

Abstract
Drugs and toxins frequently are associated with the development of various types of acute kidney disease and CKD. Although medications are a widely known cause of tubulointerstitial damage, drug-related glomerular injury is not well appreciated but nonetheless, important. Glomerular damage that occurs after exposure to medications can be caused by direct cellular injury involving the mesangial, endothelial, or visceral epithelial cells (podocytes). Examples include nodular glomerulosclerosis associated with smoking and endothelial injury with thrombotic microangiopathy from a number of medications. Podocyte injury with the development of a minimal change or FSGS lesion has also been described with various medications. Glomerulopathies may also be associated with drug-induced immune-mediated processes. Through various pathways, drugs may promote the formation of a number of antibodies, which may, ultimately, affect the glomerulus. Examples include lupus-like renal lesions and ANCA-related pauci-immune vasculitis. It is critical to recognize these conditions early, because in many patients, there is improvement in renal parameters on stopping the offending medication.
Keywords: glomerular disease, GN, drugs, nephrotoxicity
Medications are thought to be a common cause of kidney disease. However, establishing that a drug has caused an adverse event is fraught with challenges (1,2). There are several reasons for this uncertainty:
The published literature describing drug-associated kidney disease is mostly composed of case reports and small case series.
Drug-associated adverse events may be subclinical, and clinical manifestations are heterogeneous in the affected patient population
Establishing a link between drug and disease may be confounded by additional exposures.
Although hypotheses exist as to how some drugs may cause glomerular disease, no definitive biomarkers exist to distinguish between a patient with idiopathic disease (e.g., systemic lupus or small vessel vasculitis) and a patient with drug-induced disease.
The history of drug exposure is subject to recall bias.
The duration of exposure needed to produce a drug reaction may vary by agent and patient.
Criteria have been proposed to establish a link between drug exposure and the development of an adverse event. In his review on drug-induced vasculitis, Merkel (2) adapts the approach of predecessors by incorporating an assessment of the quality of literature (Table 1). With these criteria in mind, it ultimately depends on clinicians to establish the diagnosis of drug-associated glomerular disease. This is an important distinction that may affect treatment decisions and prevent future disease relapse.
Table 1.
Methodology for establishing a link between a drug and an adverse event
| Methods to link specific drugs with an adverse drug reaction |
| (1) Exclusion of other agents |
| (2) Withdrawal of culprit drug |
| (3) Rechallenge |
| (4) Singularity of drug (i.e., no other potential offending agent used) |
| (5) Consistent pattern of adverse drug reaction |
| (6) Quantitation of drug level (when possible/pertinent) |
| Degrees of certainty for causality of a drug-associated event |
| (1) Causative—ideally would involve a diagnostic test that is specific for a drug-associated event |
| (2) Probable—consistent with a drug event but lacking specific objective evidence for the link between drug and event |
| (3) Possible—the event can be neither confirmed nor excluded as an adverse drug event |
| (4) Coincidental—additional investigation reveals another cause of the event |
| (5) Negative—additional investigation excludes the association (e.g., drug never taken) |
| Quality of evidence for causality of a drug-associated event |
| (1) Excellent—prospective, controlled trials; large case-control series; animal models; large number of reports |
| (2) Good evidence—large case series; separate case reports with consistent pattern of disease and good quality |
| (3) Fair evidence—individual case reports of good quality |
| (4) Poor evidence—individual case reports of poor quality |
Drugs and toxins are well established causes of renal tubulointerstitial injury; however, drug-induced glomerular disease (Figure 1) is also an important concern for clinicians. The glomerular insult associated with drug exposure can be broadly classified into two specific forms: (1) direct cellular toxicity and (2) immune-mediated injury. Direct glomerular cell injury involving the mesangial, endothelial, and visceral epithelial cells is an important form of drug-induced disease. In this setting, some of the lesions described include nodular glomerulosclerosis from smoking, thrombotic microangiopathy (TMA) from numerous medications, minimal change disease (MCD), and FSGS. Drug-induced glomerulopathies from immune-mediated causes consist of lupus-like renal lesions, ANCA-related pauci-immune vasculitis, secondary membranous nephropathy (MN), and MCD.
Figure 1.
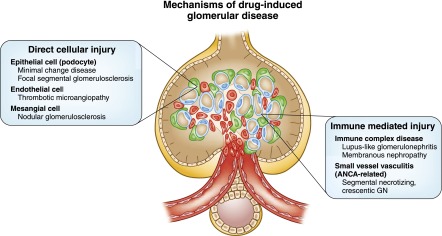
Mechanisms of drug-induced glomerular disease.
This Moving Points feature is comprised of two articles covering drug-induced glomerular injury. In the first article, Markowitz et al. (3) tackle the topic of direct drug and toxin injury to the three cellular compartments of the glomerulus—mesangial, endothelial, and visceral epithelial cells. A little-recognized cause of glomerular injury is heavy cigarette smoking. Although smoking is speculated to cause vascular injury and significant arteriosclerosis in the renal and intrarenal vessels, the resulting glomerular lesion is one that resembles diabetic nodular glomerulosclerosis. There are some differences, the most notable of which is the absence of underlying diabetes mellitus. In the past, many patients were likely inappropriately labeled as having diabetic nephropathy. However, through the work by Markowitz et al. (4) and others (5–7), it is now accepted that idiopathic nodular glomerulosclerosis is a lesion related to heavy cigarette smoking along with hypertension and vascular disease. It is more appropriate to call this renal lesion smoking-associated nodular glomerulosclerosis (8). Importantly, smoking cessation seems to reduce the likelihood of progression to ESRD (4).
TMA is a severe form of endothelial injury that occurs systemically and within the renal parenchyma (9). Many medications and toxins have been observed to cause systemic TMA, which is marked by microangiopathic hemolytic anemia and thrombocytopenia with variable end organ injury as well as renal-limited TMA (10). A drug-induced etiology should always be considered when confronted with this diagnosis. The most common culprits include antiangiogenesis drugs, chemotherapeutic agents, IFN, antiplatelet agents (thienopyridines), calcineurin inhibitors, and quinine (11–14). It is of interest that drugs promote this form of endothelial injury through a diverse number of mechanisms, including direct drug-induced endothelial damage, induction of autoantibodies to ADAMTS-13, and antiplatelet antibodies (as seen with quinine) (10–12). In a small number of patients, ADAMTS-13 deficiency or complement factor H mutations are present, with drugs being the trigger that unmasks the TMA (13). Finally, disturbed angiogenesis through inhibition of the vascular endothelial growth factor pathway may be the most common drug-induced cause of TMA (14).
Drug-induced podocytopathy can occur in a number of situations. IFN therapy is associated with podocyte injury that can manifest as nephrotic syndrome and histologic lesions, including MCD or FSGS, of both the collapsing and noncollapsing variety (15). Pamidronate in high doses can cause direct podocyte injury with impaired cell energetics, disrupted cytoskeleton, or altered cell signaling (16). Chronic lithium exposure, although predominantly associated with tubulointerstitial injury, can be associated with MCD and less commonly, FSGS. It is not clear if this is because of primary podocyte injury or a phenomenon secondary to hyperfiltration. Nonsteroidal anti-inflammatory drugs (NSAIDs) cause a diverse range of renal syndromes (17). MCD is the most common glomerular lesion observed with NSAIDs, which may be because of shunting of AA metabolites into pathways that alter immune function and promote podocyte injury (18). Patients treated with sirolimus can develop proteinuria (sometimes nephrotic range) with a de novo FSGS lesion seen on biopsy (19). Androgenic anabolic steroids are frequently abused by weightlifters and bodybuilders. Both collapsing and noncollapsing FSGS complicate the course of patients exposed to these agents, which manifest clinically as nephrotic proteinuria and AKI (20).
In the second paper, Hogan et al. (21) describe the pathogenic mechanisms underlying immune-mediated injury from drug-induced autoimmunity (DIA). DIA is an idiosyncratic (type B) reaction, which is generally unpredictable and unrelated to the mechanism of action of the drug unlike the type A reaction that is drug dependent and dose related. Starting with the report of lupus-like symptoms with sulfadiazine in 1953, >100 drugs have been implicated in DIA. The classic drugs associated with DIA (procainamide and hydralazine) have now been supplanted with newer drugs, such as TNF-α blockers, and drugs of abuse, such as levamisole-adulterated cocaine. The mechanism of glomerular injury is thought to be from the activation of the adaptive immune system by the drug or metabolites. In DIA from procainamide or hydralazine, autoantibodies are produced in a majority of patients, which could lead to clinical symptoms. The exact sequence of events is not known, but slow acetylators seem to be more prone to develop DIA, implying that high levels of exposure to the drug may be important. In the case of anti–TNF-α agents, a cytokine switch paradigm has been proposed, where a switch from a Th1 to a Th2 cytokine production leads to autoantibody production (22).
DIA is a rare phenomenon occurring in <1% of patients exposed to the drug, leading to manifestations of lupus or vasculitis. Moreover, major organ involvement, particularly kidney involvement, is even rarer and occurs in about 5% of patients with DIA. Fortunately, most of the symptoms abate after withdrawing the medication. In patients where major organ injury is present, immunosuppression may be needed to quell the inflammation and prevent permanent damage (23). Although certain clinical symptoms may predominate with certain drug classes (e.g., cutaneous lesions and serositis with hydralazine, skin vasculitis with necrosis with levamisole-adulterated cocaine, and cutaneous and systemic symptoms with anti–TNF-α inhibitors), it is important to note that there is not a classic syndrome ascribed to any one particular drug class. Similarly, although antihistone antibodies are common (especially with procainamide) and antinative DNA antibodies and low complement levels are not commonly seen, these serologic profiles are not consistent in DIA. From a diagnostic and therapeutic standpoint, it is sometimes difficult to ascribe a drug as being directly causative versus unmasking a preexisiting autoimmune syndrome. One observation that may provide a clue to DIA is the simultaneous presence of antibodies associated with lupus (antinuclear antibody, antihistone antibody, and antiphosholipid antibody) along with ANCA (antimyeloperoxidase antibody) in the patient with an autoimmune syndrome (23).
MN is the other form of DIA. Drugs used to treat rheumatoid arthritis, including pencillamine and gold salts, were associated with MN. These drugs are rarely used nowadays. At this time, drug-induced MN is rare and has been reported with organic mercurials in skin-lightening creams, the newer rheumatoid arthritis drug adalimumab, and NSAIDs, including celecoxib (25).
We hope that CJASN readers will find this Moving Points feature discussing the role of drugs in causing glomerular pathology a valuable resource. It is crucial that nephrologists are familiar with the glomerulopathies that develop in patients exposed to certain drugs. It will allow rapid diagnosis and therapy, including drug withdrawal and other therapies that target the mechanism of drug-induced injury.
Disclosures
None.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Irey NS: Teaching monograph. Tissue reactions to drugs. Am J Pathol 82: 613–647, 1976 [PMC free article] [PubMed] [Google Scholar]
- 2.Merkel PA: Drug-induced vasculitis. Rheum Dis Clin North Am 27: 849–862, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Markowitz, Bomback, Perazella: Clin J Am Soc Nephrol 7: 1292–1300, 2015 [Google Scholar]
- 4.Markowitz GS, Lin J, Valeri AM, Avila C, Nasr SH, D’Agati VD: Idiopathic nodular glomerulosclerosis is a distinct clinicopathologic entity linked to hypertension and smoking. Hum Pathol 33: 826–835, 2002 [DOI] [PubMed] [Google Scholar]
- 5.Alpers CE, Biava CG: Idiopathic lobular glomerulonephritis (nodular mesangial sclerosis): A distinct diagnostic entity. Clin Nephrol 32: 68–74, 1989 [PubMed] [Google Scholar]
- 6.Kuppachi S, Idris N, Chander PN, Yoo J: Idiopathic nodular glomerulosclerosis in a non-diabetic hypertensive smoker—case report and review of literature. Nephrol Dial Transplant 21: 3571–3575, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Li W, Verani RR: Idiopathic nodular glomerulosclerosis: A clinicopathologic study of 15 cases. Hum Pathol 39: 1771–1776, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Nasr SH, D’Agati VD: Nodular glomerulosclerosis in the nondiabetic smoker. J Am Soc Nephrol 18: 2032–2036, 2007 [DOI] [PubMed] [Google Scholar]
- 9.George JN, Nester CM: Syndromes of thrombotic microangiopathy. N Engl J Med 371: 654–666, 2014 [DOI] [PubMed] [Google Scholar]
- 10.Zakarija A, Bennett C: Drug-induced thrombotic microangiopathy. Semin Thromb Hemost 31: 681–690, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Glezerman I, Kris MG, Miller V, Seshan S, Flombaum CD: Gemcitabine nephrotoxicity and hemolytic uremic syndrome: Report of 29 cases from a single institution. Clin Nephrol 71: 130–139, 2009 [DOI] [PubMed] [Google Scholar]
- 12.Hunt D, Kavanagh D, Drummond I, Weller B, Bellamy C, Overell J, Evans S, Jackson A, Chandran S: Thrombotic microangiopathy associated with interferon beta. N Engl J Med 370: 1270–1271, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orvain C, Augusto JF, Besson V, Marc G, Coppo P, Subra JF, Sayegh J: Thrombotic microangiopathy due to acquired ADAMTS13 deficiency in a patient receiving interferon-beta treatment for multiple sclerosis. Int Urol Nephrol 46: 239–242, 2014 [DOI] [PubMed] [Google Scholar]
- 14.Izzedine H, Massard C, Spano JP, Goldwasser F, Khayat D, Soria JC: VEGF signalling inhibition-induced proteinuria: Mechanisms, significance and management. Eur J Cancer 46: 439–448, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Markowitz GS, Nasr SH, Stokes MB, D’Agati VD: Treatment with IFN-alpha, -beta, or -gamma is associated with collapsing focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 5: 607–615, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perazella MA, Markowitz GS: Bisphosphonate nephrotoxicity. Kidney Int 74: 1385–1393, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Brezin JH, Katz SM, Schwartz AB, Chinitz JL: Reversible renal failure and nephrotic syndrome associated with nonsteroidal anti-inflammatory drugs. N Engl J Med 301: 1271–1273, 1979 [DOI] [PubMed] [Google Scholar]
- 18.Pirani CL, Valeri A, D’Agati V, Appel GB: Renal toxicity of nonsteroidal anti-inflammatory drugs. Contrib Nephrol 55: 159–175, 1987 [DOI] [PubMed] [Google Scholar]
- 19.Letavernier E, Bruneval P, Mandet C, Duong Van Huyen JP, Péraldi MN, Helal I, Noël LH, Legendre C: High sirolimus levels may induce focal segmental glomerulosclerosis de novo. Clin J Am Soc Nephrol 2: 326–333, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Herlitz LC, Markowitz GS, Farris AB, Schwimmer JA, Stokes MB, Kunis C, Colvin RB, D’Agati VD: Development of focal segmental glomerulosclerosis after anabolic steroid abuse. J Am Soc Nephrol 21: 163–172, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hogan, Markowitz, Radhakrishnan: Clin J Am Soc Nephrol 7: 1301–1311, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubin RL: Drug-induced lupus. Expert Opin Drug Saf 14: 361–378, 2015 [DOI] [PubMed] [Google Scholar]
- 23.Xiao X, Chang C: Diagnosis and classification of drug-induced autoimmunity (DIA). J Autoimmun 48-49: 66–72, 2014 [DOI] [PubMed] [Google Scholar]
- 24.Wiik A: Clinical and laboratory characteristics of drug-induced vasculitic syndromes. Arthritis Res Ther 7: 191–192, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Izzedine H, Launay-Vacher V, Bourry E, Brocheriou I, Karie S, Deray G: Drug-induced glomerulopathies. Expert Opin Drug Saf 5: 95–106, 2006 [DOI] [PubMed] [Google Scholar]