

Abstract
Plasma gelsolin (pGSN) functions as part of the “extracellular actin-scavenging system,” but its potential to improve host defense against infection has not been studied. In a mouse model of primary pneumococcal pneumonia, recombinant human pGSN (rhu-pGSN) caused enhanced bacterial clearance, reduced acute inflammation, and improved survival. In vitro, rhu-pGSN rapidly improved lung macrophage uptake and killing of bacteria (Streptococcus pneumoniae, Escherichia coli, and Francisella tularensis). pGSN triggers activating phosphorylation (Ser1177) of macrophage nitric oxide synthase type III (NOS3), an enzyme with important bactericidal functions in lung macrophages. rhu-pGSN failed to enhance bacterial killing by NOS3−/− macrophages in vitro or bacterial clearance in NOS3−/− mice in vivo. Prophylaxis with immunomodulators may be especially relevant for patients at risk for secondary bacterial pneumonia, e.g., after influenza. Treatment of mice with pGSN challenged with pneumococci on postinfluenza day 7 (the peak of enhanced susceptibility to secondary infection) caused a ∼15-fold improvement in bacterial clearance, reduced acute neutrophilic inflammation, and markedly improved survival, even without antibiotic therapy. pGSN is a potential immunomodulator for improving lung host defense against primary and secondary bacterial pneumonia.
Keywords: pneumonia, innate immunity, lung macrophages, gelsolin, nitric oxide
bacterial pneumonia remains a major cause of morbidity and mortality (21). The secondary pneumonias that occur after influenza or other severe injuries are especially problematic clinically (19, 23, 26). One approach to the problem might be to enhance innate immunity and host resistance to infection. The resident lung macrophage is a “first-responder” phagocyte, ingesting and killing inhaled bacteria, an important function confirmed by greater susceptibility to infection and diminished bacterial clearance after their depletion experimentally (8, 11) or by their impairment after relevant clinical risk factors such as recent influenza infection (27).
We have investigated whether a normal human blood protein, plasma gelsolin (pGSN), can enhance innate defenses against pneumonia. pGSN is secreted into extracellular fluids; it differs from the ubiquitous cytoplasmic isoform by an additional NH2-terminal sequence of 25 amino acids. Blood levels of pGSN are 200–300 μg/ml, placing it among the most abundant single proteins in plasma. pGSN functions as part of an “extracellular actin-scavenging system,” binding and facilitating removal of potentially inflammatory actin released by injured cells (17). The discovery that pGSN binds and inactivates lipid, peptide, and small molecule mediators of inflammation suggests another role (3, 12, 22). pGSN depletion due to actin exposure at local sites of injury could enable mediators generated there to modulate host defense and repair. However, the large reservoir of circulating pGSN would bind and inhibit mediators escaping into the circulation, preventing them from exerting organ damage. Loss of such systemic protection would explain the correlation between critical pGSN depletion and adverse outcomes of severe primary injuries, such as acute respiratory distress syndrome, sepsis, major trauma, and prolonged hyperoxia. This inflammation localization hypothesis may explain why pGSN repletion in a variety of animal injury models has consistently reduced inflammatory complications and, in some cases, promoted survival (6, 16, 24). The mechanisms for these beneficial effects of pGSN are likely multiple, but they indicate its potential as a modulator of acute, innate immune responses.
We report here the new finding that pGSN also has direct benefits on macrophage-mediated antimicrobial and host defense functions. We found that pGSN enhances the ability of lung macrophages to kill bacteria in vitro, in large part through a nitric oxide (NO) synthase type III (NOS3)-dependent mechanism (28). We also used a model of primary pneumococcal infection that approximates the frequent challenge to lung defenses of small numbers by bacteria and then studied a model of postviral susceptibility to bacterial pneumonia. We found that pGSN improved resistance in mouse models of both primary infection and postinfluenza secondary pneumonia. Since pGSN depletion occurs in acute pneumonia in humans (18), pGSN repletion may offer a therapeutic approach to enhance resistance to common and serious lung infections.
MATERIALS AND METHODS
Mouse models of pneumococcal pneumonia.
Normal 8- to 12-wk-old male and female C57BL/6 mice were obtained from Charles River Laboratories (Wilmington, MA) or Jackson Laboratories (Bar Harbor, ME). C57BL/6 mice genetically deficient in NOS3 (25) were obtained from Jackson Laboratories. All animals were housed in sterile microisolator cages in a barrier facility and showed no evidence of spontaneous infection. Prior approval for all experimentation was obtained from the Harvard Medical Area Standing Committee on Animals.
Primary pneumococcal pneumonia was modeled as previously reported (1, 28). Pneumonia was induced by intranasal instillation of ∼105 colony-forming units (CFU) of Streptococcus pneumoniae type 3 into mice under anesthesia with ketamine (120 mg/kg ip) plus xylazine (16 mg/kg ip); actual CFU administered were quantitated by CFU assay and used subsequently to calculate percent clearance. For analysis at 4 or 24 h of lung inflammation and bacterial clearance, bronchoalveolar lavage was performed and analyzed as previously described (1, 28). In experiments to assess survival from primary pneumonia, mice were instilled intranasally with a single 25-μl suspension containing a higher dose of S. pneumoniae (∼3 × 105 CFU), and survival was followed for periods as reported in results. pGSN treatments to assess effects on 24-h bacterial clearance were administered 2 h before and 8 and 20 h after bacterial challenge with 10 mg·mouse−1·dose−1 sc (400 mg/kg). For these studies, recombinant human pGSN (rhu-pGSN) synthesized and purified as previously reported (16) or similar pGSN (provided by BioAegis Therapeutics, Morristown, NJ) with equivalent biological effects was used.
To model postinfluenza secondary pneumonia, mice were instilled intranasally on day 0 with a single 25- or 50-μl suspension containing influenza virus [H1N1 A/PR8/34; 1 plaque-forming unit (PFU)], under general anesthesia with ketamine (120 mg/kg ip) plus xylazine (16 mg/kg ip). Influenza-treated animals routinely lost weight and then recovered by day 7, when the secondary infection was administered. On day 7 after initial influenza infection, mice were anesthetized and then instilled intranasally with ∼500 CFU of S. pneumoniae serotype 3. pGSN was administered at 400 mg/kg sc daily starting 1 day before pneumococcal challenge. Subsequent analyses were performed as described for the primary pneumonia model described above.
Bacteria.
S. pneumoniae serotype 3 and Escherichia coli (catalog nos. 6303 and 19138, respectively, American Type Culture Collection, Rockville, MD) and Francisella tularensis LVS strain were cultured overnight on 5% sheep blood-supplemented agar petri dishes (catalog no. 90001-282, VWR, West Chester, PA) and prepared and quantified elsewhere (1, 28).
Phagocytosis and bacterial killing assay.
Macrophage binding, internalization, and killing of internalized bacteria were measured using CFU assays of cell samples lysed by 10-fold excess H2O (pH 10, 3 min) (1, 28). The B6 cell line was created by retroviral immortalization of alveolar macrophages from normal male C57BL/6 mice, as previously reported (29); a similar cell line derived by immortalization of alveolar macrophages from NOS3-deficient (NOS3−/−) mice was also used. Effects of pGSN were tested by including agents or their vehicle solutions in the assay buffers.
Western blot analysis and ELISA.
Western blot analysis was performed on macrophage lysates lysis buffer 1% NP-40 with protease and phosphatase inhibitors using the protocols described elsewhere (13). The endothelial cell line bEnd.3 (CRL-2299, American Type Culture Collection) was used as a positive control for (“endothelial”) NOS3. Antibodies for NOS3, Akt, and phosphorylation-state-specific isoforms include NOS3 rabbit polyclonal antibody (C-20, Santa Cruz Biotechnology) and phosphorylated (Ser1177) endothelial NOS (C9C3) rabbit monoclonal antibody, phosphorylated (Ser473) Akt (D9E) rabbit monoclonal antibody, Akt rabbit polyclonal antibody, and β-actin (13E5) rabbit monoclonal antibody, all from Cell Signaling Technologies. Polyclonal antibodies to pGSN were developed as previously reported (15, 16). After incubation with peroxidase-conjugated goat anti-rabbit IgG or goat anti-mouse IgG (Pierce), labeling was detected using chemiluminescence.
Statistical analysis.
Data were analyzed using Prism software (GraphPad Software). Differences in Kaplan-Meier survival curves were analyzed using a log-rank (Mantel-Cox) test. For other measurements, differences between groups were examined by ANOVA with correction for multiple groups. Data are presented as means ± SD. Results were considered significant at P < 0.05.
RESULTS
pGSN improves macrophage bacterial killing in vitro.
Since lung macrophages mediate initial innate resistance to bacteria, we investigated pGSN effects on their antimicrobial function. We used a murine lung macrophage-derived cell line (29) in an assay that measures bacterial binding, internalization, and killing of internalized bacteria by quantitation of CFU at each step (1). Using this assay under serumless conditions to approximate the low-protein alveolar milieu, we preincubated macrophages with rhu-pGSN at concentrations that approximate normal plasma levels for 1 h prior to challenge with S. pneumoniae serotype 3 at a 10:1 bacteria-to-macrophage ratio. We found no effect on bacterial binding (data not shown). In contrast, pGSN enhanced internalization of bound bacteria and also markedly increased the rate of killing of internalized bacteria (Figs. 1, A and B). The enhanced killing was seen even when the pGSN was washed away prior to bacterial challenge, consistent with a direct effect on the alveolar macrophage, rather than opsonization of bacteria. Similar results were seen when E. coli and F. tularensis bacteria were used (Fig. 1, C–F).
Fig. 1.

Plasma gelsolin (pGSN) enhances macrophage uptake and killing of bacteria. A and B: addition of recombinant human pGSN (rhu-pGSN, 125 or 250 μg/ml) to immortalized B6 lung macrophages in vitro results in dose-dependent increases in internalization (A; phagocytosis, calculated as percentage of total bound bacteria, see materials and methods) and killing of ingested S. pneumoniae (B; survival, calculated as percentage of total internalized bacteria). Values are means ± SD (n ≥ 5). *P < 0.01. C–F: E. coli (C and D, internalization and killing, respectively) and F. tularensis LVS strain (E and F) produced results similar to A and B. Values are means ± SD of 2 replicates per group (n ≥ 3 trials). *P < 0.01.
pGSN improves lung host defense against pneumococcal pneumonia in vivo.
We pretreated mice with subcutaneous injections of rhu-pGSN, which gradually (over 6 h) raises pGSN levels (16). After 2 h, animals were challenged with intranasal instillation of S. pneumoniae (∼105 CFU). After 24 h, we measured bacterial clearance (CFU assay) and lung inflammation (cell yield and differential) in bronchoalveolar lavage samples. pGSN-treated animals showed substantially better clearance of bacteria (survival of fewer live bacteria) and less inflammation than vehicle-treated controls (Fig. 2, A and B). To test the effect of lung-only delivery, we exposed mice to aerosolized pGSN followed by bacterial challenge. We observed persistence of rhu-pGSN in the lungs for several hours, an increase in clearance of bacteria from lungs, and diminished lung neutrophilia (Fig. 2, C–E). Even without antibiotic treatment, mice receiving pGSN showed markedly greater survival in this model of primary pneumococcal pneumonia (Fig. 2F).
Fig. 2.

pGSN enhances resistance to pneumococcal pneumonia. A and B: 24 h after intranasal inoculation of S. pneumoniae [∼105 colony-forming units (CFU)], lung samples from mice treated with pGSN (10 mg·mouse−1·dose−1 sc) contain fewer live bacteria (bacterial survival) and show less acute inflammation [bronchoalveolar lavage (BAL) neutrophils] than lung samples from vehicle-treated (control) mice. Values are means ± SD from ≥3 independent experiments (n ≥ 12). *P < 0.01 vs. control. C: after aerosol exposure to rhu-pGSN, Western blot analysis of lung lavage samples shows detectable levels of rhu-pGSN for up to 7 h (2 μg protein loaded/lane); normal levels of endogenous murine pGSN (mu-pGSN) were also detected but were unchanged. D and E: aerosol exposure to pGSN followed by S. pneumoniae inoculation causes enhanced bacterial clearance, as measured by diminished bacterial survival in lungs at 24 h (D, 30-min exposure, n ≥ 12) and reflected in decreased neutrophilia in lavage samples [E, 30-min exposure, n ≥ 12, summary of 3 independent experiments; data from 15-min (15′) exposure are from a single trial using 3 replicates]. F: systemic (subcutaneous) treatment with pGSN results in substantially better survival from pneumonia. Values are means ± SD of 3 independent experiments (n ≥ 12). *P < 0.01.
NOS3 mediates pGSN beneficial effects in macrophages.
Mechanisms for killing phagocytosed bacteria include production of reactive oxygen species (e.g., superoxide produced by NADPH oxidase) and reactive nitrogen intermediates (e.g., NO produced by NOS). We recently reported that NOS3, constitutively expressed in murine and human alveolar macrophages, functions in lung macrophage host defense against bacterial pneumonia (28). We tested the role of NOS3 in the ability of pGSN to enhance macrophage antibacterial function. We compared effects of pGSN in vitro on wild-type or NOS3−/− macrophages. Absence of NOS3 eliminated the beneficial enhancement of bacterial killing caused by addition of pGSN (Fig. 3A). Studies of NOS3 in endothelial cell biology have identified rapid activation via phosphorylation of NOS3, e.g., at Ser1177 (13), and this activation occurs via specific signaling cascades, i.e., via phosphorylated inositol-3 kinase and protein kinase B, also known as Akt (4, 9). To investigate the pathway in macrophages, we exposed macrophages to pGSN and performed Western blot analysis on lysates after 1–4 h. The analysis showed rapid phosphorylation of Akt and NOS3 in response to pGSN (Fig. 3B). Using the primary pneumococcal pneumonia model, we found that NOS3−/− mice failed to show the enhanced bacterial clearance seen in wild-type mice (Fig. 3C).
Fig. 3.
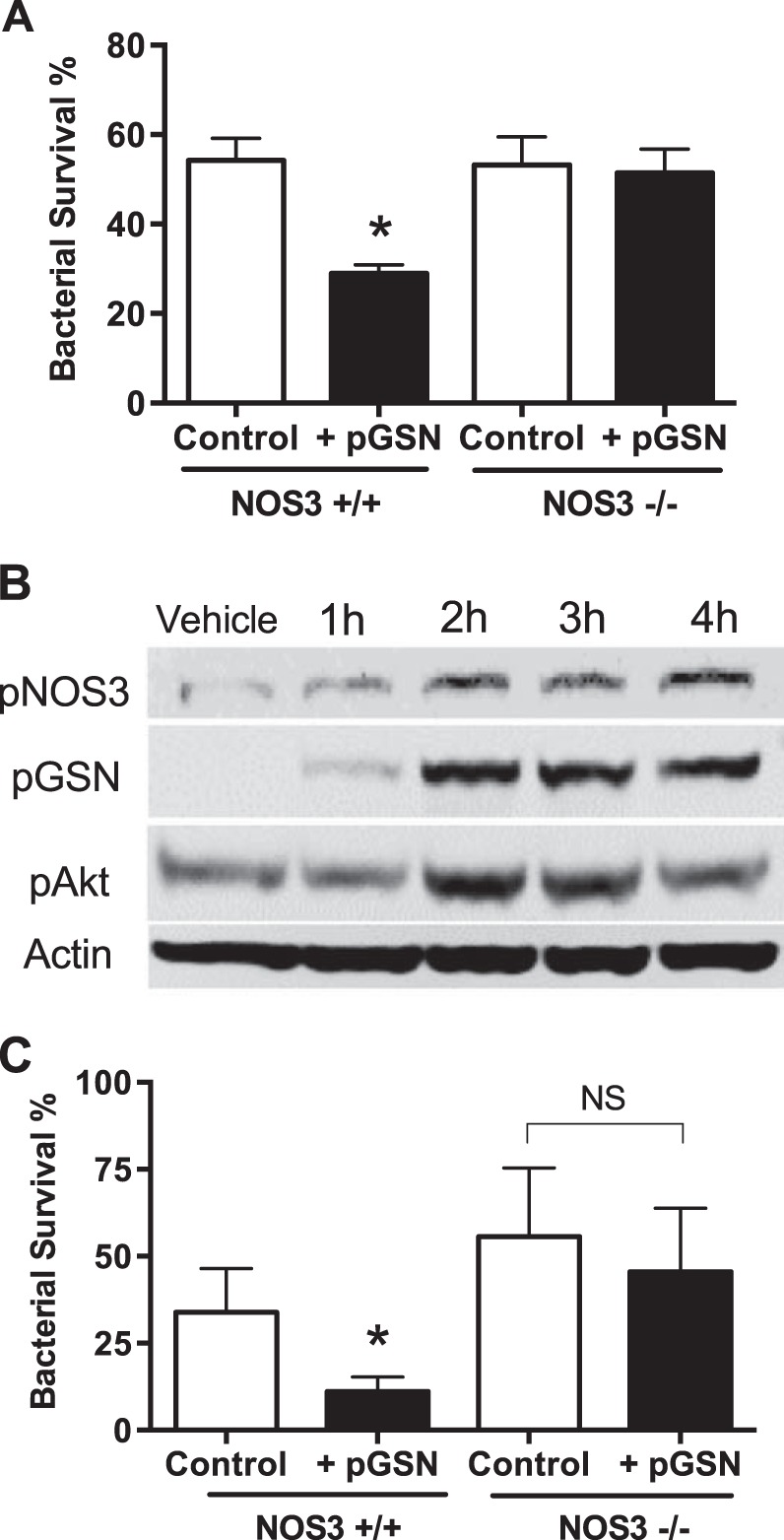
pGSN enhances host defense by effects on macrophage nitric oxide synthase type III (NOS3). A: macrophages deficient in NOS3 (NOS3−/−) no longer show improved in vitro bacterial killing (decreased bacterial survival) seen in wild-type phagocytes after pGSN treatment (2 replicates per group, n ≥ 3 trials). B: lysates of macrophages (80 μg protein loaded/lane) incubated with rhu-pGSN show increased phosphorylation at sites associated with functional activation of Akt (∼55 kDa, Ser473) and NOS3 (∼140 kDa, Ser1177), as well as internalization of pGSN (∼90 kDa). Data are representative of 3 independent experiments. C: in vivo, clearance of S. pneumoniae at 24 h is enhanced by pGSN treatment in wild-type control mice, but no effect is seen in NOS3−/− mice (n = 6–8/group, summary of 2 trials). Values are means ± SD. *P < 0.01.
pGSN improves outcomes in secondary postinfluenza bacterial pneumonia.
Secondary bacterial pneumonia is a frequent complication of seasonal or epidemic viral influenza. We tested pGSN for potential benefit in a murine model of enhanced susceptibility to secondary pneumococcal pneumonia after nonlethal influenza challenge. In mice infected with low-dose inocula of influenza (PR8, 1 PFU) or S. pneumoniae alone (100 or 500 CFU), survival was 100%. In contrast, mortality was 100% in mice receiving similarly small bacterial challenges on postinfluenza day 7. The magnitude and timing of enhanced susceptibility are similar to those reported previously (11, 27) but are, nonetheless, remarkable: for comparison, in normal mice (without prior influenza), Arredouani et al. (1) reported 100% survival with 104 CFU (data not shown) and only partial mortality at higher inocula, e.g., ∼50% with 105 CFU.
With this model, bacterial clearance measurements at 24 h show extraordinary bacterial growth in the postinfluenza lung, consistent with marked impairment of innate host defenses (19, 20). pGSN treatment markedly improved bacterial clearance and reduced acute inflammation at 24 h after bacterial inoculation (Fig. 4, A and B). Mice treated with a lower dose of pGSN showed ∼50% survival and recovered from initial weight loss (Fig. 4, C and D), while a higher-dose trial showed 100% survival (Figs. 4, D and E). In both cases, the improved outcome took place without antibiotic treatment.
Fig. 4.

pGSN enhances resistance to postinfluenza pneumococcal pneumonia. The model uses, in sequence, low doses of influenza (1 plaque-forming unit) and S. pneumoniae (300–500 colony-forming units), which allow 100% survival when used alone. A and B: pGSN improves clearance of the large number of bacteria at 24 h (A) and also results in diminished neutrophilia, quantified as percentage or total number of polymorphonuclear leukocytes (PMNs, B). Veh, vehicle. Values are means ± SD (n = 6/group, 1 trial). *P < 0.01. C–F: pretreatment with pGSN results in a dose-dependent, partial (C and D, 10 mg·mouse−1·day−1; n ≥ 13/group, sum of 2 trials) or total (E and F, 40 mg·mouse−1·day−1; n = 8/group, 1 trial) improvement in survival (C and E) and in recovery of initial weight loss (D and F). Values are means ± SD. *P < 0.01.
DISCUSSION
We sought to identify new approaches to enhancing innate immunity to bacterial pneumonia by investigating whether pGSN could act to augment lung host defense. We conclude that pGSN can mediate greater host resistance to pneumonia, in large part, via effects on the constitutively expressed NOS3 in lung macrophages. Pharmacological administration of pGSN led to improved bacterial clearance and improved survival from secondary pneumococcal pneumonia. These observations identify a novel host defense mechanism and a potential therapeutic benefit for pGSN, an endogenous protein that has undergone phase I clinical testing in limited human studies (5).
Our findings complement previous work demonstrating that pGSN administration improves the survival of animals subjected to peritoneal bacterial infection (7, 16). However, those outcomes were ascribed solely to protective effects of pGSN against systemic inflammation. These prior results suggest that some modulation of inflammatory responses might also contribute to the enhanced survival observed with pGSN treatment, along with effects on improved bacterial clearance. Indeed, the ability of pGSN to augment phagocytic uptake suggests additional effects on signaling or receptor(s) function.
The mechanism by which pGSN may act includes priming or activating macrophage NOS3 (28), which can then contribute to improved killing of ingested bacteria by direct bactericidal mechanisms, either alone or in combination with superoxide (10); pGSN might also act to mediate salutary intracellular signaling events (14). Additional pathways are also suggested by the observation that pGSN caused enhanced internalization of bacteria, an effect not observed in the presence of other activators of macrophage NOS3 (28). Although our understanding of the mechanisms by which pGSN stimulates macrophage function is rudimentary, our finding that pGSN results in the phosphorylation of NOS3 to activate it for bactericidal action is consistent with pGSN's enhancement of bacterial uptake. Membrane polyphosphoinositides are important intermediates for phagocytosis and protein kinase B activation, which is upstream from NOS3 phosphorylation (2). Although externally disposed membrane receptors for pGSN have not been identified, pGSN has been shown to act as a carrier of ligands that are released by and/or interact with macrophage receptors (3, 12, 22).
We consider that the translational potential of these findings may be most relevant for the problem of secondary pneumonia, specifically the increased susceptibility to bacterial lung infections that follows influenza. Treatment before the bacterial challenge is useful for proof-of-principle in the primary pneumonia model, but this clearly does not mirror likely clinical usage. In contrast, pretreatment, as used in the secondary pneumonia model, represents a realistic scenario for short-term, prophylactic immunomodulatory therapy. This might benefit patients identified to be at high risk of secondary pneumonia, e.g., hospitalized individuals with severe flu, after seasonal or pandemic influenza. Such translation is more than a theoretical possibility, since recombinant pGSN administration has been shown to be therapeutically beneficial in animals (present study) and feasible and safe in humans (5).
GRANTS
This work was supported by National Institutes of Health Grants HL-11578 and ES-00002 (to L. Kobzik) and HL-19429 (to T. P. Stossel).
DISCLOSURES
T. P. Stossel's employer, Brigham and Women's Hospital, has licensed pending and issued patent applications regarding pGSN to BioAegis Therapeutics. T. P. Stossel is a consultant for BioAegis Therapeutics, from which he has been granted stock options.
AUTHOR CONTRIBUTIONS
Z.Y. and T.T.-Y.C. performed the experiments; Z.Y., T.T.-Y.C., T.P.S., and L.K. analyzed the data; Z.Y., T.T.-Y.C., T.P.S., and L.K. interpreted the results of the experiments; Z.Y. and L.K. prepared the figures; Z.Y., T.P.S., and L.K. edited and revised the manuscript; Z.Y., T.T.-Y.C., T.P.S., and L.K. approved the final version of the manuscript; L.K. developed the concept and designed the research; L.K. drafted the manuscript.
REFERENCES
- 1.Arredouani M, Yang Z, Ning Y, Qin G, Soininen R, Tryggvason K, Kobzik L. The scavenger receptor MARCO is required for lung defense against pneumococcal pneumonia and inhaled particles. J Exp Med 200: 267–272, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bohdanowicz M, Grinstein S. Role of phospholipids in endocytosis, phagocytosis, and macropinocytosis. Physiol Rev 93: 69–106, 2013. [DOI] [PubMed] [Google Scholar]
- 3.Bucki R, Kulakowska A, Byfield FJ, Zendzian-Piotrowska M, Baranowski M, Marzec M, Winer JP, Ciccarelli NJ, Górski J, Drozdowski W, Bittman R, Janmey PA. Plasma gelsolin modulates cellular response to sphingosine 1-phosphate. Am J Physiol Cell Physiol 299: C1516–C1523, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chambliss KL, Shaul PW. Estrogen modulation of endothelial nitric oxide synthase. Endocrinol Rev 23: 665–686, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Chan W, Lau L, Kwok K, Law W, Ho J, Chu K, Poon R. A randomized, double-blind, placebo-controlled, ascending-dose trial of the pharmacokinetics and safety of intravenous infusion of recombinant human plasma gelsolin in acutely ill patients with decreased plasma gelsolin levels (Abstract). Am J Respir Crit Care Med A5601: A5601, 2011. [Google Scholar]
- 6.Christofidou-Solomidou M, Scherpereel A, Solomides CC, Christie JD, Stossel TP, Goelz S, Dinubile MJ. Recombinant plasma gelsolin diminishes the acute inflammatory response to hyperoxia in mice. J Investig Med 50: 54–60, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Cohen TS, Bucki R, Byfield FJ, Ciccarelli NJ, Rosenberg B, Dinubile MJ, Janmey PA, Margulies SS. Therapeutic potential of plasma gelsolin administration in a rat model of sepsis. Cytokine 54: 235–238, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dockrell DH, Whyte MK, Mitchell TJ. Pneumococcal pneumonia: mechanisms of infection and resolution. Chest 142: 482–491, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duckles SP, Miller VM. Hormonal modulation of endothelial NO production. Eur J Physiol 459: 841–851, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol 2: 820–832, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Ghoneim HE, Thomas PG, McCullers JA. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J Immunol 191: 1250–1259, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goetzl EJ, Lee H, Azuma T, Stossel TP, Turck CW, Karliner JS. Gelsolin binding and cellular presentation of lysophosphatidic acid. J Biol Chem 275: 14573–14578, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez E, Kou R, Lin AJ, Golan DE, Michel T. Subcellular targeting and agonist-induced site-specific phosphorylation of endothelial nitric-oxide synthase. J Biol Chem 277: 39554–39560, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Hernansanz-Agustín P, Izquierdo-Álvarez A, García-Ortiz A, Ibiza S, Serrador JM, Martínez-Ruiz A. Nitrosothiols in the immune system: signaling and protection. Antioxid Redox Signal 18: 288–308, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee PS, Sampath K, Karumanchi SA, Tamez H, Bhan I, Isakova T, Gutierrez OM, Wolf M, Chang Y, Stossel TP, Thadhani R. Plasma gelsolin and circulating actin correlate with hemodialysis mortality. J Am Soc Nephrol 20: 1140–1148, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee PS, Waxman AB, Cotich KL, Chung SW, Perrella MA, Stossel TP. Plasma gelsolin is a marker and therapeutic agent in animal sepsis. Crit Care Med 35: 849–855, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Lee WM, Galbraith RM. The extracellular actin-scavenger system and actin toxicity. N Engl J Med 326: 1335–1341, 1992. [DOI] [PubMed] [Google Scholar]
- 18.Lind SE, Smith DB, Janmey PA, Stossel TP. Depression of gelsolin levels and detection of gelsolin-actin complexes in plasma of patients with acute lung injury. Am Rev Respir Dis 138: 429–434, 1988. [DOI] [PubMed] [Google Scholar]
- 19.McCullers JA. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat Rev Microbiol 12: 252–262, 2014. [DOI] [PubMed] [Google Scholar]
- 20.Metzger DW, Sun K. Immune dysfunction and bacterial coinfections following influenza. J Immunol 191: 2047–2052, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mizgerd JP. Lung infection—a public health priority. PLos Med 3: e76, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Osborn TM, Dahlgren C, Hartwig JH, Stossel TP. Modifications of cellular responses to lysophosphatidic acid and platelet-activating factor by plasma gelsolin. Am J Physiol Cell Physiol 292: C1323–C1330, 2007. [DOI] [PubMed] [Google Scholar]
- 23.Rice TW, Rubinson L, Uyeki TM, Vaughn FL, John BB, Miller RR 3rd, Higgs E, Randolph AG, Smoot BE, Thompson BT. Critical illness from 2009 pandemic influenza A virus and bacterial coinfection in the United States. Crit Care Med 40: 1487–1498, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rothenbach PA, Dahl B, Schwartz JJ, O'Keefe GE, Yamamoto M, Lee WM, Horton JW, Yin HL, Turnage RH. Recombinant plasma gelsolin infusion attenuates burn-induced pulmonary microvascular dysfunction. J Appl Physiol 96: 25–31, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA 93: 13176–13181, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shrestha S, Foxman B, Weinberger DM, Steiner C, Viboud C, Rohani P. Identifying the interaction between influenza and pneumococcal pneumonia using incidence data. Sci Transl Med 5: 191ra184, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-γ during recovery from influenza infection. Nat Med 14: 558–564, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Yang Z, Huang YC, Koziel H, De Crom R, Ruetten H, Wohlfart P, Thomsen RW, Kahlert JA, Sørensen HT, Józefowski S, Colby A, Kobzik L. Female resistance to pneumonia identifies lung macrophage nitric oxide synthase-3 as a therapeutic target. eLife October 15: 3, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou H, Imrich A, Kobzik L. Characterization of immortalized MARCO and SR-AI/II-deficient murine alveolar macrophage cell lines. Part Fibre Toxicol 5: 7, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]