

Abstract
Transforming growth factor β-activated kinase 1 (TAK1) is critical for survival of many KRAS mutated colorectal cancer cells, and TAK1 inhibition with 5Z-7-oxozeaenol has been associated with oxidative stress leading to tumor cell killing. When SW 620 and HCT 116 human colon cancer cells were treated with 5 µM 5Z-7-oxozeaenol, cell viability, growth, and clonogenic survival were significantly decreased. Consistent with TAK1 inhibition being causally related to thiol-mediated oxidative stress, 10 mM N-acetylcysteine (NAC) partially reversed the growth inhibitory effects of 5Z-7-oxozeaenol. In addition, 5Z-7-oxozeaenol also increased steady-state levels of H2DCFDA oxidation as well as increased levels of total glutathione (GSH) and glutathione disulfide (GSSG). Interestingly, depletion of GSH using buthionine sulfoximine did not significantly potentiate 5Z-7-oxozeaenol toxicity in either cell line. In contrast, pre-treatment of cells with auranofin (Au) to inhibit thioredoxin reductase activity significantly increased levels of oxidized thioredoxin as well as sensitized cells to 5Z-7-oxozeaenol-induced growth inhibition and clonogenic cell killing. These results were confirmed in SW 620 murine xenografts, where treatment with 5Z-7-oxozeaenol or with Au plus 5Z-7-oxozeaenol significantly inhibited growth, with Au plus 5Z-7-oxozeaenol trending toward greater growth inhibition compared to 5Z-7-oxozeaenol alone. These results support the hypothesis that thiol-mediated oxidative stress is causally related to TAK1-induced colon cancer cell killing. In addition, these results support the hypothesis that thioredoxin metabolism is a critical target for enhancing colon cancer cell killing via TAK1 inhibition and could represent an effective therapeutic strategy in patients with these highly resistant tumors.
Keywords: TAK1, KRAS, Colorectal cancer, Thioredoxin, Treatment resistance
Graphical abstract
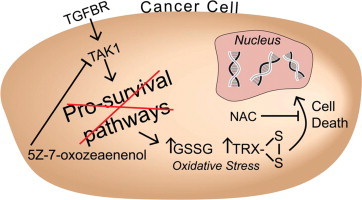
Introduction
Up to 60% of patients with colorectal cancer will develop metastatic disease [1]. The treatment of metastatic colon cancer depends, in part, on KRAS mutation status. Colon cancers that are wild-type KRAS can be treated with monoclonal antibodies such as cetuximab and panitumumab against the epidermal growth factor receptor (EGFR), and a proportion of individuals will have enhanced survival because of these treatments [2–4]. Unfortunately, tumors harboring KRAS mutations are unresponsive to these anti-EGFR therapies. Furthermore, even those tumors which are initially KRAS wild-type often develop resistance to EGFR blockade [5,6]. In these cases, patients have limited treatment options.
Transforming growth factor β-activated kinase 1 (TAK1) has recently been identified as a pro-survival signaling pathway for KRAS mutant colon cancer cell lines. Inhibition of TAK1 has been shown to impair growth and is suggested to increase steady-state levels of reactive oxygen species (ROS) in KRAS mutant cells; however the causal role of oxidative stress in the cell killing induced by 5Z-7-oxozeaenol has not been established [7–9]. 5Z-7-oxozeaenol is a commercially available irreversible inhibitor of TAK1 and ATPase activity. Within 20 min of treatment, over 90% of TAK1 is irreversibly bound by 5Z-7-oxozeaenol [8]. Since KRAS activation is thought to increase steady-state levels of pro-oxidants in cancer cells [9], and modest increases in ROS are thought to enhance pro-survival pathways such as TAK1 making cells resistant to oxidative stress [10], we reasoned that TAK1 inhibition in KRAS mutant colon cancer cells would render them unable to adapt to endogenous metabolic oxidative stress associated with the cancer cell phenotype. This would lead to increased pro-oxidant levels and enhanced oxidative stress-induced cell killing.
In the current study, we tested the hypothesis that inhibition of TAK1 with 5Z-7-oxozeaenol would enhance steady-state levels of H2DCFDA oxidation and induce cell killing by a mechanism involving oxidative stress in KRAS mutant colon cancer cell lines. We found that 5Z-7-oxozeaenol led to increased levels of H2DCFDA oxidation, increased levels of total and oxidized glutathione, and clonogenic cell killing. Furthermore, both increases in H2DCFDA oxidation and clonogenic cell killing mediated by 5Z-7-oxozeaenol were inhibited by the thiol antioxidant, N-acetylcysteine (NAC). Interestingly, depletion of glutathione with buthionine sulfoximine did not significantly enhance cell killing by TAK1 inhibition. In contrast, inhibition of thioredoxin reductase with auranofin significantly enhanced cell killing mediated by 5Z-7-oxozeaenol. These results support the hypothesis that TAK1-induced KRAS mutant colon cancer cell killing is causally related to thiol-mediated oxidation. This work also suggests that thioredoxin metabolism is a critical target for enhancing colon cancer cell killing with TAK1 inhibition and may represent an effective treatment strategy for these highly resistant cancers.
Materials and methods
Cell lines, media, and culture conditions
HCT 116, a colorectal adenocarcinoma cell line isolated from a primary tumor, and SW 620, a colorectal carcinoma cell line isolated from a lymph node metastasis, were obtained from ATCC (Manassas, VA; products CCL-247 and CCL-227, respectively). Both are KRAS mutant. HCT 116 cells were maintained in Dulbecco's Modified Eagle Medium (Gibco/Life Technologies, Grand Island, NY) and SW 620 were maintained in Leibovitz's L-15 Medium (Gibco/Life Technologies, Grand Island, NY), both supplemented with 10% FBS and 100 U/mL penicillin/streptomycin. Both cell lines were maintained at 37 °C and 5% CO2. Routine subculture was performed by washing with 1× PBS and detaching cells with TrypLE Express (Life Technologies, Grand Island, NY).
Drug treatment
Cells were treated with 5Z-7-oxozeaenol (TAK1 inhibitor) (Cat. no. O9890, Sigma-Aldrich, St. Louis, MO). A stock solution of Z-7-oxozeaenol inhibitor in DMSO was added to achieve a final concentration of 5 µM in media. DMSO (0.1%) was used as the vehicle control in all cases. Injectable N-acetylcysteine (NAC) (Acetadote, Cumberland Pharmaceuticals, Nashville, TN) was added 1 h following treatment with TAK1 inhibitor to achieve a final concentration of 10 mM. Buthionine sulfoximine (BSO) dissolved in PBS (Sigma-Aldrich, St. Louis, MO) was added 8 h prior to the addition of TAK1 inhibitor to a final concentration of 100 µM. Cells were treated with auranofin dissolved in absolute ethanol (Enzo Life Sciences, Farmingdale, NY) 3 h prior to treatment with TAK1 inhibitor to achieve a final concentration of 500 nM (0.1% ethanol). Treatments were added directly to the complete media specific for each cell line.
Cell viability assay
The effect of TAK1 inhibition on cell viability and the ability of NAC to counteract that effect was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were seeded at a density of 5×104 in a 96-well plate and incubated at 37 °C. After 24 h, cells were treated with TAK1 inhibitor, with and without subsequent NAC treatment. After 24 h of incubation, 100 µL of 0.5 mg/mL MTT (Sigma-Aldrich, St. Louis, MO) was added to each well for 4 h, following which the media with MTT was removed, cells were washed with PBS, and 100 µL DMSO was added to each well as the solubilizing agent. A microplate reader was used to read absorbance at 540 nm, and cell viability was determined by dividing the average absorbance of treated cells by untreated cells.
Growth curves
The effect of TAK1 inhibition, glutathione depletion, and thioredoxin reductase inhibition on cell growth was determined as follows. Cells were plated at time 0. Time points for treatment varied by drug and were as follows: at time 16 h, BSO was added; at 21 h, auranofin; at 24 h, TAK1 inhibitor; at 25 h, NAC. Cell counts were obtained 48, 72, and 96 h following plating. Cells were detached with TrypLE Express and then viable cells were counted using the Countess Automated Cell Counter (Life Technologies, Grand Island, NY).
Clonogenic survival assay
The ability of individual clones to proliferate following treatment with TAK1 inhibitor, BSO, or auranofin was measured by clonogenic cell survival assay. Briefly, cells were plated in 6-well dishes, treatment was applied as specified in each experiment: at time 16 h, BSO; at 21 h, auranofin; at 24 h, TAK1 inhibitor; at 25 h, NAC. At 48 h, cells were rinsed with PBS, detached with TrypLE Express, counted, and then plated in media without drug treatment at low density (100–500 cells per dish). Clones were allowed to grow for 14 days, at which point they were fixed with 70% ethanol and stained with 0.5% w/v crystal violet in methanol. Colonies of greater than 50 cells were counted. Cloning efficiency was calculated as the number of colonies per plate divided by the number of cells plated and data from drug treated dishes was normalized to the respective sham treated control.
Flow cytometry
Alterations in non-specific intracellular oxidation reactions caused TAK1 inhibition, and the ability of NAC to suppress these changes, was measured by following the oxidation of the fluorescent probe 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (Carboxy-H2DCFDA, C-400, Molecular Probes/Life Technologies, Grand Island, NY). Following 24 h of treatment with TAK1 inhibitor, with and without subsequent NAC treatment, cells were washed with PBS twice and then incubated for 15 min at 37 °C with 10 µg/mL Carboxy-H2DCFDA. Carboxy-H2DCFDA is nonfluorescent and membrane permeable; its ester groups undergo cleavage by intracellular esterases, followed by non-specific pro-oxidant mediated oxidation of the oxidation sensitive bond to yield the fluorescent product dichlorofluorescein (DCF). The oxidation insensitive probe Carboxy-DCFDA (C-369, Molecular Probes/Life Technologies, Grand Island, NY) was used as a control in each experiment to ensure that increased DCF fluorescence was due to changes in dye oxidation, rather than to changes in dye uptake, efflux, or ester cleavage. Fluorescence was detected with a Beckton Dickinson LSR II flow cytometer using 488 nm excitation and 525 nm emission. Results of the Mean Florescence Intensity (MFI) of 10,000 cells obtained with the oxidation sensitive probe were normalized to the MFI of the respective treatment group labeled with the oxidation insensitive probe, to show that changes in dye oxidation (and not dye uptake, ester cleavage, or efflux) were being detected.
Glutathione assay
Cells were plated at a density of 2×106 in 150 mm dishes. At 16 h, BSO was applied; at 24 h, TAK1 inhibitor; and at 25 h, NAC. At 48 h, cells were washed in cold PBS and then scraped into 200 µL of 5% 5-sulfosalicylic acid (Sigma-Aldrich, St. Louis, MO) in water. Total GSH content was measured as described previously [11]. Glutathione disulfide (GSSG), the oxidized form of GSH, was measured by adding 20 µL of a 1:1 mixture of 2-vinylpyridine and ethanol per 100 µL of sample, incubating for 2 h, and assaying as previously described [12]. Glutathione determinations were normalized to the protein content using the BCA protein assay (Thermo Fisher Scientific, Waltham, MA). Overall values were normalized to vehicle control.
Thioredoxin reductase activity
Thioredoxin reductase (TrxR) activity was determined in cell homogenates spectrophotometrically using previously described methods (CSO170 Sigma-Aldrich, St. Louis, MO) [13]. Enzymatic activity was assessed by determining the difference between the time-dependent increase in absorbance at 412 nm in the presence of the TrxR activity inhibitor from total activity. One activity unit equaled 1 µmol/L 5′-thionitrobenzoic acid formed/(min mg protein). Lowry assay was used to determine protein concentrations.
Thioredoxin Western
Oxidized and reduced forms of thioredoxin (Trx) were separated based on the method described previously [13,14]. Cells were plated in 100 mm dishes and treated with auranofin at 21 h, with TAK1 inhibitor at 24 h, and with NAC at 25 h. At 48 h, cells were harvested by scraping into G-lysis buffer containing 50 mM iodoacetic acid, incubating at 37° for 30 min, then removing excess iodoacetic acid with desalting spin columns (illustra MicroSpin G-25 columns, GE Healthcare, Pittsburgh, PA). Equal amounts of total protein prepared in a non-denaturing, non-reducing buffer were loaded into a 15% non-denaturing, non-reducing gel that was then electroblotted to nitrocellulose membrane. Blots were probed for Trx using anti-Trx-1 primary antibody (Cell Signaling, Boston, MA). Goat anti-rabbit HRP-conjugated IgG (BD Transduction Laboratories, San Diego, CA) was used for the secondary antibody. Chemiluminescent detection was performed with Super Signal West Pico Chemiluminescent Reagent (Pierce, Rockford, IL) on a Typhoon FLA 7000 fluorescent detection system. Band densities were calculated with ImageJ software as previously described [15].
Tumor xenograft growth in a murine model
Athymic, female nu/nu nude mice 6–8 weeks old were purchased from Harlan Laboratories (Indianapolis, Indiana) and housed in the Animal Care Facility at the University of Iowa. All procedures conformed to National Institutions of Health established guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Iowa. 2×106 SW 620 cells were injected subcutaneously into mice hind limbs. Following 11 days of tumor growth, mice were randomly assigned to one of four treatment groups: vehicle control (sterile 2.5% DMSO in vegetable oil); 5Z-7-oxozeaenol (ENZO Life Science ALX-380-267-M005) 15 mg/kg, dissolved in sterile DMSO (final concentration 2.5%) and vegetable oil; auranofin 1.6 mg/kg, dissolved in sterile DMSO and vegetable oil; and a combination of 5Z-7-oxozeaenol and auranofin. The drugs were administered via intraperitoneal injection for five consecutive days, followed by 2 days of auranofin only, and then four more consecutive days of combination treatment. Tumor volume was measured daily by calipers by an individual blinded to the treatment group, and tumor volumes calculated.
Statistical analysis
For in vitro work, statistical analysis was done using GraphPad Prism version 6.0 for (GraphPad Software, La Jolla, CA). To determine differences between 3 or more means, one-way ANOVA with least significant difference posttests were performed. Error bars represent the standard error of the mean. All statistical analysis was performed at the P <0.05 level of significance.
For tumor volume in the murine model, linear mixed effects regression models were used to estimate and compare group-specific tumor growth curves. Pairwise comparisons were performed to identify specific group differences in the growth curves. All tests were two-sided and carried out at the 5% level of significance. Analyses were performed with SAS version 9.3 (SAS Institute Inc., Cary, NC).
Results
Cell viability, growth, and clonogenic survival are impaired by TAK1 inhibition but restored with NAC, suggesting a role for oxidative stress in TAK1-mediated tumor cell death
Treatment with the TAK1 inhibitor 5Z-7-oxozeaenol has been shown to increase apoptosis in several KRAS-mutated colorectal cancer cells [7], though the mechanism is not entirely understood. To investigate this, we characterized the effects of TAK1 inhibition on viability, cell growth, and the ability of human colon cancer cells to form clonal populations. Following 24 h of treatment with the TAK1 inhibitor, cell viability was measured using the MTT assay. HCT 116 and SW 620 cells treated with TAK1 inhibitor had significantly decreased viability compared to vehicle control cells, which was restored by treating with NAC 1 h after addition of 5Z-7-oxozeaenol (Fig. 1A).
Fig. 1.
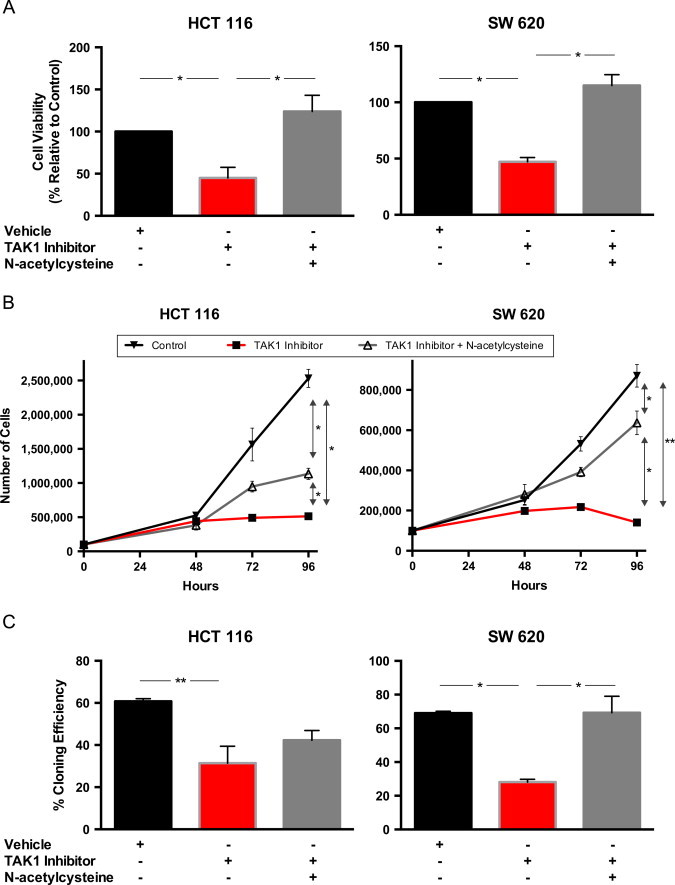
TAK1 inhibition decreased cell viability, growth, and clonogenic survival in HCT 116 and SW 620 cells; N-acetylcysteine, a thiol antioxidant, reversed these changes. Cells were treated with 5 µM 5Z-7-oxozeaenol and then 10 mM N-acetylcysteine. Vehicle=0.1% DMSO. A. Cell viability after 24 h of treatment was measured by MTT. (B) Cells were plated at time 0 and treatment applied at 24 h; cell counts were obtained at 48, 72, and 96 h after plating to determine cell growth rate. (C) After 24 h of treatment, cells were detached using TrypLE Express and plated at low density. 14 days later, colonies were fixed, stained, and counted to determine the rate of clonogenic survival. Data are mean±SEM of at least 3 separate experiments. *P <0.01, **P <0.05.
Similar effects on cell growth and clonogenic survival were seen following treatment with TAK1 inhibitor and again these growth inhibitory effects were reversed by NAC (Fig. 1B and C). The addition of NAC restored clonogenic survival in both cell lines, however, the results did not reach statistical significance in HCT 116; SW 620 clonogenic survival was completely restored to that of controls. NAC's ability to abrogate the toxic effects of TAK1 inhibition suggests that thiol-mediated oxidative stress may be important to the mechanism by which cell viability, growth, and clonogenic survival are impaired.
TAK1 inhibition leads to an increase in steady-state levels of pro-oxidants and an upregulation of glutathione metabolism
Relative to the oxidation insensitive analog of the dye, H2DCFDA oxidation was significantly increased in both HCT 116 and SW 620 cells treated with 5Z-7-oxozeaenol. This effect was abrogated by treatment with NAC (Fig. 2A). These results show that TAK1 inhibition is accompanied by an increase in non-specific intracellular pro-oxidants that can be inhibited by NAC. Furthermore, these results are consistent with the hypothesis that there is a causal relationship between TAK1-induced cell killing and thiol-mediated oxidative stress.
Fig. 2.
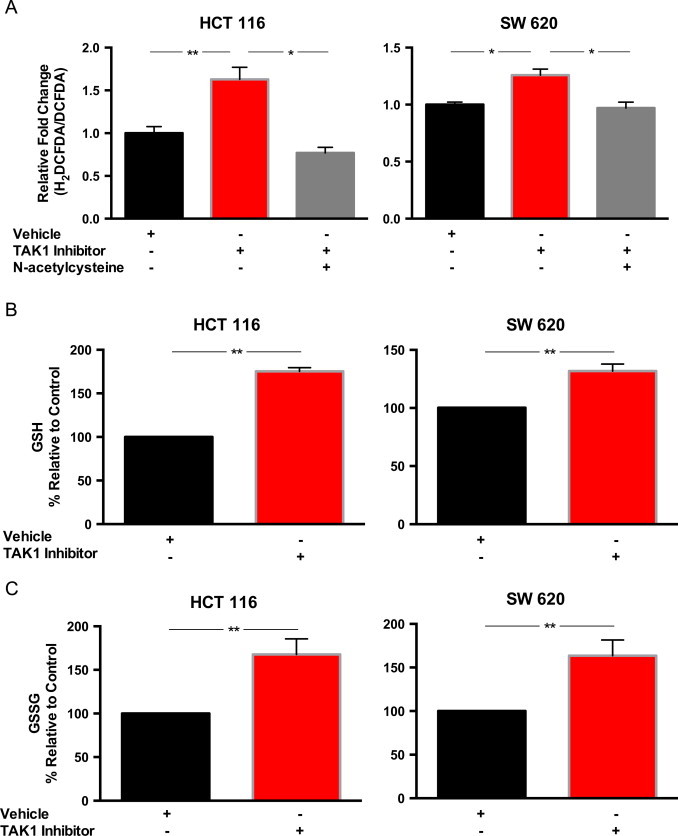
Oxidative stress is increased with TAK1 inhibition, and cells respond to TAK1 inhibition with increased intracellular glutathione. Cells were treated with 5 µM 5Z-7-oxozeaenol and then 10 mM N-acetylcysteine. Vehicle=0.1% DMSO. (A) Flow cytometry was used to analyze intracellular reactive oxygen species generation after 24 h of treatment using H2DCFDA (redox-sensitive probe) and DCFDA (redox-insensitive probe). Data normalized to vehicle control and presented as fold change of mean fluorescence intensity for H2DCFDA-stained cells/DCFDA-stained cells. (B, C) After 24 h treatment with TAK1 inhibitor, cells were scrape-harvested and reduced glutathione (GSH), (B) and oxidized glutathione (GSSG), (C) levels were assayed. Normalized to vehicle control. Errors represent ±1 SEM of at least 3 separate experiments. *P <0.01, **P <0.05.
As glutathione is the major soluble thiol antioxidant, we examined whether this increase in pro-oxidants was accompanied by changes in intracellular glutathione (GSH) levels. We examined both the total cellular GSH as well as the amount of glutathione disulfide (GSSG). In both HCT 116 and SW 620 cell lines, total GSH as well as GSSG were significantly increased when cells were exposed to TAK1 inhibitor (Fig. 2B and C). The increase in total GSH and GSSG suggests greater amounts of both synthesis and oxidation of glutathione, which is in accordance with the increased H2DCFDA oxidation demonstrated by flow cytometry causing oxidative stress.
Depleting glutathione did not potentiate the effects of TAK1 inhibition
Given the protective effects of the thiol antioxidant NAC as well as the increase in glutathione when cells were treated with 5Z-7-oxozeaenol, we hypothesized that glutathione depletion would render cells more vulnerable to the effects of the TAK1 inhibitor. Cells were pre-treated with BSO, an irreversible inhibitor of γ-glutamylcysteine synthetase, the rate-limiting enzyme for glutathione synthesis. We first confirmed that intracellular glutathione was significantly decreased in both HCT 116 and SW 620 cells treated with BSO (Fig. 3A). Next, we assayed cell growth rate and clonogenic survival in cells pretreated with BSO. Surprisingly, BSO treatment caused no significant differences in growth rate (Fig. 3B) or clonogenic survival (Fig. 3C) between cells treated with 5Z-7-oxozeaenol and those pretreated with BSO followed by 5Z-7-oxozeaenol. For both cell lines, treatment with BSO alone did not significantly impair either growth or clonogenic survival. From these data, we concluded that alterations in glutathione metabolism were not central to the mechanism by which TAK1 inhibition caused growth inhibitory effects in colon cancer cells.
Fig. 3.
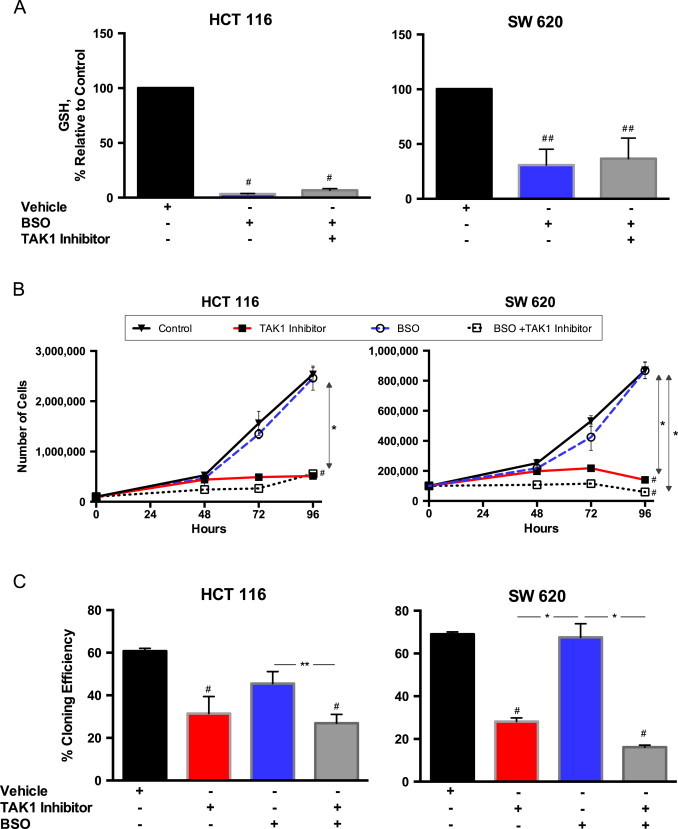
Depleting intracellular glutathione had little effect on cell growth and clonogenic survival when coupled with TAK1 inhibition. Cells were pretreated with 100 µM BSO for 8 h to deplete intracellular glutathione, then treated with 5 µM 5Z-7-oxozeaenol. (A) After 24 h of treatment, cells were scrape harvested and total intracellular glutathione levels, shown as normalized to vehicle control, were assayed. (B) Cells were plated at time 0 and treatment applied at 24 h; cell counts were obtained at 48, 72, and 96 h after plating to determine cell growth rate. (C) After 24 h of treatment, cells were detached using TrypLE Express and plated at low density. 14 days later, colonies were fixed, stained, and counted to determine the rate of clonogenic survival. Values represent mean±1 SEM of at least 3 separate experiments. #P-value<0.01 versus control. *P-value<0.01. **P-value<0.05.
Auranofin inhibits thioredoxin reductase (TrxR) and, when combined with 5Z-7-oxozeaenol, leads to an increase in the ratio of oxidized to total thioredoxin
To study the other major intracellular thiol-dependent pathway that may be responsible for protection from 5Z-7-oxozeaenol, cells were treated with auranofin to inhibit TrxR. TrxR is critical for regenerating the reduced form of Trx necessary for reducing protein thiols and driving the catalytic decomposition of hydroperoxides by peroxiredoxins [16,17]. TrxR also initiates signaling pathways that affect gene regulation related to protecting cells from oxidative stress [17]. As such, its role in cell defense against oxidative stress has been suggested to represent a significant therapeutic target during cancer therapy [18].
We confirmed that auranofin significantly inhibited TrxR activity in both HCT 116 and SW 620 cells (Fig. 4A). We then evaluated the effects of auranofin treatment on the redox state of Trx-1. SW 620 cells were treated as previously described and then analyzed for oxidized and reduced Trx by Western blot (Fig. 4B). Combination treatment with auranofin and 5Z-7-oxozeaenol led to a significant increase in the ratio of oxidized to total Trx-1 as compared to either 5Z-7-oxozeaenol or auranofin alone (Fig. 4B and C).
Fig. 4.
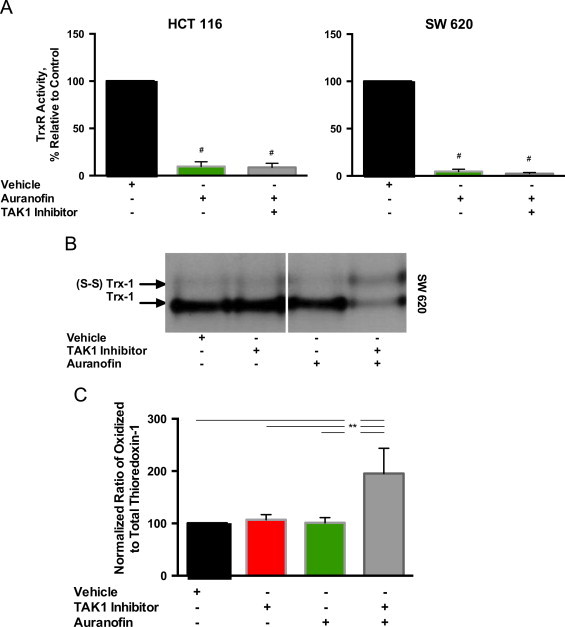
Auranofin inhibits thioredoxin reductase (TrxR) activity and, when combined with the TAK1 inhibitor, increases the ratio of oxidized to total thioredoxin in SW 620 cells. SW 620 cells were pretreated with 500 nM auranofin, then with 5 µM 5Z-7-oxozeaenol. (A) Cells were scrape harvested and thioredoxin reductase activity was assayed and normalized to vehicle control. (B) SW 620 cells were scrape harvested. Thioredoxin-1 western blot was performed showing oxidized [(S-S) trx] and total thioredoxin (trx). (C) Semiquantitative analysis of Western blots for oxidized to total thioredoxin. Values represent the mean±1 SEM from 3 separate experiments. #P-value<0.01 versus control. **P-value<0.05.
Inhibition of thioredoxin metabolism potentiates the effects of TAK1 inhibition
When coupled with 5Z-7-oxozeaenol, auranofin led to significantly impaired growth (Fig. 5A) and dramatically reduced clonogenic survival (Fig. 5B) as compared to the TAK1 inhibitor alone in both HCT 116 and SW 620 cells. In contrast to BSO, auranofin alone had a toxic effect on both cell lines, yielding a statistically significantly slower rate of growth and clonogenic survival as compared to control (Fig. 5A and B). These results support the hypothesis that HCT 116 and SW 620 cells are sensitive to the disruption of Trx metabolism, and inhibition of TrxR activity significantly sensitizes colon cancer cell lines to TAK1 inhibition.
Fig. 5.
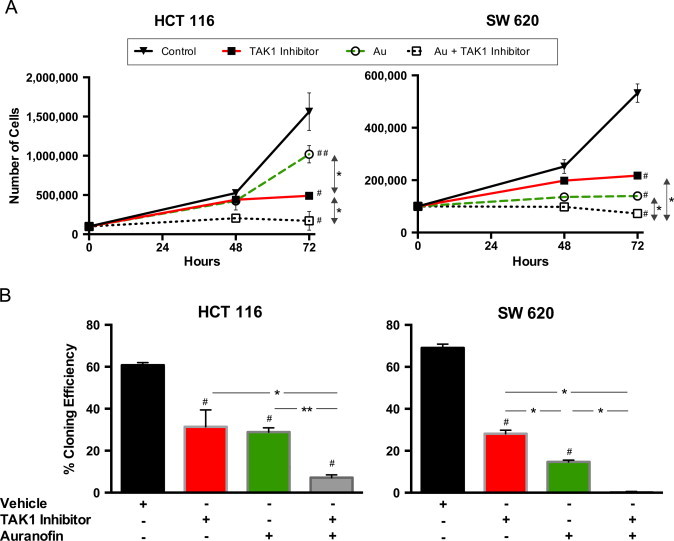
Auranofin inhibits thioredoxin reductase (TrxR) activity and enhances toxicity of TAK1 inhibition in both HCT 116 and SW 620 cells. SW 620 cells were pretreated with 500 nM auranofin, then with 5 µM 5Z-7-oxozeaenol. (A) Cells were plated at time 0 and treatment applied at 24 h; cell counts were obtained at 48 and 72 h after plating to determine cell growth rate. (B) After 24 h of treatment, cells were detached using TrypLE Express and plated at low density. 14 days later, colonies were fixed, stained, and counted to determine the rate of clonogenic survival. Errors represent ±1 SEM of at least 3 separate experiments. #P-value<0.01 versus control. ##P-value<0.05 versus control. *P-value<0.01. **P-value<0.05.
Auranofin plus TAK1 inhibitor significantly slows growth of human colon cancer tumor xenografts
SW 620 xenografts were grown for 11 days in athymic nude mice and were treated with (1) vehicle control (sterile DMSO in vegetable oil), (2) 5Z-7-oxozeaenol (15 mg/kg, dissolved in sterile DMSO and vegetable oil), (3) auranofin (1.6 mg/kg, dissolved in sterile DMSO and vegetable oil), or (4) the combination of 5Z-7-oxozeaenol and auranofin (Fig. 6). The drugs were administered as noted via intraperitoneal injection for five consecutive days. This was followed by 2 days of treatment with auranofin only, and then four additional consecutive days of administration of both drugs. Mice in both the TAK1 inhibitor and the combination groups lost a significant amount of weight during treatment; however they quickly regained weight both during the 2 days that TAK1 was not given and at the completion of therapy (data not shown). Auranofin alone did not significantly slow tumor growth as compared to the control group. Mice treated with TAK1 inhibitor or with TAK1 inhibitor plus auranofin demonstrated significantly slowed tumor growth compared to control. The mice treated with the combination of TAK1 inhibitor plus auranofin showed the slowest tumor growth, however this did not reach statistical significance as compared to TAK1 inhibitor alone (P=0.07).
Fig. 6.
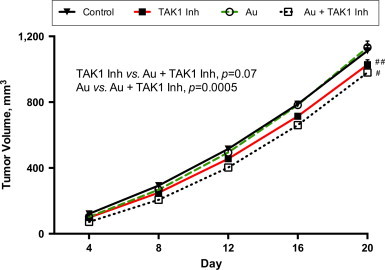
The combination of auranofin plus TAK1 inhibitor yields the slowest growth of hind-limb human colon cancer xenografts. 2×106 SW 620 cells were injected into the hind limbs of nude mice. After 11 days of tumor growth, mice were randomly assigned to treatment with intraperitoneal injection of vehicle control, TAK1 inhibitor (15 mg/kg), auranofin (1.6 mg/kg), or TAK1 inhibitor plus auranofin. Tumor size was measured with calipers and tumor volume calculated. N=7 per group; #P-value<0.01 versus control; ##P-value<0.05 versus control.
Discussion
Approximately 30–50% of colon cancers harbor KRAS mutations, which are predictive of poor treatment responses to drugs targeting EGFR thereby limiting therapeutic options for these patients [4,19,20]. Recent research has stimulated an interest in TAK1 as a potential new therapeutic target for KRAS mutated tumors [7]. In the current study, cancer cell growth inhibitory properties of TAK1 inhibition were determined in KRAS mutant colon cancer cells. The results demonstrate that treatment with the TAK1 inhibitor 5Z-7-oxozeaenol decreases cancer cell viability, cell growth rate, and clonogenic cell survival. These effects were all reversed by adding a non-specific thiol antioxidant, NAC, which for the first time supported a causal relationship between thiol-mediated oxidative stress and the growth inhibitory and cytotoxic effects of TAK1 inhibition on KRAS mutant colon cancer cells.
Other investigators have suggested that TAK1 regulates ROS levels in endothelial cells [21,22]. We have confirmed the fact that H2DCFDA oxidation is increased with TAK1 inhibition in KRAS mutant colon cancer cell lines as well as extending this to show NAC mitigate the effects of TAK1 inhibition on non-specific H2DCFDA oxidation. These novel results continue to support a causal relationship between TAK1 inhibition-induced thiol-mediated oxidative stress and cancer cell growth inhibition.
We have now also extended the TAK1 inhibition studies in colon cancer cells to include an analysis of redox state as determine by alterations intracellular GSH/GSSG. GSH is one of the major thiol redox buffers in cells responsible for ROS detoxification and is associated with cancer cell resistance to chemotherapy [23]. While GSH and GSSG levels were found to be significantly increased following treatment with 5Z-7-oxozeaenol, consistent with TAK1 inhibition inducing oxidative stress; GSH depletion using BSO did not sensitize the colon cancer cell lines to 5Z-7-oxozeaenol suggesting that GSH-independent thiol metabolic pathways were important in colon cancer cell resistance to TAK1 inhibition.
We next interrogated thioredoxin-dependent metabolism using the TrxR inhibitor, auranofin [24]. In contrast to the results seen with GSH depletion, growth inhibition and clonogenic cell killing following exposure to 5Z-7-oxozeaenol was significantly enhanced in colon cancer cells treated with auranofin as was oxidation of Trx. Auranofin is well-tolerated in humans at blood levels in the high nM to low µM range and is currently used for the treatment of rheumatoid arthritis [18,25,26]. Therefore the current results showing auranofin-mediated inhibition of TrxR activity sensitizing colon cancer cells to TAK1 inhibition suggest a rapidly translatable biochemical strategy for improving the efficacy of 5Z-7-oxozeaenol in vivo.
In order to test the efficacy of TAK1 inhibitors in vivo, SW 620 cells were grown as xenografts in nude mice and treated with the combination of auranofin and 5Z-7-oxozeaenol. Fig. 6 shows that treatment with 5Z-7-oxozeaenol or treatment with auranofin plus 5Z-7-oxozeaenol significantly inhibited xenograft growth although the effect of auranofin was not as dramatic in vivo as in vitro. This could be the result of drug availability in the solid tumors governed by tissue perfusion or other complexities of the tumor microenvironment such as O2 concentration. However, these results do continue to support the hypothesis derived from the in vitro data that thiol-mediated oxidative stress is causally related to TAK1-induced KRAS mutant colon cancer cell killing.
TAK1 has been shown to be critical for survival in KRAS-mutated colon cancer cells [7]. KRAS mutations are associated with higher steady-state levels of pro-oxidants derived from signaling pathways that have been demonstrated to be critical to inducing pro-survival pathways in cancer cells [27]. However, there is a delicate balance between pro-oxidant levels and KRAS activation of cellular metabolic pathways for scavenging pro-oxidants (such as O2•− and H2O2) that is necessary to inhibit metabolic oxidative stress, allowing for pro-oxidant-mediated growth stimulation. Because of this delicate balance, it is perhaps not surprising that drug treatments that result in an over-abundance of ROS are able to selectively kill KRAS mutated cells [9]. Consistent with the notion that KRAS mutant cells need to activate TAK1 for protection against metabolic oxidative stress, it has been shown by us and others that TAK1 inhibition can increase steady-state levels of pro-oxidants in colon cancer cells lines and in murine small intestine epithelium [22,28,29]. Finally, our results support the speculation that TAK1 inhibition combined with inhibition of thioredoxin metabolism may represent a significant target for potentiating the effects of chemotherapeutic agents in KRAS mutant colon cancers [15,16,18].
Conflicts of interest
The authors do not have any conflicts of interest to disclose.
Role of funding source
This work was supported by the American Surgical Association Foundation Fellowship (JJM); NIH grant T32CA148062 (JEH, JJM); Radiation and Free Radical Research Core in the Holden Comprehensive Cancer Center at the University of Iowa core laboratory support through NIH P30 CA086862. DRS and MAF were supported by R01 CA182804, R21 CA161182, and the Carver Trust Research Program of Excellence in Redox Biology and Medicine. FED was supported by RO1 CA115438.
Acknowledgments
The authors would like to thank Dr. Michael McCormick and the Radiation and Free Radical Research Core for glutathione and thioredoxin reductase analysis. The authors also thank Drs. Adam Case and Trenton Place for technical assistance with the flow cytometry analysis of pro-oxidants.
References
- 1.Yoo P.S., Lopez-Soler R.I., Longo W.E., Cha C.H. Liver resection for metastatic colorectal cancer in the age of neoadjuvant chemotherapy and bevacizumab. Clin. Colorectal Cancer. 2006;6(3):202–207. doi: 10.3816/CCC.2006.n.036. 17026789 [DOI] [PubMed] [Google Scholar]
- 2.National Comprehensive Cancer Network, 2014. Available from: http://www.nccn.org/professionals/physician_gls/pdf/colon.pdf.
- 3.Vale C.L., Tierney J.F., Fisher D., Adams R.A., Kaplan R., Maughan T.S. Does anti-EGFR therapy improve outcome in advanced colorectal cancer? A systematic review and meta-analysis. Cancer Treat. Rev. 2012;38(6):618–625. doi: 10.1016/j.ctrv.2011.11.002. 22118887 [DOI] [PubMed] [Google Scholar]
- 4.Karapetis C.S., Khambata-Ford S., Jonker D.J., O’Callaghan C.J., Tu D., Tebbutt N.C. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008;359(17):1757–1765. doi: 10.1056/NEJMoa0804385. 18946061 [DOI] [PubMed] [Google Scholar]
- 5.Diaz L.A., Jr., Williams R.T., Wu J., Kinde I., Hecht J.R., Berlin J. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486(7404):537–540. doi: 10.1038/nature11219. 22722843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Misale S., Yaeger R., Hobor S., Scala E., Janakiraman M., Liska D. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–536. doi: 10.1038/nature11156. 22722830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh A., Sweeney M.F., Yu M., Burger A., Greninger P., Benes C. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell. 2012;148(4):639–650. doi: 10.1016/j.cell.2011.12.033. 22341439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu J., Powell F., Larsen N.A., Lai Z., Byth K.F., Read J. Mechanism and in vitro pharmacology of TAK1 inhibition by (5Z)-7-Oxozeaenol. ACS Chem. Biol. 2013;8(3):643–650. doi: 10.1021/cb3005897. 23272696 [DOI] [PubMed] [Google Scholar]
- 9.Shaw A.T., Winslow M.M., Magendantz M., Ouyang C., Dowdle J., Subramanian A. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc. Natl. Acad. Sci. USA. 2011;108(21):8773–8778. doi: 10.1073/pnas.1105941108. 21555567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sayin V.I., Ibrahim M.X., Larsson E., Nilsson J.A., Lindahl P., Bergo M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014;6(221) doi: 10.1126/scitranslmed.3007653. 24477002 221ra15 [doi: [DOI] [PubMed] [Google Scholar]
- 11.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal. Biochem. 1969;27(3):502–522. doi: 10.1016/0003-2697(69)90064-5. 4388022 [DOI] [PubMed] [Google Scholar]
- 12.Griffith O.W. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal. Biochem. 1980;106(1):207–212. doi: 10.1016/0003-2697(80)90139-6. 7416462 [DOI] [PubMed] [Google Scholar]
- 13.Holmgren A., Fagerstedt M. The in vivo distribution of oxidized and reduced thioredoxin in Escherichia coli. J. Biol. Chem. 1982;257(12):6926–6930. 7045097 [PubMed] [Google Scholar]
- 14.Watson W.H., Pohl J., Montfort W.R., Stuchlik O., Reed M.S., Powis G. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J. Biol. Chem. 2003;278(35):33408–33415. doi: 10.1074/jbc.M211107200. 12816947 [DOI] [PubMed] [Google Scholar]
- 15.Scarbrough P.M., Mapuskar K.A., Mattson D.M., Gius D., Watson W.H., Spitz D.R. Simultaneous inhibition of glutathione- and thioredoxin-dependent metabolism is necessary to potentiate 17AAG-induced cancer cell killing via oxidative stress. Free Radic. Biol. Med. 2012;52(2):436–443. doi: 10.1016/j.freeradbiomed.2011.10.493. 22100505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simons A.L., Parsons A.D., Foster K.A., Orcutt K.P., Fath M.A., Spitz D.R. Inhibition of glutathione and thioredoxin metabolism enhances sensitivity to perifosine in head and neck cancer cells. J. Oncol. 2009;2009:519563. doi: 10.1155/2009/519563. 19746172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smart D.K., Ortiz K.L., Mattson D., Bradbury C.M., Bisht K.S., Sieck L.K. Thioredoxin reductase as a potential molecular target for anticancer agents that induce oxidative stress. Cancer Res. 2004;64(18):6716–6724. doi: 10.1158/0008-5472.CAN-03-3990. 15374989 [DOI] [PubMed] [Google Scholar]
- 18.Fath M.A., Ahmad I.M., Smith C.J., Spence J., Spitz D.R. Enhancement of carboplatin-mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011;17(19):6206–6217. doi: 10.1158/1078-0432.CCR-11-0736. 21844013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amado R.G., Wolf M., Peeters M., Van Cutsem E., Siena S., Freeman D.J. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008;26(10):1626–1634. doi: 10.1200/JCO.2007.14.7116. 18316791 [DOI] [PubMed] [Google Scholar]
- 20.De Roock W., De Vriendt V., Normanno N., Ciardiello F., Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12(6):594–603. doi: 10.1016/S1470-2045(10)70209-6. 21163703 [DOI] [PubMed] [Google Scholar]
- 21.Morioka S., Inagaki M., Komatsu Y., Mishina Y., Matsumoto K., Ninomiya-Tsuji J. TAK1 kinase signaling regulates embryonic angiogenesis by modulating endothelial cell survival and migration. Blood. 2012;120(18):3846–3857. doi: 10.1182/blood-2012-03-416198. 22972987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Acuña U.M., Wittwer J., Ayers S., Pearce C.J., Oberlies N.H., DE Blanco E.J. Effects of (5Z)-7-oxozeaenol on the oxidative pathway of cancer cells. Anticancer Res. 2012;32(7):2665–2671. 22753724 [PMC free article] [PubMed] [Google Scholar]
- 23.Simons A.L., Ahmad I.M., Mattson D.M., Dornfeld K.J., Spitz D.R. 2-deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 2007;67(7):3364–3370. doi: 10.1158/0008-5472.CAN-06-3717. 17409446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L., Fath M.A., Scarbrough P.M., Watson W.H., Spitz D.R. Combined inhibition of glycolysis, the pentose cycle, and thioredoxin metabolism selectively increases cytotoxicity and oxidative stress in human breast and prostate cancer. Redox Biol. 2015;4:127–135. doi: 10.1016/j.redox.2014.12.001. 25560241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gottlieb N.L. Pharmacology of auranofin: overview and update. Scand. J. Rheumatol. Suppl. 1986;63:19–28. 3110942 [PubMed] [Google Scholar]
- 26.Marzano C., Gandin V., Folda A., Scutari G., Bindoli A., Rigobello M.P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic. Biol. Med. 2007;42(6):872–881. doi: 10.1016/j.freeradbiomed.2006.12.021. 17320769 [DOI] [PubMed] [Google Scholar]
- 27.Weinberg F., Hamanaka R., Wheaton W.W., Weinberg S., Joseph J., Lopez M. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA. 2010;107(19):8788–8793. doi: 10.1073/pnas.1003428107. 20421486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kajino-Sakamoto R., Omori E., Nighot P.K., Blikslager A.T., Matsumoto K., Ninomiya-Tsuji J. TGF-beta-activated kinase 1 signaling maintains intestinal integrity by preventing accumulation of reactive oxygen species in the intestinal epithelium. J. Immunol. 2010;185(8):4729–4737. doi: 10.4049/jimmunol.0903587. 20855879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Omori E., Inagaki M., Mishina Y., Matsumoto K., Ninomiya-Tsuji J. Epithelial transforming growth factor beta-activated kinase 1 (TAK1) is activated through two independent mechanisms and regulates reactive oxygen species. Proc. Natl. Acad. Sci. USA. 2012;109(9):3365–3370. doi: 10.1073/pnas.1116188109. 22331902 [DOI] [PMC free article] [PubMed] [Google Scholar]