

Significance
Glycosphingolipids are a heterogeneous group of membrane lipids formed through the covalent linkage of a glycan moiety to ceramide. Genetic evidence suggests that aberrant glycosphingolipid metabolism plays an important role in several neuromuscular diseases. Here, we investigated whether alterations in glycosphingolipids contribute to neurodegeneration in amyotrophic lateral sclerosis (ALS). We show that ALS patients and model mice display disease-related changes in spinal cord glycosphingolipids levels and in the enzymes that regulate their metabolism. Importantly, we demonstrate that inhibition of glycosphingolipid synthesis in ALS model mice exacerbated disease progression, whereas administration of GM3, a subtype of glycosphingolipids, slowed it, thus implicating glycosphingolipids as potentially important participants in ALS pathogenesis and potential targets for future drug development.
Keywords: motor neuron disease, SALS, FALS, mouse models neurological disease
Abstract
Recent genetic evidence suggests that aberrant glycosphingolipid metabolism plays an important role in several neuromuscular diseases including hereditary spastic paraplegia, hereditary sensory neuropathy type 1, and non-5q spinal muscular atrophy. Here, we investigated whether altered glycosphingolipid metabolism is a modulator of disease course in amyotrophic lateral sclerosis (ALS). Levels of ceramide, glucosylceramide, galactocerebroside, lactosylceramide, globotriaosylceramide, and the gangliosides GM3 and GM1 were significantly elevated in spinal cords of ALS patients. Moreover, enzyme activities (glucocerebrosidase-1, glucocerebrosidase-2, hexosaminidase, galactosylceramidase, α-galactosidase, and β-galactosidase) mediating glycosphingolipid hydrolysis were also elevated up to threefold. Increased ceramide, glucosylceramide, GM3, and hexosaminidase activity were also found in SOD1G93A mice, a familial model of ALS. Inhibition of glucosylceramide synthesis accelerated disease course in SOD1G93A mice, whereas infusion of exogenous GM3 significantly slowed the onset of paralysis and increased survival. Our results suggest that glycosphingolipids are likely important participants in pathogenesis of ALS and merit further analysis as potential drug targets.
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder characterized by selective loss of motor neurons (MNs) within the CNS. Although our understanding of the genetic basis of ALS has advanced greatly in recent years (1), the adverse biological processes that converge on the neuromuscular axis to drive both MN death and neuropathological features in additional cell types remain largely unknown. Glycosphingolipids (GSLs) are a heterogeneous group of membrane lipids formed through the covalent linkage of a glycan moiety to ceramide (Cer; see SI Appendix, Fig. S1 for an overview of GSL metabolism). Glucosylceramide (GlcCer) and galactosylceramide (GalCer) are GSLs with a single sugar residue: glucose and galactose respectively. The successive addition of galactose and sialic acid moieties to GlcCer results in the synthesis of gangliosides (e.g., GM3, GM2, and GM1) (2). GSLs are especially abundant in the CNS and have bioactive roles in metabolism, growth factor signaling, oligodendrocyte differentiation, neuroinflammation, angiogenesis, and pathways of cell death (2–9)—all of which are thought to participate in ALS disease pathogenesis.
Several lines of evidence suggest that aberrant changes in GSL homeostasis may contribute to disease pathogenesis in ALS. Evidence includes the detection of unique gangliosides (10), high titer serum auto-antibodies to GM2 and GM1 (11, 12), and elevated GM2 levels within the motor cortex of ALS patients (13). Furthermore, a number of neuromuscular diseases are associated with mutations in genes that regulate the metabolism of Cer and GSLs. For example, hereditary sensory neuropathy type I (HSNT1), a disease that features dorsal root ganglion cell and MN degeneration, is attributed to mutations in serine palmitoyltransferase long chain base subunit-1 (SPTLC1), the rate-limiting enzyme in Cer synthesis. Notably, certain mutations in SPTLC1 associated with HSNT1 result in elevated levels of Cer and GlcCer (14). In addition, mutations in acid ceramidase (ASHA1), an enzyme that mediates the hydrolysis of Cer, are linked to forms of spinal muscular atrophy that are not caused by the more frequent mutations in the survival motor neuron 1 gene (non-5q SMA) (15). Moreover, hereditary spastic paraplegia (HSP), a disease with corticospinal tract and in some cases, spinal cord MN degeneration, is attributed to mutations in GM2 synthase (16) and the nonlysosomal glucosylceramidase, GBA2 (17). Last, patients with adult-onset Tay-Sachs disease (GM2 gangliosidosis), a disease triggered by a deficiency in hexosaminidase (HEX), a lysosomal enzyme that hydrolyzes GM2 to GM3, have been reported in some instances to display a disease phenotype that closely mimics ALS (18–20). Interestingly, HEX mRNA is up-regulated within MNs of transgenic mice expressing the mutant human SOD1 protein (i.e., SOD1G93A mice), a familial model of ALS (21, 22). Collectively, these findings suggest that titration of GSLs is important to maintaining neuromuscular system homeostasis. Similar to other metabolic aspects thought to influence ALS disease progression (23), GSL bioactivity in the neuromuscular system is likely exerted through an inverted U-shaped dose–response curve (SI Appendix, Fig. S1). Healthy (or eustatic) GSL levels are achieved in the central, optimal range of the curve, whereas GSL levels that promote features of disease (or cacostasis) are associated with either end of the curve (i.e., instances of GSL insufficiency or excess). Here, we investigated whether disrupted GSL homeostasis contributes to neurodegeneration in ALS. We show that altered GSL metabolism is a manifestation of sporadic and familial ALS and that modulation of GSL levels in SOD1G93A mice significantly affects disease course.
Results
GSL Levels Are Increased in the Spinal Cords of ALS Patients.
To determine whether GSL levels are modified in the spinal cords of ALS patients, we examined cervical spinal tissue samples collected at autopsy from six male ALS patients and six age-matched male controls. In contrast to earlier studies that examined total GSL levels in the whole spinal cord (10), we analyzed gray matter (GM) and ventral white matter (VWM) samples separately, because regional variation in total ganglioside levels within segments of spinal cord have previously been reported (24). We also measured individual molecular isoforms of GSLs because of their distinct biological functions (25). Lipids analyzed include ceramide (Cer), sphingomyelin (SPM), glucosylceramide (GlcCer), galactosylceramide (GalCer), lactosylceramide (LacCer), globotriaosylceramide (GL3), and the gangliosides GM3 and GM1 (SI Appendix, Fig. S1). Total levels of Cer, cerebroside (GlcCer + GalCer), LacCer, and GM1 were significantly elevated up to twofold (P < 0.01) in ALS patient GM tissue samples vs. control (SI Appendix, Fig. S2). Subsequent analysis of GSL isoforms showed that significant (see Fig. 1 for P values) elevations in Cer, SPM, GlcCer, GalCer, LacCer, GL3, GM3, and GM1 were present in both GM and VWM tissue samples of ALS patients (Fig. 1 and SI Appendix, Table S1). Collectively, these finding demonstrate that a number of lipids derived from Cer are elevated in the spinal cords of ALS patients, particularly in the GM.
Fig. 1.
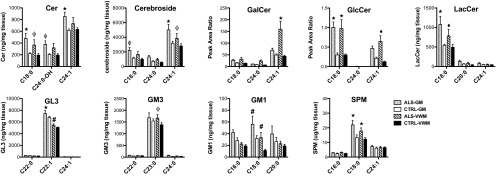
GSL accumulation is a neuropathological feature of disease in ALS patients. Major isoforms of Cer, Cerebroside, GalCer, GlcCer, LacCer, GL3, GM3, GM1, and SPM are significantly elevated (#P < 0.05, ϕP < 0.01, ♦P < 0.001, *P < 0.0001) in cervical spinal cord homogenates of ALS patients. Statistical comparisons shown are made to control (CTRL) tissue subtype. Minor isoform data and comparisons between different subtypes are reported in SI Appendix, Table S1. Error bars represent ± SEM.
SOD1G93A Mice Display Complex Changes in GSL Levels.
To determine whether ALS mice also display disease related changes in GSLs, spinal cord samples were collected from male WT and ALS mice (n = 7/group) at SYMP (symptom onset), ES (onset of partial paralysis), and MB (full paralysis/meets euthanization criteria) stages (23). Only in MB SOD1G93A mice (i.e., at a similar disease phase to the human samples) did we observe a significant (P < 0.05) increase in Cer (C24:0-OH isoform) levels vs. WT controls. In contrast, in SYMP and ES SOD1G93A mice, a significant (P < 0.0001) decrease in C24:1 Cer levels was detected. Fluctuations in cerebroside levels mirrored changes in Cer, with SYMP and ES displaying significant (P < 0.05) declines (for C24:0 and C24:1 isoforms) and MB mice showing a significant (P < 0.001) increase relative to SYMP mice (Fig. 2 and SI Appendix, Table S2). Subsequent cerebroside analysis (i.e., peak area ratio of GlcCer to GalCer) showed that levels of GlcCer nearly doubled (P < 0.0001) in MB SOD1G93A mice, which is very similar to what was seen human ALS tissue. Interestingly, similar fluctuations (i.e., an initial significant decline in SYMP and ES mice followed by a resurgence in MB mice relative to SYMP mice) were observed for almost all GSLs downstream of GlcCer including LacCer, GL3 and GM1, with the only exception being GM3. Total levels of GM3 were significantly elevated at all phases of disease (SI Appendix, Fig. S3) and specific isoforms displayed progressive accumulation (Fig. 2 and SI Appendix, Table S2). Disease-related fluctuations in GalCer and SPM, additional lipids derived from Cer (Fig. 1), were similar to several of the GSLs analyzed with levels (for major isoforms) being significantly (P < 0.0001) depressed in SYMP and ES mice followed by a rebound in MB mice (Fig. 2).
Fig. 2.

ALS mice display complex changes in GSL levels within the spinal cord. Major isoforms of Cer, Cerebroside, GalCer, GlcCer, LacCer, GL3, GM3, GM1, and SPM are significantly altered (#P < 0.05, ϕP < 0.01, ♦P < 0.001, *P < 0.0001) in ALS mice vs. WT controls. Minor isoform data and comparisons between different phases of ALS disease are reported in SI Appendix, Table S2. Error bars represent ± SEM.
Enzymes Involved in Modulating GSL Levels Are Altered in ALS Patients and SOD1G93A Mice.
Next, we asked whether GSL accumulation within the spinal cords of ALS patients was due to impaired enzymatic action either within lysosomes or other cytoplasmic organelles. Measurement of enzymatic activities at acidic (pH 4.5) and neutral (pH 7.0) pH in the cervical spinal cord GM and VWM was performed for each ALS patient and age-matched control for the following enzymes: GBA1, GBA2, HEX, galactosylceramidase (GALC), α-galactosidase (α-GAL), and β-galactosidase (β-GAL).
Average activities of all of the enzymes examined (irrespective of the pH used in the measurements) were not significantly reduced, suggesting that GSL accumulation is not due to reduced enzyme activity. However, acidic GBA1 activity was dramatically reduced in the GM of two of the six ALS patients compared with controls (Fig. 3A). Interestingly, perhaps as a compensatory response to GSL accumulation, the average activity of all of the enzymes in the GSL pathway was significantly increased (by 67–343%) in ALS vs. control samples. In the ALS GM samples of patients, significant elevations in acidic GBA2 (67%; P < 0.05), GALC (145%; P < 0.05), α-GAL (328%; P < 0.01), and β-GAL (345%; P < 0.01) enzyme activities and in neutral GBA2 (126%; P < 0.01), HEX (323%; P < 0.05), GALC (96%; P < 0.05), α-GAL (214%; P < 0.01), and β-GAL (343%; P < 0.01) activities were detected (Fig. 3). In the VWM samples from ALS patients, acidic GBA1 (90%; P < 0.01) and HEX (144%; P < 0 0.05) activity were also higher (Fig. 3A).
Fig. 3.

GSL metabolism is altered in the spinal cords of ALS patients and mice. (A) Lysosomal (pH 4.5) and cytoplasmic (pH = 7.0) GBA1, GBA2, HEX, GALC, α-GAL, and β-GAL activities in the cervical spinal cord homogenates of ALS patients and age-matched controls. Statistical comparisons are relative to CTRL tissue subtype (#P < 0.05, ϕP < 0.01). (B) Lysosomal enzyme activities measured in SYMP, ES, and MB ALS mice compared with WT controls. Groups not connected by the same letter are significantly different (P < 0.01). Error bars represent ± SEM.
We also compared the activities of the enzymes at both acidic and neutral pH in the spinal cords of ALS mice at various phases of disease (SYMP, ES, and MB; n = 6/phase) to those in WT controls (n = 6). In contrast to what was observed in ALS patients, significant reductions (P < 0.01) in the activities of GBA1 (pH 7), GBA2 (pH 4.5, pH 7), and GALC (pH 4.5, pH 7) were observed in the spinal cords of ALS mice during the ES and MB phases of disease (Fig. 3B). The activities of α-GAL and β-GAL were not significantly altered during the course of the disease in ALS mice. However, it should be stressed that the differences in ALS patients were primarily limited to GM samples, whereas the measurements in mice encompassed pooled GM + WM. Similar to ALS patients, neutral and acidic HEX enzyme activities were significantly elevated (P < 0.01) in the spinal cords of ES and MB ALS mice vs. controls (Fig. 3B).
Inhibition of Glucosylceramide Synthase Significantly Accelerates Disease Progression in SOD1G93A Mice.
To determine the functional significance of GlcCer accumulation in MB SOD1G93A mice, we chronically treated ALS mice (starting at P50, n =17 sex per group) with GENZ-667161 (60 mg/kg per day), an inhibitor of GlcCer synthase (GCS). We recently reported that GENZ-667161 lowers brain GlcCer levels in a mouse model of neuronopathic Gaucher disease, a lysosomal storage disorder that features lipid accumulation due to a deficiency in GBA1 (26). We also tested the impact of GENZ-642347 (360 mg/kg per day starting at P50, n =16 sex per group), another inhibitor of GCS that is equally effective at lowering GlcCer levels in the peripheral tissues but does not cross the blood–brain barrier (27). ALS mice treated with GENZ-642347 showed no measureable changes in disease course (i.e., body mass, symptom onset, paralysis onset. or survival; SI Appendix, Fig. S4). In contrast, inhibition of GCS within the CNS of ALS mice with GENZ-667161 significantly accelerated disease progression. Loss of body mass was observed in male mice administered GENZ-667161 starting at the earlier age of 87 d (Fig. 4A; P < 0.01). Symptom onset in either sex was not altered, but onset of paralysis was significantly accelerated (P < 0.0001) relative to untreated same-sex control mice (104 vs. 118 d of age in males and 108.5 vs. 128.5 d in females). Overall survival was also adversely impacted with lifespan significantly shortened (P < 0.0001) by 11 and 11.5 d in male (112 vs. 123 d) and female (121 vs. 132.5 d) mice treated with GENZ-667161 (Fig. 4A). These findings suggest that accumulation of GlcCer within the spinal cord of ALS mice during the MB phase of disease may be a compensatory response to slow the disease course.
Fig. 4.

Modulation of GSL levels impacts disease progression in ALS mice. (A) Inhibition of GSL synthesis within the CNS accelerates disease progression in ALS mice. ALS mice treated with GENZ-667161 (161) (inhibitor of glucosylceramide synthase with CNS penetrance) showed significant changes in the rate of body weight loss (*P < 0.01), the median onset of end stage (males = 104 vs. 118 d, females = 100.5 vs. 128.5 d; P < 0.0001), and median survival (males = 112 vs. 123 d, females = 121 vs. 132.5 d; P < 0.0001) vs. untreated ALS sibling/sex-matched counterparts. (B) The onset of paralysis was significantly (P < 0.0095) delayed by 8 d, and survival was significantly (P < 0.0473) extended by 6 d in ALS mice that received continuous intracerebroventricular infusion of GM3 (12 μg/d) vs. mice infused with vehicle (artificial cerebrospinal fluid) (treatment initiated at day 90).
Intracerebroventricular Infusion of GM3 Significantly Slows Disease Course in SOD1G93A Mice.
Because we observed a significant increase in HEX activity and GM3 levels in the spinal cords of SOD1G93A mice (and ALS patients), we sought to determine what the impact of further increases in these two measures would have on disease course in ALS mice. Although CNS delivery of recombinant adeno-associated viral (AAV) vectors encoding human HEX (n =18/sex) at P1 significantly increased (P < 0.01) enzyme activity within the spinal cords of SOD1G93A mice, no notable alterations in disease phenotype was observed compared with sex-matched control mice (n = 18/sex) treated with an empty vector (AAV-Null) (SI Appendix, Fig. S5). In a follow-up experiment, we examined the impact of directly increasing GM3 levels, rather than rely on HEX-mediated metabolism of GM2 to GM3. Mice were chronically infused with either GM3 (12 µg/d) or vehicle (artificial cerebrospinal fluid) into the lateral ventricles of male SOD1G93A mice starting at 90 d of age (n = 10/group). Infusion of GM3 significantly delayed the onset of paralysis by 8 d (112 vs. 104 d, P < 0.0095) and significantly extended (albeit modestly) survival by 6 d (122 vs. 116 d, P < 0.04; Fig. 4B). These findings suggest that the observed elevations in HEX activity and the resultant higher GM3 levels observed in ALS patients and mice may be a compensatory response to slow the disease course.
Discussion
Emerging evidence indicates that aberrant Cer and GSL metabolism has an important role in the pathogenesis of a number of neuromuscular diseases including HSP, HSN1, and non-5q SMA (14–17, 28). Here, we show that alterations in GSLs are also a feature of ALS and provide evidence to suggest that these changes likely influence the rate of disease progression. A previous investigation of disease-related changes in gangliosides, a subtype of GSL, in ALS patients revealed no major quantitative differences. However, in that study, the entire intact spinal cord was examined for changes in total ganglioside levels (10). In contrast, our analysis of cervical spinal cord gray and white matter samples showed that the major isoforms of Cer, GalCer, GlcCer, LacCer, GM3, GM1, GL3, and SPM were significantly elevated in ALS patients. These findings corroborate an earlier study that evaluated Cer and SPM levels in human ALS spinal cords (29). Importantly, we also showed that GSL-related pathology in ALS patients was not due to a reduction in the activity of the enzymes that mediate their degradation (as is observed in lysosomal storage diseases); acidic enzymatic activities of GBA1, GBA2, HEX, GALC, α-GAL, and β-GAL were all significantly increased in ALS spinal cord samples. Furthermore, for a number of these enzymes (i.e., GBA2, HEX, GALC, α-GAL, and β-GAL), neutral enzyme activities were also significantly elevated, suggesting that increased GSL hydrolysis also occurred at the plasma membrane or within organelles that mediate their synthesis. The complex question of what causes Cer and GSLs to accumulate (and in which organelles) is unknown and warrants further investigation. However, an aberrant increase in the de novo synthesis of Cer (leading to increased GSL levels) is an unlikely explanation for these observations, given that in fully differentiated cells (i.e., neurons), GSL levels (50–90%) are predominately derived from salvage pathways rather than from de novo synthesis (30), Further, the expression of palmitoyltransferase long chain base subunit-2 (SPTLC2), the rate-limiting enzyme in Cer synthesis, is reduced more than threefold in the MNs of ALS patients (31).
It is difficult to determine whether GSL accumulation during the terminal phase of disease contributes to disease pathogenesis or if it is part of a compensatory response to slow disease progression. At sufficient levels, LacCer would be expected to promote disease, as it is a significant mediator of neuroinflammation and apoptosis (8). Interestingly, LacCer activation of microglia is mediated in part through stimulation of the NF-κB signaling pathway (32), which was recently reported to play an important role in microglia-induced MN death in ALS (33). Moreover, accumulation of GL3 and GalCer would also be expected to exacerbate disease. For example, lysosomal GL3 accumulation in Fabry disease, a manifestation of reduced α-GAL activity, results in cerebral vascular dysfunction (34), which is also a pathological feature of ALS (35). Similarly, impairment in GalCer metabolism due to mutations in GALC results in Krabbe disease, a fatal disorder that features demyelination and severe motor impairment (36).
Alternatively, the increased flux of Cer in the GlcCer pathway may initiate the synthesis of neurotrophic gangliosides that promote neurite outgrowth and axonal health (5) to slow the rate of ALS disease pathogenesis. Indeed, in our current study, GM3 levels were significantly elevated in the spinal cords of ALS patients and SOD1G93A mice. An increase in GM3 may also potentially slow disease by promoting oligodendrocyte differentiation (6), which is adversely affected in ALS mice (37), or by inhibiting the activation of matrix metalloproteinase-9 (38), which is expressed at high levels in those MNs most vulnerable in ALS (39). Interestingly, gene expression analysis studies carried out in ALS mice have shown that HEX (an enzyme that metabolizes GM2 to GM3) mRNA level is up-regulated in MNs before and during overt signs of disease (21, 22). We confirmed these observations (SI Appendix, Fig. S6) and also showed that corresponding HEX enzyme activity was increased nearly threefold in the spinal cords of SOD1G93A mice. Similarly, we also detected a significant increase in HEX activity in the spinal cords of ALS patients. However, we found that further increasing Hex activity to 10-fold above WT levels in SOD1G93A mice through adeno-associated virus-mediated gene transfer did not impact the disease course. This result may have been due to a limited availability of the endogenous substrate (GM2) for hydrolysis by HEX. Consistent with this supposition, intracerebroventricular infusion of exogenous GM3 in SOD1G93A mice modestly delayed the onset of paralysis and improved survival outcome. It is difficult to predict if increasing GM3 levels would impact human ALS, given that baseline GM3 levels are nearly fivefold higher in humans compared with mice, and we also detected significant species differences in the major isoforms of GM3 (SI Appendix, Fig. S7). Nevertheless, assessing the effect of specific GM3 isoforms in newly developed in vitro human cell models of ALS (40–42) has the potential to validate the merit of altering GM3 levels to treat the human condition.
Characterization of GSL levels in SOD1G93A mouse spinal cords at various phases of disease revealed complex changes in Cer and GSL levels. Cer and GSL levels in moribund ALS mice were similar to ALS patients at autopsy in that their spinal cords had significantly elevated levels of specific isoforms of Cer, GlcCer, and GM3 vs. WT controls. In contrast, during the early symptomatic and end stage phases of disease, we found that Cer and the majority of derived GSLs (GalCer, LacCer, GL3, and GM1) were significantly lower in ALS vs. WT mice. This finding could be due to reduced Cer synthesis, which seems likely given that SPTLC (isoforms 1, 2, and 3) and 3-ketodihydrosphingosine reductase (FVT1; second step in Cer synthesis) mRNA levels were significantly reduced in ALS mouse spinal cords (SI Appendix, Fig. S8). Alternatively, diminished Cer levels may be due to reduced entry of Cer from salvage pathways following hydrolysis of various Cer-derived lipids. Consistent with this possibility, both GBA and GALC enzyme activities were significantly reduced in ALS mice. Whether the level of reduction in Cer levels during the earlier phases of disease contributes to disease pathogenesis or is part of a compensatory response is unknown. If a threshold level of reduction is reached, then a number of relevant ALS disease features including demyelination and Wallerian degeneration may be either initiated or exacerbated (43–45). Consistent with this notion, mutations in SPTLC1 and FVT1 that cause a complete loss of enzyme activity are associated with HSN1 (46) and bovine spinal muscular atrophy (28). Alternatively, the level of reduction in Cer synthesis may be a compensatory response to avert synthesis of toxic lipids derived from Cer with reduced cerebroside and ganglioside synthesis being a tolerable consequence.
Although the genetic evidence cited above clearly shows that complete inhibition of de novo Cer synthesis is detrimental to MN survival, it is also evident that titration of Cer (i.e., its metabolism and flux along various lipid-generating pathways) is also critical to MN survival. For example, certain mutations in SPTLC that lead to increased enzyme activity and Cer are also associated with HSHN1 (14). Moreover, mutations in ASAH1 (an enzyme mediating lysosomal hydrolysis of Cer) effecting a 70% reduction in activity are a known cause of non-5q SMA (15). Near complete (90–95%) loss of ASAH1 enzyme activity causes Farber disease, a fatal lysosomal storage disease of young children (47). The wide phenotypic spectrum associated with mutations in ASAH1 suggests that relatively small alterations in Cer flux through various biological pathways can profoundly affect MN survival. The importance of Cer homeostasis is also supported by zebrafish experiments showing that morpholino-based oligonucleotide-mediated knockdown of the ASAH1 ortholog leads to a marked loss of MN axonal branching and increased apoptosis in the spinal cord (15). Moreover, a twofold increase in expression of neutral ceramidase (ASAH2) in astrocytes bearing mutant SOD1 suggests that nonlysosomal aberrant Cer metabolism may also generate intercellular reactions leading to ALS (48).
Therefore, we hypothesize that during the course of ALS, accumulation of toxic agents derived from Cer becomes favored, triggering two homeostatic adjustments: (i) an initial reduction in Cer synthesis, as implicated in ALS mice and patients; and (ii) an increase in Cer flux down the GlcCer metabolic pathway, also observed in ALS mice and patients. The shunting of Cer to the GlcCer metabolic pathway precludes entry of Cer into other biological pathways that are ultimately toxic to MNs. In support, an increase in GlcCer synthase (GCS) activity (i.e., GlcCer accumulation) is a known negative regulator of Cer-induced apoptosis (49), and we found in our current study that disease was significantly accelerated in SOD1G93A mice treated with a GCS inhibitor.
Overall, our study provides previously unidentified insights into the neuropathological roles played by GSLs and their corresponding enzymes in ALS. Similar to our previous work (23, 50), our present study underscores in metabolic terms that multiple pathological aspects of ALS are still unexplored. A greater understanding of the biological pathways that regulate Cer flux following inhibition of GCS in ALS mice has the potential to lead to the identification of toxic entities that are targetable for drug development.
Materials and Methods
Transgenic male ALS mice that highly express the mutant SOD1G93A were divided equally among groups. Mutant SOD1G93A gene copy number and protein expression were confirmed by PCR and Western blot analysis, respectively. Animals were housed under light:dark (12:12 h) cycles and provided with food and water ad libitum. At 75 d of age, food pellets were place on the cage floor to facilitate food consumption by motor-affected mice. Mice were scored into three phases: symptomatic (SYMP) = abnormal hind limb splay, median age = 82 d; end stage (ES) = onset of limb paralysis (typically hind limb), median age = 103 d; and moribund (MB) = unable to right themselves within 30 s when placed on their backs, median age = 122 d. All procedures followed protocols approved by Genzyme’s Institutional Animal Care and Use Committees. For further information regarding lipid analysis, enzyme activity measurements, and modulation of GSL levels through administration of small molecules and exogenous GM3, please see SI Appendix, Materials and Methods.
Supplementary Material
Acknowledgments
We thank Jonathan Fidler, Thomas Tamsett, Elyse Bourque, Cecilia Bastos, and Robin Ziegler for technical assistance. Human tissue was obtained from the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders at the University of Maryland (Contract HHSN275200900011C; Ref. N01-HD-9-0011).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1508767112/-/DCSupplemental.
References
- 1.Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17(1):17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu RK, Bieberich E, Xia T, Zeng G. Regulation of ganglioside biosynthesis in the nervous system. J Lipid Res. 2004;45(5):783–793. doi: 10.1194/jlr.R300020-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Nordström V, et al. Neuronal expression of glucosylceramide synthase in central nervous system regulates body weight and energy homeostasis. PLoS Biol. 2013;11(3):e1001506. doi: 10.1371/journal.pbio.1001506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao H, et al. Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes. 2007;56(5):1210–1218. doi: 10.2337/db06-0719. [DOI] [PubMed] [Google Scholar]
- 5.Inokuchi J. Neurotrophic and neuroprotective actions of an enhancer of ganglioside biosynthesis. Int Rev Neurobiol. 2009;85:319–336. doi: 10.1016/S0074-7742(09)85022-8. [DOI] [PubMed] [Google Scholar]
- 6.Yim SH, Farrer RG, Hammer JA, Yavin E, Quarles RH. Differentiation of oligodendrocytes cultured from developing rat brain is enhanced by exogenous GM3 ganglioside. J Neurosci Res. 1994;38(3):268–281. doi: 10.1002/jnr.490380305. [DOI] [PubMed] [Google Scholar]
- 7.Prokazova NV, Samovilova NN, Gracheva EV, Golovanova NK. Ganglioside GM3 and its biological functions. Biochem. Biokhim. 2009;74(3):235–249. doi: 10.1134/s0006297909030018. [DOI] [PubMed] [Google Scholar]
- 8.Chatterjee S, Pandey A. The Yin and Yang of lactosylceramide metabolism: Implications in cell function. Biochim Biophys Acta. 2008;1780(3):370–382. doi: 10.1016/j.bbagen.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 9.Ishibashi Y, Kohyama-Koganeya A, Hirabayashi Y. New insights on glucosylated lipids: Metabolism and functions. Biochim Biophys Acta. 2013;1831(9):1475–1485. doi: 10.1016/j.bbalip.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 10.Dawson G, Stefansson K. Gangliosides of human spinal cord: Aberrant composition of cords from patients with amyotrophic lateral sclerosis. J Neurosci Res. 1984;12(2-3):213–220. doi: 10.1002/jnr.490120209. [DOI] [PubMed] [Google Scholar]
- 11.Salazar-Grueso EF, Routbort MJ, Martin J, Dawson G, Roos RP. Polyclonal IgM anti-GM1 ganglioside antibody in patients with motor neuron disease and variants. Ann Neurol. 1990;27(5):558–563. doi: 10.1002/ana.410270517. [DOI] [PubMed] [Google Scholar]
- 12.Stevens A, Weller M, Wiethölter H. A characteristic ganglioside antibody pattern in the CSF of patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1993;56(4):361–364. doi: 10.1136/jnnp.56.4.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rapport MM, Donnenfeld H, Brunner W, Hungund B, Bartfeld H. Ganglioside patterns in amyotrophic lateral sclerosis brain regions. Ann Neurol. 1985;18(1):60–67. doi: 10.1002/ana.410180111. [DOI] [PubMed] [Google Scholar]
- 14.Dawkins JL, Hulme DJ, Brahmbhatt SB, Auer-Grumbach M, Nicholson GA. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet. 2001;27(3):309–312. doi: 10.1038/85879. [DOI] [PubMed] [Google Scholar]
- 15.Zhou J, et al. Spinal muscular atrophy associated with progressive myoclonic epilepsy is caused by mutations in ASAH1. Am J Hum Genet. 2012;91(1):5–14. doi: 10.1016/j.ajhg.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boukhris A, et al. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am J Hum Genet. 2013;93(1):118–123. doi: 10.1016/j.ajhg.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin E, et al. Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am J Hum Genet. 2013;92(2):238–244. doi: 10.1016/j.ajhg.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitsumoto H, et al. Motor neuron disease and adult hexosaminidase A deficiency in two families: Evidence for multisystem degeneration. Ann Neurol. 1985;17(4):378–385. doi: 10.1002/ana.410170413. [DOI] [PubMed] [Google Scholar]
- 19.Jellinger K, Anzil AP, Seemann D, Bernheimer H. Adult GM2 gangliosidosis masquerading as slowly progressive muscular atrophy: Motor neuron disease phenotype. Clin Neuropathol. 1982;1(1):31–44. [PubMed] [Google Scholar]
- 20.Argov Z, Navon R. Clinical and genetic variations in the syndrome of adult GM2 gangliosidosis resulting from hexosaminidase A deficiency. Ann Neurol. 1984;16(1):14–20. doi: 10.1002/ana.410160105. [DOI] [PubMed] [Google Scholar]
- 21.Olsen MK, et al. Disease mechanisms revealed by transcription profiling in SOD1-G93A transgenic mouse spinal cord. Ann Neurol. 2001;50(6):730–740. doi: 10.1002/ana.1252. [DOI] [PubMed] [Google Scholar]
- 22.Lobsiger CS, Boillée S, Cleveland DW. Toxicity from different SOD1 mutants dysregulates the complement system and the neuronal regenerative response in ALS motor neurons. Proc Natl Acad Sci USA. 2007;104(18):7319–7326. doi: 10.1073/pnas.0702230104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fidler JA, et al. Disease progression in a mouse model of amyotrophic lateral sclerosis: The influence of chronic stress and corticosterone. FASEB J. 2011;25(12):4369–4377. doi: 10.1096/fj.11-190819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vorwerk CK. Ganglioside patterns in human spinal cord. Spinal Cord. 2001;39(12):628–632. doi: 10.1038/sj.sc.3101232. [DOI] [PubMed] [Google Scholar]
- 25.Hannun YA, Obeid LM. Many ceramides. J Biol Chem. 2011;286(32):27855–27862. doi: 10.1074/jbc.R111.254359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cabrera-Salazar MA, et al. Systemic delivery of a glucosylceramide synthase inhibitor reduces CNS substrates and increases lifespan in a mouse model of type 2 Gaucher disease. PLoS ONE. 2012;7(8):e43310. doi: 10.1371/journal.pone.0043310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McEachern KA, et al. A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher disease. Mol Genet Metab. 2007;91(3):259–267. doi: 10.1016/j.ymgme.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Krebs S, Medugorac I, Röther S, Strässer K, Förster M. A missense mutation in the 3-ketodihydrosphingosine reductase FVT1 as candidate causal mutation for bovine spinal muscular atrophy. Proc Natl Acad Sci USA. 2007;104(16):6746–6751. doi: 10.1073/pnas.0607721104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cutler RG, Pedersen WA, Camandola S, Rothstein JD, Mattson MP. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann Neurol. 2002;52(4):448–457. doi: 10.1002/ana.10312. [DOI] [PubMed] [Google Scholar]
- 30.Gillard BK, Clement RG, Marcus DM. Variations among cell lines in the synthesis of sphingolipids in de novo and recycling pathways. Glycobiology. 1998;8(9):885–890. doi: 10.1093/glycob/8.9.885. [DOI] [PubMed] [Google Scholar]
- 31.Kirby J, et al. Phosphatase and tensin homologue/protein kinase B pathway linked to motor neuron survival in human superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain. 2011;134(Pt 2):506–517. doi: 10.1093/brain/awq345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Won JS, Singh AK, Singh I. Lactosylceramide: A lipid second messenger in neuroinflammatory disease. J Neurochem. 2007;103(Suppl 1):180–191. doi: 10.1111/j.1471-4159.2007.04822.x. [DOI] [PubMed] [Google Scholar]
- 33.Frakes AE, et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81(5):1009–1023. doi: 10.1016/j.neuron.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitsias P, Levine SR. Cerebrovascular complications of Fabry’s disease. Ann Neurol. 1996;40(1):8–17. doi: 10.1002/ana.410400105. [DOI] [PubMed] [Google Scholar]
- 35.Zhong Z, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. 2008;11(4):420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki K. Globoid cell leukodystrophy (Krabbe’s disease): Update. J Child Neurol. 2003;18(9):595–603. doi: 10.1177/08830738030180090201. [DOI] [PubMed] [Google Scholar]
- 37.Kang SH, et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16(5):571–579. doi: 10.1038/nn.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang XQ, Sun P, Paller AS. Ganglioside GM3 inhibits matrix metalloproteinase-9 activation and disrupts its association with integrin. J Biol Chem. 2003;278(28):25591–25599. doi: 10.1074/jbc.M302211200. [DOI] [PubMed] [Google Scholar]
- 39.Kaplan A, et al. Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron. 2014;81(2):333–348. doi: 10.1016/j.neuron.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haidet-Phillips AM, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29(9):824–828. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Re DB, et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 2014;81(5):1001–1008. doi: 10.1016/j.neuron.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kiskinis E, et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell. 2014;14(6):781–795. doi: 10.1016/j.stem.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheikh KA, et al. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc Natl Acad Sci USA. 1999;96(13):7532–7537. doi: 10.1073/pnas.96.13.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohmi Y, et al. Gangliosides are essential in the protection of inflammation and neurodegeneration via maintenance of lipid rafts: Elucidation by a series of ganglioside-deficient mutant mice. J Neurochem. 2011;116(5):926–935. doi: 10.1111/j.1471-4159.2010.07067.x. [DOI] [PubMed] [Google Scholar]
- 45.Rajesh M, Kolmakova A, Chatterjee S. Novel role of lactosylceramide in vascular endothelial growth factor-mediated angiogenesis in human endothelial cells. Circ Res. 2005;97(8):796–804. doi: 10.1161/01.RES.0000185327.45463.A8. [DOI] [PubMed] [Google Scholar]
- 46.Bejaoui K, et al. Hereditary sensory neuropathy type 1 mutations confer dominant negative effects on serine palmitoyltransferase, critical for sphingolipid synthesis. J Clin Invest. 2002;110(9):1301–1308. doi: 10.1172/JCI16450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sugita M, Dulaney JT, Moser HW. Ceramidase deficiency in Farber’s disease (lipogranulomatosis) Science. 1972;178(4065):1100–1102. doi: 10.1126/science.178.4065.1100. [DOI] [PubMed] [Google Scholar]
- 48.Ferraiuolo L, et al. Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain. 2011;134(Pt 9):2627–2641. doi: 10.1093/brain/awr193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kohyama-Koganeya A, et al. Drosophila glucosylceramide synthase: A negative regulator of cell death mediated by proapoptotic factors. J Biol Chem. 2004;279(34):35995–36002. doi: 10.1074/jbc.M400444200. [DOI] [PubMed] [Google Scholar]
- 50.Dodge JC, et al. Metabolic signatures of amyotrophic lateral sclerosis reveal insights into disease pathogenesis. Proc Natl Acad Sci USA. 2013;110(26):10812–10817. doi: 10.1073/pnas.1308421110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.