

Abstract
Nitrite is a storage reservoir of nitric oxide that is readily reduced to nitric oxide under pathological conditions. Previous studies have demonstrated that nitrite levels are significantly reduced in cardiovascular disease states, including peripheral vascular disease. We investigated the cytoprotective and proangiogenic actions of a novel, sustained-release formulation of nitrite (SR-nitrite) in a clinically relevant in vivo swine model of critical limb ischemia (CLI) involving central obesity and metabolic syndrome. CLI was induced in obese Ossabaw swine (n = 18) by unilateral external iliac artery deployment of a full cross-sectional vessel occlusion device positioned within an endovascular expanded polytetrafluoroethylene-lined nitinol stent-graft. At post-CLI day 14, pigs were randomized to placebo (n = 9) or SR-nitrite (80 mg, n = 9) twice daily by mouth for 21 days. SR-nitrite therapy increased nitrite, nitrate, and S-nitrosothiol in plasma and ischemic skeletal muscle. Oxidative stress was reduced in ischemic limb tissue of SR-nitrite- compared with placebo-treated pigs. Ischemic limb tissue levels of proangiogenic growth factors were increased following SR-nitrite therapy compared with placebo. Despite the increases in cytoprotective and angiogenic signals with SR-nitrite therapy, new arterial vessel formation and enhancement of blood flow to the ischemic limb were not different from placebo. Our data clearly demonstrate cytoprotective and proangiogenic signaling in ischemic tissues following SR-nitrite therapy in a very severe model of CLI. Further studies evaluating longer-duration nitrite therapy and/or additional nitrite dosing strategies are warranted to more fully evaluate the therapeutic potential of nitrite therapy in peripheral vascular disease.
Keywords: nitric oxide, metabolic syndrome, angiogenesis, peripheral arterial disease, Ossabaw swine, endothelial nitric oxide synthase, nitrite
peripheral arterial disease (PAD) is caused by atherosclerosis, resulting in obstructions in the arteries and limiting blood supply to organs other than the heart. PAD is an important public health problem, with a total prevalence of ∼5 million people in the United States alone (41). A major symptomatic clinical manifestation of PAD is critical limb ischemia (CLI). CLI is defined as reduced blood flow to the lower limb resulting in pain, even at rest, with or without associated ulcers or gangrene (2). Patients with CLI have a 6-mo risk of major amputation of 25–40% and an annual mortality of 20% (18). Surgical and catheter-based revascularization strategies are favored approaches for CLI patients. However, because of the severe vascular dysfunction associated with CLI, many patients are poor candidates, or have no option, for revascularization. Additionally, even after successful revascularization, stenosis or failure of the grafts often reoccurs, resulting in a need for development of new treatments.
Metabolic syndrome (MetS) is characterized by abdominal obesity, dyslipidemia, hypercholesterolemia, hypertension, and hyperglycemia and currently afflicts one-third of the US adult population. Factors associated with MetS significantly impair vascular reactivity by disrupting endothelial and smooth muscle function, leading to severe vascular complications (35). Patients with MetS have increased risk of vascular damage, leading to PAD and CLI (20).
Ossabaw swine represent a clinically relevant animal model for MetS and are ideal for investigation of cardiovascular disease states. Over the past 400 years, these feral animals have adapted to the harsh conditions of Georgia's Ossabaw Island, often with extended periods of starvation, by feeding heavily when food is readily available and storing a greater proportion of body fat than any other breed of swine or terrestrial mammal (27). Thus, Ossabaw swine are selectively and genetically predisposed to obesity and offer a unique natural animal model for MetS when fed a high-cholesterol atherogenic diet. After consuming the atherogenic diet, Ossabaw swine develop central obesity, insulin resistance, glucose intolerance, dyslipidemia, hypertension, and atherosclerosis (3–5).
Nitrite, an oxidative breakdown product of nitric oxide (NO), is traditionally viewed simply as a biomarker of NO formation in biological systems that is devoid of biological activity. More recently, this view has changed with the discovery that nitrite is a physiologically important storage reservoir of NO that is readily reduced to NO under pathological conditions including ischemia (15, 30). Nitrite has been shown to be cytoprotective in numerous animal models of ischemic injury, including myocardial ischemia-reperfusion injury (19, 31, 38), heart failure (6), cerebral vasospasm (36), and hindlimb ischemia (6, 10, 19, 26). In a recent phase II PAD clinical trial (i.e., SONIC), sodium nitrite therapy was shown to be safe and well tolerated (32). A major limitation for the clinical use of nitrite therapy for chronic disease states is the very short (i.e., 25–35 min) half-life of the currently available sodium nitrite formulations (24). A longer half-life is likely required to generate protective levels of NO and NO-mediated signaling. Given the robust preclinical data regarding the cytoprotective effects of nitrite therapy in PAD (1, 7), we sought to evaluate the effects of a novel, sustained-release nitrite (SR-nitrite) formulation in a clinically relevant model of CLI and MetS.
METHODS
Human CLI and control skeletal muscle collection.
After removal of the affected extremity, the amputation specimen was transferred to a separate sterile table, and elliptical 8 × 4 cm muscle biopsies of the anterior tibialis and gastrocnemius muscles were performed. For above-knee amputations, an additional biopsy of the vastus medialis was performed. All patients consented to the use of their amputated tissue for research purposes and signed a Health Insurance Portability and Accountability Act release for use of their medical records. Patients were segregated into normal perfusion or CLI according to pulse examination and objective imaging criteria.
Animals.
Castrated male Ossabaw swine, 6–8 mo of age, were obtained from Indiana University. Although female swine display a more rapid development of dyslipidemia and atherosclerosis when fed a cholesterol-enriched high-fat diet, castrated males were utilized for this study (13, 21). Animals were handled in compliance with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals” and according to the guidelines of the Emory University Institutional Animal Care and Use Committee, which approved the study.
Ossabaw swine diet and feeding.
For 6 mo, Ossabaw swine were fed an atherogenic obese diet (1 kg/day) consisting of 2% cholesterol, 17% coconut oil, 2.5% corn oil, and 0.7% sodium cholate (KT324, Purina Test Diet).
Sodium nitrite therapy.
SR-nitrite tablets were generously supplied by TheraVasc (Cleveland, OH). Animals received placebo (n = 9) or SR-nitrite (80 mg, n = 9) by mouth twice daily. Treatment was initiated at 14 days following induction of CLI and continued for 21 days (Fig. 1).
Fig. 1.

Critical limb ischemia (CLI) protocol. Obese Ossabaw swine were subjected to 35 days of CLI. Sustained-release (SR)-nitrite therapy was initiated at a dose of 80 mg by mouth twice a day beginning 14 days after induction of CLI. Vascular angiographic images were obtained at baseline, immediately post-CLI, and on day 35. Animals were euthanized after 35 days of CLI, and skeletal muscle was collected for molecular and pharmacokinetic analyses. RSNO, S-nitrosothiol.
CLI model.
Obese male swine (n = 18) were subjected to CLI as depicted in Fig. 1. Nine pigs were originally assigned to the placebo group and nine to the SR-nitrite group in a blinded and randomized fashion. During the course of the 35-day CLI protocol, three pigs in the placebo group and two in SR-nitrite group died as a result of complications of CLI and obesity/MetS. Therefore, there were six pigs in the placebo group and seven in the SR-nitrite group at day 35. Animals were sedated with ketamine (15 mg/kg) in combination with xylazine (1 mg/kg) and, to aid induction, were endotracheally intubated and maintained on isoflurane in oxygen. The preoperative analgesic buprenorphine (0.15 mg for each pig) was administered along with the perioperative antibiotic cefazolin (1,000 mg). Postoperative analgesics, carprofen (10 mg) and buprenorphine (0.15–0.3 mg), were given immediately after the procedure. On postoperative day 1, additional analgesics, buprenorphine (0.3 mg) in combination with carprofen (100 mg), were administered. Heart rate, respiration, peripheral capillary O2 saturation, end-tidal CO2, fluid volume, and body temperature were monitored throughout the procedure. Activated clotting time was measured with a coagulation-monitoring system (Hemachron) after placement of the arterial sheath and administration of heparin.
Under sterile conditions and with fluoroscopic guidance, an 8-French introducer (Maximum Hemostasis Introducer 23-cm ACT Sheath, St. Jude Medical, St. Paul, MN) was inserted into the right external carotid artery, advanced into the abdominal aorta, and placed just above the aortoiliac bifurcation. Heparin (300 U/kg) was administered, and activated clotting time was monitored throughout the procedure to ensure clotting times of ≥250 s. A percutaneous guide wire (0.035 × 230 cm; Rosen Starter, Boston Scientific, Natick, MA) was positioned in the right external iliac artery. A 7-French delivery guide catheter (Mach 1 MP1, Boston Scientific) containing a self-expanding endoluminal endoprosthesis consisting of an expanded polytetrafluoroethylene (ePTFE) lining with an external nitinol stent (6 mm × 15 cm; VIABAHN, W. L. Gore and Associates, Flagstaff, AZ) was advanced, positioned within the right external iliac artery, and deployed. Contrast arteriography was performed to evaluate vessel patency. After placement of the ePTFE-lined endoprosthesis, a self-expanding nitinol mesh occlusion device (8 × 7 mm; AMPLATZER Vascular Plug II, St. Jude Medical) was loaded onto the delivery guide catheter, advanced, positioned within the right external iliac artery, and deployed within the proximal portion of the ePTFE-lined stent. An arteriogram was performed to verify occlusion. The guide wire, guide catheter, and introducer sheath were removed, and the left external carotid artery was ligated. The incision was closed, the animal was allowed to recover, and buprenorphine (0.05 mg/kg) was administered for analgesia. The ePTFE-lined endoprosthesis served to inhibit recanalization of the artery by collateral vessels, typically observed in animal models of CLI.
Euthanasia and tissue collection.
After all in vivo measurements were completed, heparin (200 U/kg iv) was injected and allowed to circulate for ∼5 min. Animals were euthanized while under deep inhalant anesthesia (isoflurane, 5%). The gastrocnemius muscle was collected from control and CLI limbs.
Measurement of NO metabolites.
Nitrite, nitrate, S-nitrosothiol (RSNO), and NO-heme concentrations were quantified as previously described (6). Immediately after euthanasia, ischemic and control skeletal muscles were removed and flash-frozen using liquid nitrogen. Tissue was homogenized in PBS (10 ml/mg of tissue). In some cases, differences in sample size are due to a limited quantity of adequate tissue samples required for these complex assays.
Angiography.
Hindlimb vasculature was visualized during recordings using undiluted contrast (Oxilan 300, Guerbet, Bloomington, IN). Arteriographs of the uninstrumented left and occluded right iliac arteries were obtained at baseline and on day 35.
Ankle-brachial index.
An automated blood pressure machine with a 2-inch cuff was used to measure limb blood pressure before CLI, immediately post-CLI, and at the end of the experiment. Animals were sedated using isoflurane in oxygen by mask to effect, and a pressure cuff was secured to each limb, rapidly inflated to ∼30 mmHg above the anticipated systolic pressure, and then deflated slowly for determination of systolic blood pressure in each of the forelimbs and hindlimbs. Ankle-brachial index (ABI) was calculated for each side of the animal by dividing the systolic blood pressure measured at the hindlimb (ankle) by that measured at the contralateral hindlimb. In some cases, we were unable to obtain reproducible or reliable cuff readings for ABI data; in these cases, ABI was not reported. Therefore, the sample size reported for the ABI data is variable.
cGMP RIA.
Skeletal muscle cGMP concentrations were quantified as previously described (6).
Western blot analysis.
Gastrocnemius muscle was used for Western blot analysis, performed as described previously (25). The following primary antibodies were used: VEGF (Abcam), CD31 (Novus Biologicals), Ser1177-phosphorylated endothelial NO synthase (eNOS; Cell Signaling), and eNOS, Thr495-phosphorylated eNOS, and GAPDH (Santa Cruz Biotechnology).
Real-time PCR.
Skeletal muscle mRNA was quantified using TaqMan primers (Life Technologies) as previously described (34).
Quantitative angiographic score.
Collateral vessel growth in the control and CLI limbs was assessed using a grid overlay of 2-mm squares (44). Quantitative vessel analysis was performed on angiographic images of the control and CLI limbs distal from the ePTFE-lined endoprosthesis at post-CLI day 35. The angiogenic score was calculated as percentage of the number of contrast-opacified vessels crossing the squares divided by the total number of vessels. Sample size was limited to five per group because of technical complications during angiographic imaging as well as the high mortality rate in the Ossabaw swine subjected to CLI.
ELISAs.
VEGF, CD31, and von Willebrand factor were assayed using ELISA kits according to the manufacturer's recommendations (MyBioSource, San Diego, CA).
Determination of protein carbonyl content.
Protein carbonyl content of skeletal muscle was measured as described previously (25).
Measurement of malondialdehyde levels.
Malondialdehyde (MDA) levels were measured in skeletal muscle as previously described (25).
Statistical analysis.
Values are means ± SE. Differences in data between the groups were compared using Prism 4 (GraphPad Software) with Student's unpaired, two-tailed t-test when only two groups were compared. P < 0.05 was considered statistically significant.
RESULTS
CLI patients exhibit NO deficiency.
The NO intermediates (nitrite, nitrosothiols, and NO-heme), as well as cGMP and cGMP-dependent protein kinase-1 (PKG-1), were measured in gastrocnemius, anterior tibialis, and vastus medialis tissue obtained postamputation from normal and CLI patients (Fig. 2). Patient data for the control and CLI patients are presented in Table 1. We observed significant (P < 0.01) reductions in skeletal muscle nitrite (Fig. 2A), RSNO (Fig. 2B), and NO-heme (Fig. 2C) levels in CLI patients compared with age-matched controls. Skeletal muscle cGMP levels were also significantly (P < 0.01) attenuated in CLI patients compared with controls (Fig. 2D). Furthermore, PKG-1 protein expression and mRNA levels were significantly (P < 0.01) reduced in CLI tissues compared with controls (Fig. 2, G and H).
Fig. 2.

CLI patients exhibit nitric oxide (NO) deficiency in the ischemic limb. A–E: nitrite, RSNO, NO-heme, cGMP, and nitrate in gastrocnemius muscle from CLI patients and controls. F: representative Western blot of gastrocnemius cGMP-dependent protein kinase 1 (PKG-1). G and H: PKG-1 RNA message and PKG-1 protein expression. Numbers inside circles denote number of patients.
Table 1.
Patient information for control and CLI patients who underwent extremity amputations
| Group |
||
|---|---|---|
| Control (n = 4) | CLI (n = 16) | |
| Age, yr | 64.5 ± 13.44 | 68.1 ± 14.24 |
| Male, % | 0 | 63 |
| Diabetes, % | 50 | 50 |
| Hypertension, % | 50 | 63 |
| Congestive heart failure, % | 0 | 25 |
| Current/former smoker, % | 0 | 31 |
Values for age are means ± SD. Skeletal muscle biopsies were obtained from anterior tibialis and gastrocnemius muscles. CLI, critical limb ischemia.
Obese Ossabaw swine exhibit eNOS dysfunction and are NO-deficient.
Western blot analysis of soleus muscle obtained from lean and obese swine revealed no change in total eNOS protein expression (Fig. 3B). However, there was a significant increase in phosphorylation at the eNOS inhibitory site, Thr495, in the obese swine (Fig. 3C). There was no difference in phosphorylation status at the eNOS activation site, Ser1177, between groups (Fig. 3D). Asymmetric dimethylarginine (ADMA) is structurally similar to l-arginine and inhibits NO synthase, resulting in decreased NO production and endothelial dysfunction. The obese swine displayed elevated circulating ADMA levels compared with the lean group (Fig. 3E). Nitrite, a marker for NO bioavailability, was significantly (P < 0.05) decreased in plasma and skeletal muscle of the obese animals compared with the lean controls (Fig. 3F), providing further evidence of NO insufficiency in the obese Ossabaw animals.
Fig. 3.

Obese Ossabaw swine display dysfunctional endothelial NO synthase (eNOS) and reduced NO levels. A: representative Western blot of eNOS in lean and obese Ossabaw skeletal muscle. B–D: total eNOS, Thr495-phosphorylated eNOS, and Ser1177-phosphorylated eNOS protein expression. E and F: plasma asymmetric dimethylarginine (ADMA) and plasma and skeletal muscle nitrite levels in lean and obese swine. Numbers inside circles denote number of animals.
Pharmacokinetics of SR-nitrite in CLI.
Beginning at post-CLI day 14, SR-nitrite (80 mg) or placebo was administered by mouth twice daily for 21 days (Fig. 4A). We did not observe a significant change in plasma nitrite or nitrate in the placebo-treated group during the 35-day CLI experimental protocol. In contrast, we observed an increase in circulating nitrite levels following SR-nitrite therapy, with a statistically significant (P < 0.05) difference between the SR-nitrite- and placebo-treated groups at day 35. We failed to observe statistically significant increases in circulating nitrite levels at days 21 and 28. This is possibly due to immediate reduction of nitrite to NO and NO scavenging in the circulation due to severe ischemia and oxidative stress in the setting of CLI. Additionally, plasma nitrate levels were markedly increased following SR-nitrite administration at days 21 (P < 0.01), 28 (P < 0.01), and 35 (P < 0.05; Fig. 2C). Furthermore, we observed an increase in skeletal muscle tissue nitrite (Fig. 4B) and nitrate (Fig. 4D) levels following SR-nitrite therapy, as evidenced by a fourfold increase (P < 0.01) in nitrite in the CLI limb compared with the placebo-treated group. We also examined RSNO concentrations in plasma and skeletal muscle. At day 35, plasma RSNO levels were significantly (P < 0.05) greater in the nitrite- than placebo-treated group. We observed a 3.9-fold increase in tissue RSNO concentration in the CLI limb of the nitrite- compared with placebo-treated swine (Fig. 4E). These data indicate that nitrite levels are increased in the ischemic limb and result in increased NO bioavailability. Safety of augmented nitrite levels following therapy was validated by stable circulating methemoglobin levels in the SR-nitrite-treated group at day 35. There was no difference in percent methemoglobin between the SR-nitrite- and placebo-treated groups (3.1 ± 0.9% vs. 2.1 ± 0.5%, P = NS).
Fig. 4.

SR-nitrite pharmacokinetics. A and B: plasma nitrite at baseline, day 21, day 28, and day 35 and skeletal muscle nitrite at day 35 in SR-nitrite- and placebo-treated control and CLI limbs. C and D: plasma nitrate at baseline, day 21, day 28, and day 35 and skeletal muscle nitrate at day 35 in SR-nitrite- and placebo-treated control and CLI limbs. E and F: plasma RSNO at baseline and day 35 and skeletal muscle RSNO at day 35 in control and ischemic limbs. Numbers inside circles denote number of animals. *P < 0.05 vs. placebo.
Nitrite therapy fails to improve extremity blood flow in CLI.
Measurement of blood flow revealed an immediate reduction in ABI to 0 following the CLI procedure. This reduction gradually improved during the 35-day CLI protocol but continued to demonstrate a persistent ischemic state. ABI (Fig. 5) revealed no significant change in blood flow of the ischemic limb comparing the SR-nitrite- vs. placebo-treated group.
Fig. 5.
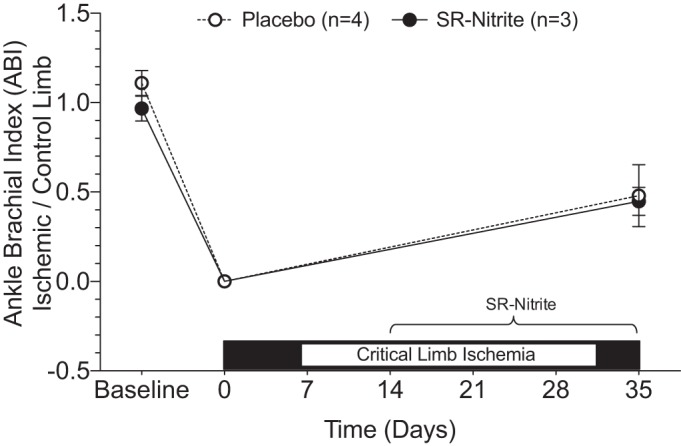
Nitrite therapy does not improve ankle-brachial index (ABI) in an animal model of CLI. Ischemic/control limb relative ankle-brachial index at baseline, immediately post-CLI, and at day 35.
Contrast angiography.
Representative, magnified-view contrast angiographic images of Ossabaw swine peripheral vasculature obtained at baseline, immediately after induction of CLI, and at post-CLI day 35 are shown in Fig. 6A.
Fig. 6.

Contrast angiographic images and angiographic score at post-CLI day 35. A: magnified-view contrast angiographic images of swine peripheral vasculature at baseline, immediately post-CLI, and at day 35 post-CLI after interventional deployment of a self-expanding endoluminal endoprosthesis consisting of an expanded polytetrafluoroethylene (ePTFE) lining with an external nitinol stent (black arrow) and a self-expanding nitinol mesh occlusion device (white arrow) into the right external iliac artery. B: hindlimb vessel density shown as angiographic score of magnified-view contrast angiographic images of control and CLI limbs distal from the ePTFE-lined endoprosthesis at CLI day 35. Numbers inside circles denote number of animals. **P < 0.01 vs. placebo-treated control limb.
Hindlimb vessel density was determined from the angiographic score of the magnified-view contrast angiographic images for the control and CLI limb distal from the ePTFE-lined endoprosthesis at post-CLI day 35. Quantitative angiographic analysis (Fig. 6B) revealed a significant reduction in perfusion of the CLI limb (P < 0.01) in the placebo-treated group compared with the control limb; however, no significant change (P = NS) was observed in the SR-nitrite-treated animals between the control and CLI limb at post-CLI day 35. Angiographic assessment of hindlimb perfusion at day 35 revealed no significant changes in the CLI limb of SR-nitrite-treated animals compared with the placebo-treated group.
Nitrite therapy promotes angiogenic signaling in the ischemic limb.
Quantitative PCR and Western blot analysis performed at post-CLI day 35 demonstrate increases in known angiogenic signals following SR-nitrite therapy (Fig. 7). VEGF mRNA (Fig. 7A) was increased by approximately twofold in the ischemic limb following SR-nitrite treatment (P < 0.05 vs. placebo). Platelet endothelial cell adhesion molecule-1 (or CD31) is a marker for endothelial cell expansion, and we did not observe an increase in CD31 mRNA in the CLI limb of SR-nitrite- compared with placebo-treated animals (P = 0.51; Fig. 7B). Representative Western blots for VEGF and CD31 are shown in Fig. 7C. VEGF protein expression trended higher (P = 0.051) in the SR-nitrite- than placebo-treated CLI limb, while SR-nitrite failed to alter VEGF protein expression in the control limb (Fig. 7D). CD31 protein expression was significantly (P < 0.05) increased in the CLI limb of the SR-nitrite- group compared with the placebo-treated group (Fig. 7E). Skeletal muscle protein levels of basic fibroblast growth factor (Fig. 7F) were unchanged in control and CLI limbs of placebo- and SR-nitrite-treated animals. However, we did observe a significant (P < 0.05) increase in skeletal muscle von Willebrand factor in the CLI limb of the SR-nitrite-treated group (Fig. 7G). Collectively, these data suggest that SR-nitrite therapy did result in increased angiogenic activity in ischemic hindlimb skeletal muscle.
Fig. 7.

Nitrite therapy promotes angiogenesis during CLI. A and D: message RNA and protein expression of VEGF in control and CLI limb skeletal muscle. B and E: message RNA and protein expression of CD31 (platelet endothelial cell adhesion molecule) in control and ischemic limb skeletal muscle. C: representative immunoblots of VEGF and CD31 expression in ischemic skeletal muscle. F and G: basic fibroblast growth factor and von Willebrand factor levels in skeletal muscle obtained at day 35. Numbers inside circles denote number of animals. *P < 0.01 vs. SR-nitrite-treated control limb.
SR-nitrite attenuates skeletal muscle oxidative stress and activates eNOS following CLI.
One of the pathological consequences of chronic tissue ischemia is increased oxidative stress and cellular injury. Since NO is known to modulate redox status and mitigate oxidative stress, we evaluated the extent of oxidative stress in control and CLI skeletal muscle at post-CLI day 35. Skeletal muscle levels of carbonyl and MDA are shown in Fig. 8. Carbonyl levels (Fig. 8A) were unchanged in the control limb following CLI. However, carbonyl levels were significantly (P < 0.05) elevated in the CLI limb compared with the control limb in placebo-treated animals. Treatment with SR-nitrite significantly (P < 0.05) attenuated carbonyl levels in the CLI limb compared with placebo treatment. Similar results for MDA are shown in Fig. 8B. No changes in MDA levels were observed in the control limb following CLI. MDA levels were significantly (P < 0.05 vs. control) increased in the CLI limb of placebo-treated animals compared with the control limb. SR-nitrite failed to significantly attenuate MDA levels in the CLI limb compared with placebo treatment. These studies confirm previous findings that nitrite and NO significantly attenuate oxidative stress during pathological states (23, 42). Recent studies have shown that nitrite therapy activates eNOS and that a reduction in oxidative stress also recouples eNOS to its more functional dimerized form (6, 11, 25). We observed that SR-nitrite therapy resulted in improved eNOS functionality in the ischemic skeletal muscle, as evidenced by increased Ser1177 phosphorylation of eNOS compared with placebo treatment (Fig. 8, C and E). There was no change in total eNOS expression between groups (Fig. 8, C and D).
Fig. 8.

Nitrite therapy reduces elevated oxidative stress and activates eNOS in the CLI limb. A and B: skeletal muscle carbonyl protein content and malondialdehyde (MDA) levels. C: representative immunoblots of eNOS protein expression in ischemic skeletal muscle. D and E: total and Ser1177-phosphorylated eNOS expression in placebo- and SR-nitrite-treated animals. Numbers inside circles denote number of animals. *P < 0.05 vs. placebo-treated control. **P < 0.01 vs. SR-nitrite-treated control.
DISCUSSION
Effective new treatments for CLI are critically important, given the high incidence of major amputation and mortality in this population (22). Increasing blood flow, tissue perfusion, and oxygen and nutrient delivery via new vessel growth or collateralization could promote tissue regeneration and/or delay or prevent tissue necrosis and the development of conditions such as gangrene. Since ∼50% of CLI patients are poor candidates for revascularization because of unavailable anatomy or conduit (37), we sought a pharmacological approach to enhance perfusion, attenuate skeletal muscle injury, and reduce necrosis in a severe model of CLI in the setting of obesity and MetS. In the present study we evaluated the effects of delayed therapy with a novel, SR-nitrite formulation on severe CLI in a clinically relevant porcine model of obesity and MetS. Our data clearly indicate that oral administration of SR-nitrite is well tolerated, increases circulating and ischemic zone tissue levels of nitrite and other NO intermediates, reduces oxidative stress in ischemic tissue, and triggers proangiogenic and cytoprotective tissue signaling in the setting of severe CLI. However, we failed to observe significant increases in ischemic zone blood flow or vascularity following SR-nitrite treatment in CLI.
Previous studies of limb ischemia have been primarily limited to murine and rabbit model systems, in which CLI is introduced via surgical ligation of the femoral artery (16, 40). This procedure often results in substantial angiogenic and arteriogenic responses, with complete restoration of hindlimb blood flow in a relatively short time frame. This response to surgical occlusion of a peripheral artery is inconsistent with the pathophysiology of PAD or CLI in humans (12, 26, 43). Therefore, we sought to create a more clinically relevant model of CLI in the setting of obesity/MetS. Angiographic analysis and ABI measurements immediately following CLI induction clearly demonstrate complete occlusion of the external iliac artery and undetectable ankle blood pressure. At post-CLI day 35, angiography revealed a nearly complete absence of significant collateral vessel growth at the location of the stent.
It is well established that blood vessels require continuous production of NO (29), and reductions in NO bioavailability significantly impact vascular function, resulting in vasoconstriction, thrombosis, atherogenesis, and hypertension (17). Initial experiments demonstrated significant reductions in NO bioavailability and NO-mediated signaling in human gastrocnemius muscle of CLI tissue compared with healthy control tissue. Böger et al. (8) observed a correlation between gradual reduction of NO synthesis and increased symptoms in PAD patients. NO deficiency, likely due to the combination of NO scavenging and NO synthase dysregulation, results in profound reductions in NO bioavailability and vascular dysfunction. In addition to being a potent vasodilator, antiapoptotic agent, and antioxidant, NO has also been shown to mediate angiogenesis and promote blood flow in response to tissue ischemia (14, 33). Specifically, NO therapy with sodium nitrite increases ischemic hindlimb blood flow and vascular density in murine models of PAD, including the db/db diabetic mouse model (26). Although no large-animal studies of treatment of CLI with NO-based therapeutics have been reported, a recent phase II clinical trial for safety and efficacy in which PAD patients were treated with oral sodium nitrite reported the drug to be well tolerated (32). High-dose sodium nitrite resulted in improved endothelial function, as seen by changes in flow-mediated vasodilation, along with improvements in overall physical functioning and pain, in diabetic patients (32).
In the current study we investigated a SR-nitrite formulation that was administered orally. Nitrite is metabolized fairly rapidly in vivo under conditions of tissue ischemia, and the SR-nitrite formulation utilized in the current study was selected to provide more continuous nitrite delivery to the ischemic limb. Safety was validated by stable circulating methemoglobin levels in the SR-nitrite-treated group. Pharmacokinetic analysis revealed a steady and significant rise in circulating nitrite, as well as nitrite delivery to the ischemic limb following treatment. Importantly, RSNO levels in the plasma and ischemic limb indicate that nitrite was converted to NO, as evidenced by increases in NO-bound protein. In the placebo-treated group, there was a significant reduction in RSNO and a trending decrease in nitrite levels in skeletal muscle of the ischemic compared the control limb. This reduction in NO intermediates in the porcine CLI model is consistent with our finding that NO levels are significantly attenuated in skeletal muscle from CLI patients.
The occlusion of large vessels presents a challenging scenario for a pharmacological agent, where the ischemic tissue requires extensive vessel formation to prevent necrosis. Preclinical studies using murine models of hindlimb ischemia have shown NO to be a proangiogenic factor (9, 28, 45). In the current study we evaluated whether nitrite therapy had an impact on collateralization and revascularization of larger vessels. Analysis of gastrocnemius skeletal muscle revealed a trending, but not statistically significant, increase in vessel density in the ischemic limb of the SR-nitrite-treated group. ABI measurements confirm that there was no improvement in ischemic limb blood pressure following SR-nitrite therapy. However, analysis of the angiogenic growth factor VEGF and markers for endothelial cell proliferation (CD31 and von Willebrand factor) indicated that SR-nitrite therapy did, in fact, promote signaling for vessel growth.
A primary limitation of this study is the extreme severity of CLI that is induced following implantation of a covered stent in combination with an AMPLATZER occluder in the external iliac artery. This procedure results in complete occlusion of the external iliac artery and initial absence of blood flow. The extreme reductions in hindlimb perfusion coupled with MetS result in an extremely severe model for testing therapies. Another potential limitation of our study is the delayed administration of SR-nitrite therapy at post-CLI day 14 and the relatively short duration of SR-nitrite therapy. Although therapy would have likely been more beneficial if administered at the time of CLI induction, we initiated SR-nitrite therapy in a clinically relevant timeline. Similarly, if we had administered the SR-nitrite beyond day 35, we may have observed improvements in hindlimb perfusion and ABI.
Upregulation of endothelial proliferation factors, such as VEGF, may not have resulted in arterial vessel formation and improved blood flow due to the severe model and delayed SR-nitrite therapy. It is not entirely surprising that sustained VEGF signaling observed in these studies (day 35) failed to result in endothelial proliferation. Recent studies suggest that prolonged elevations in VEGF-A can act as a negative regulator via a splicing mechanism to inhibit angiogenic signaling (39).
Finally, our data reveal that circulating and ischemic tissue levels of nitrite were significantly increased at 21 days following initial dosing, but we failed to observe statistically significant increases in plasma nitrite at the earlier time points. It is unclear exactly what circulating or tissue levels are required to exert cytoprotective and proangiogenic effects in animal models or humans. Therefore, it is conceivable that our dosing regimen did not result in therapeutic nitrite levels during the entire treatment period. Our dosing rationale was based on a phase II clinical trial to treat patients with PAD (32).
While the present study provides important new insights into nitrite therapy in CLI, additional studies are clearly required to more fully evaluate the therapeutic potential of SR-nitrite and other nitrite formulations in severe PAD and CLI to determine the potential efficacy of this NO-based therapeutic in patients with peripheral vascular disease.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants 1R01 HL-092141, 1R01 HL-093579, 1U24 HL-094373, and 1P20 HL-113452 (to D. J. Lefer) and 1K08 HL-119592 (to L. P. Brewster) and in part by National Center for Advancing Translational Sciences Clinical and Translational Science Award Program Grant UL1 TR-000454 (to L. P. Brewster) and American Heart Association Grant 13IRG14740001 (to L. P. Brewster). Generous funding was provided by the Carlyle Fraser Heart Center at Emory University Midtown Hospital, the Emory University Department of Surgery, and the LSU Medical School Alumni Association.
DISCLOSURES
D. J. Lefer is on the scientific advisory board of Theravasc, Inc., which is currently developing sodium nitrite for the treatment of cardiovascular diseases. D. J. Lefer is a participant of a pending US patent filed on 14 October 2003 (patent no. 60/511244) regarding the use of sodium nitrite in cardiovascular disease. D. J. Lefer is a participant of a pending US patent filed on 15 November 2007 (patent no. 61/003150) regarding the use of nitrite salts in chronic ischemia.
AUTHOR CONTRIBUTIONS
D.J.P., J.M.B., L.P.B., C.C.C., I.I.P., T.T.G., and D.J.L. developed the concept and designed the research; D.J.P., J.M.B., K.N.I., L.P.B., J.W.C., Y.T., C.C.C., I.I.P., T.T.G., and D.J.L. performed the experiments; D.J.P., J.M.B., K.N.I., and D.J.L. analyzed the data; D.J.P., J.M.B., I.I.P., and D.J.L. interpreted the results of the experiments; D.J.P., J.M.B., and D.J.L. prepared the figures; D.J.P., J.M.B., and D.J.L. drafted the manuscript; D.J.P., J.M.B., L.P.B., J.W.C., T.T.G., and D.J.L. edited and revised the manuscript; D.J.P., J.M.B., K.N.I., L.P.B., J.W.C., C.C.C., I.I.P., T.T.G., and D.J.L. approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Shashi Bhushan, Hiroyuki Otsuka, Scott Robinson, Kevin Brown, Michael Sweet, Jacob Vinten-Johansen, Susan L. Schmarkey, and Amy Souder for their expert technical assistance. We are grateful to Theravasc (Cleveland, OH) for the generous supply of SR-nitrite and W. L. Gore and Associates (Flagstaff, AZ) for providing the GORE VIABAHN endoprosthesis.
REFERENCES
- 1.Amin A, Choi SK, Osman-Elazeik Y, Badr El-Din NK, Kevil CG, Navar LG, Kadowitz P, Trebak M, Matrougui K. Sodium nitrite therapy rescues ischemia-induced neovascularization and blood flow recovery in hypertension. Pflügers Arch 464: 583–592, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Annex BH. Therapeutic angiogenesis for critical limb ischaemia. Nat Rev Cardiol 10: 387–396, 2013. [DOI] [PubMed] [Google Scholar]
- 3.Bell LN, Lee L, Saxena R, Bemis KG, Wang M, Theodorakis JL, Vuppalanchi R, Alloosh M, Sturek M, Chalasani N. Serum proteomic analysis of diet-induced steatohepatitis and metabolic syndrome in the Ossabaw miniature swine. Am J Physiol Gastrointest Liver Physiol 298: G746–G754, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bender SB, Tune JD, Borbouse L, Long X, Sturek M, Laughlin MH. Altered mechanism of adenosine-induced coronary arteriolar dilation in early-stage metabolic syndrome. Exp Biol Med (Maywood) 234: 683–692, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berwick ZC, Dick GM, Moberly SP, Kohr MC, Sturek M, Tune JD. Contribution of voltage-dependent K+ channels to metabolic control of coronary blood flow. J Mol Cell Cardiol 52: 912–919, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhushan S, Kondo K, Polhemus DJ, Otsuka H, Nicholson CK, Tao YX, Huang H, Georgiopoulou VV, Murohara T, Calvert JW, Butler J, Lefer DJ. Nitrite therapy improves left ventricular function during heart failure via restoration of nitric oxide-mediated cytoprotective signaling. Circ Res 114: 1281–1291, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bir SC, Pattillo CB, Pardue S, Kolluru GK, Shen X, Giordano T, Kevil CG. Nitrite anion therapy protects against chronic ischemic tissue injury in db/db diabetic mice in a NO/VEGF-dependent manner. Diabetes 63: 270–281, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Böger RH, Bode-Böger SM, Thiele W, Junker W, Alexander K, Frolich JC. Biochemical evidence for impaired nitric oxide synthesis in patients with peripheral arterial occlusive disease. Circulation 95: 2068–2074, 1997. [DOI] [PubMed] [Google Scholar]
- 9.Brevetti LS, Chang DS, Tang GL, Sarkar R, Messina LM. Overexpression of endothelial nitric oxide synthase increases skeletal muscle blood flow and oxygenation in severe rat hind limb ischemia. J Vasc Surg 38: 820–826, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Bryan NS, Calvert JW, Gundewar S, Lefer DJ. Dietary nitrite restores NO homeostasis and is cardioprotective in endothelial nitric oxide synthase-deficient mice. Free Radic Biol Med 45: 468–474, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87: 840–844, 2000. [DOI] [PubMed] [Google Scholar]
- 12.Cao R, Brakenhielm E, Pawliuk R, Wariaro D, Post MJ, Wahlberg E, Leboulch P, Cao Y. Angiogenic synergism, vascular stability and improvement of hind-limb ischemia by a combination of PDGF-BB and FGF-2. Nat Med 9: 604–613, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Christoffersen B, Golozoubova V, Pacini G, Svendsen O, Raun K. The young Gottingen minipig as a model of childhood and adolescent obesity: influence of diet and gender. Obesity (Silver Spring) 21: 149–158, 2013. [DOI] [PubMed] [Google Scholar]
- 14.Cooke JP, Losordo DW. Nitric oxide and angiogenesis. Circulation 105: 2133–2135, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO 3rd, Gladwin MT. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med 9: 1498–1505, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Couffinhal T, Silver M, Zheng LP, Kearney M, Witzenbichler B, Isner JM. Mouse model of angiogenesis. Am J Pathol 152: 1667–1679, 1998. [PMC free article] [PubMed] [Google Scholar]
- 17.Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation 109: III27–III32, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Dormandy JA, Rutherford RB. Management of peripheral arterial disease (PAD). TASC Working Group TransAtlantic Inter-Society Consensus (TASC). J Vasc Surg 31: S1–S296, 2000. [PubMed] [Google Scholar]
- 19.Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet SF, Wang X, Kevil CG, Gladwin MT, Lefer DJ. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest 115: 1232–1240, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorter PM, Olijhoek JK, van der Graaf Y, Algra A, Rabelink TJ, Visseren FL, Group SS. Prevalence of the metabolic syndrome in patients with coronary heart disease, cerebrovascular disease, peripheral arterial disease or abdominal aortic aneurysm. Atherosclerosis 173: 363–369, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Jacobsson L. Comparison of experimental hypercholesterolemia and atherosclerosis in male and female mini-pigs of the Gottingen strain. Artery 16: 105–117, 1989. [PubMed] [Google Scholar]
- 22.Jude EB, Oyibo SO, Chalmers N, Boulton AJ. Peripheral arterial disease in diabetic and nondiabetic patients: a comparison of severity and outcome. Diabetes Care 24: 1433–1437, 2001. [DOI] [PubMed] [Google Scholar]
- 23.Kanner J, Harel S, Granit R. Nitric oxide as an antioxidant. Arch Biochem Biophys 289: 130–136, 1991. [DOI] [PubMed] [Google Scholar]
- 24.Kevil CG, Kolluru GK, Pattillo CB, Giordano T. Inorganic nitrite therapy: historical perspective and future directions. Free Radic Biol Med 51: 576–593, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao YX, Dugas TR, Kelley EE, Elrod JW, Huang PL, Wang R, Lefer DJ. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc Natl Acad Sci USA 111: 3182–3187, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar D, Branch BG, Pattillo CB, Hood J, Thoma S, Simpson S, Illum S, Arora N, Chidlow JH Jr, Langston W, Teng X, Lefer DJ, Patel RP, Kevil CG. Chronic sodium nitrite therapy augments ischemia-induced angiogenesis and arteriogenesis. Proc Natl Acad Sci USA 105: 7540–7545, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lassaletta AD, Chu LM, Robich MP, Elmadhun NY, Feng J, Burgess TA, Laham RJ, Sturek M, Sellke FW. Overfed Ossabaw swine with early stage metabolic syndrome have normal coronary collateral development in response to chronic ischemia. Basic Res Cardiol 107: 243, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lloyd PG, Yang HT, Terjung RL. Arteriogenesis and angiogenesis in rat ischemic hindlimb: role of nitric oxide. Am J Physiol Heart Circ Physiol 281: H2528–H2538, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Loscalzo J, Welch G. Nitric oxide and its role in the cardiovascular system. Prog Cardiovasc Dis 38: 87–104, 1995. [DOI] [PubMed] [Google Scholar]
- 30.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discovery 7: 156–167, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Milsom AB, Patel NS, Mazzon E, Tripatara P, Storey A, Mota-Filipe H, Sepodes B, Webb AJ, Cuzzocrea S, Hobbs AJ, Thiemermann C, Ahluwalia A. Role for endothelial nitric oxide synthase in nitrite-induced protection against renal ischemia-reperfusion injury in mice. Nitric Oxide Biol Chem 22: 141–148, 2010. [DOI] [PubMed] [Google Scholar]
- 32.Mohler ER 3rd, Hiatt WR, Gornik HL, Kevil CG, Quyyumi A, Haynes WG, Annex BH. Sodium nitrite in patients with peripheral artery disease and diabetes mellitus: safety, walking distance and endothelial function. Vasc Med 19: 9–17, 2014. [DOI] [PubMed] [Google Scholar]
- 33.Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest 101: 2567–2578, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicholson CK, Lambert JP, Chow CW, Lefer DJ, Calvert JW. Chronic exercise downregulates myocardial myoglobin and attenuates nitrite reductase capacity during ischemia-reperfusion. J Mol Cell Cardiol 64: 1–10, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olijhoek JK, van der Graaf Y, Banga JD, Algra A, Rabelink TJ, Visseren FL, Group SS. The metabolic syndrome is associated with advanced vascular damage in patients with coronary heart disease, stroke, peripheral arterial disease or abdominal aortic aneurysm. Eur Heart J 25: 342–348, 2004. [DOI] [PubMed] [Google Scholar]
- 36.Pluta RM, Dejam A, Grimes G, Gladwin MT, Oldfield EH. Nitrite infusions to prevent delayed cerebral vasospasm in a primate model of subarachnoid hemorrhage. JAMA 293: 1477–1484, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Powell RJ, Simons M, Mendelsohn FO, Daniel G, Henry TD, Koga M, Morishita R, Annex BH. Results of a double-blind, placebo-controlled study to assess the safety of intramuscular injection of hepatocyte growth factor plasmid to improve limb perfusion in patients with critical limb ischemia. Circulation 118: 58–65, 2008. [DOI] [PubMed] [Google Scholar]
- 38.Rassaf T, Totzeck M, Hendgen-Cotta UB, Shiva S, Heusch G, Kelm M. Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ Res 114: 1601–1610, 2014. [DOI] [PubMed] [Google Scholar]
- 39.Saito T, Takeda N, Amiya E, Nakao T, Abe H, Semba H, Soma K, Koyama K, Hosoya Y, Imai Y, Isagawa T, Watanabe M, Manabe I, Komuro I, Nagai R, Maemura K. VEGF-A induces its negative regulator, soluble form of VEGFR-1, by modulating its alternative splicing. FEBS Lett 587: 2179–2185, 2013. [DOI] [PubMed] [Google Scholar]
- 40.Seifert FC, Banker M, Lane B, Bagge U, Anagnostopoulos CE. An evaluation of resting arterial ischemia models in the rat hind limb. J Cardiovasc Surg 26: 502–508, 1985. [PubMed] [Google Scholar]
- 41.Selvin E, Erlinger TP. Prevalence of and risk factors for peripheral arterial disease in the United States: results from the National Health and Nutrition Examination Survey, 1999–2000. Circulation 110: 738–743, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Sindler AL, Fleenor BS, Calvert JW, Marshall KD, Zigler ML, Lefer DJ, Seals DR. Nitrite supplementation reverses vascular endothelial dysfunction and large elastic artery stiffness with aging. Aging Cell 10: 429–437, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takeshita S, Zheng LP, Brogi E, Kearney M, Pu LQ, Bunting S, Ferrara N, Symes JF, Isner JM. Therapeutic angiogenesis. A single intraarterial bolus of vascular endothelial growth factor augments revascularization in a rabbit ischemic hind limb model. J Clin Invest 93: 662–670, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vincent KA, Shyu KG, Luo Y, Magner M, Tio RA, Jiang C, Goldberg MA, Akita GY, Gregory RJ, Isner JM. Angiogenesis is induced in a rabbit model of hindlimb ischemia by naked DNA encoding an HIF-1alpha/VP16 hybrid transcription factor. Circulation 102: 2255–2261, 2000. [DOI] [PubMed] [Google Scholar]
- 45.Yu J, deMuinck ED, Zhuang Z, Drinane M, Kauser K, Rubanyi GM, Qian HS, Murata T, Escalante B, Sessa WC. Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proc Natl Acad Sci USA 102: 10999–11004, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]