

Abstract
Calcium, phosphate, and magnesium are multivalent cations that are important for many biologic and cellular functions. The kidneys play a central role in the homeostasis of these ions. Gastrointestinal absorption is balanced by renal excretion. When body stores of these ions decline significantly, gastrointestinal absorption, bone resorption, and renal tubular reabsorption increase to normalize their levels. Renal regulation of these ions occurs through glomerular filtration and tubular reabsorption and/or secretion and is therefore an important determinant of plasma ion concentration. Under physiologic conditions, the whole body balance of calcium, phosphate, and magnesium is maintained by fine adjustments of urinary excretion to equal the net intake. This review discusses how calcium, phosphate, and magnesium are handled by the kidneys.
Keywords: renal physiology, calcium, cell and transport physiology, electrolytes, ion, channel
Introduction
Imbalances of calcium, phosphorus, and magnesium result in a number of serious clinical complications, including arrhythmias, seizures, and respiratory difficulties. The kidney plays a critical role in regulating serum levels of these ions. Regulation of calcium, phosphate, and magnesium occurs in different parts of the nephron and involves a number of different channels, transporters, and pathways. Below we describe the mechanisms governing renal control of these ions.
Calcium
The total amount of calcium in the human body ranges from 1000 to 1200 g. Approximately 99% of body calcium resides in the skeleton; the other 1% is present in the extracellular and intracellular spaces. Although >99% of the total body calcium is located in bone, calcium is a critical cation in both the extracellular and intracellular spaces. Approximately 1% of the calcium in the skeleton is freely exchangeable with calcium in the extracellular fluid compartment. Serum calcium concentration is held in a very narrow range in both spaces. Calcium serves a vital role in nerve impulse transmission, muscular contraction, blood coagulation, hormone secretion, and intercellular adhesion (1,2).
Gastrointestinal Absorption of Calcium
Calcium balance is tightly regulated by the concerted action of calcium absorption in the intestine, reabsorption in the kidney, and exchange from bone, which are all under the control of the calciotropic hormones that are released upon a demand for calcium (Figure 1A). In healthy adults, approximately 800–1000 mg of calcium should be ingested daily. This amount will vary depending on the amount of dairy product consumed. When 1 g of calcium is ingested in the diet, approximately 800 mg is excreted in the feces and 200 mg in the urine. Approximately 400 mg of the usual 1000 mg dietary calcium intake is absorbed by the intestine, and calcium loss by way of intestinal secretions is approximately 200 mg/d. Therefore, a net absorption of calcium is approximately 200 mg/d (20%) (3). Although serum calcium levels can be maintained in the normal range by bone resorption, dietary intake is the only source by which the body can replenish stores of calcium in bone. Calcium is absorbed almost exclusively within the duodenum, jejunum, and ileum. Each of these intestinal segments has a high absorptive capacity for calcium, with their relative calcium absorption being dependent on the length of each respective intestinal segment and the transit time of the food bolus (3).
Figure 1.
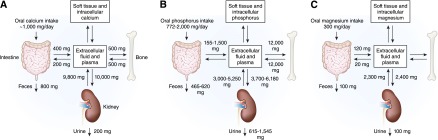
Calcium, phosphate, and magnesium flux between body compartments. Calcium (A), phosphate (B), and magnesium (C) balance is a complex process involving bone, intestinal absorption of dietary calcium, phosphate, and magnesium, and renal excretion of calcium, phosphate, and magnesium.
There are two routes for the absorption of calcium across the intestinal epithelium: the paracellular pathway (i.e., between the cells) and the transcellular route (i.e., through the cell) (Figure 2A). The paracellular pathway is passive, and it is the predominant route of calcium absorption when the lumen concentration of calcium is high. The paracellular route is indirectly influenced by calcitriol [1,25(OH)2D] because it is capable of altering the structure of intracellular tight junctions by activation of protein kinase C, making the tight junction more permeable to the movement of calcium. However, 1,25(OH)2D mainly controls the active absorption of calcium. Calcium moves down its concentration gradient through a calcium channel into the apical section of the microvillae. Because the luminal concentration of calcium is usually much higher than the intracellular concentration of calcium, a large concentration gradient favors the passive movement of calcium. Calcium is rapidly and reversibly bound to the calmodulin-actin-myosin I complex. Calcium moves to the basolateral area of the cell by way of microvesicular transport. As the calmodulin-actin-myosin I complex becomes saturated with calcium, the concentration gradient becomes less favorable, which slows down calcium absorption. 1,25(OH)2D exerts influence on the intestinal epithelial cells to increase their synthesis of calbindin. Calcium binds to calbindin, thereby unloading the calcium-calmodulin complexes, which then remove calcium from the microvilli region. This decrease in calcium concentration again favors the movement of calcium into the microvilli. As the calbindin-calcium complex dissociates, the free intracellular calcium is actively extruded from the cell sodium-calcium (Na-Ca) exchanger (4,5).
Figure 2.
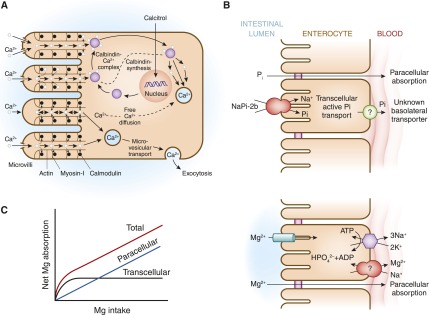
Intestinal pathways for calcium, phosphorus, and magnesium absorption. (A) Proposed pathways for calcium (Ca) absorption across the intestinal epithelium. Two routes exist for the absorption of Ca across the intestinal epithelium: the paracellular pathway and the transcellular route. (B) Proposed pathways for phosphorus (Pi) absorption across the intestinal epithelium. NaPi2b mediates active transcellular transport of Pi. A paracellular pathway is also believed to exist. (C) Proposed pathways for magnesium (Mg) absorption across the intestinal epithelium. Apical absorption is mediated by the TRPM6/TRPM7 channel, whereas basolateral exit occurs by an Mg exchanger that is yet to be fully defined. A paracellular pathway is also believed to exist. TRPM, transient receptor potential melastatin.
Renal Regulation of Calcium Balance
Total serum calcium consists of ionized, protein bound, and complexed fractions (approximately 48%, 46%, and 7%, respectively). The complexed calcium is bound to molecules such as phosphate and citrate. The ultrafilterable calcium equals the total of the ionized and complexed fractions. Normal total serum calcium is approximately 8.9–10.1 mg/dl (about 2.2–2.5 mmol/l). Calcium can be bound to albumin and globulins. For each 1.0-g/dl decrease in serum albumin, total serum calcium decreases by 0.8 mg/dl. For each 1.0-g/dl decrease in serum globulin fraction, total serum calcium decreases by 0.12 mg/dl. Acute alkalosis decreases the ionized calcium. Because both hydrogen ions and calcium are bound to serum albumin, in the presence of metabolic alkalosis, bound hydrogen ions dissociate from albumin, freeing up the albumin to bind with more calcium and thereby decreasing the freely ionized portion of the total serum calcium. For every 0.1 change in pH, ionized calcium changes by 0.12 mg/dl (6).
In humans who have a GFR of 170 liters per 24 hours, roughly 10 g of calcium is filtered per day. The amount of calcium excreted in the urine usually ranges from 100 to 200 mg per 24 hours; hence, 98%–99% of the filtered load of calcium is reabsorbed by the renal tubules. Approximately 60%–70% of the filtered calcium is reabsorbed in the proximal convoluted tubule, 20% in the loop of Henle, 10% by the distal convoluted tubule, and 5% by the collecting duct. The terminal nephron, although responsible for the reabsorption of only 5%–10% of the filtered calcium load, is the major site for regulation of calcium excretion (1) (Figure 3A).
Figure 3.
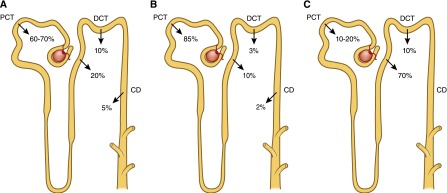
Schematic illustration of the reabsorption of calcium, phosphorus, and magnesium by different segments of the nephron. (A) Calcium is filtered at the glomerulus, with the ultrafilterable fraction of plasma calcium entering the proximal tubule. Within the proximal convoluted tubule and the proximal straight tubule, 60%–70% of the filtered calcium has been reabsorbed. No reabsorption of calcium occurs within the thin segment of the loop of Henle. The cortical segments of the loop of Henle reabsorb about 20% of the initially filtered load of calcium. Approximately 10% of the filtered calcium is reabsorbed in the distal convoluted tubule, with another 3%–10% of filtered calcium reabsorbed in the connecting tubule. (B) The majority (approximately 85%) of phosphate reabsorption occurs in the proximal convoluted tubule. Approximately 10% of Pi reabsorption occurs in the loop of Henle, 3% occurs in the distal convoluted tubule, and 2% in the collecting duct via unidentified pathways. (C) Approximately 10%–30% of the filtered magnesium is absorbed in the proximal tubule, 40%–70% of filtered magnesium is absorbed in the thick ascending limb, and the remaining 5%–10% of magnesium is reabsorbed in the distal convoluted tubule. CD, collecting duct; DCT, distal convoluted tubule; PCT, proximal convoluted tubule.
The reabsorption of calcium in the proximal convoluted tubule parallels that of sodium and water. Proximal tubular calcium reabsorption is thought to occur mainly by passive diffusion and solvent drag. This is based on the observation that the ratio of calcium in the proximal tubule fluid to that in the glomerular filtrate is 1:1.2. The passive paracellular pathways account for approximately 80% of calcium reabsorption in this segment of the nephron. A small but significant component of active calcium transport is observed in the proximal tubules. The active transport of calcium proceeds in a two-step process, with calcium entry from the tubular fluid across the apical membrane and exit though the basolateral membrane. This active transport is generally considered to constitute 10%–15% of total proximal tubule calcium reabsorption and it is mainly regulated by parathyroid hormone (PTH) and calcitonin (7).
No reabsorption of calcium occurs within the thin segment of the loop of Henle (Figure 3A). In the thick ascending limb of the loop of Henle, 20% of the filtered calcium is reabsorbed largely by the cortical thick ascending limb, through both transcellular and paracellular routes. In the thick ascending limb, the bulk of calcium reabsorption proceeds through the paracellular pathway and is proportional to the transtubular electrochemical driving force. The apical Na+-K+-2Cl− cotransporter NKCC2 and the renal outer medullary potassium K+ (ROMK) channel generate the “driving force” for paracellular cation transport. Whereas NaCl reabsorption through NKCC2 is electroneutral (NKCC2 translocates one Na+, one K+, and two Cl− ions from the lumen into the cell), apical potassium represents the rate-limiting step of this process and potassium ions back-diffuse into the lumen through the ROMK channels. Na+ and Cl− accumulated inside the cell are then transported into the bloodstream through basolateral Na+-K+-ATPase and Cl− channels, respectively. Overall, these processes yield a net cellular reabsorption of NaCl and the generation of a lumen-positive transepithelial potential difference, which drives nonselective calcium reabsorption through the paracellular route (8) (Figure 4). Calcium transport in the thick ascending limb of the loop of Henle is also influenced by the calcium-sensing receptor (CaSR) (7–10), which is localized in the basolateral membrane. How CaSR controls the calcium reabsorption in the thick ascending limb is now better understood. Using microdissected, in vitro microperfused rat cortical thick ascending limb, Loupy et al. showed that an acute inhibition of the CaSR does not alter NaCl reabsorption or the transepithelial potential difference but increased the permeability to calcium of the paracellular pathway (11). The tight junction in the thick ascending limb expresses several claudins, including claudin-14, claudin-16, and claudin-19. A normal expression of claudin-16 and claudin-19 is required for a normal absorption of divalent cations in this tubular segment. Toka et al. reported that the disruption of CaSR decreases the abundance of the claudin-14 mRNA and increases that of the claudin-16 mRNA (12). A treatment by cinacalcet increases the abundance of claudin-14 mRNA, and in cell culture models overexpression of claudin-14, decreases the paracellular permeability to calcium (8,12). Calciotropic hormones, such as PTH and calcitonin, stimulate active cellular calcium absorption in the cortical thick ascending limb (7).
Figure 4.

Model of calcium and magnesium absorption by the thick ascending limb of Henle. Calcium absorption proceeds through both an active, transcellular pathway and by a passive paracellular pathway. Only transport pathways relevant to calcium absorption are shown. Basal absorption is passive and is driven by the ambient electrochemical gradient for calcium. The apical Na+-K+-2Cl− cotransporter and the renal outer medullary potassium K+ channel generate the “driving force” for paracellular cation transport. Calciotropic hormones, such as parathyroid hormone and calcitonin, stimulate active calcium absorption in cortical thick ascending limbs. Inhibition of Na-K-2Cl cotransport by loop diuretics or in Bartter’s syndrome decreases the transepithelial voltage, thus diminishing passive calcium absorption. In the model of magnesium absorption by thick ascending limb of Henle, 40%–70% of filtered magnesium is absorbed in the thick ascending limb by a paracellular pathway, mostly enhanced by lumen-positive transepithelial voltage. The apical Na-K-2Cl cotransporter mediates apical absorption of Na, K, and Cl. The apical renal outer medullary K channel mediates apical recycling of K back to the tubular lumen and generates lumen-positive voltage. Cl channel Kb mediates Cl exit through the basolateral membrane. Here Na,K-ATPase also mediates Na exit through the basolateral membrane and generates the Na gradient for Na absorption. The tight junction proteins claudin-16 and claudin-19 play a prominent role in magnesium absorption. The calcium-sensing receptor was also recently determined to regulate magnesium transport in this segment: upon stimulation, magnesium transport is decreased. CaSR, calcium-sensing receptor.
In contrast with the proximal tubule and the thick ascending limb of the loop of Henle, the distal tubule reabsorbs calcium exclusively via the transcellular route. The distal convoluted tubule absorbs 5%–10% of the filtered calcium. Calcium absorption in this segment is active because it proceeds against a chemical and an electrical gradient. This active process can be divided into three steps. The first step requires calcium influx across the apical membrane. The transient receptor potential vanilloid 5 has been identified as the responsible protein in this process. The second step is the diffusion of calcium through the cytosol. During this process, calbindin-D28k binds intracellular calcium transported via transient receptor potential vanilloid 5 and shuttles it through the cytosol toward the basolateral membrane where calcium is extruded via sodium-calcium exchanger NCX1 and the plasma membrane calcium-ATPase PMCA1b, which is the final step in this process (13–15). Figure 5 is a cell model of the three-step process of transcellular calcium transport.
Figure 5.

Model of calcium and magnesium absorption by distal convoluted tubules. Calcium entry across the plasma membrane proceeds through calcium channels with basolateral exit occurring through a combination of the plasma membrane ATPase and Na+-Ca+ exchanger. Calcium absorption is entirely transcellular. Calciotropic hormones such as parathyroid hormone and calcitonin stimulate calcium absorption. Calcitriol [1,25(OH)2D] stimulates calcium absorption through the activation of nuclear transcription factors. Inhibition of the apical NaCl cotransporter by thiazide diuretics or in Gitelman’s syndrome indirectly stimulates calcium absorption. In the model of magnesium absorption by distal convoluted tubules, approximately 5%–10% of magnesium is reabsorbed in the distal convoluted tubule mainly by active transcellular transport mediated by TRPM6. The absorbed magnesium is then extruded via a recently identified magnesium/sodium exchanger across the basolateral membrane. The apical K channel Kv1.1 potentiates TRPM6-mediated magnesium absorption by establishing favorable luminal potential. In addition, the basolateral K channel Kir4.1 and the γ-subunit of Na,K-ATPase also regulate magnesium reabsorption.
Hormonal and Other Factors Regulating Renal Calcium Handling
PTH.
Many physiologic, pharmacologic, and pathologic factors influence renal calcium absorption (Table 1). The most important regulator is PTH, which stimulates calcium absorption. PTH is a polypeptide secreted from the parathyroid gland in response to a decrease in the plasma concentration of ionized calcium. Therefore, the major physiologic role of the parathyroid gland is to regulate calcium homeostasis. PTH acts to increase the plasma concentration of calcium in three ways: (1) it stimulates bone resorption, (2) it enhances intestinal calcium and phosphate absorption by promoting the formation within the kidney of 1,25(OH)2D, and (3) it augments active renal calcium absorption. These effects are reversed by small changes in the serum calcium concentration that lower PTH secretion.
Table 1.
Factors that alter renal regulation of calcium
| Increase Calcium Absorption | Decrease Calcium Absorption |
| Hyperparathyroidism | Hypoparathyroidism |
| Calcitriol | Low calcitriol levels |
| Hypocalcemia | Hypercalcemia |
| Volume contraction | Extracellular fluid expansion |
| Metabolic alkalosis | Metabolic acidosis |
| Thiazides diuretics | Loop diuretics |
PTH secretion is tightly regulated on a transcriptional and post-transcriptional level dependent on the concentration of extracellular calcium. In fact, PTH gene transcription is increased by hypocalcemia, glucocorticoids, and estrogen. Hypercalcemia also can increase the intracellular degradation of PTH (16). PTH release is increased by hypocalcemia, adrenergic agonists, dopamine, and prostaglandin E2 (16). Changes in serum calcium are sensed by the CaSR, which is localized in the cell membrane of the parathyroid cells. The receptor permits variations in the plasma calcium concentration to be sensed by the parathyroid gland, leading to desired changes in PTH secretion (16).
Vitamin D.
Vitamin D3 (cholecalciferol) is a fat-soluble steroid that is present in the diet and also can be synthesized in the skin from 7-dehydrocholestrol in the presence of ultraviolet light. The hepatic enzyme 25-hydroxylase catalyzes the hydroxylation of vitamin D at the 25 position, resulting in the formation of 25-hydroxyvitamin D or calcidiol. 25-Hydroxyvitamin D produced by the liver enters the circulation and travels to the kidney, bound to vitamin D binding protein. In the kidney, tubular cells contain two enzymes (1α-hydroxylase and 24α-hydroxylase) that can further hydroxylate calcidiol, producing 1,25(OH)2D, the most active form of vitamin D, or 24,25-dihydroxyvitamin D, an inactive metabolite (17). Hence, the vitamin D hormonal system consists of multiple forms, ranging from cutaneous precursors or dietary components to the most active metabolite, 1,25(OH)2D, which acts upon the target organ receptors to maintain calcium homeostasis and bone health. However, the serum concentration of 25(OH)D, which is the precursor form of the biologically active vitamin D, is the best indicator of the overall vitamin D storage or status (18). The 1,25(OH)2D enters the circulation and is transported to the small intestine, where it enhances intestinal calcium absorption. The most important endocrine effect of 1,25(OH)2D in the kidney is a tight control of its own homeostasis through simultaneous suppression of 1α-hydroxylase and stimulation of 24-hydroxylase. An intact 1,25(OH)2D–vitamin D receptor system is critical for both basal and PTH-induced osteoclastogenesis. Mature osteoclasts release calcium and phosphorus from the bone, maintaining the appropriate levels of the two minerals in the plasma (17,18).
Serum Calcium.
Hypercalcemia is associated with an increase in urinary calcium excretion as a consequence of an increase in the filtered load and a decrease in the tubular reabsorption of calcium. Although hypercalcemia can decrease GFR by renal vasoconstriction, which tends to offset the increase in filtered load, hypercalcemia also causes a decline in tubular reabsorption of calcium by both PTH-dependent and -independent effects. Hypocalcemia decreases renal calcium excretion by decreasing the filtered load and enhancing the tubular reabsorption of calcium (1,17).
Extracellular Fluid.
Expansion of the extracellular fluid is associated with an increase in sodium, chloride, and calcium excretion, whereas reciprocal effects are seen with volume contraction. The mechanisms of this effect are interrelated with the effects of sodium reabsorption and compensatory changes that occur as a result of volume expansion (1).
Metabolic Acidosis.
Acute and chronic metabolic acidosis can be associated with an increase in calcium excretion, independent of PTH changes. The calciuria may, in part, be due to the mobilization of calcium from bone, as the hydrogen ion is buffered in the skeleton; however, direct effects of acidosis on tubular calcium resorption also play a role (1,7).
Diuretics.
Loop diuretics decrease calcium absorption as a result of inhibition of the transport of sodium chloride at the NKCC2 transporter in the ascending loop of Henle. Thiazide diuretics, which act in the distal tubule, are associated with hypocalciuria (1,7). Two main mechanisms have been proposed to explain the effect of thiazides on calcium excretion: (1) increased proximal sodium and water reabsorption due to volume depletion, and (2) increased distal calcium reabsorption at the thiazide-sensitive site in the distal convoluted tubule.
Clinical Consequences of Alterations in Calcium Balance
Hypocalcemia.
The main causes of hypocalcemia can be categorized as follows: (1) lack of PTH (e.g., hereditary or acquired hypoparathyroidism), (2) lack of vitamin D (e.g., dietary deficiency or malabsorption, inadequate sunlight, and defective metabolism such as in liver and kidney disease), and (3) increased calcium complexation (e.g., bone hunger syndrome, rhabdomyolysis, acute pancreatitis, tumor lysis syndrome) (19). Low serum calcium stimulates both PTH synthesis and release. Both hypocalcemia and PTH increase the activity of the 1α-hydroxylase enzyme in the proximal tubular cells of the nephron, which increases the synthesis of 1,25(OH)2D. PTH increases bone resorption by osteoclast. In addition, PTH and 1,25(OH)2D stimulate calcium reabsorption in the distal convoluted tubule. 1,25(OH)2D increases the fractional absorption of dietary calcium by the gastrointestinal tract. All of these mechanisms aid in returning the serum calcium to normal levels.
Hypercalcemia.
Important causes of hypercalcemia include excess PTH production (e.g., primary hyperparathyroidism), excess 1,25(OH)2D (e.g., vitamin D intoxication, sarcoidosis), increased bone resorption (e.g., metastatic osteolytic tumors, humoral hypercalcemia, immobilization, Paget’s disease), increased intestinal absorption of calcium (e.g., milk-alkali syndrome), decreased renal excretion of calcium (e.g., thiazides), and impaired bone formation (e.g., adynamic bone disease). Elevated serum calcium inhibits PTH synthesis and release. Decreases in PTH and hypercalcemia are known to decrease the activity of the 1α-hydroxylase enzyme, which, in turn, decrease the synthesis of 1,25(OH)2D. Hypercalcemia also stimulates the C cells in the thyroid gland that increase synthesis of calcitonin. Bone resorption by osteoclasts is blocked by the increased calcitonin and decreased PTH. The decrease in 1,25(OH)2D decreases gastrointestinal tract absorption of dietary calcium. Low levels of PTH and 1,25(OH)2D also inhibit calcium reabsorption in the distal convoluted tubule, which increases renal calcium excretion. Finally, in the setting of hypercalcemia, activation of the basolateral CaSr inhibits ROMK channels, which are important contributors to the potassium recycling into the lumen of the thick ascending limb of the loop of Henle. This effect of hypercalcemia limits the rate of Na+-K+-2CL− cotransport by decreasing the availability of luminal K+ and therefore, dissipating the lumen-positive transepithelial voltage. The end result is that the CaSR activation diminishes paracellular sodium, calcium, and magnesium transport, producing a phenotype similar to Bartter’s syndrome. The signaling pathways underpinning the inhibitory effects of CaSR activation on NKCC2 and ROMK activities involve, at least in part, production of cytochrome P450 metabolites and/or of prostaglandins (8–10). All of these effects tend to return serum calcium to normal levels. Main therapy for hypercalcemia includes saline and loop diuretics that increase renal excretion of calcium and bisphosphonates, which inhibit bone resorption (19).
Phosphorus
Daily Phosphorus Balance
At steady state, oral phosphorus intake is balanced by phosphate (Pi) excretion in the urine and feces (Figure 1B). Daily phosphorus intake varies between 700 and 2000 mg, depending on consumption of phosphorus-rich foods, such as dairy products. After absorption, phosphorus is transported across cell membranes as phosphate (31 mg/l elemental phosphorus=1 mmol/l phosphate). Phosphate in the plasma or extracellular fluid undergoes one of three fates: transport into cells, deposition in bone or soft tissue, or elimination predominantly by the kidneys. Within the body, the majority of phosphorus stores are in the bone. Although serum phosphate (Pi) levels constitute <1% of total body phosphorus stores, maintenance of serum Pi within a relatively narrow range (2.5–4.5 mg/dl in adulthood) is crucial for several important cellular processes, including energy metabolism, bone formation, signal transduction, or as a constituent of phospholipids and nucleic acids (20). Maintenance of serum phosphate within the normal range depends on a complex interplay between absorption of phosphate in the gut, exchange with bone stores, shifts between intracellular and intravascular compartments, and renal excretion.
Gastrointestinal Absorption of Phosphate
Although the kidneys are the major regulators of phosphate homeostasis, serum levels of phosphate are also altered by intestinal Pi absorption mediated by the type IIb NaPi cotransporters Npt2b (21) (Figure 2B). Npt2b is regulated by dietary phosphate intake as well as 1,25(OH)2D. In the rat, phosphate reabsorption is greatest in the duodenum and the jejunum, with very little occurring in the ileum. By contrast, in the mouse, Pi is absorbed along the entire intestine with the highest levels of Pi reabsorption occurring in the ileum. The human pattern of intestinal phosphate reabsorption is thought to resemble that found in the rat.
Renal Regulation of Phosphate Balance
The kidney plays a key role in phosphate homeostasis. In normal adults, between 3700 and 6100 mg/d of phosphorus is filtered by the glomerulus (Figure 1B). Net renal excretion of phosphorus is between 600 and 1500 mg/d, which means that between 75% and 85% of the daily filtered load is reabsorbed by the renal tubules.
Maintenance of normal serum phosphorus levels is primarily achieved through a tightly regulated process of Pi reabsorption from the glomerular filtrate. Within the nephron, approximately 85% of phosphate reabsorption occurs within the proximal tubule (Figure 3B). The remainder of the nephron plays a minor role in Pi regulation and the transporters involved have yet to be identified (22).
Within the proximal tubule, Pi transport from the ultrafiltrate across the proximal tubule epithelium is an energy-dependent process that requires sodium (23). The three renal sodium phosphate cotransporters, Npt2a, Npt2c, and PiT-2, are all positioned in the apical brush border membrane of renal proximal tubule cells and use the energy derived from the transport of sodium down its gradient to move inorganic phosphate from the luminal filtrate into the cell (Figure 6). The amount of phosphate reabsorbed from the filtrate is determined by the abundance of the cotransporters in the apical membrane of proximal tubule cells and not by any alterations in the rate or affinity of Pi transport by post-transcriptional modifications (23). Thus, hormones or dietary factors that alter phosphate reabsorption in the kidney do so by changing the abundance of the sodium phosphate cotransporters in the apical membrane of renal proximal tubule cells. An increase in the brush border levels of the sodium phosphate cotransporters abundance results in increased phosphate absorption from the urine, whereas a decrease in cotransporter abundance leads to phosphaturia. Transport of Pi from the renal proximal tubule to the peritubular capillaries occurs via an unknown basolateral transporter (24).
Figure 6.

Model of phosphate reabsorption in the renal proximal tubule. Pi is reabsorbed via three sodium phosphate cotransporters: Npt2a, Npt2c and PiT-2. In humans, Npt2a and Npt2c are believed to play the most important role in phosphate reabsorption. The sodium phosphate cotransporters, which are positioned in the apical membrane of renal proximal tubule cells, use energy derived from the movement of sodium down its gradient to move Pi from the filtrate to the cell interior. The amount of phosphate reabsorbed is dependent on the abundance of the sodium phosphate cotransporters in the apical brush border membrane and hormones such as parathyroid hormone and fibroblast growth factor-23 decrease Pi reabsorption by decreasing the abundance of the sodium phosphate cotransporters in the brush border. Movement of phosphate from the interior of renal proximal tubular cells to the peritubular capillaries occurs via an unknown transporter.
Npt2a and Npt2c are encoded by members of the SLC34 solute carrier family (25), of which Npt2a is encoded by SLC34A1 (26), whereas Npt2c is encoded by SLC34A3 (27). PiT-2 (SLC20A2) is encoded by a member of the SLC20 family (28). Within the proximal tubule in rats and mice, the abundance of Npt2a gradually decreases along the proximal tubule, whereas Npt2c and PiT-2 are expressed mainly in the first (S1) segment (29). Work from knockout studies in rodents has shown that in mice, Npt2a is responsible for the majority (approximately 70%) of the renal regulation of phosphate transport (29). By contrast, linkage analyses have found that in humans, Npt2a and Npt2c may contribute equally to phosphate reabsorption (30,31). The importance of PiT-2 in the renal control of Pi in humans remains unclear (20).
There are a number of functional differences between the three cotransporters (Figure 6). Both Npt2a and Npt2c preferentially transport divalent phosphate. However, Npt2a is electrogenic, transporting three sodium ions into the cell for every one phosphate ion, whereas Npt2c is electroneutral, transporting two sodium ions for every one phosphate ion (32,33). PiT-2, on the other hand, although electrogenic like NaPi2a, preferentially transports monovalent phosphate (23). Changes in pH also affect the transporters differently. Npt2a and Npt2c show a doubling of Pi transport between pH 6.5 and 8, whereas the transport rate of PiT-2 is constant over this range (34,35).
Renal control of phosphate reabsorption is regulated by a number of hormonal and metabolic factors (Table 2) that are discussed below in more detail. These factors change phosphate reabsorption from the ultrafiltrate by changing the abundance of the three sodium phosphate cotransporters in the brush border membrane of the proximal tubule. It should be noted that the time course of response to various stimulatory or inhibitory factors differs between the three cotransporters. In general, dietary or hormonal changes result in relatively rapid (minutes to hours) insertion or removal of Npt2a from the brush border membrane, whereas regulation of Npt2c and PiT-2 occurs more slowly (hours to days) (22).
Table 2.
Factors that alter renal regulation of phosphate
| Increase Phosphate Absorption | Decrease Phosphate Absorption |
| Low-phosphate diet | Parathyroid hormone |
| 1,25-Vitamin D3 | Phosphatonins (e.g., FGF23) |
| Thyroid hormone | High-phosphate diet |
| Metabolic acidosis | |
| Potassium deficiency | |
| Glucocorticoids | |
| Dopamine | |
| Hypertension | |
| Estrogen |
FGF23, fibroblast growth factor-23.
Dietary Factors Regulating Renal Phosphate Handling
In animals with normal renal function, ingestion of phosphorus-containing foods leads to removal of Npt2a, Npt2c, and PiT-2 from the proximal tubule brush border membrane, thereby decreasing phosphate reabsorption from the ultrafiltrate. By contrast, dietary Pi restriction leads to insertion of the sodium phosphate cotransporters in the proximal tubule brush border membrane, increasing phosphate reabsorption.
Potassium deficiency leads to an increase in phosphate excretion in the urine despite a paradoxical increase in the abundance of Npt2a in the proximal tubule brush border membrane that should increase phosphate reabsorption. Potassium deficiency leads to changes in the brush border membrane lipid composition that are thought to inhibit Npt2a activity (36).
Hormonal and Other Factors Regulating Renal Phosphate Handling
PTH.
PTH causes decreased renal reabsorption of phosphate and phosphaturia by decreasing the abundance of Npt2a, Npt2c, and PiT-2 in the renal proximal tubule brush border membrane (Figure 7). In response to PTH, Npt2a is removed rapidly (within minutes), whereas the decrease in apical membrane abundance of Npt2c and PiT-2 takes hours (27,37,38). The sodium phosphate cotransporter response to PTH involves several kinases, including protein kinases A and C and mitogen-activated protein kinase extracellular signal-regulated kinase 1/2, as well as a myosin motor (myosin VI) (39–41).
Figure 7.

Schematic of PTH-induced homeostatic mechanisms to correct hyperphosphatemia as GFR falls. As GFR falls, serum phosphorus levels increase, which stimulates release of PTH from the parathyroid glands. This in turn decreases the brush border abundance of Npt2a and Npt2c in the renal proximal tubule, leading to increased urinary excretion of phosphorus. PTH, parathyroid hormone.
Fibroblast Growth Factor-23.
Fibroblast growth factor-23 (FGF23) is produced in osteoblasts in response to increases in serum Pi. To exert its physiologic effects on the proximal tubule, FGF23 requires the presence of a cofactor, Klotho, which is produced in the kidney and activates FGF receptor 1 (42). FGF23 reduces the expression and activity of the sodium phosphate cotransporters in the renal proximal tubule and is also thought to decrease the activity of the intestinal sodium phosphate cotransporter. FGF23 also reduces serum levels of calcitriol by decreasing the renal expression of 1α-hydroxylase, which is the rate-limiting step in calcitriol synthesis, and increasing renal expression of 24-hydroxylase, which is required for calcitriol degradation (43) (Figure 8). In addition, FGF23 suppresses PTH synthesis, although the parathyroid glands are believed to become resistant to FGF23 as kidney disease progresses.
Figure 8.

Schematic of the role of FGF23 in reducing hyperphosphatemia as GFR falls. As GFR falls, serum phosphorus levels increase, leading to increased FGF23 secretion from bone. FGF23 binds Klotho, a required cofactor, and the FGF23/Klotho complex binds and activates FGFR1c, leading to decreased renal tubular expression of Npt2a and Npt2c. In addition, activation of FGFR1c by the FGF23/Klotho complex results in decreased 1α-hydroxylase activity and increased 24α-hydroxylase activity, which leads to decreased levels of 1,25(OH)2D and decreased Pi absorption from the gut. FGF23, fibroblast growth factor-23; FGFR1c, FGF23 receptor 1c.
1,25(OH)2D.
Calcitriol is believed to increase phosphate reabsorption in the proximal tubule (44) but the effects are confounded by the fact that changes in 1,25(OH)2D also alter plasma calcium and PTH levels.
Glucocorticoids.
Increased glucocorticoid levels lead to decreased proximal tubule synthesis and abundance of Npt2a as well as changes in brush border membrane lipid composition, which is thought to modulate sodium phosphate cotransporter activity (45).
Estrogen.
Estrogen causes phosphaturia by decreasing the abundance of Npt2a in the proximal tubule without altering Npt2c levels (46). Estrogen also increases FGF23 synthesis (47).
Thyroid Hormone.
Increased levels of thyroid hormone increase phosphate absorption by increasing proximal tubule transcription and expression of Npt2a (48). The Npt2a gene contains a thyroid response element and transcription of Npt2a mRNA is regulated by 3,5,3-tri-iodothyronine (49).
Dopamine.
Dopamine leads to phosphaturia by inducing internalization of Npt2a from the proximal tubule brush border membrane. Dopamine-mediated internalization of Npt2a is dependent on a scaffolding protein (sodium-hydrogen exchanger regulatory factor 1) because dopamine does not induce phosphaturia in sodium-hydrogen exchanger regulatory factor 1 knockout mice (50).
Metabolic Acidosis.
Metabolic acidosis stimulates phosphaturia, which helps remove acid from the blood because phosphate serves as a titratable acid (51). By contrast, metabolic alkalosis increases renal phosphate absorption (52). In mice, acidosis increases proximal tubule brush border membrane abundance of Npt2a and Npt2c, suggesting that phosphaturia results from inhibition of sodium phosphate cotransporter activity rather than changes in the levels of these proteins.
Hypertension.
An acute increase in BP leads to decreased renal phosphate reabsorption by inducing removal of Npt2a from the proximal tubule brush border membrane microvilli to subapical endosomes (53).
Clinical Consequences of Alterations in Phosphorus Balance
Hypophosphatemia.
Hypophosphatemia can be caused by a shift of phosphorus from extracellular fluid into cells, decreased intestinal absorption of phosphate, or increased renal excretion of phosphate (54). Hypophosphatemia is also seen in rare genetic disorders that decrease renal sodium phosphate cotransporter activity or increase PTH or FGF23 levels (55). Hypophosphatemia is generally asymptomatic but serum phosphate levels <1.5 mg/dl can cause anorexia, confusion, rhabdomyolysis, paralysis, and seizures. Respiratory depression in particular is associated with serum levels <1 mg/dl (56). Mild hypophosphatemia is treated with oral supplementation, whereas severe hypophosphatemia requires intravenous repletion (55).
Hyperphosphatemia.
As the GFR falls, free serum calcium levels fall and serum phosphorus increases (21). The decrease in free serum calcium and increase in serum phosphorus stimulate the parathyroid glands to produce PTH, which decreases the abundance of Npt2a and Npt2c in the renal proximal tubule, leading to increased Pi excretion in the urine that in turn lowers serum Pi levels (Figure 6). Hyperphosphatemia also stimulates production of FGF23, which decreases levels of Npt2a and Npt2c in the kidney, resulting in increased urinary excretion of phosphate (Figure 7). FGF23 also decreases production of 1,25(OH)2D, which decreases intestinal Pi absorption, further decreasing serum Pi levels (21). As GFR continues to fall, however, these compensatory mechanisms fail, leading to hyperphosphatemia.
Acute elevations in serum phosphorus levels caused by oral sodium phosphate bowel purgatives (e.g., used in preparation for colonoscopy) can cause acute phosphate nephropathy (57). Acute phosphate nephropathy is characterized by AKI leading to chronic renal failure and biopsy findings of tubular injury and tubular and interstitial calcium phosphate deposits.
Hyperphosphatemia is strongly associated with increased cardiovascular morbidity and mortality and increases the risk of calciphylaxis (58). Current treatment of hyperphosphatemia involves the use of oral phosphate binders that block phosphorus absorption from food.
Secondary Hyperparathyroidism
As mentioned above, under physiologic circumstances, PTH maintains serum calcium, increases phosphorus excretion, and stimulates production of 1,25(OH)2D. However, as kidney function declines, PTH rises. A decrease in GFR causes an increase in serum phosphorus and FGF23 levels, which, consequently, decreases 1α-hydroxylase activity and 1,25(OH)2D synthesis. The decrease in serum concentrations of 1,25(OH)2D decreases serum calcium levels and secondary hyperparathyroidism ensues (16). Although FGF23 is known to suppress PTH in a nonuremic milieu, the parathyroid gland becomes resistant to FGF23 with progressive kidney disease, favoring PTH release and worsening of secondary hyperparathyroidism (59). Hence, the lack of 1,25(OH)2D and increased FGF23 allow increased PTH production to continue (Figure 9).
Figure 9.

Pathogenesis of secondary hyperparathyroidism in CKD. Progressive loss of renal mass impairs renal phosphate excretion, which causes an increase in serum phosphorus. Abnormalities in serum phosphorus homeostasis stimulate FGF23 from bone. Higher serum FGF23 levels in addition to decreased renal mass cause a quantitative decrease in synthesis of 1,25(OH)2D. High serum FGF23 levels decrease the activity of the 1α-hydroxylase enzyme. 1,25(OH)2D deficiency decreases intestinal absorption of calcium, leading to hypocalcemia, which is augmented by the direct effect of hyperphosphatemia. Hypocalcemia and hyperphosphatemia stimulate PTH release and synthesis. The lack of 1,25(OH)2D, which would ordinarily feed back to inhibit the transcription of prepro-PTH and exert an antiproliferative effect on parathyroid cells, allows the increased PTH production to continue. Current therapeutic methods used to decrease PTH release in CKD include correction of hyperphosphatemia, maintenance of normal serum calcium levels, administration of 1,25(OH)2D analogs orally or intravenously, and administration of a CaSR agonist (e.g., cinacalcet).
The management of secondary hyperparathyroidism in dialysis patients principally involves the administration of some combination of the following: phosphate binders, vitamin D analogs, and calcimimetics. The CaSR is one of the main factors regulating PTH synthesis and secretion; therefore, this target offers the potential to suppress PTH by complementary mechanism to vitamin D analogs. Calcimimetics are agents that allosterically increase the sensitivity of the CaSR in the parathyroid gland to calcium (22).
Magnesium
Regulation of Magnesium Homeostasis
Magnesium is the second most abundant intracellular divalent cation. Magnesium serves several important functions in the human body, including intracellular signaling, serving as a cofactor for protein and DNA synthesis, oxidative phosphorylation, cardiovascular tone, neuromuscular excitability, and bone formation (60–64).
The total body magnesium content in adults is approximately 24 g, 99% of which is intracellular, stored predominantly in bone, muscle, and soft tissue, with only 1% in the extracellular space. Normal total serum magnesium concentration is in the range of 0.7–1.1 mmol/l, 1.4–2.2 mEq/l, or 1.7–2.6 mg/dl. Sixty percent of serum magnesium exists in the ionized, free, physiologically active form, which is important for its physiologic functions. Ten percent of serum magnesium exists complexed to serum anions. Thirty percent of serum magnesium is albumin-bound (60–64).
As detailed below, serum magnesium concentration is regulated by the dynamic balance and interplay between intestinal and renal transport and bone exchange (Figure 1C).
Intestinal Magnesium Absorption
Typical magnesium ingestion is approximately 300 mg/d (65). Intestinal absorption can range from 25% when eating magnesium-rich diets to 75% when eating magnesium-depleted diets. Approximately 120 mg of magnesium is absorbed and 20 mg is lost in gastrointestinal secretions, amounting to a net daily intake of 100 mg/d. Intestinal magnesium absorption occurs via a saturable transcellular pathway and a nonsaturable paracellular passive pathway (65,66) (Figure 2C). The majority of magnesium is absorbed by the small intestine and, to a lesser extent, by the colon. Transcellular magnesium absorption is permitted by the transient receptor potential melastatin (TRPM) cationic channels TRPM6 and TRPM7. Mutations in TRMP6 result in hypomagnesemia with secondary hypocalcemia (67,68). TRMP7 also plays an important role in intestinal magnesium absorption because heterozygote TRMP7-deficient mice develop hypomagnesemia due to decreased intestinal magnesium absorption, whereas renal magnesium excretion is low to compensate for the decreased intestinal magnesium absorption (69). When dietary magnesium intake is normal, transcellular transport mediates 30% of intestinal magnesium absorption. This fraction increases when dietary magnesium intake is lower (70–72). When dietary magnesium is higher, then the majority of intestinal magnesium absorption occurs via the paracellular pathway, which is regulated by proteins comprising the tight junction, including claudins, occludin, and zona-occludens-1 (73–76). Tight junction assembly and function can be modulated by a number of signaling molecules that alter the phosphorylation state of the tight junctional proteins and the ionic permeability of the paracellular pathway. Paracellular transport depends on the transepithelial electrical voltage, which is approximately 5 mV lumen-positive with respect to blood. In addition, luminal magnesium concentrations range between 1.0 and 5.0 mmol/l compared with serum magnesium concentrations of between 0.70 and 1.10 mmol/l, which also provides a transepithelial chemical concentration gradient favoring absorption.
Proton pump inhibitors, especially after long-term use, have been associated with hypomagnesemia. Impaired intestinal magnesium absorption, rather than renal absorption, seems to mediate the effect of proton pump inhibitors. Potential mediators include increased intestinal magnesium secretion, decreased active transcellular magnesium absorption due to decreased TRPM6 activity secondary to decreased intestinal acidity, or decreased paracellular magnesium absorption (77,78).
Renal Regulation of Magnesium
Assuming a normal GFR, the kidney filters approximately 2000–2400 mg of magnesium per day. This takes into account the fact that only 70% of total serum magnesium (30% is protein-bound) is available for glomerular filtration. Under normal conditions, 96% of filtered magnesium is reabsorbed in the renal tubules by several coordinated transport processes and magnesium transporters detailed below (9,12,79–84).
Proximal Tubule.
As shown in Figure 3C, 10%–30% of the filtered magnesium is absorbed in the proximal tubule. Although the exact mechanisms are not known, magnesium is believed to be absorbed via a paracellular pathway aided by a chemical gradient generated by Na gradient–driven water transport that increases intraluminal magnesium as well as lumen-positive potential.
Thick Ascending Limb.
A paracellular pathway in the thick ascending limb absorbs 40%–70% of filtered magnesium, mostly enhanced by lumen-positive transepithelial voltage, in which claudin-16 and claudin-19 play an important role. The NKCC2 cotransporter mediates apical absorption of Na, K, and Cl. The apical ROMK mediates apical recycling of K back to the tubular lumen and generation of lumen-positive voltage. The Cl channel ClC-Kb mediates Cl exit through the basolateral membrane. Na, K,-ATPase also mediates Na exit through the basolateral membrane and generates the Na gradient for Na absorption. The tight junction proteins claudin-16 and claudin-19 play a prominent role in magnesium absorption. The CaSR has also been determined to regulate magnesium transport in this segment: upon stimulation, magnesium transport is decreased. Basolateral receptor activation inhibits apical K channels and possibly Na-2C1-K cotransport in the rat thick ascending limb (85,86). This inhibition would be expected to diminish transepithelial voltage and, in turn, passive transport of magnesium within the cortical thick ascending limb.
Bartter’s syndrome is caused by mutations in NKCC2, or ROMK, or ClC-Kb, or Barttin (an essential β-subunit for ClC-Ka and ClC-Kb chloride channels), and/or CaSR. However, hypomagnesemia is not present in all patients with Bartter’s syndrome.
Mutations in CLDN16 or CLDN19 encoding the tight junction proteins claudin-16 and claudin-19 result in increased urinary magnesium excretion, hypomagnesemia, increased urinary calcium excretion, and nephrocalcinosis (autosomal recessive familial hypomagnesemia with hypercalciuria and nephrocalcinosis).
By contrast, a recent study indicates a different phenotype for mutations in claudin-10 (87). Claudin-10 determines paracellular sodium permeability and its loss leads to hypermagnesemia and nephrocalcinosis. In isolated perfused thick ascending limb tubules of claudin-10–deficient mice, paracellular permeability of sodium is decreased and the relative permeability of calcium and magnesium is increased. Moreover, furosemide-inhibitable transepithelial voltage is increased, leading to a shift from paracellular sodium transport to paracellular hyperabsorption of calcium and magnesium. These data identify claudin-10 as a key factor in control of cation selectivity and transport in the thick ascending limb, and deficiency in this pathway as a cause of nephrocalcinosis.
Loop diuretics inhibit chloride (Cl−) absorption by NKCC2 and also decrease basolateral Cl− efflux. This results in loss of lumen-positive potential, thereby decreasing the driving force for paracellular magnesium reabsorption via claudin-16 and claudin-19.
Distal Convoluted Tubule.
The remaining 5%–10% of magnesium is reabsorbed in the distal convoluted tubule mainly by active transcellular transport mediated by TRPM6. The apical K channel Kv1.1 potentiates TRPM6-mediated magnesium absorption by establishing favorable luminal potential. EGF increases magnesium transport through TRPM6. Inhibitors of EGF receptors such as cetuximab or panitumumab, utilized for treatment of EGF responsive cancers, are able to induce hypomagnesemia by interfering with magnesium transport in the kidney (88).
Basolateral K channel Kir4.1 and the γ-subunit of Na,K-ATPase also increase magnesium reabsorption by generating a sodium gradient, making it possible for the thiazide-sensitive NCC (thiazide-sensitive Na-Cl Cotransporter) to facilitate sodium transport from the apical lumen to the cytosol.
The absorbed magnesium is then extruded via a recently identified magnesium/sodium exchanger SLC41A1 family (89–92) across the basolateral membrane (93). Interestingly, mutations in SLC41A1 result in a nephronophthisis-like phenotype (93). In addition, CNNM2 (cyclin M2) has also been identified as a gene involved in renal Mg2+ handling in patients of two unrelated families with unexplained dominant hypomagnesemia. In the kidney, CNNM2 was predominantly found along the basolateral membrane of distal tubular segments involved in Mg2+ reabsorption (94).
In addition to TRMP6, the importance of these transporters in the thick ascending limb and distal convoluted tubule as well as the tight junction proteins in the thick ascending limb to regulation of magnesium reabsorption is reflected by the fact that mutations of these proteins result in various forms of genetic syndromes of hypomagnesemia (Table 3).
Table 3.
Factors that alter renal regulation of magnesium
| Increase Magnesium Absorption | Decrease Magnesium Absorption |
| Dietary magnesium restriction | Hypermagnesemia |
| Parathyroid hormone | Metabolic acidosis |
| Glucagon | Hypercalcemia |
| Calcitonin | Phosphate depletion |
| Vasopressin | Potassium depletion |
| Aldosterone | Diuretics (loop and thiazide) |
| Insulin | Antibiotics (aminoglycosides) |
| Amiloride | Antifungals (amphotericin B) |
| Metabolic alkalosis | Antivirals (foscarnet) |
| EGF | Chemotherapy agents (cisplatin) |
| Immunosuppressants (tacrolimus, cyclosporine, rapamycin) | |
| EGF receptor antagonists | |
| FHHNC caused by mutations in claudin-16 and claudin-19 | |
| HSH caused by mutations in TRPM6 | |
| Bartter’s syndrome caused by mutations in NKCC2 (type 1), ROMK (type II), ClC-Kb (type III), or CaSR (type V) | |
| Dominant hypomagnesemia caused by mutations of the FXYD2 gene, HNF1B, or CNNM2 | |
| Isolated dominant hypomagnesemia caused by mutations of Kv1.1 | |
| Isolated recessive hypomagnesemia caused by mutations of pro-EGF | |
| Gitelman’s syndrome caused by mutations of NCC | |
| EAST/SeSAME caused by mutations in Kir4.1 |
FHHNC, familial hypomagnesemia with hypercalciuria and nephrocalcinosis; HSH, hypomagnesemia with secondary hypocalcemia; TRPM6, transient receptor potential melastatin 6; NKCC2, Na+-K+-2Cl− cotransporter; ROMK, renal outer medullary potassium K+; CLC-Kb, Cl channel Kb; CaSR, calcium-sensing receptor; FXYD2, sodium/potassium-transporting ATPase gamma chain (a protein that in humans is encoded by the FXYD2 gene); HFN1B, hepatocyte nuclear factor 1 beta; CNNM2, cyclin M2; NCC, thiazide-sensitive Na-Cl Cotransporter; EAST (epilepsy, ataxia, sensorineural deafness, and tubulopathy)/SeSAME (seizures, sensineural deafness, ataxia, mental retardation, and electrolyte imbalance).
Mutations in PCBD1 (pterin-4α-carbinolamine dehydratase/dimerization cofactor of hepatocyte NF 1 homeobox A) were recently shown to cause hypomagnesemia secondary to renal magnesium wasting (95). PCBD1 is a dimerization cofactor for the transcription factor HNF1B. PCDB1 binds HNF1B to costimulate the FXYD2 (sodium/potassium-transporting ATPase gamma chain [a protein that in humans is encoded by the FXYD2 gene]) promoter, which increases renal tubular magnesium reabsorption in the distal convoluted tubule.
Bone.
At least 50% of the total body magnesium content resides in bone as hydroxyapatite crystals (96). Dietary magnesium restriction causes decreased bone magnesium content (97). Hypomagnesemia in rodents induces osteopenia with accelerated bone turnover, decreased bone volume, and decreased bone strength (98–101). Although the bone magnesium stores are dynamic, the transporters that mediate magnesium flux in and out of bone have not yet been determined.
Clinical Consequences of Alterations in Magnesium Balance
Hypomagnesemia.
Hypomagnesemia is usually defined as serum magnesium<0.7 mmol/L, 1.4 mEq/L, or 1.7 mg/dl. Biochemical hypomagnesemia is common, with a prevalence of up to 15% in the general population and up to 65% in patients in the intensive care units (60–64,102–109). The causes of hypomagnesemia are listed in Table 3.
Hypomagnesemia can be secondary to impaired intestinal magnesium absorption or increased urinary magnesium excretion secondary to various hormones or drugs that inhibit magnesium reabsorption. At the clinical level, the assessment of magnesium stores and cause of magnesium deficiency continues to be a real challenge. Simultaneous measurements of serum and urine magnesium may help differentiate the cause of hypomagnesemia.
Although proton pump inhibitors most likely cause impaired intestinal magnesium absorption, most of the other drugs associated with hypomagnesemia impair renal tubular magnesium reabsorption by direct or indirect inhibition of magnesium reabsorption in the thick ascending limb or the distal convoluted tubule (102,110,111).
Hypomagnesemia is associated with hypokalemia, which is mediated by stimulation of the ROMK channel resulting in increased potassium excretion. Hypomagnesemia is also associated with hypocalcemia secondary to impaired PTH release and PTH resistance. Clinical manifestations of hypomagnesemia include weakness and fatigue, muscle cramps, tetany, numbness, seizures, and arrhythmias.
Hypermagnesemia.
Hypermagnesemia is caused by ingestion and increased intestinal absorption of Epsom salts and magnesium-containing cathartics, antacids, laxative abuse, and enemas. In addition, overzealous intravenous or intramuscular injection of magnesium for treatment of preeclampsia can also result in hypermagnesemia.
Hypermagnesemia is associated with nausea, vomiting, neurologic impairment, hypotension, and electrocardiography changes including prolonged QRS, PR, and QT intervals. At higher levels due to intoxication, complete heart block, respiratory paralysis, coma, and shock can occur.
Maintenance of normal serum levels of calcium, phosphorus, and magnesium depends on a complex interplay between absorption from the gut, exchange from bone stores, and renal regulation. Renal reabsorption of calcium, phosphorus, and magnesium occurs in several different parts of the nephron and involves a number of channels, transporters, and paracellular pathways, some of which remain to be defined. The importance of the kidney in maintaining normal calcium, phosphorus, and magnesium homeostasis can be seen in renal failure in which abnormalities of calcium, phosphorus, and magnesium levels are very common clinical findings.
Disclosures
M.L. has received grant support (unrelated to this publication) from the National Institutes of Health, US Department of Veterans Affairs, Intercept, Daiichi Sankyo, Abbott, Genzyme, and Novartis.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Kumar R: Calcium metabolism. In: The Principles and Practice of Nephrology, edited by Jacobson HR, Striker GE, Klahr S, 2nd Ed., St. Louis, MO, Mosby-Year Book, 1995, pp 964–971 [Google Scholar]
- 2.Hebert SC, Brown EM: The scent of an ion: Calcium-sensing and its roles in health and disease. Curr Opin Nephrol Hypertens 5: 45–53, 1996 [PubMed] [Google Scholar]
- 3.Johnson JA, Kumar R: Renal and intestinal calcium transport: Roles of vitamin D and vitamin D-dependent calcium binding proteins. Semin Nephrol 14: 119–128, 1994 [PubMed] [Google Scholar]
- 4.Kumar R: Calcium transport in epithelial cells of the intestine and kidney. J Cell Biochem 57: 392–398, 1995 [DOI] [PubMed] [Google Scholar]
- 5.Keller J, Schinke T: The role of the gastrointestinal tract in calcium homeostasis and bone remodeling. Osteoporos Int 24: 2737–2748, 2013 [DOI] [PubMed] [Google Scholar]
- 6.Friedman PA, Gesek FA: Cellular calcium transport in renal epithelia: Measurement, mechanisms, and regulation. Physiol Rev 75: 429–471, 1995 [DOI] [PubMed] [Google Scholar]
- 7.Felsenfeld A, Rodriguez M, Levine B: New insights in regulation of calcium homeostasis. Curr Opin Nephrol Hypertens 22: 371–376, 2013 [DOI] [PubMed] [Google Scholar]
- 8.Riccardi D, Brown EM: Physiology and pathophysiology of the calcium-sensing receptor in the kidney. Am J Physiol Renal Physiol 298: F485–F499, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrè S, Hoenderop JG, Bindels RJ: Sensing mechanisms involved in Ca2+ and Mg2+ homeostasis. Kidney Int 82: 1157–1166, 2012 [DOI] [PubMed] [Google Scholar]
- 10.Houillier P: Calcium-sensing in the kidney. Curr Opin Nephrol Hypertens 22: 566–571, 2013 [DOI] [PubMed] [Google Scholar]
- 11.Loupy A, Ramakrishnan SK, Wootla B, Chambrey R, de la Faille R, Bourgeois S, Bruneval P, Mandet C, Christensen EI, Faure H, Cheval L, Laghmani K, Collet C, Eladari D, Dodd RH, Ruat M, Houillier P: PTH-independent regulation of blood calcium concentration by the calcium-sensing receptor. J Clin Invest 122: 3355–3367, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toka HR, Al-Romaih K, Koshy JM, DiBartolo S, 3rd, Kos CH, Quinn SJ, Curhan GC, Mount DB, Brown EM, Pollak MR: Deficiency of the calcium-sensing receptor in the kidney causes parathyroid hormone-independent hypocalciuria. J Am Soc Nephrol 23: 1879–1890, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lambers TT, Bindels RJ, Hoenderop JG: Coordinated control of renal Ca2+ handling. Kidney Int 69: 650–654, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Hoenderop JG, Nilius B, Bindels RJ: Molecular mechanism of active Ca2+ reabsorption in the distal nephron. Annu Rev Physiol 64: 529–549, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Mensenkamp AR, Hoenderop JG, Bindels RJ: Recent advances in renal tubular calcium reabsorption. Curr Opin Nephrol Hypertens 15: 524–529, 2006 [DOI] [PubMed] [Google Scholar]
- 16.Goodman WG, Quarles LD: Development and progression of secondary hyperparathyroidism in chronic kidney disease: Lessons from molecular genetics. Kidney Int 74: 276–288, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Dusso AS, Brown AJ, Slatopolsky E: Vitamin D. Am J Physiol Renal Physiol 289: F8–F28, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Holick MF: Vitamin D deficiency. N Engl J Med 357: 266–281, 2007 [DOI] [PubMed] [Google Scholar]
- 19.Drüeke TB, Lacour B: Disorders of calcium, phosphate, and magnesium metabolism. In: Comprehensive Clinical Nephrology, edited by Feehally J, Floege J, Johnson RJ, 3rd Ed., Philadelphia, Mosby/Elsevier, 2007, pp 123–140 [Google Scholar]
- 20.Biber J, Hernando N, Forster I: Phosphate transporters and their function. Annu Rev Physiol 75: 535–550, 2013 [DOI] [PubMed] [Google Scholar]
- 21.Marks J, Debnam ES, Unwin RJ: Phosphate homeostasis and the renal-gastrointestinal axis. Am J Physiol Renal Physiol 299: F285–F296, 2010 [DOI] [PubMed] [Google Scholar]
- 22.Blaine J, Weinman EJ, Cunningham R: The regulation of renal phosphate transport. Adv Chronic Kidney Dis 18: 77–84, 2011 [DOI] [PubMed] [Google Scholar]
- 23.Forster IC, Hernando N, Biber J, Murer H: Proximal tubular handling of phosphate: A molecular perspective. Kidney Int 70: 1548–1559, 2006 [DOI] [PubMed] [Google Scholar]
- 24.Azzarolo AM, Ritchie G, Quamme GA: Some characteristics of sodium-independent phosphate transport across renal basolateral membranes. Biochim Biophys Acta 1064: 229–234, 1991 [DOI] [PubMed] [Google Scholar]
- 25.Murer H, Forster I, Biber J: The sodium phosphate cotransporter family SLC34. Pflugers Arch 447: 763–767, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Magagnin S, Werner A, Markovich D, Sorribas V, Stange G, Biber J, Murer H: Expression cloning of human and rat renal cortex Na/Pi cotransport. Proc Natl Acad Sci U S A 90: 5979–5983, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Segawa H, Kaneko I, Takahashi A, Kuwahata M, Ito M, Ohkido I, Tatsumi S, Miyamoto K: Growth-related renal type II Na/Pi cotransporter. J Biol Chem 277: 19665–19672, 2002 [DOI] [PubMed] [Google Scholar]
- 28.Villa-Bellosta R, Ravera S, Sorribas V, Stange G, Levi M, Murer H, Biber J, Forster IC: The Na+-Pi cotransporter PiT-2 (SLC20A2) is expressed in the apical membrane of rat renal proximal tubules and regulated by dietary Pi. Am J Physiol Renal Physiol 296: F691–F699, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Segawa H, Onitsuka A, Furutani J, Kaneko I, Aranami F, Matsumoto N, Tomoe Y, Kuwahata M, Ito M, Matsumoto M, Li M, Amizuka N, Miyamoto K: Npt2a and Npt2c in mice play distinct and synergistic roles in inorganic phosphate metabolism and skeletal development. Am J Physiol Renal Physiol 297: F671–F678, 2009 [DOI] [PubMed] [Google Scholar]
- 30.Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS: Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci U S A 95: 5372–5377, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tenenhouse HS, Martel J, Gauthier C, Segawa H, Miyamoto K: Differential effects of Npt2a gene ablation and X-linked Hyp mutation on renal expression of Npt2c. Am J Physiol Renal Physiol 285: F1271–F1278, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Bacconi A, Virkki LV, Biber J, Murer H, Forster IC: Renouncing electroneutrality is not free of charge: Switching on electrogenicity in a Na+-coupled phosphate cotransporter. Proc Natl Acad Sci U S A 102: 12606–12611, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Virkki LV, Forster IC, Biber J, Murer H: Substrate interactions in the human type IIa sodium-phosphate cotransporter (NaPi-IIa). Am J Physiol Renal Physiol 288: F969–F981, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Ravera S, Virkki LV, Murer H, Forster IC: Deciphering PiT transport kinetics and substrate specificity using electrophysiology and flux measurements. Am J Physiol Cell Physiol 293: C606–C620, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Virkki LV, Biber J, Murer H, Forster IC: Phosphate transporters: A tale of two solute carrier families. Am J Physiol Renal Physiol 293: F643–F654, 2007 [DOI] [PubMed] [Google Scholar]
- 36.Breusegem SY, Takahashi H, Giral-Arnal H, Wang X, Jiang T, Verlander JW, Wilson P, Miyazaki-Anzai S, Sutherland E, Caldas Y, Blaine JT, Segawa H, Miyamoto K, Barry NP, Levi M: Differential regulation of the renal sodium-phosphate cotransporters NaPi-IIa, NaPi-IIc, and PiT-2 in dietary potassium deficiency. Am J Physiol Renal Physiol 297: F350–F361, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Déliot N, Hernando N, Horst-Liu Z, Gisler SM, Capuano P, Wagner CA, Bacic D, O’Brien S, Biber J, Murer H: Parathyroid hormone treatment induces dissociation of type IIa Na+-P(i) cotransporter-Na+/H+ exchanger regulatory factor-1 complexes. Am J Physiol Cell Physiol 289: C159–C167, 2005 [DOI] [PubMed] [Google Scholar]
- 38.Millar AL, Jackson NA, Dalton H, Jennings KR, Levi M, Wahren B, Dimmock NJ: Rapid analysis of epitope-paratope interactions between HIV-1 and a 17-amino-acid neutralizing microantibody by electrospray ionization mass spectrometry. Eur J Biochem 258: 164–169, 1998 [DOI] [PubMed] [Google Scholar]
- 39.Lederer ED, Khundmiri SJ, Weinman EJ: Role of NHERF-1 in regulation of the activity of Na-K ATPase and sodium-phosphate co-transport in epithelial cells. J Am Soc Nephrol 14: 1711–1719, 2003 [DOI] [PubMed] [Google Scholar]
- 40.Mahon MJ, Cole JA, Lederer ED, Segre GV: Na+/H+ exchanger-regulatory factor 1 mediates inhibition of phosphate transport by parathyroid hormone and second messengers by acting at multiple sites in opossum kidney cells. Mol Endocrinol 17: 2355–2364, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Blaine J, Okamura K, Giral H, Breusegem S, Caldas Y, Millard A, Barry N, Levi M: PTH-induced internalization of apical membrane NaPi2a: Role of actin and myosin VI. Am J Physiol Cell Physiol 297: C1339–C1346, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berndt T, Kumar R: Phosphatonins and the regulation of phosphate homeostasis. Annu Rev Physiol 69: 341–359, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Hasegawa H, Nagano N, Urakawa I, Yamazaki Y, Iijima K, Fujita T, Yamashita T, Fukumoto S, Shimada T: Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int 78: 975–980, 2010 [DOI] [PubMed] [Google Scholar]
- 44.Kurnik BR, Hruska KA: Mechanism of stimulation of renal phosphate transport by 1,25-dihydroxycholecalciferol. Biochim Biophys Acta 817: 42–50, 1985 [DOI] [PubMed] [Google Scholar]
- 45.Levi M, Shayman JA, Abe A, Gross SK, McCluer RH, Biber J, Murer H, Lötscher M, Cronin RE: Dexamethasone modulates rat renal brush border membrane phosphate transporter mRNA and protein abundance and glycosphingolipid composition. J Clin Invest 96: 207–216, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Faroqui S, Levi M, Soleimani M, Amlal H: Estrogen downregulates the proximal tubule type IIa sodium phosphate cotransporter causing phosphate wasting and hypophosphatemia. Kidney Int 73: 1141–1150, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cannata-Andía JB, Carrillo-López N, Naves-Díaz M: Estrogens and bone disease in chronic kidney disease: Role of FGF23. Curr Opin Nephrol Hypertens 19: 354–358, 2010 [DOI] [PubMed] [Google Scholar]
- 48.Alcalde AI, Sarasa M, Raldúa D, Aramayona J, Morales R, Biber J, Murer H, Levi M, Sorribas V: Role of thyroid hormone in regulation of renal phosphate transport in young and aged rats. Endocrinology 140: 1544–1551, 1999 [DOI] [PubMed] [Google Scholar]
- 49.Ishiguro M, Yamamoto H, Masuda M, Kozai M, Takei Y, Tanaka S, Sato T, Segawa H, Taketani Y, Arai H, Miyamoto K, Takeda E: Thyroid hormones regulate phosphate homoeostasis through transcriptional control of the renal type IIa sodium-dependent phosphate co-transporter (Npt2a) gene. Biochem J 427: 161–169, 2010 [DOI] [PubMed] [Google Scholar]
- 50.Weinman EJ, Biswas R, Steplock D, Douglass TS, Cunningham R, Shenolikar S: Sodium-hydrogen exchanger regulatory factor 1 (NHERF-1) transduces signals that mediate dopamine inhibition of sodium-phosphate co-transport in mouse kidney. J Biol Chem 285: 13454–13460, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nowik M, Picard N, Stange G, Capuano P, Tenenhouse HS, Biber J, Murer H, Wagner CA: Renal phosphaturia during metabolic acidosis revisited: Molecular mechanisms for decreased renal phosphate reabsorption. Pflugers Arch 457: 539–549, 2008 [DOI] [PubMed] [Google Scholar]
- 52.Biber J, Hernando N, Forster I, Murer H: Regulation of phosphate transport in proximal tubules. Pflugers Arch 458: 39–52, 2009 [DOI] [PubMed] [Google Scholar]
- 53.Magyar CE, Zhang Y, Holstein-Rathlou NH, McDonough AA: Proximal tubule Na transporter responses are the same during acute and chronic hypertension. Am J Physiol Renal Physiol 279: F358–F369, 2000 [DOI] [PubMed] [Google Scholar]
- 54.Felsenfeld AJ, Levine BS: Approach to treatment of hypophosphatemia. Am J Kidney Dis 60: 655–661, 2012 [DOI] [PubMed] [Google Scholar]
- 55.Carpenter TO: The expanding family of hypophosphatemic syndromes. J Bone Miner Metab 30: 1–9, 2012 [DOI] [PubMed] [Google Scholar]
- 56.Gravelyn TR, Brophy N, Siegert C, Peters-Golden M: Hypophosphatemia-associated respiratory muscle weakness in a general inpatient population. Am J Med 84: 870–876, 1988 [DOI] [PubMed] [Google Scholar]
- 57.Markowitz GS, Perazella MA: Acute phosphate nephropathy. Kidney Int 76: 1027–1034, 2009 [DOI] [PubMed] [Google Scholar]
- 58.Kendrick J, Kestenbaum B, Chonchol M: Phosphate and cardiovascular disease. Adv Chronic Kidney Dis 18: 113–119, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, Sirkis R, Naveh-Many T, Silver J: The parathyroid is a target organ for FGF23 in rats. J Clin Invest 117: 4003–4008, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whang R, Hampton EM, Whang DD: Magnesium homeostasis and clinical disorders of magnesium deficiency. Ann Pharmacother 28: 220–226, 1994 [DOI] [PubMed] [Google Scholar]
- 61.Konrad M, Schlingmann KP, Gudermann T: Insights into the molecular nature of magnesium homeostasis. Am J Physiol Renal Physiol 286: F599–F605, 2004 [DOI] [PubMed] [Google Scholar]
- 62.Topf JM, Murray PT: Hypomagnesemia and hypermagnesemia. Rev Endocr Metab Disord 4: 195–206, 2003 [DOI] [PubMed] [Google Scholar]
- 63.Ayuk J, Gittoes NJ: How should hypomagnesaemia be investigated and treated? Clin Endocrinol (Oxf) 75: 743–746, 2011 [DOI] [PubMed] [Google Scholar]
- 64.Agus ZS: Hypomagnesemia. J Am Soc Nephrol 10: 1616–1622, 1999 [DOI] [PubMed] [Google Scholar]
- 65.Quamme GA: Recent developments in intestinal magnesium absorption. Curr Opin Gastroenterol 24: 230–235, 2008 [DOI] [PubMed] [Google Scholar]
- 66.Fine KD, Santa Ana CA, Porter JL, Fordtran JS: Intestinal absorption of magnesium from food and supplements. J Clin Invest 88: 396–402, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schlingmann KP, Weber S, Peters M, Niemann Nejsum L, Vitzthum H, Klingel K, Kratz M, Haddad E, Ristoff E, Dinour D, Syrrou M, Nielsen S, Sassen M, Waldegger S, Seyberth HW, Konrad M: Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet 31: 166–170, 2002 [DOI] [PubMed] [Google Scholar]
- 68.Walder RY, Landau D, Meyer P, Shalev H, Tsolia M, Borochowitz Z, Boettger MB, Beck GE, Englehardt RK, Carmi R, Sheffield VC: Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat Genet 31: 171–174, 2002 [DOI] [PubMed] [Google Scholar]
- 69.Ryazanova LV, Rondon LJ, Zierler S, Hu Z, Galli J, Yamaguchi TP, Mazur A, Fleig A, Ryazanov AG: TRPM7 is essential for Mg(2+) homeostasis in mammals. Nat Commun 1: 109, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Milla PJ, Aggett PJ, Wolff OH, Harries JT: Studies in primary hypomagnesaemia: Evidence for defective carrier-mediated small intestinal transport of magnesium. Gut 20: 1028–1033, 1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Karbach U: Cellular-mediated and diffusive magnesium transport across the descending colon of the rat. Gastroenterology 96: 1282–1289, 1989 [DOI] [PubMed] [Google Scholar]
- 72.Jüttner R, Ebel H: Characterization of Mg2+ transport in brush border membrane vesicles of rabbit ileum studied with mag-fura-2. Biochim Biophys Acta 1370: 51–63, 1998 [DOI] [PubMed] [Google Scholar]
- 73.Anderson JM, Van Itallie CM: Physiology and function of the tight junction. Cold Spring Harb Perspect Biol 1: a002584, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shen L, Weber CR, Raleigh DR, Yu D, Turner JR: Tight junction pore and leak pathways: A dynamic duo. Annu Rev Physiol 73: 283–309, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Amasheh S, Fromm M, Günzel D: Claudins of intestine and nephron - A correlation of molecular tight junction structure and barrier function. Acta Physiol (Oxf) 201: 133–140, 2011 [DOI] [PubMed] [Google Scholar]
- 76.Günzel D, Yu AS: Claudins and the modulation of tight junction permeability. Physiol Rev 93: 525–569, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lameris AL, Hess MW, van Kruijsbergen I, Hoenderop JG, Bindels RJ: Omeprazole enhances the colonic expression of the Mg(2+) transporter TRPM6. Pflugers Arch 465: 1613–1620, 2013 [DOI] [PubMed] [Google Scholar]
- 78.Hess MW, Hoenderop JG, Bindels RJ, Drenth JP: Systematic review: Hypomagnesaemia induced by proton pump inhibition. Aliment Pharmacol Ther 36: 405–413, 2012 [DOI] [PubMed] [Google Scholar]
- 79.Bindels RJ: 2009 Homer W. Smith Award: Minerals in motion: From new ion transporters to new concepts. J Am Soc Nephrol 21: 1263–1269, 2010 [DOI] [PubMed] [Google Scholar]
- 80.Quamme GA: Molecular identification of ancient and modern mammalian magnesium transporters. Am J Physiol Cell Physiol 298: C407–C429, 2010 [DOI] [PubMed] [Google Scholar]
- 81.Ferrè S, Hoenderop JG, Bindels RJ: Insight into renal Mg2+ transporters. Curr Opin Nephrol Hypertens 20: 169–176, 2011 [DOI] [PubMed] [Google Scholar]
- 82.Tyler Miller R: Control of renal calcium, phosphate, electrolyte, and water excretion by the calcium-sensing receptor. Best Pract Res Clin Endocrinol Metab 27: 345–358, 2013 [DOI] [PubMed] [Google Scholar]
- 83.Haisch L, Almeida JR, Abreu da Silva PR, Schlingmann KP, Konrad M: The role of tight junctions in paracellular ion transport in the renal tubule: Lessons learned from a rare inherited tubular disorder. Am J Kidney Dis 57: 320–330, 2011 [DOI] [PubMed] [Google Scholar]
- 84.Haisch L, Konrad M: Impaired paracellular ion transport in the loop of Henle causes familial hypomagnesemia with hypercalciuria and nephrocalcinosis. Ann N Y Acad Sci 1258: 177–184, 2012 [DOI] [PubMed] [Google Scholar]
- 85.Hebert SC: Extracellular calcium-sensing receptor: Implications for calcium and magnesium handling in the kidney. Kidney Int 50: 2129–2139, 1996 [DOI] [PubMed] [Google Scholar]
- 86.Wang WH, Lu M, Hebert SC: Cytochrome P-450 metabolites mediate extracellular Ca(2+)-induced inhibition of apical K+ channels in the TAL. Am J Physiol 271: C103–C111, 1996 [DOI] [PubMed] [Google Scholar]
- 87.Breiderhoff T, Himmerkus N, Stuiver M, Mutig K, Will C, Meij IC, Bachmann S, Bleich M, Willnow TE, Müller D: Deletion of claudin-10 (Cldn10) in the thick ascending limb impairs paracellular sodium permeability and leads to hypermagnesemia and nephrocalcinosis. Proc Natl Acad Sci U S A 109: 14241–14246, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Petrelli F, Borgonovo K, Cabiddu M, Ghilardi M, Barni S: Risk of anti-EGFR monoclonal antibody-related hypomagnesemia: Systematic review and pooled analysis of randomized studies. Expert Opin Drug Saf 11[Suppl 1]: S9–S19, 2012 [DOI] [PubMed] [Google Scholar]
- 89.Goytain A, Quamme GA: Functional characterization of human SLC41A1, a Mg2+ transporter with similarity to prokaryotic MgtE Mg2+ transporters. Physiol Genomics 21: 337–342, 2005 [DOI] [PubMed] [Google Scholar]
- 90.Kolisek M, Launay P, Beck A, Sponder G, Serafini N, Brenkus M, Froschauer EM, Martens H, Fleig A, Schweigel M: SLC41A1 is a novel mammalian Mg2+ carrier. J Biol Chem 283: 16235–16247, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mandt T, Song Y, Scharenberg AM, Sahni J: SLC41A1 Mg(2+) transport is regulated via Mg(2+)-dependent endosomal recycling through its N-terminal cytoplasmic domain. Biochem J 439: 129–139, 2011 [DOI] [PubMed] [Google Scholar]
- 92.Kolisek M, Nestler A, Vormann J, Schweigel-Röntgen M: Human gene SLC41A1 encodes for the Na+/Mg²+ exchanger. Am J Physiol Cell Physiol 302: C318–C326, 2012 [DOI] [PubMed] [Google Scholar]
- 93.Hurd TW, Otto EA, Mishima E, Gee HY, Inoue H, Inazu M, Yamada H, Halbritter J, Seki G, Konishi M, Zhou W, Yamane T, Murakami S, Caridi G, Ghiggeri G, Abe T, Hildebrandt F: Mutation of the Mg2+ transporter SLC41A1 results in a nephronophthisis-like phenotype. J Am Soc Nephrol 24: 967–977, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stuiver M, Lainez S, Will C, Terryn S, Günzel D, Debaix H, Sommer K, Kopplin K, Thumfart J, Kampik NB, Querfeld U, Willnow TE, Němec V, Wagner CA, Hoenderop JG, Devuyst O, Knoers NV, Bindels RJ, Meij IC, Müller D: CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia. Am J Hum Genet 88: 333–343, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ferrè S, de Baaij JH, Ferreira P, Germann R, de Klerk JB, Lavrijsen M, van Zeeland F, Venselaar H, Kluijtmans LA, Hoenderop JG, Bindels RJ: Mutations in PCBD1 cause hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol 25: 574–586, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Elin RJ: Assessment of magnesium status. Clin Chem 33: 1965–1970, 1987 [PubMed] [Google Scholar]
- 97.Alfrey AC, Miller NL, Trow R: Effect of age and magnesium depletion on bone magnesium pools in rats. J Clin Invest 54: 1074–1081, 1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lai CC, Singer L, Armstrong WD: Bone composition and phosphatase activity in magnesium deficiency in rats. J Bone Joint Surg Am 57: 516–522, 1975 [PubMed] [Google Scholar]
- 99.Burnell JM, Liu C, Miller AG, Teubner E: Effects of dietary alteration of bicarbonate and magnesium on rat bone. Am J Physiol 250: F302–F307, 1986 [DOI] [PubMed] [Google Scholar]
- 100.Boskey AL, Rimnac CM, Bansal M, Federman M, Lian J, Boyan BD: Effect of short-term hypomagnesemia on the chemical and mechanical properties of rat bone. J Orthop Res 10: 774–783, 1992 [DOI] [PubMed] [Google Scholar]
- 101.Kenney MA, McCoy H, Williams L: Effects of magnesium deficiency on strength, mass, and composition of rat femur. Calcif Tissue Int 54: 44–49, 1994 [DOI] [PubMed] [Google Scholar]
- 102.Lameris AL, Monnens LA, Bindels RJ, Hoenderop JG: Drug-induced alterations in Mg2+ homoeostasis. Clin Sci (Lond) 123: 1–14, 2012 [DOI] [PubMed] [Google Scholar]
- 103.Dimke H, Monnens L, Hoenderop JG, Bindels RJ: Evaluation of hypomagnesemia: Lessons from disorders of tubular transport. Am J Kidney Dis 62: 377–383, 2013 [DOI] [PubMed] [Google Scholar]
- 104.Whang R, Ryder KW: Frequency of hypomagnesemia and hypermagnesemia. Requested vs routine. JAMA 263: 3063–3064, 1990 [PubMed] [Google Scholar]
- 105.Thienpont LM, Dewitte K, Stöckl D: Serum complexed magnesium—A cautionary note on its estimation and its relevance for standardizing serum ionized magnesium. Clin Chem 45: 154–155, 1999 [PubMed] [Google Scholar]
- 106.Sanders GT, Huijgen HJ, Sanders R: Magnesium in disease: A review with special emphasis on the serum ionized magnesium. Clin Chem Lab Med 37: 1011–1033, 1999 [DOI] [PubMed] [Google Scholar]
- 107.Huijgen HJ, Soesan M, Sanders R, Mairuhu WM, Kesecioglu J, Sanders GT: Magnesium levels in critically ill patients. What should we measure? Am J Clin Pathol 114: 688–695, 2000 [DOI] [PubMed] [Google Scholar]
- 108.Külpmann WR, Gerlach M: Relationship between ionized and total magnesium in serum. Scand J Clin Lab Invest Suppl 224: 251–258, 1996 [DOI] [PubMed] [Google Scholar]
- 109.Dewitte K, Dhondt A, Giri M, Stöckl D, Rottiers R, Lameire N, Thienpont LM: Differences in serum ionized and total magnesium values during chronic renal failure between nondiabetic and diabetic patients: A cross-sectional study. Diabetes Care 27: 2503–2505, 2004 [DOI] [PubMed] [Google Scholar]
- 110.van Angelen AA, Glaudemans B, van der Kemp AW, Hoenderop JG, Bindels RJ: Cisplatin-induced injury of the renal distal convoluted tubule is associated with hypomagnesaemia in mice. Nephrol Dial Transplant 28: 879–889, 2013 [DOI] [PubMed] [Google Scholar]
- 111.Dimke H, van der Wijst J, Alexander TR, Meijer IM, Mulder GM, van Goor H, Tejpar S, Hoenderop JG, Bindels RJ: Effects of the EGFR inhibitor erlotinib on magnesium handling. J Am Soc Nephrol 21: 1309–1316, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]