC. GAPDH was used for normalization of the real-time PCR data (error bars = standard error, *p = 0.041). b Representative western blot of SOST protein levels in cultured normal chondrocytes after treatment with 5 μM 5-AzadC and a bar graph showing relative SOST protein expression normalized to β-actin in 5-AzadC-treated normal chondrocytes (n = 3) (error bars = standard error, *p = 0.009). c DNA methylation and unmethylation status of the SOST promoter in cultured normal chondrocytes after treatment with 5 μM 5-AzadC. d DNA methylation values in CpG-rich region of the SOST promoter in cultured normal chondrocytes (n = 3) after treatment with 5 μM 5-AzadC by quantitative methylation-specific PCR (error bars = standard error, *p = 0.032)
DNA methylation contributes to regulation of SOST expression by impairing the binding affinity of Smad 1/5/8 transcription factors in its promoter
BMP-2 has been reported to play a significant role in the regulation of SOST expression in bone. To gain insight into the molecular mechanisms underlying SOST expression in OA chondrocytes, we examined the effect of BMP-2 on SOST expression in normal and OA chondrocytes. We found that BMP-2 treatment resulted in significant induction of SOST expression in OA chondrocytes (p = 0.004), but not in normal chondrocytes (Fig. 4a and b). However, SOST expression levels were upregulated after BMP-2 treatment in 5-AzadC-treated normal chondrocytes (Fig. 4c) (p = 0.001). To further investigate the intracellular signaling pathway involved in BMP-2-induced SOST expression, normal, OA, BMP-2 and/or 5-AzadC-treated chondrocytes were subjected to ChIP assay using an antibody against Smad-1/5/8 and we tested whether Smads bind to the SOST promoter via Smad binding elements. We found that the SOST promoter contains a conserved Smad binding site in the CpG island located upstream of the TSS and that Smad1/5/8 binding was enhanced in OA compared to normal chondrocytes (p = 0.05) and in BMP-2-treated OA compared to untreated chondrocytes (p = 0.05) (Fig. 5a and c). Moreover, stronger binding of Smad1/5/8 was observed in 5-AzadC-treated normal chondrocytes compared to untreated (Fig. 5b) (p = 0.05) and this affinity was significantly increased in BMP-2/5-AzadC-treated normal chondrocytes compared to 5-AzadC-treated, BMP-2-treated and untreated chondrocytes (Fig. 5d and e) (p = 0.05). No difference was observed between BMP-2-treated and untreated normal chondrocytes (Fig. 5c).
Fig. 4.
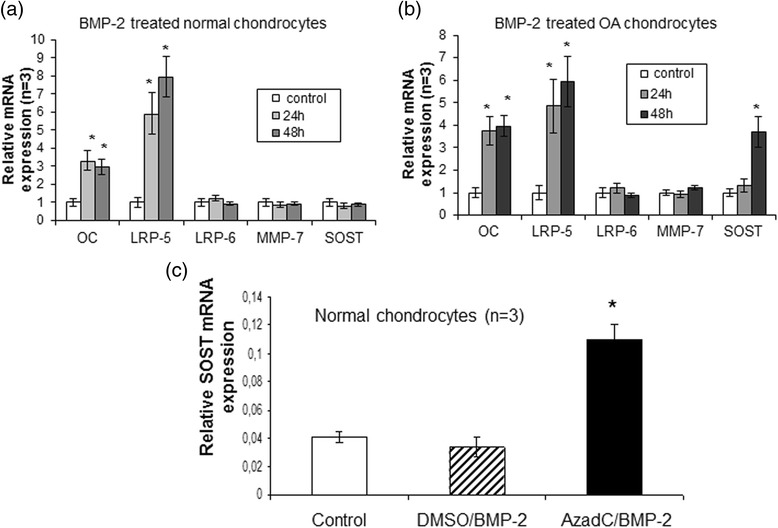
Effect of bone morphogenic protein 2 (BMP-2) and 5-AzadC treatment on SOST expression. a and b Efects of BMP-2 on SOST mRNA expression levels in cultured normal (n = 3) and osteoarthritis (OA) chondrocytes (n = 3). Osteocalcin and LRP-5 used as positive controls that upregulated by BMP-2 and LRP-6 and MMP-7 used as negative controls that not regulated by BMP-2(error bars = standard error, *p = 0.004). c Detection of SOST mRNA expression levels by real time PCR after BMP-2 treatment in cultured normal chondrocytes (n = 3) with or without 5-AzadC. GAPDH was used for normalization of the real-time PCR data (error bars = standard error, *p = 0.001 versus control and DMSO/BMP-2 treatment). MMP matrix metalloproteinase, LRP low-density lipoprotein receptor-related protein
Fig. 5.
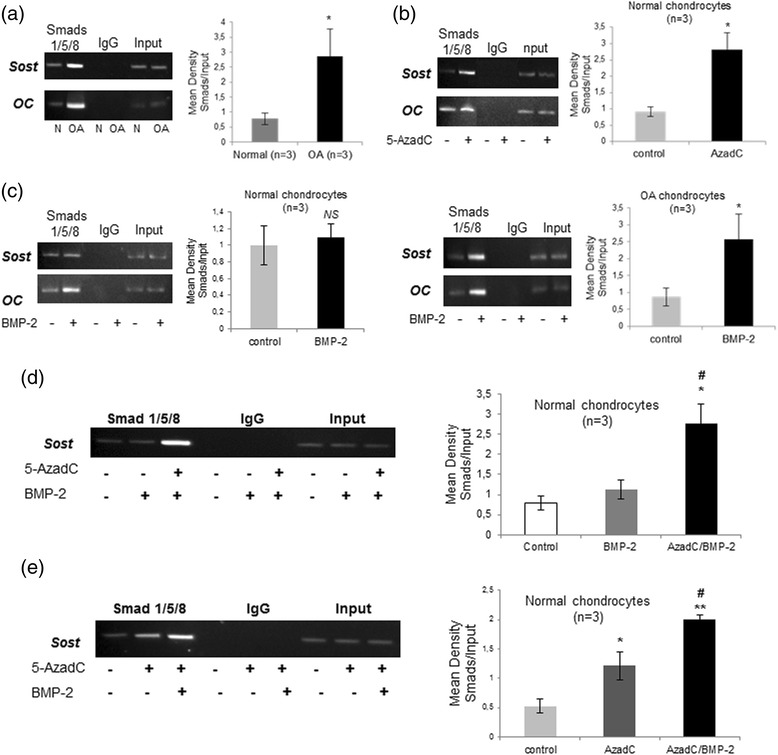
Occupancy of the SOST promoter by Smad1/5/8 by chromatin immunoprecipitation (ChIP) analysis. a Representative gel of Smad1/5/8 binding on the SOST promoter in cultured normal and osteoarthritis (OA) chondrocytes and densitometric analysis of the band intensity in cultured normal (n = 3) and OA chondrocytes (n = 3) (error bars = standard error, *p = 0.05 versus normal chondrocytes). b Representative gel of Smad1/5/8 binding on the SOST promoter in 5-AzadC- treated and untreated normal chondrocytes and densitometric analysis of the band intensity in three different samples (n = 3) (error bars = standard error, *p = 0.05 versus control) c Representative gel of Smad1/5/8 binding on the SOST promoter in bone morphogenic protein 2 (BMP-2)-treated and untreated normal and OA chondrocytes and densitometric analysis of the band intensity in normal (n = 3) and OA samples (n = 3) (error bars = standard error, *p = 0.05 versus control, NS = not significant). d Representative gel of Smad1/5/8 binding on the SOST promoter after BMP-2 treatment in cultured normal chondrocytes with or without 5-AzadC and densitometric analysis of the band intensity in three different samples (n = 3). (error bars = standard error, *p = 0.05 versus control and #p = 0.05 versus BMP-2 treatment). Input chromatin used as positive control and IgG as negative control. e Representative gel of Smad1/5/8 binding on the SOST promoter after 5-AzadC treatment in cultured normal chondrocytes with or without BMP-2 and densitometric analysis of the band intensity in three different samples (n = 3) (error bars = standard error, *p = 0.05 versus control, #p = 0.05 versus control and **p = 0.05 versus 5-AzadC treatment)
Discussion
SOST is a potent Wnt antagonist and a key regulator of bone metabolism [9, 29]. OA is characterized by changes in bone matrix composition and metabolism, however, the role of SOST in OA pathogenesis is not well known. In the present study, we found that SOST is upregulated in OA chondrocytes compared to normal, in agreement with recent studies demonstrating that SOST is expressed by articular chondrocytes and not only by bone cells in end-stage OA [9, 30, 31]. However, Roudier et al. showed that there is no difference in SOST expression between normal and OA cartilage but a strong SOST staining only in chondrocyte clusters that are often observed in damaged OA articular cartilage [32]. Our results showed an association between SOST expression and OA, however further investigation is necessary to determine whether upregulation of SOST expression in OA chondrocytes is a causal factor in OA pathogenesis or a result of the OA process.
It is known that DNA methylation patterns change with increasing age and age-dependent hypomethylation may contribute to pathological processes such as OA, an age-related disease [33]. In an attempt to investigate the role of DNA methylation on SOST expression in OA, we investigated the methylation status of the SOST gene and observed, for the first time, that the SOST promoter was hypermethylated in normal chondrocytes and hypomethylated in OA chondrocytes, suggesting the involvement of epigenetic mechanisms in the regulation of SOST expression in OA. However, additional functional studies are needed to clarify whether these DNA methylation changes in the SOST promoter are biologically relevant. In general, changes in DNA methylation have been shown to have an impact on OA pathology, as several studies have demonstrated a different methylation profile between OA and normal cartilage [34–36].
Further evidence for a relationship between DNA methylation and gene expression was obtained after treatment of chondrocytes with 5-AzadC, a potent inhibitor of DNA methylation. We found that 5-AzadC treatment in normal chondrocytes resulted in upregulation of SOST’s expression through altered methylation status of the SOST promoter, suggesting that this region of the promoter influences SOST expression in chondrocytes. Previous studies have demonstrated that DNA methylation contributes to the regulation of SOST’s expression in human osteocytes and bone cells [18, 25, 26, 37], suggesting a common molecular mechanism of SOST gene expression in different cell types.
Besides DNA methylation, other factors, such as growth factors and hormones can modulate SOST expression. BMP-2 is a growth factor which plays an important role in cartilage and bone homeostasis and it has been demonstrated that BMP-2 contributes to the regulation of SOST expression [16–18]. We found that treatment with BMP-2 resulted in significant increase in SOST expression in OA chondrocytes but not in normal. However, SOST expression was upregulated after BMP-2 treatment in 5-AzadC-treated normal chondrocytes, suggesting that DNA methylation may impair the binding of transcriptional factors, especially Smad binding in the SOST promoter. By using the ChIP assay, we demonstrated the existence of a Smad binding site in the CpG region located upstream of the TSS and found that the binding affinity was decreased in the methylated promoter, as the CpG dinucleotide, which were shown to be methylated, were located at/or near the Smad binding site. In addition, we observed that BMP-2 induced SOST expression in 5-AzadC-treated normal chondrocytes, which correlated with stronger binding affinity of Smads in the SOST promoter, suggesting that ΒΜΡ-2 and the methylation status of the SOST promoter regulate SOST transcriptional levels. In a recent study, it was demonstrated that BMPs stimulate SOST expression in human bone cells by a mechanism involving BMPR1A receptor and the downstream Smad-dependent pathway, suggesting a direct influence of BMPs on SOST transcriptional levels [18]. Moreover, Thillainadesan et al. showed an association between DNA methylation status and Smad binding, as they demonstrated that transforming growth factor (TGF)-β signaling activation resulted in DNA demethylation of p15ink4b gene and subsequently increased recruitment of SMAD2/3 on p15ink4b gene promoter [38].
SOST has become an attractive target for the treatment of osteoporosis and other skeletal diseases associated with low bone mineral density and increased fracture risk [31, 39]. Preclinical studies have demonstrated that antisclerostin therapy results in increased bone formation and bone mass in animal models [40–42]. Moreover, recent human clinical trials with sclerostin-neutralizing monoclonal antibody (Scl-Ab) therapy have shown beneficial effects on bone formation and resorption markers in healthy men and postmenopausal women [43]. However, in OA the findings in preclinical studies using antisclerostin therapy have been disappointing, as genetic absence of sclerostin or antisclerostin therapy with monoclonal antibody had no impact on articular cartilage remodeling in animals with age-dependent OA or post-traumatic OA, respectively [32]. On the other hand, a recent study showed that plasma and synovial fluid SOST levels are inversely associated with radiographic severity in knee OA, suggesting SOST as a protective factor in OA and a possible biochemical marker of knee OA for reflecting the degenerative process of primary knee OA [44]. Based on the above reports and our results it can be suggested that SOST may play a role in OA pathogenesis but its impact on cartilage biology and extracellular matrix degradation may be less powerful compared to its major regulatory role in bone mass.
Conclusion
Our novel data strongly suggest that BMP-2 signaling modulates SOST transcription in OA through changes in Smad 1/5/8 binding affinity to the CpG region located upstream of the TSS in the SOST gene, pointing towards the involvement of DNA methylation in SOST expression in OA.
Acknowledgements
The present work was supported by the research financed project, Epigenetic regulation in osteoarthritis through DNA methylation, from the Hellenic Society of Orthopaedic Surgery and Traumatology.
Abbreviations
- 5-AzadC
5’-Aza-2-deoxycytidine
- BMP-2
bone morphogenetic protein 2
- bp
base pairs
- ChΙP
chromatin immunoprecipitation
- DMEM/F-12
Dulbecco’s Modified Eagles Medium/Ham’s F-12
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- MSP
methylation-specific polymerase chain reaction
- OA
osteoarthritis
- PCR
polymerase chain reaction
- qMSP
quantitative methylation-specific polymerase chain reaction
- SBE
Smad binding element
- SOST
sclerostin
- TSS
transcript start site
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
IP conceived of the study, designed and performed the experiments, analysed the data and drafted the manuscript. FK participated in data interpretation and helped to revise the manuscript. KNM participated in the study design, provided cartilage samples and clinical evaluation of patients and helped to draft the manuscript. AT conceived of the study, conducted data analysis and results interpretation, and drafted and revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Ioanna Papathanasiou, Email: ioanna_papathanasiou@yahoo.gr.
Fotini Kostopoulou, Email: fotini.kostopoulou@gmail.com.
Konstantinos N. Malizos, Email: malizos@med.uth.gr
Aspasia Tsezou, Email: atsezou@med.uth.gr.
References
- 1.Suri P, Morgenroth DC, Hunter DJ. Epidemiology of osteoarthritis and associated comorbidities. Pm R. 2012;4:10–9. doi: 10.1016/j.pmrj.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 2.Johnson VL, Hunter DJ. The epidemiology of osteoarthritis. Best Pract Res Clin Rheumatol. 2014;28:5–15. doi: 10.1016/j.berh.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Hunter DJ, Felson DT. Osteoarthritis. BMJ. 2006;332:639–42. doi: 10.1136/bmj.332.7542.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertrand J, Cromme C, Umlauf D, Frank S, Pap T. Molecular mechanisms of cartilage remodelling in osteoarthritis. Int J Biochem Cell Biol. 2010;42:1594–601. doi: 10.1016/j.biocel.2010.06.022. [DOI] [PubMed] [Google Scholar]
- 5.Tchetina EV. Developmental mechanisms in articular cartilage degradation in osteoarthritis. Arthritis. 2011;2011:683970. doi: 10.1155/2011/683970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldring MB, Goldring SR. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann NY Acad Sci. 2010;1192:230–7. doi: 10.1111/j.1749-6632.2009.05240.x. [DOI] [PubMed] [Google Scholar]
- 7.Roach HI, Aigner T. DNA methylation in osteoarthritic chondrocytes: a new molecular target. Osteoarthritis Cartilage. 2007;15:128–37. doi: 10.1016/j.joca.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 8.Goldring MB. Chondrogenesis, chondrocyte differentiation, and articular cartilage metabolism in health and osteoarthritis. Ther Adv Musculoskelet Dis. 2012;4:269–85. doi: 10.1177/1759720X12448454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan BY, Fuller ES, Russell AK, Smith SM, Smith MM, Jackson MT, et al. Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthritis Cartilage. 2011;19:874–85. doi: 10.1016/j.joca.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 10.Atkins GJ, Rowe PS, Lim HP, Welldon KJ, Ormsby R, Wijenayaka AR, et al. Sclerostin is a locally acting regulator of late-osteoblast/preosteocyte differentiation and regulates mineralization through a MEPE-ASARM-dependent mechanism. J Bone Miner Res. 2011;26:1425–36. doi: 10.1002/jbmr.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li F, Song N, Tombran-Tink J, Niyibizi C. Pigment epithelium derived factor suppresses expression of Sost/sclerostin by osteocytes: Implication for its role in bone matrix mineralization. J Cell Physiol. 2014;230:1243–9. doi: 10.1002/jcp.24859. [DOI] [PubMed] [Google Scholar]
- 12.Blom AB, van Lent PL, van der Kraan PM, van den Berg WB. To seek shelter from the WNT in osteoarthritis? WNT-signaling as a target for osteoarthritis therapy. Curr Drug Targets. 2010;11:620–9. doi: 10.2174/138945010791011901. [DOI] [PubMed] [Google Scholar]
- 13.Piters E, Culha C, Moester M, Van Bezooijen R, Adriaensen D, Mueller T, et al. First missense mutation in the SOST gene causing sclerosteosis by loss of sclerostin function. Hum Mutat. 2010;31:E1526–43. doi: 10.1002/humu.21274. [DOI] [PubMed] [Google Scholar]
- 14.Bhadada SK, Rastogi A, Steenackers E, Boudin E, Arya A, Dhiman V, et al. Novel SOST gene mutation in a sclerosteosis patient and her parents. Bone. 2013;52:707–10. doi: 10.1016/j.bone.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22:6267–76. doi: 10.1093/emboj/cdg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moester MJ, Papapoulos SE, Lowik CW, van Bezooijen RL. Sclerostin: current knowledge and future perspectives. Calcif Tissue Int. 2010;87:99–107. doi: 10.1007/s00223-010-9372-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paszty C, Turner CH, Robinson MK. Sclerostin: a gem from the genome leads to bone-building antibodies. J Bone Miner Res. 2010;25:1897–904. doi: 10.1002/jbmr.161. [DOI] [PubMed] [Google Scholar]
- 18.Delgado-Calle J, Arozamena J, Perez-Lopez J, Bolado-Carrancio A, Sanudo C, Agudo G, et al. Role of BMPs in the regulation of sclerostin as revealed by an epigenetic modifier of human bone cells. Mol Cell Endocrinol. 2013;369:27–34. doi: 10.1016/j.mce.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Steinert AF, Proffen B, Kunz M, Hendrich C, Ghivizzani SC, Noth U, et al. Hypertrophy is induced during the in vitro chondrogenic differentiation of human mesenchymal stem cells by bone morphogenetic protein-2 and bone morphogenetic protein-4 gene transfer. Arthritis Res Ther. 2009;11:R148. doi: 10.1186/ar2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shu B, Zhang M, Xie R, Wang M, Jin H, Hou W, et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J Cell Sci. 2011;124:3428–40. doi: 10.1242/jcs.083659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leupin O, Kramer I, Collette NM, Loots GG, Natt F, Kneissel M, et al. Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J Bone Miner Res. 2007;22:1957–67. doi: 10.1359/jbmr.070804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keller H, Kneissel M. SOST is a target gene for PTH in bone. Bone. 2005;37:148–58. doi: 10.1016/j.bone.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 23.Kamiya N, Kobayashi T, Mochida Y, Yu PB, Yamauchi M, Kronenberg HM, et al. Wnt inhibitors Dkk1 and Sost are downstream targets of BMP signaling through the type IA receptor (BMPRIA) in osteoblasts. J Bone Miner Res. 2010;25:200–10. doi: 10.1359/jbmr.090806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perez-Campo FM, Sanudo C, Delgado-Calle J, Arozamena J, Zarrabeitia MT, Riancho JA. A Sclerostin super-producer cell line derived from the human cell line SaOS-2: a new tool for the study of the molecular mechanisms driving Sclerostin expression. Calcif Tissue Int. 2014;95:194–9. doi: 10.1007/s00223-014-9880-5. [DOI] [PubMed] [Google Scholar]
- 25.Delgado-Calle J, Sanudo C, Bolado A, Fernandez AF, Arozamena J, Pascual-Carra MA, et al. DNA methylation contributes to the regulation of sclerostin expression in human osteocytes. J Bone Miner Res. 2012;27:926–37. doi: 10.1002/jbmr.1491. [DOI] [PubMed] [Google Scholar]
- 26.Reppe S, Noer A, Grimholt RM, Halldorsson BV, Medina-Gomez C, Gautvik VT, et al. Methylation of bone SOST, its mRNA, and serum sclerostin levels correlate strongly with fracture risk in postmenopausal women. J Bone Miner Res. 2015;30:249–56. doi: 10.1002/jbmr.2342. [DOI] [PubMed] [Google Scholar]
- 27.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–82. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 28.Parrish RR, Day JJ, Lubin FD. Direct bisulfite sequencing for examination of DNA methylation with gene and nucleotide resolution from brain tissues. Curr Protoc Neurosci. 2012: Chapter 7:Unit 7 24. [DOI] [PMC free article] [PubMed]
- 29.Honasoge M, Rao AD, Rao SD. Sclerostin: recent advances and clinical implications. Curr Opin Endocrinol Diabetes Obes. 2014;21:437–46. doi: 10.1097/MED.0000000000000114. [DOI] [PubMed] [Google Scholar]
- 30.Karlsson C, Dehne T, Lindahl A, Brittberg M, Pruss A, Sittinger M, et al. Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthritis Cartilage. 2010;18:581–92. doi: 10.1016/j.joca.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 31.Lewiecki EM. Role of sclerostin in bone and cartilage and its potential as a therapeutic target in bone diseases. Ther Adv Musculoskelet Dis. 2014;6:48–57. doi: 10.1177/1759720X13510479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roudier M, Li X, Niu QT, Pacheco E, Pretorius JK, Graham K, et al. Sclerostin is expressed in articular cartilage but loss or inhibition does not affect cartilage remodeling during aging or following mechanical injury. Arthritis Rheum. 2013;65:721–31. doi: 10.1002/art.37802. [DOI] [PubMed] [Google Scholar]
- 33.Richardson B. Impact of aging on DNA methylation. Ageing Res Rev. 2003;2:245–61. doi: 10.1016/S1568-1637(03)00010-2. [DOI] [PubMed] [Google Scholar]
- 34.Rushton MD, Reynard LN, Barter MJ, Refaie R, Rankin KS, Young DA, et al. Characterization of the cartilage DNA methylome in knee and hip osteoarthritis. Arthritis Rheumatol. 2014;66:2450–60. doi: 10.1002/art.38713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeffries MA, Donica M, Baker LW, Stevenson ME, Annan AC, Humphrey MB, et al. Genome-wide DNA methylation study identifies significant epigenomic changes in osteoarthritic cartilage. Arthritis Rheumatol. 2014;66:2804–15. doi: 10.1002/art.38762. [DOI] [PubMed] [Google Scholar]
- 36.Moazedi-Fuerst FC, Hofner M, Gruber G, Weinhaeusel A, Stradner MH, Angerer H, et al. Epigenetic differences in human cartilage between mild and severe OA. J Orthop Res. 2014;32:1636–45. doi: 10.1002/jor.22722. [DOI] [PubMed] [Google Scholar]
- 37.Delgado-Calle J, Riancho JA, Klein-Nulend J. Nitric oxide is involved in the down-regulation of SOST expression induced by mechanical loading. Calcif Tissue Int. 2013;94:414–22. doi: 10.1007/s00223-013-9821-8. [DOI] [PubMed] [Google Scholar]
- 38.Thillainadesan G, Chitilian JM, Isovic M, Ablack JN, Mymryk JS, Tini M, et al. TGF-beta-dependent active demethylation and expression of the p15ink4b tumor suppressor are impaired by the ZNF217/CoREST complex. Mol Cell. 2012;46:636–49. doi: 10.1016/j.molcel.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 39.Clarke BL. Anti-sclerostin antibodies: Utility in treatment of osteoporosis. Maturitas. 2014;78:199–204. doi: 10.1016/j.maturitas.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2009;24:578–88. doi: 10.1359/jbmr.081206. [DOI] [PubMed] [Google Scholar]
- 41.Ominsky MS, Vlasseros F, Jolette J, Smith SY, Stouch B, Doellgast G, et al. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res. 2010;25:948–59. doi: 10.1002/jbmr.14. [DOI] [PubMed] [Google Scholar]
- 42.Agholme F, Li X, Isaksson H, Ke HZ, Aspenberg P. Sclerostin antibody treatment enhances metaphyseal bone healing in rats. J Bone Miner Res. 2010;25:2412–8. doi: 10.1002/jbmr.135. [DOI] [PubMed] [Google Scholar]
- 43.Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26:19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 44.Mabey T, Honsawek S, Tanavalee A, Wilairatana V, Yuktanandana P, Saetan N, et al. Plasma and synovial fluid sclerostin are inversely associated with radiographic severity of knee osteoarthritis. Clin Biochem. 2014;47:547–51. doi: 10.1016/j.clinbiochem.2014.03.011. [DOI] [PubMed] [Google Scholar]