

Significance
Spondylolysis is a crack in part of a vertebra that occurs in 3–6% of the general population. The cracked vertebra sometimes slips forward over the vertebra below it, resulting in spondylolisthesis and lower-back pain. Although inherited spondylolysis has long been described, the genetic etiology of these disorders remains unclear. Studies of families with autosomal-dominant mutations provide a unique means to investigate the pathogenesis of spondylolysis, which can also be used as biomarkers, even during the asymptomatic period. This research identified two novel missense mutations in independent families that were located at the conserved Stas domain. Functional analyses demonstrated that sulfate uptake activities of mutant SLC26A2 were significantly reduced. This study suggests that the pathogenesis of chondrodysplasia is associated with dysplastic spondylolysis.
Keywords: dysplastic spondylolysis, spondylolisthesis, lumbosacral spine, solute carrier family 26 sulfate transporter, whole-exome sequencing
Abstract
Spondylolysis is a fracture in part of the vertebra with a reported prevalence of about 3–6% in the general population. Genetic etiology of this disorder remains unknown. The present study was aimed at identifying genomic mutations in patients with dysplastic spondylolysis as well as the potential pathogenesis of the abnormalities. Whole-exome sequencing and functional analysis were performed for patients with spondylolysis. We identified a novel heterozygous mutation (c.2286A > T; p.D673V) in the sulfate transporter gene SLC26A2 in five affected subjects of a Chinese family. Two additional mutations (e.g., c.1922A > G; p.H641R and g.18654T > C in the intron 1) in the gene were identified by screening a cohort of 30 unrelated patients with the disease. In situ hybridization analysis showed that SLC26A2 is abundantly expressed in the lumbosacral spine of the mouse embryo at day 14.5. Sulfate uptake activities in CHO cells transfected with mutant SLC26A2 were dramatically reduced compared with the wild type, confirming the pathogenicity of the two missense mutations. Further analysis of the gene–disease network revealed a convergent pathogenic network for the development of lumbosacral spine. To our knowledge, our findings provide the first identification of autosomal dominant SLC26A2 mutations in patients with dysplastic spondylolysis, suggesting a new clinical entity in the pathogenesis of chondrodysplasia involving lumbosacral spine. The analysis of the gene–disease network may shed new light on the study of patients with dysplastic spondylolysis and spondylolisthesis as well as high-risk individuals who are asymptomatic.
Spondylolysis is defined as a defect in the pars interarticularis of the vertebral arch (1, 2). The occurrence of spondylolysis in Asian and Caucasian populations has been reported to be about 3–6% (2, 3). Dysplastic spondylolysis (i.e., Wiltse–Newman type I) represents a type of congenital defect of vertebral pars interarticularis in the upper sacrum and lower lumbar, commonly affecting L5 (1). This type of spondylolysis is not rare, comprising 14–21% of cases in a large sample population (4). Among 605 pediatric patients who underwent surgical treatment for spondylolisthesis in multisurgeon centers from 2004 to 2007, 10% of them were affected by dysplastic spondylolysis (5). An alternative classification system was proposed with two broad categories (6): developmental spondylolysis and acquired spondylolysis. The developmental category includes disorders resulting from an inherited dysplasia of the pars, lumbar facets, disks, or vertebral endplates. This classification combines the dysplastic and isthmic categories of Wiltse–Newman. Because of the dysplasia inherent in this condition, there is often insufficient strength to withstand the forward thrust and the last free lumbar vertebra gradually slips forward on the one below, thereby resulting in spondylolisthesis [Online Mendelian Inheritance in Man (OMIM) ID 184200].
The genetic background for lumbar spondylolysis has been previously studied and extensively reviewed (7–9). Inherited spondylolysis and spondylolisthesis have been also identified in many families of different races, either in autosomal dominant (1, 3, 10–15) or autosomal recessive form (12, 15). Moreover, the reported incidence of spondylolysis and spondylolisthesis in family members of index patients was 22% (16), compared with 8% in the general population (17), suggesting it is genetically heterogeneous. Interestingly, spondylolysis was found to be very frequent in Alaskan Eskimos (18).
Spondylolysis may be present as a single entity or it may present as just one of many signs or symptoms. In the latter case, patients with spondylolysis may also show spina bifida occulta, scoliosis, spondylolisthesis, disk herniation, Scheuermann’s disease (8, 19), or a combination of several other nonskeletal related disorders. In particular, dysplastic spondylolysis with spondylolisthesis was also found, as an accompanying sign, in several patients with autosomal dominant brachydactyly (OMIM ID 113100). The genetic cause of the syndrome was identified to be the frameshift mutation insG206 in the growth differentiation factor 5 (GDF5) (also known as cartilage-derived morphogenetic protein-1, CDMP-1) (20). This gene is a member of the bone morphogenetic protein (BMP) family in the TGF-beta superfamily, which plays an important role in skeletal development.
Several other genes involving chondrocyte growth and proliferation were also linked to developmental disorders in lumbar spine. Functional variants in the Col11a1 gene, which encodes the α1 chain of type XI collagen, are associated with lumbar disk herniation (21). Mutations in this gene also cause type II Stickler syndrome (OMIM ID 604841), which includes spinal abnormalities such as spondylolisthesis, scoliosis, and hyperkyphosis (22). Moreover, three collagen IX encoded genes were also found to be common genetic risk factors for lumbar disk disease (23). In addition, a case report described lumbar disk herniation in patients with cystic fibrosis (24), which resulted from mutations in the CFTR gene.
Despite these findings, the etiology and pathogenesis of lumbar spondylolysis remain unclear. To identify the potential genetic defects of this disorder, we applied whole-exome sequencing (WES) and bioinformatics for recently recruited patients with spondylolysis and associated spondylolisthesis. In the present study, we report the identification of heterozygous mutations in the solute carrier family 26 sulfate transporter (SLC26A2) gene in patients with the disease and show the expression pattern of SLC26A2 in developing lumbosacral spine. We also present functional alterations, in terms of sulfate uptake, of these mutants in CHO cells. Furthermore, we discuss potential relations between chondrodysplasia-related genes and dysplastic spondylolysis-associated disorders.
Results
Phenotype Details.
As shown in the three-generation pedigree chart (Fig. 1A), four progenies (II-2, II-3, III-1, and III-2) of the two ancestors of the XAHT01 family were referred for evaluation of back and lower left extremity pain. None of the symptoms was attributed to sport activities. The pattern of inheritance is consistent with the autosomal dominant form. The age of onset for showing symptoms of spondylolysis and spondylolisthesis, such as lower back pain, is variable, ranging from 19 y for the proband (III-2) to 41 y for the subject II-3. Clinical manifestations and radiological examinations from each of the family members are summarized in Table 1. X-ray and computed tomography (CT) or MRI examinations showed bilateral spondylolysis of L5 in each of the affected progenies (Fig. 1 B and C). Also, displacement of the fifth lumbar vertebra over the first sacral vertebra with grade-II and grade-I spondylolisthesis was developed in subjects III-2 and II-3, respectively (Fig. 1 B and C), which caused moderate to severe pain and was treated by surgery for relieving pain. CT scans also showed spina bifida occulta of L5-S1 in subjects II-2 and III-1. The ancestor, subject I-1, was diagnosed with mild spinal stenosis in L4-L5 and L5-S1 and disk herniation of L3-S1 by X-ray and CT scans.
Fig. 1.

Pedigree and radiographic analysis. (A) Pedigree chart of a family with autosomal dominant spondylolysis. Filled symbols represent affected subjects. Squares signify male and circles, female. Arrow marks the proband. (B) Spinal CT scan of the proband III-2 shows bilateral spondylolysis (arrows) at the fifth lumbar vertebra (L5). (C) Lateral view of MRI shows grade-II spondylolisthesis of L5 (arrow) with spinal misalignment as well as hyperosteogeny. (D–F) An unrelated patient was diagnosed by CT (E) and 3D CT reconstruction (F) analysis with bilateral spondylolysis and grade-I spondylolisthesis of L4.
Table 1.
Clinical conditions in each of the XAHT-1 family members
| Subject | Sex | Age, y | Onset, y | Clinical manifestations | X-ray, CT, and MRI |
| I-1 | M | 73 | UN | Occasional lower back pain | X-ray and CT: disk herniation of L3-S1; mild spinal stenosis in L4-L5 and L5-S1 |
| I-2 | F | 68 | N/A | No | Normal |
| II-1 | F | 49 | N/A | No | Normal |
| II-2 | M | 49 | 41 | Mild lower back pain | X-ray and CT: bilateral spondylolysis of L5; spina bifida occulta S1; hypertrophy and pseudoarthrosis of right diapophysis |
| II-3 | F | 45 | 40 | Severe pain in back and left lower extremity; treated by surgery* | X-ray and CT: bilateral spondylolysis of L5; spondylolisthesis of L5-S1, grade I, mild disk herniation of L4-L5 |
| II-4 | M | 44 | N/A | No | Normal |
| III-1 | M | 26 | 25 | Mild lower back pain | X-ray and CT: bilateral spondylolysis L5 spina bifida occulta L5-S1 |
| III-2 | M | 24 | 19 | Severe back pain, varying degrees of pain radiation to the lower limb; treated by surgery | CT and MRI: bilateral spondylolysis of L5; spondylolisthesis of L5-S1, grade II |
Subject number is designated in Fig. 1A. L, lumbar; N/A, not applicable; S, sacral; UN, unknown.
Lower back pain was dramatically alleviated by surgery as evaluated using the Simplified Chinese Oswestry disability index.
Identification of Mutations in SLC26A2 by WES.
To identify potential causative mutations, we performed WES in seven members of the family except subject II-1 (Table 1). On average, we generated 64.67 Mb of aligned reads, of which 97% of reads reached ≥10× coverage per sample (Table S1). The pathogenic cause of these patients was assumed to be the same heterozygous mutation of a single gene. A total of 37,681 nonsynonymous single nucleotide variants (SNVs) (missense, nonsense, and splice site mutations) and 2,354 indels (short coding insertions or deletions) were identified from the three patients (Table S2). After the known variants (present in the normal family members, subjects I-2 and II-4, or in the databases mentioned in Materials and Methods) and nondeleterious SNVs were removed, the number of candidate genes was reduced to six. After further bioinformatics analyses, we excluded five of them (TTC24, DNAH10, ALDH7A1, SLC25A37, and PLEC) because they were either not expressed in the lumbosacral spine or they are functionally unrelated (Table S3). SLC26A2, a sulfate transporter, was prioritized as a strong candidate gene because it plays an important role in endochondral bone formation.
Table S1.
Summary of exome sequencing data for the family (XAHT01) with hereditary spondylolysis
| Sample | Total reads, Mb | Aligned reads, Mb | Percent, % | Initial bases on exon, Mb | Bases covered on exon, Mb | Sequencing depth | Effective bases on target, % | Coverage, % | 4× coverage, % | 10× coverage, % |
| I-1 | 62.20 | 61.89 | 99.69 | 50.62 | 50.55 | 70.86 | 63.40 | 99.90 | 99.40 | 97.80 |
| I-2 | 55.28 | 55.01 | 99.82 | 50.62 | 50.49 | 63.25 | 62.70 | 99.80 | 99.20 | 97.40 |
| II-1 | 63.09 | 62.77 | 99.8 | 50.62 | 50.55 | 68.4 | 60.30 | 99.90 | 99.40 | 97.90 |
| II-2 | 70.55 | 70.20 | 99.87 | 50.62 | 50.5 | 81.25 | 63.50 | 99.80 | 99.40 | 98.30 |
| II-3 | 63.64 | 63.31 | 99.81 | 50.62 | 50.55 | 72.6 | 63.00 | 99.90 | 99.40 | 97.90 |
| III-1 | 64.58 | 64.22 | 99.77 | 50.62 | 50.56 | 72.17 | 62.20 | 99.90 | 99.50 | 98.10 |
| III-2 | 75.79 | 75.30 | 99.62 | 50.62 | 50.56 | 88.7 | 64.60 | 99.90 | 99.60 | 98.50 |
| Mean | 65.02 | 64.67 | 99.76 | 50.62 | 50.53 | 73.89 | 62.00 | 99.00 | 99.00 | 97.00 |
| SD | 65.21 | 64.58 | 0.08 | 0 | 0.02 | 8.47 | 0.01 | 0.00 | 0.001 | 0.003 |
Overall, a mean of 65.02 million reads as 100-bp paired ends were generated from seven samples from the XAHT01 family. An average of 99.76% of the raw sequence reads were aligned to the reference genome (hg19) using SOAPaligner program with fewer than three mismatches (soap.genomics.org.cn/). Of these mapped reads, about 62% effective bases were classified to the target regions with an average of 73.89 folds sequencing depth. Especially, 99%, 99%, and 97% of targeted bases had more than 1-, 4-, and 10-fold coverage, respectively. SNPs and indels were then identified by SOAPsnp (soap.genomics.org.cn/) and GATK (https://www.broadinstitute.org/gatk/) for each target base with a minimum sequencing of 10 folds and quality score of 20.
Table S2.
Steps of whole-exome sequencing and pathogenesis analysis
| Step | WES analysis | Result | Note |
| 1 | Total nonsynonymous SNV/indel in patients | 37,239/2,358 | |
| 2 | SNV/indel shared by all patients | 5,526/322 | |
| 3 | SNV/indel shared by all patients (after filtering SNV/indel in normal family members) | 260/20 | |
| 4 | Novel SNV/indel (after filtering known SNVs/indel from three major databases) | 17/6 | |
| 5 | Deleterious SNV/ indel (after filtering nonpathogenic SNV/ indel by three commonly used programs (SIFT, PolyPhen-2, and MutationTaster) | 6/0 | Indels are nonframeshift |
| 6 | Determine candidate gene by comprehensive analysis | Mutation in SLC26A2 | |
| 7 | Sanger sequencing validation | Confirmed | |
| 8 | SLC26A2 expression analysis in developing lumbar | Identified | |
| 9 | Functional analysis: sulfate uptake | Decreased | |
| 10 | Gene–disease network analysis | Proposed |
All synonymous SNVs were removed. Nonsynonymous SNVs include exonic nonsynonymous, stop-gain/stop-loss, readthrough, and splicing variants. Details of the six deleterious SNVs are provided in Table S3.
Table S3.
Five variants identified by WES are excluded for the patients with dysplastic spondylolysis
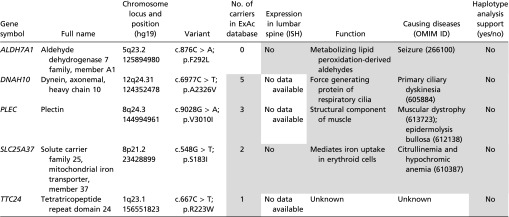 |
Data in the shaded cells are not supportive of the variants’ being a pathogenic cause of the dysplastic spondylolysis in lumbar spine. Exome Aggregation Consortium (EXAC) database (exac.broadinstitute.org/) provides exome sequences of 60,706 unrelated individuals. Function annotations are referred to GeneCards (www.genecards.org). Eurexpress (www.eurexpress.org/ee/) provides ISH images of transcriptome atlas for mouse embryo at embryonic day 14.5.
Confirmation by Sanger Sequencing.
Through the use of Sanger sequencing, we confirmed the heterozygous mutation (c.2286A > T) in exon 3 of SLC26A2 (GenBank accession no. NM_000112) of all patients (Fig. 2A), but not in the controls (subjects I-2 and II-4, Fig. 2B). This mutation changed the codon 673 GAT > GTT from aspartic acid into valine (p.D673V) in the Stas domain of the encoded protein (Fig. 2C), resulting in a missense mutation of the evolutionarily conserved residue aspartate (Fig. 2D). This mutation was predicted to be pathogenic by at least three different programs, including SIFT (affecting protein function with a score of 0.05), Polyphen-2 (probably damaging with a score of 1.00), and MutationTaster (disease causing with a score of 0.99). This mutation is a novel variant that was not found in the ESP6500, 1000 Genome, ExAc, or 1,500 in-house Chinese exome databases. Furthermore, haplotype analysis with SNPs flanking the SLC26A2 locus confirmed that it is a private variant cosegregated with the phenotype in the family (Table S4).
Fig. 2.

Mutation and protein structure analysis. (A) A heterozygous mutation c.2286A > T in SLC26A2 is confirmed in proband III-2 by bidirectional Sanger sequencing. (B) The subject II-4 serves as a normal control. (C) The encoded protein by SLC26A2 harbors 12 predicted transmembrane motifs, one sulfate transporter domain and one Stas domain. Both p.H641R and p.D673V mutations (in red) are located in the Stas domain. (D) Sequence alignment of multiple species shows the evolutionary conservation of the indicated residues. (E) A novel heterozygous mutation c.1922A > G in SLC26A2 is identified in patient XA-30 by Sanger sequencing. (F) The residue D673 (in red) is positioned in 3D structural model of the Stas domain using PyMOL Molecular Graphics System (Schrödinger L. Version 1.3r1; 2010). Data bank: PDB ID code 3LLO.
Table S4.
Haplotype analysis of variants within the p.D673V mutation-containing region at chromosome 5q31-q34
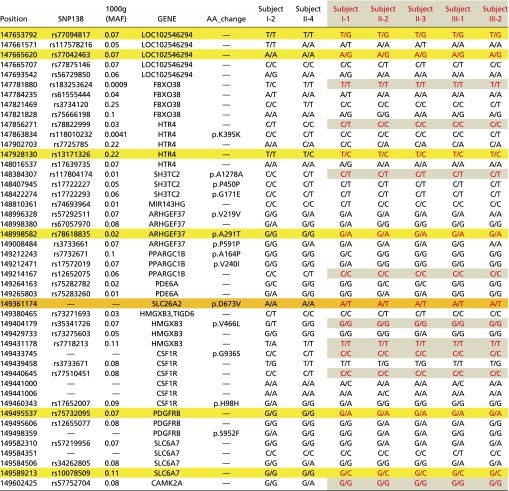 |
The variants in patients (in red and also highlighted) are segregating with the disease phenotype within about two Mb from nt 147653792 to 149602425. Multiple variants in this regain are found to segregate with the patients’ phenotypes in the family. The disease-causing locus is shaded in orange, and the flanking cosegregated loci are shaded in yellow or gray.
Additional Variants Identified in Unrelated Patients.
To examine whether additional mutations in SLC26A2 could be identified, we screened the whole coding region, intron–exon boundaries, and part of the intronic sequences of the gene with specific primers (Table S5) in a cohort of 30 unrelated patients with lumbar spondylolysis and spondylolisthesis by PCR and Sanger sequencing. As a result, we found a novel missense mutation (c.1922A > G, p.H641R, Fig. 2 C–E) in patient XA-30. This substitution was not present in the in-house Chinese exome database or in the commonly used databases as mentioned above. This patient was referred to surgical treatment because of severe lower back pain. X-ray and CT scans showed bilateral spondylolysis of L4-5, grade-I lumbar vertebra spondylolisthesis of L4, and disk herniation of L4-S1 (Fig. 1 D–F). Because the DNA of the patient’s parents was unavailable, it was not determined whether the c.1922A > G mutation was inherited or de novo.
Table S5.
Primers and oligonucleotides used for SLC26A2 amplification and mutagenesis
| Primer | Forward | Reverse | Product, bp |
| Exon 1 | TATTTGCTTTGACTTGGCTGTT | ATTGTGATTCCTCAGATCCCTTA | 1,028 |
| Exon 2A | TGACATTCTGTGATGCTCTGAT | GCAGAGGACAGCATAGTAACAA | 982 |
| Exon 2B | AAGAATCAACAGGCTGCCATAC | CCATTAGCTTCAAAGGAGGAATA | 1,026 |
| c.1922A > G | CAGTGCAACTTTCCCGTGATCCCTTGGAGCT | AGCTCCAAGGGATCACGGGAAAGTTGCACTG | Mutated |
| c.2286A > T | AGAAGTTCGCAGAGTTTATGAAGCCATTGGAA | TTCCAATGGCTTCATAAACTCTGCGAACTTCT | Mutated |
We also identified a novel variant (g.18654T > C, i.e., c.-93 T > C) in intron 1 from patient XA-12 (Fig. 3 A–C), who was treated by surgery for bilateral dysplastic spondylolysis of L4-L5 and spondylolisthesis of L5 (Fig. 3 D and E). This variant is located within the consensus motif for the transcription factor FOXA2 (5′-[AC]A[AT]T[AG]TT[GT][AG][CT]T[CT]-3′). It is known that FOXA2 is a crucial positive regulator in chondrocyte hypertrophy and that it is required for formation of intervertebral discs in mice (25). Interestingly, another flanking intronic variant (c.26+2T > C), located only 66 bp downstream, has been previously demonstrated to cause chondrodysplasia, such as multiple epiphyseal dysplasia (26). Because the size of intron 1 is larger than 16 kb, no appropriate plasmid reporter could be readily constructed for evaluation of the mutation’s effect on the transcriptional level of the gene. In addition, there was no family history or parent’s DNA for further genetic analysis. Potential pathogenicity of this variant remains a goal for future investigation.
Fig. 3.

Novel variant in intron 1 of SLC26A2 identified from the case XA-12. (A) Schematic representation of the genomic structure of SLC26A2. The variant g.18654T > C is located in intron 1, near the common “Finnish” pathogenic allele c.26+2T > C, which was previously identified in patients with diastrophic dysplasia. (B and C) Sanger sequencing of the g.18654T > C variant in the case XA-12. (D and E) Lateral view of X-ray or CT scan shows spondylolysis and grade-I spondylolisthesis of L5 (arrows).
Three-Dimensional Structure of the Stas Domain.
It has been documented that the main function of the Stas domain (Fig. 2C) of SLC26A2 is to interact with the regulatory domain of CFTR (27), which is a cAMP-regulated epithelial cell channel involved in ion transport. In the model of crystal structure of rat SLC26A5 (Fig. 2F), substitution of the negatively charged aspartate 673 by the neutral valine may affect the structural stability, as well as interaction between the Stas domain and CFTR, as shown in the 3D structural model (Fig. 2F). It should be noted that the intervening sequence (IVS) region of amino acids 566–653 between helix α1 and strand β3 was excluded from the model because its deletion was required for production of the first Stas domain crystals diffracting to high resolution (28). Therefore, it is not clear how the p.H641 substitution may affect the surrounding IVS structure.
Analysis of in Situ Hybridization Images.
To examine whether Slc26a2 is involved in lumbar development, we analyzed publicly accessible gene expression databases. The whole-mount in situ hybridization (ISH) image of a mouse embryo at embryonic day 14.5 shows that Slc26a2 is expressed not only in limb and craniofacial regions, but also in the axial skeleton in lumbosacral spine (Fig. 4A). The expression pattern of Slc26a2 is strikingly similar to that of Chad (Fig. 4B), which encodes the cartilage matrix protein chondroadherin. Also, Cftr is at least partially colocalized with Slc26a2 in the lumbosacral spine (Fig. 4C). Of note, FOXA2, the encoded transcription factor that is predicted to bind intron 1 of SLC26A2, shares a similar expression pattern with SLC26A2 in the lumbosacral spine (Fig. 4D). At postnatal day 4 of a juvenile mouse, Slc26a2 expression in the lumbosacral spine is weaker, but detectable by ISH (Fig. 3E), than the Chad expression (Fig. 3F). Expression of Cftr and FOXA2 is also detectable in a limited region (Fig. 3 G and H). At day 56 of an adult mouse, neither Slc26a2 nor Cftr expression is detectable in the lumbosacral spine (not shown).
Fig. 4.

Expression of Slc26a2 in developing skeleton. EURExpress or GenPaint images (A–D) show sagittal sections of 14.5 dpc mouse embryos processed for ISH detection of Slc26a2, Chad, Cftr, and Foxa2, respectively. The axial lumbosacral spine is indicated by arrows. Allen Spinal Column images (E–H) show cross-sections of spinal tissues from juvenile mouse at 4 d of age processed for ISH analysis of the four genes. Expression of each gene in spinal cartilage and soft tissues surrounding spinal cord is indicated by arrows. In particular, Chad is strongly expressed in spinal chondrocytes.
Sulfate Uptake Activity of p.D673V and p.H641 Mutations.
To examine whether p.D673V and p.H641R are pathogenic, the mutant plasmid DNA was transfected in the sulfate transport deficient CHO cells. Wild-type SCL26A2 plasmid DNA and pcDNA3.1 vector were also transfected as controls. After 48 h of culturing, sulfate uptake in cells transfected with the p.D673V or p.H641R mutant was found to be much lower than that in the wild-type SCL26A2 transfected cells. However, residual activities were still detectable in the p.D673V and p.H641R transfected cells compared with the negative control cells transfected with the vector alone (Fig. 5). Plasmid coding for Renilla luciferase (0.03 μg) was cotransfected for optimization of transfection efficiency. There was no significant difference in terms of luciferase activity between each of the four groups. These results suggest a significantly impaired sulfate transporter function for p.D673V and p.H641R mutations.
Fig. 5.

Functional analysis of SLC26A2 mutations. Sulfate uptake in sulfate transport-deficient CHO cells transfected with the mutant p.H641R or p.D673V, the wild-type SLC26A2, or the vector alone transfected cells (CHO). The p.D673V-transfected cells show dramatically reduced activity of sulfate uptake, and the p.H641R also shows a significant decrease compared with the wild-type SLC26A2-transfected cells. Values are presented in mean ± SD from three independent experiments in triplicate. *P < 0.05, **P < 0.01.
Discussion
Genetic defects of the hereditary spondylolysis and spondylolisthesis were first proposed several decades ago based on multiple case report studies (7, 17, 29, 30). Here, we have provided the first functional genomic evidence, to our knowledge, that autosomal dominant spondylolysis and associated dysplastic defects in the lumbosacral spine are caused by mutations in SLC26A2.
In normal cartilage, chondrocytes synthesize and deposit large amounts of sulfated proteoglycans into the extracellular matrix; SLC26A2 encodes the sulfate transporter for the uptake of inorganic sulfate that is needed for proteoglycan sulfation. Impaired activity of the transporter in chondrocytes results in intracellular sulfate depletion and insufficient synthesis of proteoglycans. Undersulfation of proteoglycans affects the composition of the extracellular matrix and leads to impaired proteoglycan deposition, which is necessary for proper endochondral bone formation (26). SLC26A2 was first identified to cause diastrophic dysplasia (DTD) by positional cloning (31). DTD is a well-characterized recessive osteochondrodysplasia with clinical features including dwarfism, spinal deformation, and specific joint abnormalities.
Furthermore, mutations in SLC26A2 were identified to cause a family of several recessive chondrodysplasias with distinct phenotypes, including in order of decreasing severity achondrogenesis 1B (ACG1B), atelosteogenesis 2 (AO2), diastrophic dysplasia (DTD, and multiple epiphyseal dysplasia (rMED). Clinical features of these disorders with hypomorphic missense mutations are summarized in Table S6 for a comparison with our cases. It seems that a genotype–phenotype correlation could be found among these disorders (32, 33). In the lethal form of chondrodysplasia, a deficient ossification in the lumbar vertebrae was observed in ACG1B (34). AO2 also showed spinal deformities, including cervical kyphosis, scoliosis, and lumbar hyperlordosis (35). In the mild form of DTD, spinal deformities, such as lordosis, kyphosis, coronal clefts of lumbar vertebrae, and particularly a decrease of the interpedicular distance were frequently observed (26). In the milder form of rMED, platyspondyly was most marked in the lower thoracic and upper lumbar spine (36, 37).
Table S6.
Missense mutations of SLC26A2 cause five different chondrodysplasias
| Disorder | OMIM ID | Mutation in protein | Protein domain | Genetic form | Phenotypes | Source |
| Achondrogenesis 1B (ACG-1B) | 600972 | L483P | Sulfate_transport | Recessive (compound heterozygote with W466X) | A severe chondrodystrophy characterized radiographically by deficient ossification in the lumbar vertebrae and absent ossification in the sacral, pubic, and ischial bones and clinically by stillbirth or early death | 34 |
| Atelosteogenesis II (AO2); De la Chapelle dysplasia (DLCD) | 256050 | T512K | Sulfate_transport | Recessive | Neonatal death dwarfism resembling atelosteogenesis; radiographic findings include cervical kyphosis, scoliosis, and lumbar hyperlordosis, etc. AO2 is similar to but more severe than DTD | 35 |
| Diastrophic dysplasia (DTD) | 222600 | Q454P | Sulfate_transport | Recessive | A girl was born with short limbs, short extremities, and clubfeet. X-ray showed severe platyspondyly, wide metaphyses, and fibular overgrowth | 49 |
| Epiphyseal dysplasia, multiple, 4 (EDM4); aka recessive multiple epiphyseal dysplasia (rMED) | 226900 | R279W | Sulfate_transport | Recessive | Multiple epiphyseal dysplasia, first symptoms usually present in childhood at age 2–14 y, waddling gait, restriction of joint mobility, and pain and stiffness in the weight-bearing joints | 35, 50, 51 |
| 226900 | C653S | Stas domain | Recessive | Dislocation of a multilayered patella, mild lumbar scoliosis, and defects of the anterior cortical rims of the vertebral endplates | 37 | |
| Spondylolysis | NA (this study) | H641R | Stas domain | Dominant (heterozygote mutation) | Spondylolysis of lumbar spine, spondylolisthesis, and lower back pain | This study |
| D673V | Stas domain | Dominant (heterozygote mutation) | Spondylolysis of lumbar spine, spondylolisthesis, and lower back pain | This study |
For a comparison with the missense mutations identified in this study, reported missense mutations of SLC26A2 in patients with four different disorders are summarized in this table. These disorders are listed in order of decreasing severity. NA, not applied.
It is interesting that the clinical abnormalities in our cases are restricted to cartilage and bone in the lumbosacral spine, although SLC26A2 is widely expressed in developing cartilage and many other tissues (31). In the dysplastic type of congenital spondylolysis and spondylolisthesis, abnormalities such as poorly developed pars interarticularis, a predisposition to cracking and separating, or spine bifida are often observed in the upper sacrum and the lower lumbar vertebrae (1). The resulting lumbar stenosis may cause L5 nerve radiculopathy as well as bowel and bladder dysfunction from compression of sacral nerve roots. Children and adolescents with dysplastic spondylolisthesis are more likely to carry greater risk of progressive deformity and subsequent neurologic injury than do patients with isthmic or acquired spondylolisthesis (4). Indeed, patients with dysplastic spondylolisthesis, such as patients II-3, III-2, XA-12, and XA-30 in this study, are notably more likely to require surgical treatment. However, future studies are needed to determine whether genetic mutations can serve as a biomarker to predict high-risk individuals who may develop high-dysplastic developmental spondylolisthesis (HDDS). Identification of HDDS allows earlier surgical stabilization to prevent slip progression, deformity, and nerve root symptoms (38).
It has been proposed that the phenotype in diastrophic dysplasia is determined by multiple thresholds of residual sulfate transporter activity (32). More severe and pleiotropic phenotypes relating to chondrodysplasias, such as lordosis, kyphosis, clefts of lumbar vertebrae, and a decrease of the interpedicular distance (26), have been identified from autosomal recessive patients with homozygous or compound mutations of SLC26A2. Therefore, it seems reasonable to assume that the heterozygous mutations of the gene identified here may represent the least severe allele. Notably, there is a precedent that states heterozygous missenses in SLC26A8, which encodes a sperm-specific anion transporter, cause male infertility (39). Mutations in several other members of the SLC26 family were also found to cause various Mendelian diseases, such as chloride diarrhea and deafness with enlargement of the vestibular aqueduct (40). Therefore, our finding that impaired sulfate transport activities underlies the pathogenesis of lumbar spondylolysis fits well with current understanding of cartilage function.
It is worthy to mention that the Stas domains of SLC26 transporters in the cytoplasmic region are found to interact with the R domain of CFTR to activate anion exchange (41). Patients with cystic fibrosis due to mutations in CFTR were also found to have bone and lumbar-associated disorders, such as vertebral fractures (42) and L5-S1 lumbar disk herniation (24). Because both SLC26A2 and CFTR are expressed in the developing lumbosacral spine, it is conceivable that mutations in the Stas domain of SLC26A2, or in the R domain of CFTR, may affect the development of lumbosacral vertebrae.
Spondylolysis is a radiographic feature that may be caused by a variety of pathogenic mechanisms. Based on gene expression and bioinformatics analyses, we explored a potential gene–disease network involving lumbar spondylolysis (Fig. S1). Except for the function of SLC26A2 and CFTR in cartilage discussed above, TGF-β signaling is also implicated in this network because mutations in GDF5 and its interacting protein BMPR1B have been identified to result spondylolysis or chrondrodysplasia defects (20, 43). These molecules or pathways can be activated by 17-β estradiol stimulation (Fig. S1), a female hormone playing an important role in cartilage and bone development. Biological functions and related disorders caused by each of these genes have been summarized in Table S7. Further studies are warranted to examine genomic defects of these candidate genes in patients with spondylolysis and spondylolisthesis.
Fig. S1.

Working model of gene-disease network in cartilage and spondylolysis. IPA analysis (www.ingenuity.com) was used to explore the relations of spondylolysis-causal genes (Table S7) that are expressed in developing lumbar (nodes in orange color) and interacting molecules (nodes in light blue). Their encoded proteins are either interconnected directly (solid line) or indirectly (dashed line) according to IPA and IntAct database. Estradiol, denoted by a green node, has been previously demonstrated to regulate bone and cartilage development through (A) anion transporter SLC26A2 and (B) GDF5 involved TGF-β pathway. Like the SLC26A2 (Fig. 4 A and E), GDF5 is also expressed in (C) developing lumbosacral region (EURExpress, www.eurexpress.org/ee/ and GenPaint, www.genepaint.org/) as well as in (D) spinal cartilage and soft tissues surrounding spinal cord of the juvenile mouse at four days of age (Allen Spinal Cord Atlas, mousespinal.brain-map.org/about.html). The axial lumbosacral region is indicated by arrows.
Table S7.
Summary of two spondylolysis-causal genes and their interacted molecules involving lumbar spine
| Gene symbol | Full name | Function | Interacting protein | Causing diseases | Pathway | Source |
| BMPR1B | Bone morphogenetic protein receptor, type IB | Activate SMAD transcriptional regulators | GDF5 | Brachydactyly, type A2; chrondrodysplasia | TGF-β pathway; and BMP signaling | 43 |
| CFTR | Cystic fibrosis transmembrane conductance regulator | Transport of chloride ions, etc. | SLC26A3 and SLC26A2, etc. | Cystic fibrosis, lumbar disk herniation | Transport of small molecules | 24 |
| GDF5 | Growth differentiation factor 5 (i.e., CDMP-1) | Bone and cartilage formation | BMPR1B | Type C brachydactyly, also spondylolysis and spondylolisthesis | TGF-β pathway | 20, 52 |
| SLC26A2 | Solute carrier family 26 (anion exchanger), member 2 | Endochondral bone formation | CFTR (predicted by homolog SLC26A3) | Spondylolysis and spondylolisthesis, also platyspondylia, epiphyseal dysplasia, etc. | Anion exchanger | This study; also 31 |
In summary, this is the first identification, to our knowledge, of mutations of SLC26A2 in patients with autosomal dominant form of spondylolysis and spondylolisthesis. Our findings provide evidence that the sulfate anion transporter SLC26A2 plays a role in the development of cartilage in lumbosacral spine. Initial gene–disease analysis may suggest a convergent network that is implicated in the pathogenesis of chondrodysplasia. Candidate gene screens may identify more novel genetic defects from patients with dysplastic spondylolysis and spondylolisthesis as well as from high-risk individuals who are asymptomatic.
Materials and Methods
Study Subjects and Clinical Evaluation.
Seven affected and unaffected family members from a nonconsanguineous pedigree (XAHT01) were recruited at Xijing hospital in Northwestern China. An additional 30 unrelated patients with spondylolysis and spondylolisthesis were consecutively recruited for mutation analysis of candidate genes. Lower back pain was evaluated by using Oswestry Disability Index (ODI, version 2.1a) that has been previously simplified and validated into a Chinese version by our institute (44). Patients with occupational and/or habitual risk factors, such as heavy manual laborers and occupational drivers, were not included. Diagnosis was made based on the Wiltse–Newman classification system (1). Informed consent was obtained from all participants. This study was approved by the ethics committees of Wenzhou Medical University and Xijing Hospital, Fourth Military Medical University [XJLL(2014)0025].
WES.
Genomic DNA was isolated from peripheral blood leukocytes (Promega). Whole-exome capture by SureSelect Human All Exon Kit (Agilent) and high-throughput sequencing by HiSeq2000 sequencer (Illumina Inc.) were conducted in-house as previously described (45, 46). The reads were aligned for SNP calling and subsequent analysis for prioritization of candidate genes (SI Materials and Methods) (47). Detected sequence variants if presented in the dbSNP, HapMap, 1000 Genome, and ESP6500, ExAC, and in-house Chinese Exome Database (1,500 Chinese Han individuals) were all removed. Deleterious SNVs were predicted by SIFT (sift.bii.a-star.edu.sg/), Polyphen-2 (genetics.bwh.harvard.edu/pph2/), and MutationTaster (www.mutationtaster.org/) programs. Candidate SNVs were validated by ABI3730 sequencer.
Sanger Sequencing and a Cohort of Samples for Validation.
Thirty unrelated patients with spondylolysis, which mostly accompany spondylolisthesis or disk herniation, were screened for mutation evaluation of SLC26A2 by PCR and Sanger sequencing. Coding regions, intron–exon junctions, and partial intronic sequences of SLC26A2 were individually amplified and sequenced by specific primers (Table S5).
Gene Expression.
To examine the expression pattern of SLC26A2 and associated genes in the developing spine skeleton, we datamined EURExpress (www.eurexpress.org), GenePaint (www.genepaint.org), and Allen Spine Column (mousespinal.brain-map.org) databases (48). These online servers offer microphotographs of complete section series of 14.5-d-postcoitum (dpc) mouse embryos, P4, and P56 mouse spinal tissues processed for ISH with nonradioactive probes. Selected image series were downloaded and compared for detailed annotations.
Mutagenesis of SLC26A2 and Cell Transfection.
Wild-type human SLC26A2 cDNA driven by CMV promoter in pReceiver vector (Genecopoeia) was mutagenized using a Mutagenesis kit (Promega) to create the c.2286A > T or c.1922A > G mutation in the coding region using specific oligonucleotides (Table S5). CHO cells (ATCC), which are sulfate transport-deficient mutants and maintained in Ham’s F-12K medium, were then transfected with 2 μg of vector alone, wild-type SLC26A2, c.2286A > T, or c.1922A > G harboring SLC26A2 using Lipofectamine 2000 (Invitrogen).
Sulfate Uptake in Transfected Cells.
To measure the rate of sulfate uptake, 2 × 105 of CHO cells were cultured and transfected with 2 μg of each of SLA26A2 construct plasmids by the lipofectamine 2000. Plasmid coding for Renilla luciferase (0.03 μg) was cotransfected for optimization of transfection efficiency. Sulfate uptake was performed as described previously (35). After the transfection for 48 h, cells were then incubated for 2 min at 37 °C in 0.7 mL of low-ionic-strength buffer containing different concentrations of Na2SO4 and a constant concentration of carrier-free Na2[35S]O4 (0.1 μM, corresponding to 150 μCi/mL). The uptake medium was removed, and the cells were washed four times with ice-cold medium containing 100 mM sucrose, 100 mM NaNO3, 1 mM MgCl2, and 10 mM Tris-Hepes, pH 7.5. Cells were then lysed in 2% SDS lysis buffer. Lysates were collected, heat-denatured, and centrifuged. Supernatants were assayed for radioactivity by scintillation spectroscopy and for protein content measurement (BCA Protein Assay; Pierce). Luciferase activity was measured using a Microplate Luminometer (Berthold Technologies).
SI Materials and Methods
For the quality control, the Cutadapt (https://pypi.python.org/pypi/cutadapt) and FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/) were used to remove 3′-/5′- adapters and low-quality reads, respectively. The clean reads were mapped to the reference human genome (hg19, genome.ucsc.edu) using the BWA (Burrows–Wheeler Aligner) program with at most two mismatches (bio-bwa.sourceforge.net). The alignment files (.bam) were generated with SAMtools (samtools.sourceforge.net) and the reads of low mapping quality (<Q30) were filtered out. Clonal duplicated reads that may be derived from PCR artifacts were removed using Picard Tools by default parameters (broadinstitute.github.io/picard/). Short read alignment and annotation visualization were performed using the MagicViewer tool (2) developed in our laboratory (bioinformatics.zj.cn/magicviewer/). The percentage of alignment of the clean reads to the exome regions was obtained using our custom Perl scripts on the base of alignment files. SNVs and indels were detected by GATK (Genome Analysis ToolKit) (www.broadinstitute.org/gatk). The average transition transversion ratios for the total SNV, known SNVs, and novel SNVs are 2.70, 2.74, and 2.32 respectively. Comprehensive annotation of all of the detected SNVs and indels were annotated by ANNOVAR (annovar.openbioinformatics.org/en/latest/), including function implication (gene region, functional effect, mRNA GenBank accession number, amino acid change, cytoband, etc.) and allele frequency in dbSNP, 1000 Genomes (www.1000genomes.org), ESP6500 (evs.gs.washington.edu/EVS/), ExAc (exac.broadinstitute.org/), and in-house Chinese Exome Database (1,500 Chinese Han individuals). Damaging missense mutations were predicted by SIFT (sift.bii.a-star.edu.sg/), PolyPhen-2 (genetics.bwh.harvard.edu/pph2/), and MutationTaster (www.mutationtaster.org/).
Supplementary Material
Acknowledgments
The authors thank the patients and family members for their participation in this study. This study was supported by Ministry of Science and Technology of China Grant 2011CB964703 (to Z.-J.L.), Program for Changjiang Scholars and Innovative Research Team in University Grant IRT13051 (to Z.-J.L.), National Natural Science Foundation of China Grant 81110114 (to Z.S.S.), and research funding from the Wenzhou Medical University and Zhejiang Province Nature Science Research (to P.Y.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Exome sequence data related to this study have been deposited in the Institute of Genomic Medicine at Wenzhou Medical University database, 122.228.158.106/20150521PANSdata.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1502454112/-/DCSupplemental.
References
- 1.Wiltse LL, Newman PH, Macnab I. Classification of spondylolisis and spondylolisthesis. Clin Orthop Relat Res. 1976;(117):23–29. [PubMed] [Google Scholar]
- 2.Standaert CJ, Herring SA. Spondylolysis: A critical review. Br J Sports Med. 2000;34(6):415–422. doi: 10.1136/bjsm.34.6.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamada A, et al. Lumbar spondylolysis in juveniles from the same family: A report of three cases and a review of the literature. Case Rep Orthop. 2013;2013:272514. doi: 10.1155/2013/272514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavalier R, Herman MJ, Cheung EV, Pizzutillo PD. Spondylolysis and spondylolisthesis in children and adolescents: I. Diagnosis, natural history, and nonsurgical management. J Am Acad Orthop Surg. 2006;14(7):417–424. doi: 10.5435/00124635-200607000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Fu KM, et al. Morbidity and mortality in the surgical treatment of six hundred five pediatric patients with isthmic or dysplastic spondylolisthesis. Spine. 2011;36(4):308–312. doi: 10.1097/BRS.0b013e3181cf3a1d. [DOI] [PubMed] [Google Scholar]
- 6.Marchetti PG, Bartolozzi PB. Classification of Spondylolisthesis As a Guideline for Treatment. 2nd Ed. Lippincott-Raven; Philadelphia: 1997. pp. 1211–1254. [Google Scholar]
- 7.Wiltse LL. The etiology of spondylolisthesis. J Bone Joint Surg Am. 1962;44-A:539–560. [PubMed] [Google Scholar]
- 8.Sakai T, Sairyo K, Suzue N, Kosaka H, Yasui N. Incidence and etiology of lumbar spondylolysis: review of the literature. J Orthop Sci. 2010;15(3):281–288. doi: 10.1007/s00776-010-1454-4. [DOI] [PubMed] [Google Scholar]
- 9.Moke L, Debeer P, Moens P. Spondylolisthesis in twins: Multifactorial etiology: A case report and review of the literature. Spine. 2011;36(11):E741–E746. doi: 10.1097/BRS.0b013e3181f9f575. [DOI] [PubMed] [Google Scholar]
- 10.Amuso SJ, Mankin HJ. Hereditary spondylolisthesis and spina bifida. Report of a family in which the lesion is transmitted as an autosomal dominant through three generations. J Bone Joint Surg Am. 1967;49(3):507–513. [PubMed] [Google Scholar]
- 11.Shahriaree H, Harkess JW. A family with spondylolisthesis. Radiology. 1970;94(3):631–633. doi: 10.1148/94.3.631. [DOI] [PubMed] [Google Scholar]
- 12.Saha MM, Bhardwaj OP, Srivastava G, Pramanick A, Gupta A. Osteopetrosis with spondylolysis—four cases in one family. Br J Radiol. 1970;43(514):738–740. doi: 10.1259/0007-1285-43-514-738. [DOI] [PubMed] [Google Scholar]
- 13.Haukipuro K, et al. Familial occurrence of lumbar spondylolysis and spondylolisthesis. Clin Genet. 1978;13(6):471–476. doi: 10.1111/j.1399-0004.1978.tb01200.x. [DOI] [PubMed] [Google Scholar]
- 14.Shahriaree H, Sajadi K, Rooholamini SA. A family with spondylolisthesis. J Bone Joint Surg Am. 1979;61(8):1256–1258. [PubMed] [Google Scholar]
- 15.Martin RP, Deane RH, Collett V. Spondylolysis in children who have osteopetrosis. J Bone Joint Surg Am. 1997;79(11):1685–1689. doi: 10.2106/00004623-199711000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Albanese M, Pizzutillo PD. Family study of spondylolysis and spondylolisthesis. J Pediatr Orthop. 1982;2(5):496–499. doi: 10.1097/01241398-198212000-00006. [DOI] [PubMed] [Google Scholar]
- 17.Rowe GG, Roche MB. The etiology of separate neural arch. J Bone Joint Surg Am. 1953;35-A(1):102–110. [PubMed] [Google Scholar]
- 18.Stewart TD. Incidence of separate neural arch in the lumbar vertebrae of Eskimos. Am J Phys Anthropol. 1931;16:51–62. [Google Scholar]
- 19.Yılmaz T, Turan Y, Gülşen I, Dalbayrak S. Co-occurrence of lumbar spondylolysis and lumbar disc herniation with lumbosacral nerve root anomaly. J Craniovertebr Junction Spine. 2014;5(2):99–101. doi: 10.4103/0974-8237.139211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Savarirayan R, et al. Broad phenotypic spectrum caused by an identical heterozygous CDMP-1 mutation in three unrelated families. Am J Med Genet A. 2003;117A(2):136–142. doi: 10.1002/ajmg.a.10924. [DOI] [PubMed] [Google Scholar]
- 21.Mio F, et al. A functional polymorphism in COL11A1, which encodes the alpha 1 chain of type XI collagen, is associated with susceptibility to lumbar disc herniation. Am J Hum Genet. 2007;81(6):1271–1277. doi: 10.1086/522377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rose PS, et al. Thoracolumbar spinal abnormalities in Stickler syndrome. Spine. 2001;26(4):403–409. doi: 10.1097/00007632-200102150-00017. [DOI] [PubMed] [Google Scholar]
- 23.Paassilta P, et al. Identification of a novel common genetic risk factor for lumbar disk disease. JAMA. 2001;285(14):1843–1849. doi: 10.1001/jama.285.14.1843. [DOI] [PubMed] [Google Scholar]
- 24.Denne C, et al. Lumbar disc herniation in three patients with cystic fibrosis: a case series. J Med Case Reports. 2011;5:440. doi: 10.1186/1752-1947-5-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maier JA, Lo Y, Harfe BD. Foxa1 and Foxa2 are required for formation of the intervertebral discs. PLoS ONE. 2013;8(1):e55528. doi: 10.1371/journal.pone.0055528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonafé L, Mittaz-Crettol L, Ballhausen D, Superti-Furga A. Diastrophic dysplasia. In: Pagon RA, et al., editors. GeneReviews. University of Washington; Seattle: 1993. [PubMed] [Google Scholar]
- 27.El Khouri E, Touré A. Functional interaction of the cystic fibrosis transmembrane conductance regulator with members of the SLC26 family of anion transporters (SLC26A8 and SLC26A9): Physiological and pathophysiological relevance. Int J Biochem Cell Biol. 2014;52:58–67. doi: 10.1016/j.biocel.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Pasqualetto E, et al. Structure of the cytosolic portion of the motor protein prestin and functional role of the STAS domain in SLC26/SulP anion transporters. J Mol Biol. 2010;400(3):448–462. doi: 10.1016/j.jmb.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 29.Ota H. [Spondylolysis: Familial occurrence and its genetic implications] Nippon Seikeigeka Gakkai Zasshi. 1967;41(8):931–941. Japanese. [PubMed] [Google Scholar]
- 30.Kleinberg S. Spondylolisthesis. Ann Surg. 1923;77(4):490–495. doi: 10.1097/00000658-192304000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hästbacka J, et al. The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell. 1994;78(6):1073–1087. doi: 10.1016/0092-8674(94)90281-x. [DOI] [PubMed] [Google Scholar]
- 32.Dwyer E, Hyland J, Modaff P, Pauli RM. Genotype-phenotype correlation in DTDST dysplasias: Atelosteogenesis type II and diastrophic dysplasia variant in one family. Am J Med Genet A. 2010;152A(12):3043–3050. doi: 10.1002/ajmg.a.33736. [DOI] [PubMed] [Google Scholar]
- 33.Macías-Gómez NM, Mégarbané A, Leal-Ugarte E, Rodríguez-Rojas LX, Barros-Núñez P. Diastrophic dysplasia and atelosteogenesis type II as expression of compound heterozygosis: First report of a Mexican patient and genotype-phenotype correlation. Am J Med Genet A. 2004;129A(2):190–192. doi: 10.1002/ajmg.a.30149. [DOI] [PubMed] [Google Scholar]
- 34.Rossi A, Bonaventure J, Delezoide AL, Cetta G, Superti-Furga A. Undersulfation of proteoglycans synthesized by chondrocytes from a patient with achondrogenesis type 1B homozygous for an L483P substitution in the diastrophic dysplasia sulfate transporter. J Biol Chem. 1996;271(31):18456–18464. doi: 10.1074/jbc.271.31.18456. [DOI] [PubMed] [Google Scholar]
- 35.Bonafé L, et al. A novel mutation in the sulfate transporter gene SLC26A2 (DTDST) specific to the Finnish population causes de la Chapelle dysplasia. J Med Genet. 2008;45(12):827–831. doi: 10.1136/jmg.2007.057158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Czarny-Ratajczak M, et al. New intermediate phenotype between MED and DD caused by compound heterozygous mutations in the DTDST gene. Am J Med Genet A. 2010;152A(12):3036–3042. doi: 10.1002/ajmg.a.33707. [DOI] [PubMed] [Google Scholar]
- 37.Hinrichs T, et al. Recessive multiple epiphyseal dysplasia (rMED) with homozygosity for C653S mutation in the DTDST gene—phenotype, molecular diagnosis and surgical treatment of habitual dislocation of multilayered patella: Case report. BMC Musculoskelet Disord. 2010;11:110. doi: 10.1186/1471-2474-11-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lamartina C, Zavatsky JM, Petruzzi M, Specchia N. Novel concepts in the evaluation and treatment of high-dysplastic spondylolisthesis. Eur Spine J. 2009;18(Suppl 1):133–142. doi: 10.1007/s00586-009-0984-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dirami T, et al. Missense mutations in SLC26A8, encoding a sperm-specific activator of CFTR, are associated with human asthenozoospermia. Am J Hum Genet. 2013;92(5):760–766. doi: 10.1016/j.ajhg.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alper SL, Sharma AK. The SLC26 gene family of anion transporters and channels. Mol Aspects Med. 2013;34(2-3):494–515. doi: 10.1016/j.mam.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ko SB, et al. Gating of CFTR by the STAS domain of SLC26 transporters. Nat Cell Biol. 2004;6(4):343–350. doi: 10.1038/ncb1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossini M, et al. Prevalence and correlates of vertebral fractures in adults with cystic fibrosis. Bone. 2004;35(3):771–776. doi: 10.1016/j.bone.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 43.Lehmann K, et al. Mutations in bone morphogenetic protein receptor 1B cause brachydactyly type A2. Proc Natl Acad Sci USA. 2003;100(21):12277–12282. doi: 10.1073/pnas.2133476100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu H, Tao H, Luo Z. Validation of the simplified Chinese version of the Oswestry Disability Index. Spine. 2009;34(11):1211–1216, discussion 1217. doi: 10.1097/BRS.0b013e31819e2b34. [DOI] [PubMed] [Google Scholar]
- 45.Wu J, Shen E, Shi D, Sun Z, Cai T. Identification of a novel Cys146X mutation of SOD1 in familial amyotrophic lateral sclerosis by whole-exome sequencing. Genet Med. 2012;14(9):823–826. doi: 10.1038/gim.2012.50. [DOI] [PubMed] [Google Scholar]
- 46.Bi C, et al. Mutations of ANK3 identified by exome sequencing are associated with autism susceptibility. Hum Mutat. 2012;33(12):1635–1638. doi: 10.1002/humu.22174. [DOI] [PubMed] [Google Scholar]
- 47.Li J, et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol Psychiatry. 2015 doi: 10.1038/mp.2015.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geffers L, Herrmann B, Eichele G. Web-based digital gene expression atlases for the mouse. Mamm Genome. 2012;23(9-10):525–538. doi: 10.1007/s00335-012-9413-3. [DOI] [PubMed] [Google Scholar]
- 49.Mégarbané A, et al. Homozygosity for a novel DTDST mutation in a child with a ‘broad bone-platyspondylic’ variant of diastrophic dysplasia. Clin Genet. 1999;56(1):71–76. doi: 10.1034/j.1399-0004.1999.560110.x. [DOI] [PubMed] [Google Scholar]
- 50.Superti-Furga A, et al. Recessively inherited multiple epiphyseal dysplasia with normal stature, club foot, and double layered patella caused by a DTDST mutation. J Med Genet. 1999;36(8):621–624. [PMC free article] [PubMed] [Google Scholar]
- 51.Czarny-Ratajczak M, et al. A mutation in COL9A1 causes multiple epiphyseal dysplasia: Further evidence for locus heterogeneity. Am J Hum Genet. 2001;69(5):969–980. doi: 10.1086/324023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Francis-West PH, et al. Mechanisms of GDF-5 action during skeletal development. Development. 1999;126(6):1305–1315. doi: 10.1242/dev.126.6.1305. [DOI] [PubMed] [Google Scholar]