

Significance
Inherited multisystemic diseases, myotonic dystrophies type 1 (DM1) and type 2 (DM2), are caused by long CUG and CCUG RNA repeats. The mutant RNA CCUG repeats should be degraded after intron excision; however, this RNA accumulates in cells, leading to pathology. Although mutant RNAs may be degraded with synthetic oligonucleotides, the identification of a cause of the increased stability of CUG and CCUG RNAs would help to improve the efficiency of their degradation. We found that the reduction of RNA helicase p68 in skeletal muscle biopsies of DM1 and DM2 patients contributes to the delay of degradation of the mutant RNAs. Our work suggests RNA helicase p68 as a therapeutic target in DM, correction of which improves degradation of the mutant RNAs, reducing DM pathology.
Keywords: myotonic dystrophy, RNA foci, p68 RNA helicase, CUG repeats
Abstract
Myotonic dystrophies type 1 (DM1) and type 2 (DM2) are neuromuscular diseases, caused by accumulation of CUG and CCUG RNAs in toxic aggregates. Here we report that the increased stability of the mutant RNAs in both types of DM is caused by deficiency of RNA helicase p68. We have identified p68 by studying CCUG-binding proteins associated with degradation of the mutant CCUG repeats. Protein levels of p68 are reduced in DM1 and DM2 biopsied skeletal muscle. Delivery of p68 in DM1/2 cells causes degradation of the mutant RNAs, whereas delivery of p68 in skeletal muscle of DM1 mouse model reduces skeletal muscle myopathy and atrophy. Our study shows that correction of p68 may reduce toxicity of the mutant RNAs in DM1 and in DM2.
Myotonic dystrophy type 1 (DM1) is a neuromuscular disease characterized by myotonia, distal muscle weakness, heart conduction defects, and, in the congenital form, a delay in myogenesis and severe cognitive abnormalities (1). DM1 is caused by expanded CTG repeats within the 3′ untranslated region of the DMPK gene (2). Myotonic dystrophy type 2 (DM2) is a late-onset disease that is caused by expanded CCTG repeats in intron 1 of the ZNF9/CNBP gene (3). Development of therapeutic approaches for DM1 or DM2 is an urgent need. Numerous data suggest that DM1 and DM2 are caused by RNA gain-of-function mechanisms (4–6). Initial studies showed that mutant RNAs mainly affect two RNA-binding proteins, CUG-binding protein 1 (CUGBP1) and muscleblind-like protein 1 (MBNL1) (7–9). CUG repeats elevate protein levels of CUGBP1 by increasing its stability (5). In addition, CUG repeats change signal transduction pathways, such as the glycogen synthase kinase 3β (GSK3β)–cyclin D3 pathway, regulating CUGBP1 activity (5, 10). CUG and CCUG repeats form double-stranded hairpin structures and sequester MBNL1 (9, 11, 12). Several other RNA-binding proteins, such as Staufen1 and two members of the DEAD-box RNA helicases family, DDX5/p68 and DDX6, are also involved in DM1 (13–15).
We showed that the mutant CUG and CCUG RNAs are very stable (16), suggesting that the activity of RNA-binding proteins regulating RNA decay is reduced in DM1 and in DM2. In this study, we tested this hypothesis by isolation and analysis of several CCUG-binding proteins. We found that the levels of one of these proteins, p68, are reduced in DM1 and DM2 biopsied muscle and that correction of p68 leads to degradation of the mutant CUG and CCUG RNAs, disintegration of RNA foci, and reduction of DM muscle pathology.
Results and Discussion
Identification of p68 Helicase as CUG/CCUG-Binding Protein, Which is Reduced During Degradation of CCUG Repeats.
Given the critical role of RNA CUG and CCUG repeats in DM1/2, we performed a careful analysis of CCUG-binding proteins with altered activities upon accumulation and degradation of the mutant CCUG repeats in tetracycline (tet)-regulated CHO cell model of DM2. In this model, CCUG100 repeats mainly increased activities of two RNA-binding proteins with the approximate molecular weights 50 and 100 kDa (Fig. 1A). Previous analysis of tet-regulated cell models expressing CUG and CCUG repeats showed that a single dose of Dox causes maximal accumulation of CUG or CCUG repeats at 17–24 h after Dox addition, whereas approximately half of the mutant RNAs is degraded at 36–48 h after Dox addition (16). Analysis of CCUG-binding proteins in tet-HeLa-CCUG100 cells at 7–24 h after Dox addition showed that CCUG repeats increase activities of two proteins with molecular weights 50 and 100 kDa similar to those observed in inducible CHO cells (Fig. 1B), whereas the activity of CCUG-binding protein with molecular weight 75 kDa is increased at 24 h. The activities of these three proteins were reduced during degradation of the mutant CCUG repeats (48 h after Dox addition) (Fig. 1B). UV cross-linking analysis of proteins from normal and DM2 myoblasts showed that activity of the 75-kDa CCUG-binding protein is also reduced in DM2 (Fig. 1C). Normal human myoblasts contain several other CCUG-binding proteins migrating in the positions 40, 50, 55, and 100 kDa. The activities of CCUG-binding proteins with molecular weights 50 and 100 kDa were increased in DM2 myoblasts, whereas the activities of proteins with approximate molecular weights 40, 55, and 75 kDa are reduced (Fig. 1C).
Fig. 1.

P68 is a CCUG-binding protein with the reduced activity in DM2. (A) Mutant CCUG repeats increase activities of 50- and 100-kDa RNA-binding proteins. UV cross-link analysis of cytoplasmic proteins with 32P-CCUG100 probe. (B) UV cross-link analysis identifies three CCUG-binding proteins with approximate molecular weights 50, 75, and 100 kDa increased by CCUG100 repeats in tet-regulated HeLa cells. (C) UV cross-link analysis of cytoplasmic proteins from normal and DM2 myoblasts with 32P-CCUG100 RNA; arrows: proteins with increased and reduced activities in DM2 myoblasts. (D) Identification of CCUG-binding proteins (shown by arrows) by UV cross-link assay in the chromatography fractions (UnoQ column; run 1) using the CCUG100 probe. The protein fractions were stained with Coomassie Blue. Five peaks with different protein composition are underlined. (E) A diagram of purification procedure for a CCUG-binding protein with molecular weight 75 kDa (see text). (F) Coomassie staining of the purified protein with molecular weight 75 kDa after SEC125 and UnoQ (run 2). Mass spectrometry showed that a CCUG-binding protein with molecular weight 75 kDa is a p68 RNA helicase. (G) UV cross-link analysis of proteins at different steps of the purification of p68—peak 5 (first step), S125 column (second step), and UnoQ (run 2; third step)—using CCUG100 probe; arrow: p68 bound to CCUG probe. HeLa whole-cell extract (WCE) and immunoprecipitated p68 (p68-IP) from the HeLa cells were examined as controls. (H) Western blot analysis with anti-p68 confirms that the purified protein with molecular weight 75 kDa is p68 RNA helicase. Antibodies to p68 recognize p68 in the HeLa WCE in peaks 2 and 5 at the first steps of purification and in peak 5 after two rounds of purification.
Because the identified proteins have altered CCUG-binding activity in DM2 myoblasts, we suggested that these proteins might be involved in the degradation of the mutant CCUG repeats. Therefore, we performed purification of the CCUG repeat binding proteins from HeLa cells expressing CCUG100 RNA, using a series of HPLC-based chromatographies. First separation of proteins in the ion exchange UnoQ column and UV cross-link analysis identified CCUG-binding proteins with molecular weights 40, 50–55, 75, and 100 kDa (Fig. 1D). The 75-kDa CCUG-binding protein was detected in peaks 1, 4, and 5. Because peak 5 contains a much smaller number of proteins than do other peaks, the protein fractions from peak 5 were subjected to further separation by chromatography on the size exclusion column, SEC125, and by the second ion exchange chromatography on the UnoQ column (run 2) (Fig. 1E). This separation yielded a significant enrichment of the CCUG-binding protein with the approximate molecular weight 75 kDa (Fig. 1 F and G). Mass spectrometry analysis revealed that this protein is the p68 RNA helicase. The identity of this protein was confirmed in western blot assay with antibodies to p68 (Fig. 1H). Taken together, we conclude that p68 RNA helicase binds to RNA CCUG repeats and that its activity is reduced in DM2 myoblasts.
Correction of p68 Levels in DM1 and DM2 Cells Dissociates CUG and CCUG Foci in Nucleus and in Cytoplasm.
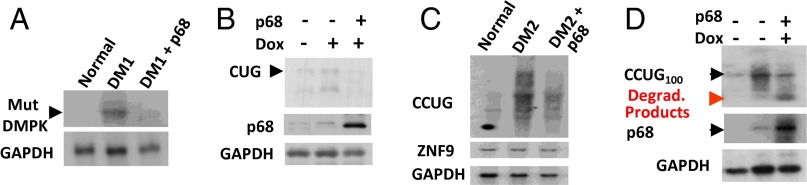
The reduction of RNA-binding activity of p68 in DM2 myoblasts suggested that p68 might also be reduced in skeletal muscle of patients with DM2. Western blot analysis showed that the levels of p68 are reduced in biopsied skeletal muscle from patients with DM2 (Fig. 2A). Recent analysis of the proteins bound to CUG repeats showed that p68 binds to CUG foci and modulates splicing activity of MBNL1 (14). It appears that p68 levels are increased in immortalized cultured DM1 myoblasts (17). However, p68 levels were reduced in DM1 biopsied muscle (Fig. 2B). CUGBP1 (used as a marker) was increased in the same muscle biopsies from patients with DM1 and DM2. It is likely that the reduction of p68 in DM1 muscle is caused by the mutant CUG repeats because p68 was also reduced in muscle of DM1 mice expressing 250 CUG repeats in the 3′ UTR of human skeletal actin (HSALR model) (18) (Fig. 2C).
Fig. 2.

Correction of p68 activity in DM1 and DM2 causes disintegration of CUG and CCUG foci. Western blot analysis of proteins from normal, DM2 (A), and DM1 (B) biopsied muscles with antibodies to p68, CUGBP1, and α-tubulin. (C) Western blot analysis of skeletal muscle proteins from WT and HSALR mice with antibodies to p68, CUGBP1, and β-actin. (D) Ectopic expression of p68 reduces the number of cells with CUG foci in DM1 myoblasts. FISH analysis of DM1 myoblasts transfected with p68-GFP using CAG probe (red). DM1 cells transfected with p68-GFP were detected by fluorescent analysis (green). Nuclei were stained with DAPI (blue). (E) p68-dependent dissociation of CUG foci depends on the length of CTG expansions. Bar graphs show the average percentage of cells with CUG foci in DM1 myoblasts from patients with 60, 300, and 500 CTG repeats transfected with GFP or p68-GFP. (F) Delivery of p68 in DM1 myoblasts with 300 CTG repeats causes dissociation of foci in both the nucleus and cytoplasm. CUG foci were counted in both the nucleus and cytoplasm (300 cells per set). (G) Ectopic expression of p68 reduces the number of CCUG foci in DM2 fibroblasts. CCUG foci (red) were detected by FISH analysis in DM2 fibroblasts transfected with p68-GFP (green). Nuclei were stained with DAPI. Original magnification is 60×. Data are presented as mean ± SEM. *P < 0.05, **P < 0.005, ***P < 0.0005.
The RNA helicases are proteins involved in dsRNA unwinding (19), allowing ribonucleases to access RNA-binding sites to facilitate RNA degradation. Given these activities of helicases, we considered p68 as a reasonable candidate to unwind ds-CUG and CCUG hairpins during decay of the mutant RNAs. Because p68 is reduced in DM1 and DM2 biopsied skeletal muscle, this deficiency of p68 might prevent unwinding of CUG and CCUG hairpins and might reduce degradation of the mutant RNAs causing their aggregation in foci. To examine this hypothesis, DM1 myoblasts with 300 CTG repeats were transfected with p68-GFP, and CUG foci were examined by FISH assay. We found that CUG foci are disintegrated in 55% of cells transfected with p68 (Fig. 2 D and E). In DM1 myoblasts with 60 CTG repeats, ectopic p68 almost completely dissociated CUG aggregates (SI Appendix, Fig. S1 A and B). However, in DM1 myoblasts with 500 CTG repeats, ectopic p68 reduced CUG foci in 40% of cells (Fig. 2E), suggesting that p68 needs additional proteins to unwind long CUG expansions in the 3′ UTR of DMPK.
The UV cross-link analysis suggested that the mechanism by which p68 reduces CUG foci might be associated with the direct binding of p68 to CUG repeats. We found that both endogenous p68 and ectopic p68, expressed in HeLa cells, bind to the expanded CUG repeats in vitro (SI Appendix, Fig. S1 C and D).
It has been suggested that the mutant DMPK mRNA form foci in the nuclei only and that after RNA foci dissociation the mutant DMPK migrates to the cytoplasm. We observed CUG-positive foci in both the nucleus and cytoplasm of DM1 myoblasts (Fig. 2D), whereas ∼30% of DM1 myoblasts did not have CUG aggregates in both compartments. Quantification of foci in DM1 myoblasts transfected with p68 showed that p68 causes the dissociation of RNA foci in the nucleus and cytoplasm of DM1 myoblasts to an equal extent (Fig. 2F). This indicates that p68 has an equally beneficial effect on CUG foci dissociation in the nucleus and cytoplasm and that foci formation in the cytoplasm is not due solely to foci dissociation in the nucleus and migration of the mutant RNA to cytoplasm. Similar to DM1 cells, ectopic expression of p68 causes disintegration of CCUG foci in DM2 fibroblasts (Fig. 2G and SI Appendix, Fig. S2) and in the Dox-inducible HeLa model of DM2 (Fig. S3).
Ectopic Expression of p68 Causes Degradation of the Mutant CUG and CCUG RNAs in DM1 and DM2 Cells.
Foci dissociation in DM1 and DM2 cells by p68 suggested that RNA foci might disintegrate due to degradation of mutant RNAs. Therefore, we compared levels of the mutant DMPK mRNA in DM1 fibroblasts after transfection with p68 using northern blot assay. We found that ectopic expression of p68 causes significant reduction of the mutant DMPK mRNA (Fig. 3A and SI Appendix, Fig. S4A). Similar reduction of the mutant CUG RNA was observed in Dox-inducible CHO cells expressing CUG914 RNA (Fig. 3B and SI Appendix, Fig. S4B). It appears that the effect of p68 on the reduction of the mutant DMPK mRNA or CUG RNA was specific because no degradation of GAPDH mRNA was observed (Fig. 3 A and B).
Fig. 3.
Ectopic expression of p68 degrades mutant RNAs in DM1 and in DM2 cells. (A) Northern blot analysis of DM1 fibroblasts transfected with p68-GFP using CAG and GAPDH probes. The levels of the mutant DMPK mRNA were quantified as ratios to the GAPDH signal (see SI Appendix, Fig. S4A). (B) Northern blot analysis of tet-inducible CHO cells expressing CUG914, transfected with p68-GFP using CAG, p68, and GAPDH probes. The CUG signal was quantified as ratio to GAPDH (SI Appendix, Fig. S4B). (C) Northern blot analysis of DM2 fibroblasts transfected with p68-GFP using CAGG, ZNF9, and GAPDH probes. CCUG signal is diffused in DM2 fibroblasts. Quantifications of the CCUG and normal ZNF9 mRNA signals are shown as ratio to GAPDH in SI Appendix, Fig. S4 C and D. (D) Northern blot analysis of inducible HeLa cells expressing CCUG100 RNA transfected with p68 using CAGG, p68, and GAPDH probes. Positions of degraded (red arrow) and undegraded (black arrow) CCUG100 RNAs are shown. Quantification of degraded CCUG100 signals as ratios to GAPDH is shown in SI Appendix, Fig. S4E.
We also analyzed the effects of ectopic expression of p68 on the degradation of the mutant CCUG repeats in DM2 fibroblasts. We found that mutant CCUG repeats migrate as a smear band on the gel (Fig. 3C). After transfection with p68, the CCUG-positive signal was significantly reduced (Fig. 3C and SI Appendix, Fig. S4C). Degradation of the mutant CCUG repeats is specific because ZNF9 and GAPDH mRNAs were not degraded (Fig. 3C and SI Appendix, Fig. S4D). p68 also degrades the mutant CCUG100 RNA in tet-regulated HeLa cells. The intensity of the CCUG100 signal was reduced in HeLa cells transfected with p68 (Fig. 3D). Respectively, transfected cells accumulated low–molecular-weight CCUG-positive RNA that likely represents products of degradation of CCUG100 RNA (Fig. 3D and SI Appendix, Fig. S4E). Based on these data, we conclude that normalization of p68 is beneficial for degradation of the mutant RNAs in DM1 and DM2.
Normalization of p68 in Skeletal Muscle of HSALR Mice Reduces Muscle Pathology.
To determine if correction of p68 might reduce DM1 muscle pathology, recombinant adeno-associated vector expressing p68-GFP (SI Appendix, Fig. S5A) was injected in Gastrocnemius (gastroc) muscle of 2-mo-old HSALR mice. At this age, the HSALR mice do not show significant muscle phenotype. By 3 mo, however, skeletal muscle in these mice is characterized by myopathy including a variability of myofiber size with central nuclei and atrophy. Muscles were analyzed 2–4 wk after injection, and expression of p68-GFP was confirmed by fluorescent analysis (SI Appendix, Fig. S5B). The UV cross-link analysis indicated that p68-GFP binds to CUG RNA in vitro (SI Appendix, Fig. S5 C and D). Recombinant p68 caused the reduction of CUG foci in muscle of HSALR mice (SI Appendix, Fig. S6). H&E staining showed significant improvement of muscle histology in HSALR mice treated with p68 (Fig. 4 and SI Appendix, Fig. S7). In control HSALR muscle treated with PBS, fibers were variable in size. However, there was a recovery of myofiber uniformity within muscle tissue treated with p68. In HSALR mice treated with PBS, at 3 mo of age the average cross-sectional area of myofibers is increased due to presence of hypertrophic fibers similar to untreated HSALR mice (10). We found that p68 treatment normalized the average area of myofibers (Fig. 4 and SI Appendix, Fig. S7A). To confirm the recovery of the fiber size in the p68-treated HSALR mice, the mean minimal Feret’s diameter was examined. We found that the minimal myofiber diameter was increased in the PBS-treated muscle of HSALR mice relatively WT muscle; however, the mean fiber diameter was normalized in the p68-treated muscle (SI Appendix, Fig. S7B). Muscles treated with p68 showed improved myofiber bundle bunching compared with muscle treated with PBS (Fig. 4). Quantification of myofiber number in similar regions of gastroc muscles from PBS- and p68-treated mice showed that p68 treatment significantly increases the total number of myofibers with reduced central nuclei (Fig. 4 and SI Appendix, Fig. S7 C and D). EM analysis showed the improvement of myofibrillar organization in the p68-treated HSALR muscle (SI Appendix, Fig. S8). These data show that normalization of p68 causes reduction of myopathy and muscle atrophy associated with DM1.
Fig. 4.

Intramuscular delivery of p68 corrects myopathy and atrophy in HSALR mice. H&E analysis of gastroc muscles of WT and HSALR mice injected with p68-GFP or with PBS. Arrows show fibers with central nuclei. Quantification of the average myofiber size, myofiber number, and central nuclei is shown in SI Appendix, Fig. S7.
Hypothetical Role of p68 in DM1/2 Pathology.
Whereas many studies are focused on the correction of toxic effects caused by the mutant CUG and CCUG repeats, the mechanisms which prevent degradation of these repeats are not well understood. Recent reports showed that RNA helicases p68/DDX5 and DDX6 bind to and remodel the mutant CUG RNA (14, 15). It has been proposed that these RNA helicases have a different effect on the mutant CUG repeats. Ectopic expression of DDX6 decreased CUG foci in DM1 cells (15). In contrast, p68 might increase the stability of CUG repeats because inhibition of p68 in tet-regulated HeLa cells by siRNA to p68/p72 reduces the number of CUG foci (14). The findings described in this paper show that the correction of p68 levels in DM1 cells reduces the number of CUG foci (Fig. 2 D and E). Therefore, we suggest that p68 and DDX6 might unwind the ds-CUG repeats within the mutant DMPK mRNA and improve its processing.
It appears that the beneficial effect of p68 in DM1 is stronger than that of DDX6 because the disintegration of CUG foci by p68 promotes degradation of the mutant DMPK mRNA (Fig. 3A and SI Appendix, Fig. S4A), whereas DDX6 disassembles CUG foci without degradation of the mutant DMPK mRNA (15). In addition, DDX6 does not reduce the stability of CCUG foci (15), whereas correction of p68 improves disintegration and degradation of both CUG and CCUG mutant RNAs (Figs. 2 and 3). The reason for the reduction of CUG foci by the inhibition of p68/p72 in the Laurent et al. paper (14) remains to be determined. It is possible that the inhibition of both p68 and p72 might have a different effect on the stability of CUG foci than has p68 alone.
Examination of the role of p68 in vivo showed that intramuscular injection of p68 in HSALR mice reduces DM1 muscle histopathology. This result is in agreement with the reduction of CUG foci in the HSALR muscle, treated with p68, but not with GFP (SI Appendix, Fig. S6).
It has been shown that the bridging integrator-1 (BIN1) protein contributes to muscle weakness in DM1 (20). The splicing of BIN1 mRNA is misregulated in DM1, leading to an increase of the isoform lacking exon 11 (20). We examined the expression of this isoform in HSALR muscle and found that the BIN1 isoform lacking exon 11 is increased in untreated HSALR mice, whereas the isoform retaining exon 11 is reduced (SI Appendix, Fig. S9). In contrast, HSALR muscle treated with p68 shows improvement of the splicing of BIN1 by reduction of the isoform lacking exon 11 and increase of the isoform retaining exon 11 (SI Appendix, Fig. S9). Because this splicing event is regulated by MBNL1, the improvement of the splicing of BIN1 in the p68-treated muscle suggests that p68 is beneficial for the correction of at least some splicing events, misregulated by MBNL1 in DM1. We hypothesize that this effect might be due to disintegration of CUG foci, which releases MBNL1 and possibly other RNA-binding proteins associated with the mutant CUG repeats. We previously suggested that several RNA-binding proteins might bind to the mutant CUG foci, including p68 and MBNL1 (21). Both p68 and DDX6 might reduce stability of CUG foci releasing sequestered RNA-binding proteins such as MBNL1. This hypothesis is supported by the findings in this study, which show that the correction of p68 is sufficient to reduce CUG foci by ∼50% in DM1 cells with 300–500 CTG repeats (Fig. 2E). Based on these data, we suggest that a synergistic effect of p68, DDX6, and MBNL1 might be needed to disintegrate the majority of CUG foci in DM1.
Further studies are necessary to determine the relationship of p68 and MBNL1 in RNA processing in DM1. It is possible that during regulation of splicing p68 promotes MBNL1 binding to its targets, as was shown by Laurent et al. (14).
The mechanism by which p68 protein is reduced in DM1 and in DM2 is unknown. Our recent study showed that the inhibitors of GSK3, correcting CUGBP1 activity in DM1 mice, significantly reduce DM1 pathology (10). We found that GSK3 inhibitor TDZD-8 restores p68 levels in HSALR mice (SI Appendix, Fig. S10). Because CUGBP1 regulates many mRNAs, we hypothesize that p68 is one of the targets of CUGBP1 and that the reduction of the repressive form of CUGBP1 by GSK3 inhibitors leads to the normalization of p68 and degradation of the mutant CUG RNA. Regardless of whether p68 is a direct or indirect target of CUGBP1, data presented in this study show that the correction of p68 helicase leads to the reduction of toxic RNAs and to correction of DM pathology.
Materials and Methods
Purification of RNA-Binding Proteins Using HPLC Techniques.
Cytoplasmic proteins were collected from the induced HeLa cells expressing CCUG100 RNA. Twenty milligrams of proteins were loaded on ion exchange column UnoQ and eluted with a gradient 0–0.5 M NaCl. Chromatography fractions were used for the electrophoretic analysis, UV cross-linking with CCUG100 probe, and for western blot assay. Fractions containing proteins of interest were combined and further separated by size exclusion and ion exchange chromatographies, as shown in Fig. 1E. The purified p75 protein was subjected to mass spectroscopy at the Baylor College of Medicine Protein Core Facility. FISH assay, Western and Northern blot analyses were performed as described (16). Detailed description of these methods is also provided in SI Appendix, Experimental Procedures.
UV cross-linking assay.
Proteins were incubated with 32P-CCUG100 or 32P-CUG123 for 30 min at room temperature, subjected to UV light, and then treated with RNase A. The RNA–protein complexes were separated by SDS/PAGE, and proteins were transferred to nitrocellulose membrane. The membranes were exposed to X-ray film and stained with Coomassie blue to verify protein loading.
Animal experiments.
The experiments with animals were approved by the Institutional Care and Use Committee at Baylor College of Medicine and Cincinnati Children's Hospital. AAV8-GFP-p68 (2.55 × 1010 GC) was administered to the right gastroc muscle of the 2-mo-old HSALR mice while the left muscle of the same mouse was injected with the same amount of AAV8-GFP or PBS. Gastroc muscles were collected 2–4 wk after injections and examined by H&E staining and by electron transmission microscopy. WT mice of the matching sex and age were used as controls. The analysis of the number of total fibers and the average fiber size in the largest area of the cross sections were determined as previously described (10). Some 5-mo-old HSALR mice were treated with TDZD-8 (at a dose of 10 mg/kg) three times a week for 1 wk.
For other methods, see SI Appendix, Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Bingwen Jin for the cloning of p68 plasmid. This research is supported by National Institutes of Health Grants AR044387, AR052791, AR064488 (to L.T.) and CA159942, DK102597, GM551888 (to N.T.); by a grant from Fondazione Malattie Miotoniche (to G.M.); and by internal development funds from Cincinnati Children’s Hospital Medical Center (to L.T. and N.T.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1422273112/-/DCSupplemental.
References
- 1.Harper PS. Myotonic Dystrophy. WB Saunders; London: 2001. [Google Scholar]
- 2.Brook JD, et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68(4):799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 3.Liquori CL, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293(5531):864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 4.Timchenko LT. Myotonic dystrophy: The role of RNA CUG triplet repeats. Am J Hum Genet. 1999;64(2):360–364. doi: 10.1086/302268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Timchenko L. Molecular mechanisms of muscle atrophy in myotonic dystrophies. Int J Biochem Cell Biol. 2013;45(10):2280–2287. doi: 10.1016/j.biocel.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Osborne RJ, Thornton CA. RNA-dominant diseases. Hum Mol Genet. 2006;15(Spec No 2):R162–R169. doi: 10.1093/hmg/ddl181. [DOI] [PubMed] [Google Scholar]
- 7.Timchenko LT, et al. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 1996a;24(22):4407–4414. doi: 10.1093/nar/24.22.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Timchenko LT, Timchenko NA, Caskey CT, Roberts R. Novel proteins with binding specificity for DNA CTG repeats and RNA CUG repeats: Implications for myotonic dystrophy. Hum Mol Genet. 1996b;5(1):115–121. doi: 10.1093/hmg/5.1.115. [DOI] [PubMed] [Google Scholar]
- 9.Miller JW, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19(17):4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones K, et al. GSK3β mediates muscle pathology in myotonic dystrophy. J Clin Invest. 2012;122(12):4461–4472. doi: 10.1172/JCI64081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mooers BH, Logue JS, Berglund JA. The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc Natl Acad Sci USA. 2005;102(46):16626–16631. doi: 10.1073/pnas.0505873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dere R, Napierala M, Ranum LP, Wells RD. Hairpin structure-forming propensity of the (CCTG.CAGG) tetranucleotide repeats contributes to the genetic instability associated with myotonic dystrophy type 2. J Biol Chem. 2004;279(40):41715–41726. doi: 10.1074/jbc.M406415200. [DOI] [PubMed] [Google Scholar]
- 13.Ravel-Chapuis A, et al. The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J Cell Biol. 2012;196(6):699–712. doi: 10.1083/jcb.201108113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laurent FX, et al. New function for the RNA helicase p68/DDX5 as a modifier of MBNL1 activity on expanded CUG repeats. Nucleic Acids Res. 2012;40(7):3159–3171. doi: 10.1093/nar/gkr1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pettersson O, et al. DDX6 regulates sequestered nuclear CUG-expanded DMPK-mRNA in dystrophia myotonica type 1. Hum Mol Genet. 2014;42(11):7186–7200. doi: 10.1093/nar/gku352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones K, et al. RNA Foci, CUGBP1, and ZNF9 are the primary targets of the mutant CUG and CCUG repeats expanded in myotonic dystrophies type 1 and type 2. Am J Pathol. 2011;179(5):2475–2489. doi: 10.1016/j.ajpath.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paul S, et al. Expanded CUG repeats Dysregulate RNA splicing by altering the stoichiometry of the muscleblind 1 complex. J Biol Chem. 2011;286(44):38427–38438. doi: 10.1074/jbc.M111.255224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mankodi A, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289(5485):1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 19.Huang Y, Liu ZR. The ATPase, RNA unwinding, and RNA binding activities of recombinant p68 RNA helicase. J Biol Chem. 2002;277(15):12810–12815. doi: 10.1074/jbc.M200182200. [DOI] [PubMed] [Google Scholar]
- 20.Fugier C, et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med. 2011;17(6):720–725. doi: 10.1038/nm.2374. [DOI] [PubMed] [Google Scholar]
- 21.Meola G, Jones K, Wei C, Timchenko LT. Dysfunction of protein homeostasis in myotonic dystrophies. Histol Histopathol. 2013;28(9):1089–1098. doi: 10.14670/HH-28.1089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.