

Significance
Genetic screening is a classic approach to identify genes acting in a biological process of interest. In mammalian cells, screens are commonly based on RNA interference (RNAi), in which a short interfering RNA (siRNA) or short-hairpin RNA (shRNA) triggers degradation of cellular messenger RNAs. RNAi approaches are prone to false-positive results because of siRNA/shRNA off-target effects and false-negative results because of siRNAs/shRNAs lacking activity. We previously established that these problems can be minimized with ultracomplex shRNA libraries. Here, we present next-generation shRNA libraries targeting the human and mouse genomes, for which we improved several features to increase shRNA activity. In a pilot screen, the new library yields complementary results to clustered regularly interspaced short palindromic repeats interference (CRISPRi), an orthogonal approach we developed recently.
Keywords: functional genomics, shRNA, genetic screen, pooled screen, microRNA
Abstract
Genetic screening based on loss-of-function phenotypes is a powerful discovery tool in biology. Although the recent development of clustered regularly interspaced short palindromic repeats (CRISPR)-based screening approaches in mammalian cell culture has enormous potential, RNA interference (RNAi)-based screening remains the method of choice in several biological contexts. We previously demonstrated that ultracomplex pooled short-hairpin RNA (shRNA) libraries can largely overcome the problem of RNAi off-target effects in genome-wide screens. Here, we systematically optimize several aspects of our shRNA library, including the promoter and microRNA context for shRNA expression, selection of guide strands, and features relevant for postscreen sample preparation for deep sequencing. We present next-generation high-complexity libraries targeting human and mouse protein-coding genes, which we grouped into 12 sublibraries based on biological function. A pilot screen suggests that our next-generation RNAi library performs comparably to current CRISPR interference (CRISPRi)-based approaches and can yield complementary results with high sensitivity and high specificity.
Functional genomics approaches in mammalian cells have the potential to dissect gene functions and to complement observational genomics approaches for the identification of disease mechanisms and therapeutic strategies. For many years, RNA interference (RNAi) was the technology of choice for loss-of-function screens in mammalian cells. Clustered regularly interspaced short palindromic repeats (CRISPR)-based screening approaches recently developed by us and others (1–4) provide a highly promising orthogonal strategy. In particular, CRISPR interference (CRISPRi) has reduced off-target effects and can reach high levels (90–100%) of knockdown (1), and CRISPR cutting can generate null alleles.
Despite the advantages of CRISPR-based strategies, there are still important uses for RNAi technology. RNAi is a single-component system that works in otherwise unengineered cells, and can thus be used in challenging biological contexts. Because CRISPRi blocks transcription at the transcription start site of endogenous genes, it does not allow selective targeting of functionally distinct coding and noncoding RNAs derived from the same primary transcript, such as splice isoforms or noncoding RNAs embedded in the introns of coding transcripts, whereas RNAi reagents can be designed for specific targeting of mature RNAs (mRNAs).
Hence, there is a continued need to improve RNAi-based screening platforms. We have previously established a quantitative framework to derive robust results from pooled screens of ultracomplex short-hairpin RNA (shRNA) libraries that target each gene with ∼25 independent shRNAs and contain thousands of negative-control shRNAs (5). Such complex libraries can be constructed using massively parallel oligonucleotide synthesis, and phenotypes in pooled populations can be determined using next-generation sequencing, as previously established by others and us (6, 7). Ultracomplex libraries minimize the rates of both false-negative results (caused by lack of shRNA activity) and false-positive results (caused by shRNA off-target effects), through the use of a rigorous statistical approach (5, 8). Our framework enables the sensitive identification of genes modulating a phenotype of interest, and of active shRNAs targeting these genes. Thus, it can provide the basis for construction and screening of double-shRNA libraries for the systematic analysis of genetic interactions between large numbers of genes to reveal cellular pathways (8).
Here, we present next-generation shRNA libraries targeting the human and mouse genomes. We tested several parameters to optimize the design of shRNAs and their expression in microRNA (miRNA) contexts. A pilot screen establishes that our next-generation RNAi platform performs comparably to our CRISPRi platform in detecting hit genes, and that results from these orthogonal approaches yield complementary results. Recently, our next-generation shRNA platform described here for the first time enabled us to identify the molecular target of a compound enhancing memory in mice, using a technically challenging flow cytometry-based screen (9).
Results
Systematic Comparison of miRNA Contexts for shRNA Expression.
Although shRNAs can be expressed in mammalian cells from RNA polymerase III promoters, such as the U6 promoter, such strategies can lead to nonspecific toxicity associated with high shRNA expression levels, possibly because of saturation of the endogenous RNAi machinery (10). For our previous double-shRNA–based approach for the quantification of genetic interactions (8), we therefore implemented an alternative strategy developed by the Elledge laboratory (11), in which shRNAs are processed from a miR-30a context embedded in an RNA polymerase II transcript.
The miR-30a context has been used successfully in many cell types. However, our comparison of the processing precision of endogenous miRNAs suggests that miR-30a processing is less precise than other human miRNAs (Fig. S1 and Dataset S1), in particular with respect to the 5′ end, which plays an important role in determining the seed region and thus the spectrum of mRNA targets. This observation raises the possibility that shRNAs expressed from other miRNA context may result in more potent and more selective gene knockdown. A systematic comparison of different miRNA contexts for shRNA expression has not been previously described. To experimentally determine the performance of a large number shRNA expression formats, we conducted a pooled screen of targeted and negative-control shRNAs expressed in different formats. We designed a pooled library targeting a set of human genes (RAB1A, SEC24A, TRAPPC8, TRAPPC11, and VPS53, all encoding trafficking factors, and RPS25, encoding a protein of the 40S ribosomal subunit), whose knockdown we had previously found to confer resistance to the toxin ricin (8). The library contained 25–50 different shRNAs targeting each gene, as well as 1,000 negative-control shRNAs designed not to target any human transcript, expressed from 79 different formats.
Fig. S1.

Cumulative distribution of the 5′ end homogeneity scores of endogenous miRNA precursors (line). Those selected in this study as templates are indicated with dots. A homogeneity score was obtained from the ratios between the counts of reads that match the most frequent 5′ end and total reads mapped to a miRBase hairpin. The ratio was calculated only when 100 or more reads were mapped to a hairpin. The fifth percentile of the ratios throughout the reference samples was chosen as a homogeneity score for a hairpin. Hairpins with ratio values calculated using 10 or more reference samples were used in this analysis. See Dataset S1 for the detailed list of scores and for the list of reference samples used for the analysis.
The formats were based on 11 different endogenous human miRNA contexts and rationally designed variations of these contexts. These variations included the use of G-U wobble base pairs to destabilize interactions between the guide and passenger strands, introduction of a “bulge” in the stem (often found in endogenous miRNAs) caused by a noncomplementary base in the middle of the passenger strand, and use of the endogenous three bases at the 3′ end of the guide, instead of the bases complementary to the shRNA target site. We also tested mutations of the loop sequence to generate a restriction enzyme cleavage site in the encoding DNA for use during sample preparation after pooled screens. Furthermore, we evaluated several miRNA-specific modifications aimed to create an optimal structure for Drosha processing: sequences were replaced or removed to generate single-stranded basal segments that are required for efficient and accurate processing (miR-7-2) (12), to make a simple duplex structure in the lower stem region for simplicity of design (miR-96, miR-125b-2, miR-130a, miR-190a, and miR-211), to convert the first nucleotide of the small RNA product into uridine, which typically appears in the first position of endogenous small RNAs (miR-100 and miR-130a), or to create a flexible loop structure (miR-125b-1 and miR-340), which enhances processing efficiency (13, 14).
We designed >100,000 oligonucleotides encoding shRNAs targeting ricin resistance genes and negative-control shRNAs expressed in the different formats (Dataset S2), cloned them into lentiviral vectors, and introduced them into human K562 cells (Fig. 1A). We then cultured the cells either in the presence or absence of ricin, after which we quantified frequencies of cells expressing each shRNA in the two populations by deep sequencing. This process enabled us to quantify a ricin-resistance phenotype (ρ) for each shRNA (5), and to calculate a P value for each gene and expression format by comparing the strength of phenotypes of shRNAs targeting each gene to the phenotypes of negative-control shRNAs for a given format.
Fig. 1.
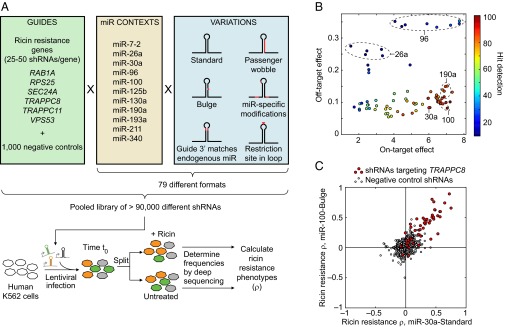
Massively parallel comparison of miRNA contexts for shRNA expression. (A) Experimental strategy to test performance of different miRNA contexts and variants for shRNA expression in a pooled genetic screen. Guides targeting genes with known ricin resistance phenotypes were selected, along with negative-control guides. These were expressed in 79 different shRNA formats, which were variations on 11 endogenous human miRNA contexts. The resulting pooled library of >100,000 different shRNAs was introduced into the human K562 cell line using lentiviral infections. The cells were grown untreated or treated with ricin, and the frequencies of cells expressing a given shRNA in these two populations were determined using deep sequencing. From these data, ricin-resistance phenotypes were calculated for all shRNAs. (B) Comparison of 79 formats with respect to three metrics derived from the pooled screen: On-target effect (x axis, a measure of phenotype strength across all ricin hit genes), off-target effects (y axis, a measure of the deviation from wild-type for negative-control shRNAs), and hit detection (heat map, a measure of the statistical significance of detecting ricin hit genes). Metrics are defined quantitatively in Materials and Methods. In several instances, formats derived from the same miRNA context show similar performance across the three metrics (dashed ovals). (C) Comparison of ricin resistance phenotypes for shRNAs targeting TRAPPC8 (red circles) and negative-control shRNAs (empty circles) expressed either in a miR-30a–standard context or a miR-100–bulge context. Phenotypes of targeted shRNAs were correlated between the two expression formats.
To evaluate the performance of the different formats, we compared three metrics: (i) the on-target strength of shRNA phenotypes, (ii) the extent of off-target effects (deduced from the phenotype distribution of negative-control shRNAs), and (iii) the confidence of calling hit genes (the P values for known ricin resistance genes). These metrics are quantitatively defined in Materials and Methods. Interestingly, we found that on-target strength and off-target effects were independent features across different expression formats (Fig. 1B). As expected, formats achieving high on-target phenotypes but low off-target effects performed best at hit gene detection (Fig. 1B). Different formats derived from the same endogenous miRNA context commonly shared performance characteristics (Fig. 1B). The on-target phenotypes achieved with independent shRNAs targeting the same genes were highly correlated for many of the well-performing expression formats (Fig. 1C and Fig. S2A).
Fig. S2.

Results from a pooled screen comparing shRNA expression formats (Fig. 1A). (A) Correlation of shRNA phenotypes for a given expression format with phenotypes obtained with the standard format miR-30a (mir30a_s) for each targeted gene. The heatmap encodes Pearson correlation coefficients R. (B) Gene-based P values for each expression format. The heatmap encodes −log10 P values.
The traditionally used miR-30a context was among the best expression formats, displaying high on-target activity, low off-target activity, and excellent hit detection performance (Fig. 1B and Fig. S2B). miR-100–based formats had even slightly higher on-target activity in this study. However, we decided to use the miR-30a context for our next-generation library, given its well-established performance in many experimental contexts. miR-100 context-based shRNA expression should be an attractive alternative for future applications that require alternative expression formats.
Variations of the miR-30a Expression Format.
We next tested the impact of modifications of the miR-30a context expression format on shRNA knockdown performance. A commonly used variation of the wild-type miR-30a context, which was present in our previous libraries, is a point mutation introducing an EcoRI site in the encoding DNA for cloning purposes (Fig. 2A). However, this point mutation also disrupts a CNNC motif recently shown to be important for effective miR-30a processing (15). We previously developed an shRNA expression vector in which this CNNC motif is restored (5). A similar strategy has been reported by others (16). We also wanted to test a pair of point mutations in the shRNA loop region that would generate a restriction site in the encoding DNA (Fig. 2A). Cleavage of this region during the preparation of samples for deep sequencing is a strategy to circumvent PCR amplification of the inverted-repeat region encoding the full-length shRNA.
Fig. 2.

Individual characterization of shRNA expression formats. (A) Variations of the miR-30a context. (Upper) A point mutation (red) in the sequence downstream of the hairpin creates an EcoRI site (underlined) in the encoding DNA. This destroys the CNNC motif (underlined in purple) shown to be important for miR-30a processing (15). (Lower) Two point mutations (red) in the hairpin loop create a HindIII site (underlined) in the encoding DNA. (B and C) An shRNA targeting GFP was expressed in different formats in a K562 cell line stably expressing GFP. Median GFP fluorescence was quantified by flow cytometry, and is normalized to GFP fluorescence in a cell line with a negative-control expression construct lacking a hairpin. The dotted line indicates the level of GFP fluorescence for the expression format we have previously used (EF1a promoter, EcoRI context, WT loop). (B) shRNA expressed from the WT context resulted in stronger knockdown compared with EcoRI context. Introduction of HindIII in the loop was not detrimental. (C) In K562 cells, expression from the SFFV promoter resulted in stronger knockdown than expression from the EF1a promoter.
We expressed these sequence variants of an shRNA targeting GFP in a K562 line stably expressing GFP and quantified GFP knockdown by flow cytometry. The wild-type miR-30a context containing the CNNC motif resulted in stronger GFP knockdown than the EcoRI version of miR-30a. Mutations in the loop region designed to introduce a HindIII site were not detrimental to knockdown performance. We therefore chose the combination of CNNC-containing miR-30a context with HindIII-modified loop for our next-generation library.
We and others have previously used the housekeeping promoter EF1a to express miRNA-context–based shRNAs. We wanted to test if a stronger promoter would result in higher shRNA activity. We selected the viral SFFV promoter, which is short and drives high levels of transcription in several cell lines. Indeed, SFFV-driven shRNA expression resulted in stronger knockdown in K562 cells (Fig. 2C). We therefore used this promoter to construct our next-generation shRNA library. However, because the SFFV promoter can be silenced in certain cell types (17), we also generated an alternative backbone vector containing the EF1a promoter instead.
Machine Learning of Sequence Rules for shRNA Activity.
It is currently not possible to precisely predict the activity of shRNA sequences. A major advance was the establishment of the so-called sensor assay, which enabled the massively parallel determination of shRNA activities (18). From data generated with the sensor assay, rules for shRNA activity were abstracted (18) and a proprietary algorithm for the prediction of shRNA activity was developed (16). Our goal was to test which rules are predictive of shRNA activity specifically in our expression context.
Our first aim was to compare the performance of 21mer shRNAs designed using the si-shRNA Selector program (19) and 22mer shRNAs designed using the Hannon laboratory shRNA retriever program (20). We constructed two libraries targeting the same set of ∼1,000 genes, each with either fifty 21mer or fifty 22mer shRNAs, and containing >1,000 matched negative-control shRNAs. The targeted genes were enriched for genes known to modulate the sensitivity to ricin. We determined ricin-resistance phenotypes in K562 cells in pooled screens to calculate gene-based P values. Although there was a correlation in the P values for hit genes derived from both libraries, the 22mer shRNA library outperformed the 21mer shRNA library (Fig. 3A). The 22mer library detected more hit genes above a 5% false-discovery rate (FDR) cut-off, as calculated using the approach by Storey and Tibshirani (21). We therefore decided to use 22mer shRNAs for our next-generation shRNA library.
Fig. 3.

A sequence score predictive of shRNA performance. (A) Comparison of 21mer vs. 22mer guide design. Two shRNA libraries targeting the same set of 1,079 genes each with 50 21mer guide strands vs. 50 22mer guide strands were used in a ricin resistance experiment. P values for each gene were calculated based the data from the two libraries. Gray line: cut-off for 5% FDR. (B) Sequence features as predictors of 22mer shRNA activity. Phenotypes of 22mer shRNAs targeting ricin hit genes were measured in a batch experiment and shRNAs were classifed as active or inactive. Features (quantitatively defined in Table S1) were target accessibility as predicted from the secondary structure stability of the mRNA context of the shRNA target, and modified versions of the sensor rules (18). (Left) Areas under the receiver operating characteristic curve (ROC AUC) for sensor rules used as quantitative metrics. Stepwise forward logistic regression was used to create an integrated sequence score predicting shRNA activity (Table S2). Features included in the sequence scores are marked by asterisks. (Right) ROC curve for the sequence score; FPR, false-positive rate; TPR, true positive rate. (C and D) Based on shRNA phenotypes in a ricin-resistance screen targeting genes with 50 shRNAs each, P values for each gene were calculated on the basis of subsets of the data; the number of shRNAs included per gene was varied. shRNA subsets were either chosen randomly 100 times, and means of −log10 of P values are shown, with error bars indicating SD, or shRNA subsets were chosen based on the highest sequence scores. (C) Results are shown for three representative genes: a strong hit (RAB1A), a moderate hit (STX16), and a nonhit (CRYAB). For the purpose of this analysis, sequence scores were created based on a dataset from which shRNAs targeting RAB1A, STX16 and CRYAB were excluded (Table S2). (D) P values calculated based on 45 shRNAs per gene are compared with P values calculated based on 10 shRNAs per gene for all 1,079 genes targeted by Library 2. Subsets were either chosen randomly (light blue) or based on their sequence score (dark blue). Sequence scores for individual shRNAs were calculated based on data subsets excluding these specific shRNAs, as described in SI Materials and Methods.
To investigate which sequence properties increased the likelihood of 22mer shRNA activity in our expression system, we applied machine-learning approaches to a training dataset of 461 individually cloned shRNAs targeting bona fide hit genes in our ricin-resistance pilot screen, which we had retested and classified as active or inactive (Materials and Methods). First, we compared base frequencies of active and inactive guide strands at each position of the guide strand, and found that an A or U at the first position of the guide strand was highly predictive of shRNA activity (P < 10−7, χ2 test) (Fig. S3A). We also investigated sequence properties of the 50 mRNA bases flanking the shRNA target site on either side, but did not find significant predictors of shRNA activity (no positions with Bonferroni-corrected P < 0.05, χ2 test).
Fig. S3.

(A) Comparison of base frequencies at each guide strand position for active and inactive shRNAs. (Upper) −Log10 of P values indicating significant differences between base frequencies in active and inactive shRNAs (χ2 test). The dotted green line indicates a Bonferroni-corrected significance level of 0.05. (Lower) Bars indicating base frequencies at each guide strand position for active and inactive shRNAs. (B) As in Fig. 3D, P values calculated based on shRNA subsets were compared with P values calculated based on 45 shRNAs per gene. (Left) Slope of the linear regression for this comparison and (Right) the Pearson correlation coefficient, both as a function of shRNA subset size. Subsets were either chosen randomly (light blue) or based on their sequence score (dark blue).
Features of active shRNAs have previously been deduced from experimental data obtained with the “sensor” assay (18), and were termed “sensor rules.” Although the design algorithms we used to create shRNAs in our primary screen library already preselected shRNAs that tended to conform to some of the sensor rules, such as thermodynamic asymmetry, several of the sensor rules still had predictive power for shRNA activity within our library (Fig. 3B). Presence of A or U in the first position of the guide strand, which we independently found to be an important predictor of shRNA activity (Fig. S3A), is also one of the sensor rules.
An additional factor that has been reported to affect shRNA activity is the accessibility of the target site within the mRNA (22). In viral genomes, the experimentally determined secondary structure of the target site is strongly anticorrelated with shRNA activity (23). However, we found no strong predictive power of either the thermodynamic stability of the mRNA segment containing the target site or of the accessibility of the target of the seed sequence only as predicted by the unafold algorithm (24) (Fig. 3B), or of secondary structure information we previously determined experimentally (25). There was also no statistically significant difference between the activity of shRNAs targeting the coding sequence or the 3′ untranslated region of target mRNAs (P > 0.2, Mann–Whitney test).
To integrate weighted information about sequence properties (Table S1) into a predictive score, which we will refer to as “sequence score,” we used stepwise forward logistic regression. The resulting sequence score (Table S2) was an excellent predictor of shRNA activity for both enriched and depleted shRNAs and only incorporated three sequence features, respectively (Fig. 3B). Taking into account additional features did not improve the predictive power of the sequence score, because many of the sequence features are correlated for shRNAs generated by the Hannon laboratory shRNA retriever program (20).
Table S1.
Quantitative definitions of variables for logistic regression
| Variable | Quantitative definition |
| Total A/U in Guide | Number of A and U in guide strand |
| A/U in Guide1–14 | Number of A and U in first 14 bases of guide strand |
| Thermodynamic asymmetry | Percentage A/U in first 14 bases of guide strand minus Percentage A/U in rest of guide strand. Note that our definition differs from the original sensor rule (which is the ratio rather than difference of percentages) to avoid division by zero. |
| Guide1 = A/U | 1 if the first position of the guide strand is A or U, otherwise 0. |
| Guide20 not A | 0 if position 20 is A, otherwise 1. |
| Guide13 = A/U or Guide14 = U | 1 if guide strand position 13 is A or U or if position 14 is U, otherwise 1. |
| Guide20 = G or Guide21 = G | 1 if positions 20 or 21 are G, otherwise 0. |
| Guide20 not C | 0 if position 20 is C, otherwise 1. |
| Guide10 = A/U | 1 if position 10 of the guide strand is A or U, otherwise 0. |
| Target accessibility | ΔG in kcal/mol predicted for the minimum energy fold of the mRNA region of the target site and the flanking 50 bases upstream and downstream, as calculated by Unafold (24). |
| Seed Target accessibility | Summed probabilities of the target mRNA bases complementary to the guide strand seed (bases 2–8) to be unpaired, based on the predicted fold of the mRNA region of the target site and the flanking 50 bases upstream and downstream, as calculated by Unafold (24). |
Table S2.
Sequence scores for shRNA activity, from forward stepwise logistic regression
| Description | Score from stepwise logistic regression |
| Sequence score | 4.69 × (Thermodynamic asymmetry) |
| +1.38 × (Guide1 = A/U) | |
| +0.99 × (Guide13 = A/U or Guide14 = U) | |
| −1.05 | |
| Sequence score (regression excluded shRNAs targeting TRAPPC11, STX16, CRYAB) | 4.75 × (Thermodynamic asymmetry) |
| +1.39 × (Guide1 = A/U) | |
| +1.09 × (Guide13 = A/U or Guide14 = U) | |
| −1.19 |
This sequence score was derived based on the activity of a limited set of shRNAs targeting hit genes and we wanted to investigate whether it would also be a useful tool to select shRNAs for increased shRNA library performance. To address this question, we analyzed data from the ricin-resistance screen of the 22mer shRNA library targeting 1,079 genes with 50 shRNAs each. We computationally created shRNA subsets and compared P values for genes calculated based on shRNA subsets of varying sizes. In Fig. 3C, results are shown for three representative genes with distinct ricin-resistance phenotypes: TRAPPC11, a strong hit gene encoding a trafficking factor, STX16, a weak hit gene encoding a syntaxin, and CRYAB, encoding a small heat shock protein, which was not a hit. When these shRNA subsets targeting these genes were created randomly, discrimination between the weak hit STX16 and the nonhit CRYAB required ∼15 or more shRNAs. However, when subsets of shRNAs were created based on the shRNAs with the highest sequence scores (calculated here for a training set of shRNAs that did not include shRNAs targeting STX16, TRAPPC11, and CRYAB), even ∼seven shRNAs were enough to clearly distinguish STX16 from CRYAB.
The trend observed for the three example genes was generally valid for all genes targeted by the library. P values calculated based on the top-scoring 10 shRNAs per gene were highly correlated with P values calculated based on 45 shRNAs per gene, and P values were consistently higher than those calculated from random subsets of 10 shRNAs per gene (Fig. 3D). This pattern was also observed for different shRNA subset sizes (Fig. S3B).
Design of Genome-Wide Human and Mouse shRNA Libraries.
We generated a set of lentiviral vectors for expression of shRNA libraries (Fig. 4A and Dataset S3) that incorporated features described above (minimal miR-30a context with the wild-type CNNC motif, and HindIII site in loop). We also included sites for the eight-cutter restriction enzyme SbfI flanking the shRNA expression cassette, to enhance our previous sample preparation strategy of size-based enrichment of genomic DNA before PCR amplification (26). To provide flexibility for use in different experimental contexts, the vector set includes alternative options for the choice of the promoter (EF1a vs. SFFV) and the fluorescent marker (mCherry vs. tagBFP).
Fig. 4.

Next-generation library design. (A) We generated a set of lentiviral expression vectors for use with the next-generation library to provide compatibility with different target cell lines and applications. Promoters: EF1a or SFFV. Fluorescent marker: mCherry or tagBFP. All vectors express the shRNA from a minimal miR-30a context that preserves the WT CNNC motif, and is embedded between SbfI sites for size fractionation and SPRI bead purification. A HindIII restriction site was introduced in the region encoding the hairpin loop. (B) Human and mouse protein-coding genes were grouped into 12 biological categories, each of which is targeted by an shRNA sublibrary that together constitute genome-wide libraries. (C) Each gene is targeted by 25 shRNAs on average. Each sublibrary also contains >1,000 negative-control shRNAs that follow the same design rules as targeted shRNAs, but have no target in the human/mouse transcriptome.
We then grouped all annotated human protein-coding genes into 12 nonoverlapping functional groups (Fig. 4B and Dataset S4), to be targeted by sublibraries of the genome-wide library, and made corresponding groups for the mouse genome (Dataset S5). We designed 25 shRNAs (on average) targeting each gene, and included 1,000 or more negative-control shRNAs in each sublibrary (Fig. 4C and Datasets S6 and S7). Negative control shRNAs were designed based on the same rules as targeted shRNAs, except that scrambled quasi-transcripts were generated, and shRNAs targeting those transcripts were selected and verified not to target actual human or mouse transcripts (see Materials and Methods for details). We previously showed that hit genes can be robustly identified by comparing the phenotype distribution of the 25 shRNAs targeting a given gene to the phenotype distribution of the negative-control shRNAs and calculating statistical significance using the Mann–Whitney u test (5).
Pilot Screen for Genes Controlling Sensitivity to a Cholera-Diphtheria Toxin.
To benchmark the performance of our next-generation library, we conducted a pilot screen for genes controlling the sensitivity of K562 cells to a cholera-diphtheria fusion toxin (CTx-DTA) (27) (Fig. 5A and Dataset S8). For this screen, we used the sublibrary targeting 2,933 human genes associated with proteostasis. We had previously carried out a genome-wide screen for CTx-DTA sensitivity in K562 cells using our recently developed CRISPRi library, which targets each human transcript with ∼10 small guide RNAs (sgRNAs) (1). Knockdown of a sizeable number of genes control sensitivity to CTx-DTA by either conferring partial resistance (i.e., cells in which these genes are knocked down enrich in the treated population) or sensitization (i.e., cells in which these genes are knocked down drop out).
Fig. 5.
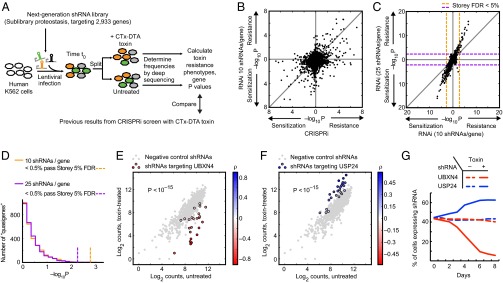
Robust detection of hit genes in a pilot screen. (A) We used the sublibrary Proteostasis of our next-generation shRNA library to screen human K562 cells for genes controlling growth and sensitivity to a cholera-diphtheria fusion toxin CTx-DTA (27). P values were calculated for each of the 2,933 genes targeted by the library. (B) P values for CTx-DTA resistance phenotypes obtained with our published CRISPRi library (1) or our next-generation RNAi library (computationally downsampled to 10 shRNAs per gene to match coverage of the CRISPRi library) agree broadly. (C) The coverage of 25 shRNAs per gene of our next-generation shRNA library detects hit genes with strongly increased statistical significance compared with a 10 shRNA per gene library. Dotted lines indicate the 5% FDR cut-off calculated using the Storey and Tibshirani approach (21). (D) Quasi-genes were generated by grouping random sets of negative-control shRNAs and calculating P values for them. The distributions of P values are not significantly different for 25 shRNAs per gene or 10 shRNAs per gene (Mann–Whitney u test). Less than 0.5% of quasi-genes pass the P value threshold corresponding to the 5% FDR calculated using the Storey and Tibshirani approach. (E–G) UBXN4, encoding a protein involved in endoplasmic reticulum-associated degradation, and USP24, encoding a ubiquitin peptidase, were two hit genes from the CTx-DTA screen. (E and F) Phenotypes of the shRNAs targeting UBXN4 and USP24 compared with the negative-control phenotypes reveals a consistent shift toward sensitization (UBXN4) or resistance (USP24). (G) Competitive growth experiment validates UBXN4 and USP24 phenotypes. K562 cells where infected with lentivirus at a multiplicity of infection of ∼0.5 expressing an shRNA targeting UBXN4 or USP24, as well as an mCherry marker that allowed monitoring of the percentage of cells expressing shRNAs. Cells were either treated with CTx-DTA toxin or grown untreated. The percentage of each cell expressing shRNAs was monitored over 8 d. UBXN4 knockdown sensitized to the toxin, whereas USP24 knockdown conferred resistance.
To directly compare hit genes identified by these two orthogonal technologies, we calculated P values for each gene. Because P values calculated by the Mann–Whitney u test are dependent on the sample sizes of the compared groups, we computationally down-sampled data from the shRNA screen to 10 shRNAs per gene (randomly chosen, average of three random draws) for the P value calculation. Comparison of the P values and the direction of the effect (CTx-DTA sensitization vs. desensitization) revealed a substantial overlap in the top hit genes called by CRISPRi and RNAi results (Fig. 5B and Fig. S4A). Calling hit genes at a 5% FDR [using the approach by Storey and Tibshirani (21)], the overlap in hit genes was highly significant (P < 10−19, Fisher’s exact test) and signed log10 P values of genes called as hits by both approaches showed good correlation (R = 0.91). Furthermore, the order-of-magnitude of P values were very similar between the two methods, indicating that their hit detection performance was equivalent on a per sgRNA/per shRNA basis. However, a limited number of genes were identified as strong hits by shRNA but not by CRISPRi, or vice versa. As detailed in the Discussion section, this finding is likely to reflect method-specific technical reasons that can lead to false-negative results.
Fig. S4.

(A) Overlap in hit genes called at different FDR cut-offs by our CRISPRi and next-generation RNAi screens for CTx-DTA resistance phenotypes. (B) Comparison of phenotype distributions of 25 shRNAs vs. 10 sgRNAs targeting hit genes (5% FDR cut-off), normalized by dividing by the average of the three strongest shRNA/sgRNA phenotypes for a given gene. (C) Comparison of phenotypes of negative-control shRNAs/sgRNAs. Standard deviations of the negative-control phenotypes were 0.11 for RNAi and 0.05 for CRISPRi.
To evaluate the enhanced sensitivity for hit detection achieved by the high complexity of the next-generation library, we compared the gene P values calculated on the basis of the full set of ∼25 shRNAs per gene to the P values calculated from the down-sampled dataset (Fig. 5C). P values obtained with the full-size library were consistently more statistically significant. We commonly classify genes as hits based on the FDR approach. Using the subsampled library (10 shRNAs per gene), 69 genes passed the threshold of 5% FDR. Using the full library (∼25 shRNAs per gene), 252 genes passed the 5% FDR threshold. Importantly, the increased significance of P values was for hit genes obtained with 25 shRNAs per gene was specific and did not lead to a spurious change in P values for all genes: when we created “quasi-genes” by either combining randomly selected sets of either 10 or 25 negative-control shRNA phenotypes and calculated the corresponding P values, the distribution of quasi-gene P values was essentially indistinguishable for 10 or 25 shRNAs per gene (Fig. 5D). With both 10 and 25 shRNAs per gene, the number of quasi-genes passing the theoretical 5% FDR was less than 0.5% (Fig. 5D), suggesting that the actual FDR in our hit gene set is much lower than 5%. Thus, our approach is not only highly sensitive, but also highly specific.
Inspection of the shRNA phenotypes for individual hit genes suggests that the vast majority of the 25 shRNAs targeting each gene had on-target activity, whereas strong off-target effects were not pervasive (Fig. 5 E and F). Individual shRNAs selected based on their performance in the primary screen robustly reproduced their protective or sensitizing effect in validation experiments (Fig. 5G). Across all hit genes, the distribution of relative phenotype strengths for individual shRNAs were very similar to that obtained with sgRNAs of our CRISPRi library (Fig. S4B); there was no statistically significant difference (P > 0.38, Mann–Whitney test). However, the phenotype distribution for negative-control shRNAs was broader than that for negative-control sgRNAs (Fig. S4C), supporting our previous finding that CRISPRi has inherently less off-target effects than RNAi. The complexity of our shRNA library and our data analysis framework allowed us to identify hit genes robustly even in the presence of off-target effects.
Discussion
Based on a series of empirical tests of shRNA library design features, we constructed next-generation shRNA libraries for pooled loss-of-function screens in human and mouse cells. The enhanced phenotypes achieved with the new design enables primary screens with great sensitivity (Fig. 5 B and C). A potential caveat of the enhanced on-target knockdown is that off-target effects may also be increased. Despite this caveat, we find that our next-generation libraries achieve high specificity in the detection of hit genes (Fig. 5D), suggesting that the high complexity of the libraries, in combination with our analysis framework, results in robust results even in the presence of off-targets. The large set of negative-control shRNAs in our libraries enables us to estimate the prevalence of off-target effects, and thus to compare shRNA expression formats (Fig. 1B) and shRNA design algorithms (28) with respect to their specificity. Future efforts should be directed at developing a sequence score that simultaneously increases on-target activity and reduces off-target effects. An alternative strategy is the development of pooled libraries in which a matched “C911” control (29) is included for each shRNA to unmask off-target effects driven by seed sequence matches.
The large size of our ultracomplex shRNA libraries and the resulting scale of the pooled screen can be prohibitive for some cell types. Division of the genome-wide library into 12 sublibraries will enable focused screens in such systems. In addition, the rising availability and affordability of complex oligo pool synthesis makes custom sublibraries based on our next-generation libraries an attractive option for specific research projects. Furthermore, our results suggest that libraries targeting each gene with 10 shRNAs would perform comparably to our current CRISPRi library (Fig. 5B). Ideally, such condensed libraries should be constructed by selecting 10 shRNAs per gene based on further rounds of machine learning to optimize over the performance of our current sequence score or the Sherwood algorithm in predicting top-performing shRNAs within our current library (Fig. S5).
Fig. S5.

(A) Performance of shRNAs in the next-generation shRNA library that were ranked based on their sequence score. (B) Performance of a subset of shRNAs in the next-generation shRNA library that were grouped on whether they are among the top 10 shRNAs predicted by the Sherwood algorithm or not. shRNA activity for shRNA hit genes was normalized for each gene.
Recently developed CRISPR-based approaches have great potential to become the technology of choice for loss-of-function genetic screens in mammalian cells (1–4). CRISPR cutting screens can generate complete knockout alleles, and our recently developed CRISPRi-based screening approach can generate an allelic series to investigate gene dosage effects for both essential and nonessential genes (1). An important advantage of CRISPRi over RNAi is the dramatically reduced incidence of off-target effects (1). Furthermore, CRISPRi can effectively target nuclear noncoding RNAs (1).
Because CRISPRi relies on targeting sgRNAs to a narrow window around the transcription start site (1), a potential source of false-negative results of CRISPRi screens is the misannotation of transcription start sites, or the use of more than one transcription start site for a given gene. These reasons may account for the small number of genes that we found as hits by RNAi, but not CRISPRi (Fig. 5B). Future versions of our CRISPRi library will aim to overcome this issue by incorporating more experimental data to define likely transcription start sites. Results from our next-generation RNAi library will provide test cases of genes that were false negatives in our first-generation CRISPRi library, and help to benchmark the improved performance of our next CRISPRi library.
Discrepancies between hit genes called by CRISPRi and RNAi are not necessarily an indication of technical shortcomings of either method, but can also guide new biological discoveries. This ability is because of an important difference in the mode of action of CRISPRi and RNAi; whereas CRISPRi targets entire transcription units, including alternative splice isoforms and embedded noncoding RNAs, RNAi targets mature transcripts and thus differentiates between different products processed from the same primary transcript. Parallel CRISPRi and RNAi screens can reveal novel transcript functions in cases where CRISPRi and RNAi phenotypes for reagents designed to target the same gene product differ. A possible example is the gene NFYC, encoding the gamma subunit of nuclear transcription factor Y, which was not a hit gene in the RNAi screen; however, it was a strong hit by CRISPRi. Inspection of the NFYC transcript reveals that it harbors the miRNAs miR-30e and miR-30c-1, raising the possibility that the CRISPRi repression of this transcript may result in a phenotype because of knockdown of the miRNAs, rather than the protein-coding mRNA. Further experimental work is needed to test this hypothesis. In summary, RNAi and CRISPRi can yield complementary insights and, in combination, uncover unexpected and novel biology.
Materials and Methods
Pooled Screening.
Pooled screens for ricin resistance and CTx-DTA resistance in K562 cells were carried out as previously described (1, 8). Sample preparation for deep sequencing was carried out as previously described (26), except that different primers were used for amplification and sequencing. Sequences of primers used with the next-generation shRNA libraries are provided in Table S3.
Table S3.
Sequences of oligonucleotides used with next-generation shRNA libraries
| Name | Sequence (5′ to 3′) | Purpose |
| oMK252 | CAAGCAGAAGACGGCATACGATGGACGAGCTGTACAAGTAA | Forward amplification primer, mCherry vectors |
| oMK482 | CAAGCAGAAGACGGCATACGACTGGGGCACAAaCTTAATTAAG | Forward amplification primer, tagBFP vectors |
| oMK483 | aatgatacggcgaccaccgaGATCGGAAGAGCACACGTCTGAACTCCAGTCACCTTGTActtactagtGAAttcTGACTGACC | Reverse amplification primer, Illumina Index 12 |
| oMK484 | aatgatacggcgaccaccgaGATCGGAAGAGCACACGTCTGAACTCCAGTCACGCCAATcttactagtGAAttcTGACTGACC | Reverse amplification primer, Illumina Index 6 |
| oMK485 | aatgatacggcgaccaccgaGATCGGAAGAGCACACGTCTGAACTCCAGTCACAGTTCCcttactagtGAAttcTGACTGACC | Reverse amplification primer, Illumina Index 14 |
| oMK486 | aatgatacggcgaccaccgaGATCGGAAGAGCACACGTCTGAACTCCAGTCACTAGCTTcttactagtGAAttcTGACTGACC | Reverse amplification primer, Illumina Index 10 |
| oMK487 | aatgatacggcgaccaccgaGATCGGAAGAGCACACGTCTGAACTCCAGTCACTTAGGCcttactagtGAAttcTGACTGACC | Reverse amplification primer, Illumina Index 3 |
| oMK488 | aatgatacggcgaccaccgaGATCGGAAGAGCACACGTCTGAACTCCAGTCACATCACGcttactagtGAAttcTGACTGACC | Reverse amplification primer, Illumina Index 1 |
| oMK489 | aatgatacggcgaccaccgaGATCGGAAGAGCACACGTCTGAACTCCAGTCACGAGTGGcttactagtGAAttcTGACTGACC | Reverse amplification primer, Illumina Index 23 |
| oMK490 | aatgatacggcgaccaccgaGATCGGAAGAGCACACGTCTGAACTCCAGTCACAGTCAActtactagtGAAttcTGACTGACC | Reverse amplification primer, Illumina Index 13 |
| oMK491 | CCCCTTGAAGTCCGAGGCAGTAGGCA | Deep sequencing primer |
shRNA Libraries.
Sequences of all elements of the pooled library used to evaluate shRNA expression formats are provided in Dataset S1. Sequences of all shRNA targets of the next-generation human and mouse RNAi libraries are provided as Datasets S6 and S7, respectively. shRNA names contain the Entrez Gene ID of the targeted gene (0 for negative-control shRNAs), followed by the sublibrary code as defined in Table S4, and a sequential number. Sequences of the next-generation backbone vectors are provided as Dataset S3. Inserts into this backbone between the XhoI and EcoRI sites have the format as in the following example: For the target site 5′- GGGAGATGTACTGTATTATATA-3′, the insert sequence is 5′-CTCGAGAAGAAGGTATATTGCTGTTGACAGTGAGCGAGGAGATGTACTGTATTATATATAGTGAAGCTTCAGATGTATATATAATACAGTACATCTCCCTGCCTACTGCCTCGGACTTCAAGGGGTCAGTCAGAATTC-3′. Details of the cloning strategy used to generate these vectors and the libraries are provided upon request.
Table S4.
Sublibraries of the next-generation shRNA libraries
| Species | Sublibrary name | Gene group | No. of shRNAs |
| Human | H01 | Apoptosis | 7,568 |
| Human | H02 | Cancer | 55,811 |
| Human | H03 | Drug_Targets | 35,245 |
| Human | H04 | Gene_Expression | 59,602 |
| Human | H05 | Kinase_Phosphatase_1 | 25,223 |
| Human | H06 | Kinase_Phosphatase_2 | 25,240 |
| Human | H07 | Membrane_Proteins | 61,945 |
| Human | H08 | Mitochondria | 22,948 |
| Human | H09 | Motility | 12,263 |
| Human | H10 | Other_Cancer | 15,323 |
| Human | H11 | Stress_Proteostasis | 74,295 |
| Human | H12 | Trafficking | 23,711 |
| Human | H13 | Unassigned | 95,681 |
| Mouse | M01 | Apoptosis | 6,980 |
| Mouse | M02 | Cancer | 54,316 |
| Mouse | M03 | Drug_Targets | 34,576 |
| Mouse | M04 | Gene_Expression | 49,141 |
| Mouse | M05 | Kinase_Phosphatase | 24,438 |
| Mouse | M06 | Membrane_Proteins | 54,353 |
| Mouse | M07 | Mitochondria | 21,837 |
| Mouse | M08 | Motility | 11,262 |
| Mouse | M09 | Other_Cancer | 14,390 |
| Mouse | M10 | Stress_Proteostasis | 72,058 |
| Mouse | M11 | Trafficking | 22,797 |
| Mouse | M12 | Unassigned | 157,552 |
Data Analysis and Machine Learning.
Pooled screens were analyzed using our previously established framework (5) and software gimap.ucsf.edu (26). Results for all genes for the pilot CTx-DTA screen are provided as Dataset S8. Further details on the analysis of the pooled screen of different expression formats, machine learning of the sequence score and design of the next-generation libraries are provided in SI Materials and Methods.
Datasets.
Datasets S1–S8 are available for download at kampmannlab.ucsf.edu/next-generation-shrna-libraries-datasets.
SI Materials and Methods
Evaluation of shRNA Expression Contexts.
Ricin resistance phenotypes (ρ) were determined for each shRNA in a pooled screen in K562 cells, as previously described (5, 8). For each gene in each expression format, phenotypes were compared between to the phenotypes obtained in the standard miR-30a format, and the slope of the linear fit passing through (0,0) was calculated. The on-target effect metric was calculated for each expression format as the sum of these slopes for all six genes. The off-target effect metric for each expression format was calculated as the SD of phenotypes of negative-control shRNAs. For each expression format, gene P values were calculated using the Mann–Whitney u test to compare phenotypes of the set of shRNAs targeting each ricin hit gene to the negative-control shRNA phenotypes. The hit-detection performance metric was calculated as the sum of –log10 P values for all six genes.
Machine Learning of Active shRNA Features.
Based on our published ricin-resistance screen (8), we defined a set of bona fide hit genes for ricin susceptibility, which encompassed protective genes up to an FDR of 5%, sensitizing genes up to an FDR of 2%, and genes with at least two shRNAs passing the minimal correlation cut-off of z = 0.8 in the GI map. shRNAs targeting these hit genes were defined as “active” if their phenotype determined in the batch retest was >0.05 (for protective shRNAs) or < –0.05 (for sensitizing shRNAs), and as “inactive” otherwise. We have shown for selected hit genes that phenotypic activity and on-target knockdown are generally highly correlated (8). The sequence score predictive of shRNA activity (Table S2) was derived based on this learning set using stepwise forward logistic regression; variables are defined in Table S1. For the analysis in Fig. 3C, a sequence score was derived from a subset of the training sets that did not include the genes to which the sequence score was then applied (Table S2).
Next-Generation Library Design.
For each human and mouse protein-coding gene, 50 shRNAs were predicted using the Hannon laboratory shRNA retriever program (20) and scored by the sequence score. Twenty-five shRNAs per gene were selected based on the following criteria: high sequence score, targeting of all (or as many as possible) annotated transcripts derived from the gene, absence of restriction sites used during library cloning or postscreen sample preparation, empirical knowledge of shRNA activity based on our own previous screens, shRNAs predicted by the sensor assay/Sherwood algorithm (16, 18), and absence of matches to transcripts derived from other genes. To generate negative-control shRNAs, human transcript sequences were randomly permutated and shRNAs targeting these quasi-transcripts were derived using the same strategy as for targeted shRNAs. Gene groups for sublibraries were formed based on Gene Ontology annotation and manual annotation. Each gene was assigned to a sublibrary in the order of sublibraries displayed in Fig. 4. Once assigned to a sublibrary, the gene was not assigned to following sublibraries, even if it belonged to more than one gene group. For the human kinase-phosphatase gene group, a second sublibrary with 25 separate shRNAs per gene was generated to enable screening of this gene group with up to 50 shRNAs per gene.
Datasets.
Datasets S1–S8 are available online: kampmannlab.ucsf.edu/next-generation-shrna-libraries-datasets.
Supplementary Material
Acknowledgments
We thank H. Ploegh and C. Guimaraes for CTx-DTA; M. McManus and I. Vaughn for mammalian promoter constructs; D. Wong for technical assistance; and Agilent for massively parallel oligonucleotide synthesis. This work was supported by the Howard Hughes Medical Institute (J.S.W.); National Cancer Institute/NIH Grant U01 CA168370 (to J.S.W.); National Cancer Institute/NIH Grant Pathway to Independence Award K99 CA181494 (to M.K.); Grant IBS-R008-D1 of Institute for Basic Science from the Ministry of Science, Information and Communication Technology, and Future Planning of Korea (to S.C.K., H.C., and V.N.K.); University of California, San Francisco Medical Scientist Training Program Grant T32 GM007618 (to M.A.H.); and a postdoctoral fellowship from the Leukemia and Lymphoma Society (to L.A.G.).
Footnotes
The authors declare no conflict of interest.
Data deposition: Datasets are available online at kampmannlab.ucsf.edu/next-generation-shrna-libraries-datasets.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1508821112/-/DCSupplemental.
References
- 1.Gilbert LA, et al. Genome-Scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159(3):647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera MdelC, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32(3):267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- 3.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343(6166):80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kampmann M, Bassik MC, Weissman JS. Integrated platform for genome-wide screening and construction of high-density genetic interaction maps in mammalian cells. Proc Natl Acad Sci USA. 2013;110(25):E2317–E2326. doi: 10.1073/pnas.1307002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silva JM, et al. Profiling essential genes in human mammary cells by multiplex RNAi screening. Science. 2008;319(5863):617–620. doi: 10.1126/science.1149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bassik MC, et al. Rapid creation and quantitative monitoring of high coverage shRNA libraries. Nat Methods. 2009;6(6):443–445. doi: 10.1038/nmeth.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bassik MC, et al. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell. 2013;152(4):909–922. doi: 10.1016/j.cell.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sidrauski C, et al. Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. Elife. 2015;4:e07314.10. doi: 10.7554/eLife.07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grimm D, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441(7092):537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 11.Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci USA. 2005;102(37):13212–13217. doi: 10.1073/pnas.0506306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han J, et al. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125(5):887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X, Zeng Y. The terminal loop region controls microRNA processing by Drosha and Dicer. Nucleic Acids Res. 2010;38(21):7689–7697. doi: 10.1093/nar/gkq645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeng Y, Yi R, Cullen BR. Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. EMBO J. 2005;24(1):138–148. doi: 10.1038/sj.emboj.7600491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Auyeung VC, Ulitsky I, McGeary SE, Bartel DP. Beyond secondary structure: Primary-sequence determinants license pri-miRNA hairpins for processing. Cell. 2013;152(4):844–858. doi: 10.1016/j.cell.2013.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knott SR, et al. A computational algorithm to predict shRNA potency. Mol Cell. 2014;56(6):796–807. doi: 10.1016/j.molcel.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herbst F, et al. Extensive methylation of promoter sequences silences lentiviral transgene expression during stem cell differentiation in vivo. Mol Ther. 2012;20(5):1014–1021. doi: 10.1038/mt.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fellmann C, et al. Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Mol Cell. 2011;41(6):733–746. doi: 10.1016/j.molcel.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matveeva OV, et al. Optimization of duplex stability and terminal asymmetry for shRNA design. PLoS ONE. 2010;5(4):e10180. doi: 10.1371/journal.pone.0010180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paddison PJ, et al. Cloning of short hairpin RNAs for gene knockdown in mammalian cells. Nat Methods. 2004;1(2):163–167. doi: 10.1038/nmeth1104-163. [DOI] [PubMed] [Google Scholar]
- 21.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ameres SL, Martinez J, Schroeder R. Molecular basis for target RNA recognition and cleavage by human RISC. Cell. 2007;130(1):101–112. doi: 10.1016/j.cell.2007.04.037. [DOI] [PubMed] [Google Scholar]
- 23.Tan X, et al. Tiling genomes of pathogenic viruses identifies potent antiviral shRNAs and reveals a role for secondary structure in shRNA efficacy. Proc Natl Acad Sci USA. 2012;109(3):869–874. doi: 10.1073/pnas.1119873109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markham NR, Zuker M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 2005;33(Web Server Issue):W577–W581. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014;505(7485):701–705. doi: 10.1038/nature12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kampmann M, Bassik MC, Weissman JS. Functional genomics platform for pooled screening and generation of mammalian genetic interaction maps. Nat Protoc. 2014;9(8):1825–1847. doi: 10.1038/nprot.2014.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guimaraes CP, et al. Identification of host cell factors required for intoxication through use of modified cholera toxin. J Cell Biol. 2011;195(5):751–764. doi: 10.1083/jcb.201108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu S, et al. Weak base pairing in both seed and 3′ regions reduces RNAi off-targets and enhances si/shRNA designs. Nucleic Acids Res. 2014;42(19):12169–12176. doi: 10.1093/nar/gku854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buehler E, Chen YC, Martin S. C911: A bench-level control for sequence specific siRNA off-target effects. PLoS ONE. 2012;7(12):e51942. doi: 10.1371/journal.pone.0051942. [DOI] [PMC free article] [PubMed] [Google Scholar]