

Abstract
Esophageal squamous cell carcinoma continues to heavily burden clinicians worldwide. Researchers have discovered the genomic landscape of esophageal squamous cell carcinoma, which holds promise for an era of personalized oncology care. One of the most pressing problems facing this issue is to improve the understanding of the newly available genomic data, and identify the driver-gene mutations, pathways, and networks. The emergence of a legion of novel targeted agents has generated much hope and hype regarding more potent treatment regimens, but the accuracy of drug selection is still arguable. Other problems, such as cancer heterogeneity, drug resistance, exceptional responders, and side effects, have to be surmounted. Evolving topics in personalized oncology, such as interpretation of genomics data, issues in targeted therapy, research approaches for targeted therapy, and future perspectives, will be discussed in this editorial.
Keywords: Cancer heterogeneity, Cultured tumor cells, Driver mutation, Drug side effects, Esophageal squamous cell carcinoma, Exceptional responder, High-throughput nucleotide sequencing, Neoplasm drug resistance, Personalized medicine, Xenograft model
Core tip: Esophageal squamous cell carcinoma represents a heavy burden on clinicians worldwide. Recently, researchers have discovered the genomic landscape of this cancer, which holds promise for an era of personalized oncology care. Evolving topics in personalized oncology, such as interpretation of genomics data, critical issues in targeted therapy, research approaches, and future perspectives, are discussed in this editorial.
INTRODUCTION
Esophageal cancer is the eighth most common cause of cancer-related death worldwide[1]. Esophageal squamous cell carcinoma (ESCC) remains the predominant histology. Surgery is still the mainstay of treatment throughout the world, and an up to 50% five-year survival rate and < 5% surgical mortality rate can be achieved in select Asian centers[2]. Notwithstanding, multimodal treatment may achieve a better outcome, as overall survival improves modestly[3]. Most patients with localized disease will develop metastatic disease, with a minimal effects from combination chemotherapy[4]. After disease progression on first-line chemotherapy, there is no standard second-line treatment[5]. The unsatisfactory outcome in ESCC is mainly due to late diagnosis, the aggressiveness of this cancer, and lack of effective treatment strategies[6].
Recently, tremendous progress has been made in cancer genomics and epigenomics with the advent of high-throughput techniques, such as next-generation sequencing. Three groups have reported the genetic landscape of human ESCC with whole genome sequencing and whole exome sequencing[7-9]. Genomic alterations include: (1) single nucleotide variants of many genes with a relatively significant frequency (≥ 5%), such as p53, KMT2D, Notch1/2/3, FAT1/3, Syne1, EP300, Rb1, Nfe2l2, Cdkn2a, Ajuba, Crebbp, Kdm6A, Fbxw7, MLL2/3, Pik3ca, Pten, Arid2, Pbrm1, etc; (2) copy number alterations of many genes with a relatively significant frequency (≥ 5%), such as CCND1, FGFs, CDKN2A, CDKN2B, Pik3ca, Dvl3, LRP5/6, KRas/MRas, EGFR, Akt1, Bcl2l1, Notch1/2/3, E2F1, SFRP4, SOS1/2, Birc5, Yap1, Sox2, Myc, IL7R, etc; and (3) alterations in multiple signaling pathways, such as cell cycle regulation, apoptosis regulation, DNA damage control, histone modifications, as well as RTK-Ras-MAPK-PI3K-Akt, Hippo, Notch, Wnt, and Nfe2l2/Keap1 pathways. The overall mutation pattern appears similar to that of head and neck squamous cell carcinoma[10,11], but different from that of esophageal adenocarcinoma[12,13] and lung squamous cell carcinoma[14].
In addition to these descriptive data, smoking was not found to be related with signature mutations[7], but the lack of alcohol consumption was associated with a cluster of gene mutations[9]. Viral integration was not found in the genomes of 88 subjects[9]. Trinucleotide signature analysis suggested DNA cytidine deaminase (APOBEC3B)-induced deamination was mainly responsible for mutations[8,15]. Moreover, mutations of single genes or gene clusters were associated with patient survival, for example, EP300 mutation[7,9]. Certain genes, for example, XPO1, were explored as a therapeutic target[8].
These landmark studies provided the research community with an enormous amount of information to better understand the molecular mechanisms of ESCC. This editorial is aimed to gain insights from such studies, and propose personalized and targeted therapy as a research direction in the future.
INTERPRETATION OF GENOMICS DATA
Driver genes and mutations
Currently available bioinformatics tools have been designed to prioritize gene mutations at the nucleotide, gene, pathway, and network levels. The number of nonsynonymous somatic mutations per ESCC averaged > 80. If a solid tumor ordinarily requires 5-8 hits (not necessarily 5-8 mutations) as suggested by classical epidemiologic studies, most of these mutations should be “passengers” instead of “drivers”, which can offer selective growth advantage to the tumor cell[16]. Therefore, it is critical to identify which gene mutations are cancer drivers.
As driver mutations may occur at high or low frequencies[17], it may not be safe to prioritize driver mutations according to their frequencies. However, as a clinically relevant parameter, a high frequency of a mutation does support its potential significance in carcinogenesis. In addition to mutated drivers, Epi-drivers are a class of driver genes that are not frequently mutated but aberrantly expressed in tumors through epigenetic alterations in DNA methylation or chromatin modification. Although epigenetics in ESCC has been studied for many years[18,19], it is still not clear how to differentiate epigenetic alterations that bring forth a selective growth advantage from those that do not[16]. According to Vogelstein et al[16]’s 20/20 rule, only 125 mutated-driver genes of human cancers have been discovered to date, and the number is nearing saturation. Tamborero et al[20] reported a list of 291 high-confidence cancer-driver genes and 144 candidate genes from 12 different types of cancer. Several databases have become available. For example, Network of Cancer Genes (NCG 4.0) contains 537 experimentally supported genes and 1463 candidate genes inferred using statistical methods[21]. The Candidate Cancer Gene Database contains cancer-driver genes from forward genetic screens in mice[22]. Considering tissue specificity of ESCC, there is a need to compile a cancer-driver gene list to support future research on ESCC therapy. However, it should be pointed out that cancer-driver genes may contain both driver mutations and passenger mutations in cancer. For example, APC mutations truncating the N-terminal amino acids are driver mutations, while those affecting other regions are passenger mutations. Even for the same driver gene (e.g., K-Ras), different driver mutations (e.g., mutations at codons 12, 13, and 61) have different impacts on carcinogenesis and clinical behaviors[23-25]. Because of these complexities, efforts need to be made in order to identify personalized driver genes in cancer[26].
Pathways and network
Increasing evidence suggests that dysregulation of cellular signaling pathways, rather than individual mutations, contributes to the pathogenesis of ESCC[27-29]. Driver genes usually do not work in isolation, but often function together to alter cellular processes[30]. There is a growing consensus that pathways rather than single genes are the primary target of mutations[31]. It is interesting that mutations in various components of a single pathway tend to be mutually exclusive[32]. Once driver genes or driver mutations are identified, the next step is to focus on driver pathways with genes grouped together according to the biochemical pathways that they play functional roles in. Pathway activity may be further validated by the downstream readouts, e.g., mRNA and protein expression, morphology, and function. Incorporation of immunohistochemistry data, or even proteomics data, may help in evaluation of the pathway activity[33,34].
One major challenge in analyzing genomics data of ESCC is the lack of information of esophagus-specific pathways. Pathway databases, e.g., KEGG, are fairly incomplete and lack tissue and cell specificities. Applying such pathway information in analyzing ESCC data may generate misleading outcomes. For example, using ChIP-seq analyses, Sox2-regulated genes in ESCC cells are different from those in embryonic stem cells because in ESCC, Sox2 tends to interact with p63 as opposed to Oct4 in embryonic stem cells[35]. Identifying bona fide target genes and using expression profiles of these genes to infer pathway activity in ESCC will be critical in the future[36].
Few bioinformatics methods involve a procedure for taking account of pathway interactions, i.e., pathways that are mutated in the same sample, and that are mutated together across a large subset of samples[8]. Similar to expression-based stratification, network-based stratification of tumor mutations can identify cancer subtypes to guide treatment and prognosis[37]. Categorizing ESCC into multiple subtypes according to its molecular alterations may be a practical step leading to final personalization of ESCC therapy. In fact, subtyping has been shown to be a successful approach in managing other cancers[38].
Drug selection
Selecting drugs according to genomics data has led to promising results in early studies on personalized and targeted therapy[39]. To date, most clinically approved targeted drugs are directed against kinases. Some of these have been utilized against ESCC (Table 1). Gefitinib, an epidermal growth factor receptor inhibitor, has been tested as a second-line treatment for esophageal cancer. In unselected patients it does not improve overall survival, but has palliative benefits in a subgroup of difficult-to-treat patients with a short-life expectancy[40]. Unfortunately, only a few cancer drivers have enzymatic activities that are targetable in this fashion, and whether a target is druggable becomes a research question[41]. Once a drug target is verified, drugs or experimental compounds may be developed. Several databases are available for search, including the Therapeutic Target Database[42] and DrugBank 4.0[43].
Table 1.
National clinical trials on targeted therapy of esophageal squamous cell carcinoma1
| Target | Agent | NCT number (phase) |
| EGFR | Erlotinib | NCT00045526 (II), NCT00030498 (I), NCT00397384 (I), NCT00524121 (II), NCT01013831 (I), NCT01561014 (I), NCT01752205 (III) |
| Gefitinib | NCT00093652 (I/II), NCT00258297 (II), NCT00258323 (II), NCT00268346 (II), NCT00290719 (I) | |
| Icotinib | NCT01973725 (II) | |
| Lapatinib | NCT00239200 (II), NCT01666431 (II) | |
| Nimotuzumab | NCT02272699 (II/III), NCT01232374 (II), NCT01336049 (II), NCT01402180 (II/III), NCT01486992 (II), NCT01688700 (II), NCT01993784 (I/II), NCT02011594 (II), NCT02034968 (II), NCT02041819 (II) | |
| Panitumumab | NCT01077999 (II), NCT01262183 (II), NCT01627379 (III) | |
| PF00299804 | NCT01608022 (II) | |
| Cetuximab | NCT02123381 (II), NCT00109850 (II), NCT00165490 (II), NCT00381706 (II), NCT00397384 (I), NCT00397904 (II), NCT00425425 (I/II), NCT00445861 (I/II), NCT00509561 (II/III), NCT00544362 (I/II), NCT00655876 (III), NCT00757549 (0), NCT00815308 (II), NCT01034189 (II), NCT01107639 (III) | |
| IGF1R | Cixutumumab | NCT01142388 (II) |
| PI3K | BKM120 | NCT01626209 (I), NCT01806649 (II) |
| BYL719 | NCT01822613 (I/II) | |
| Rigosertib | NCT01807546 (II) | |
| HDAC | Entinostat | NCT00020579 (I) |
| Vorinostat | NCT00537121 (I), NCT01249443 (I) | |
| HER3 | LJM716 | NCT01598077 (I), NCT01822613 (I/II) |
| VEGFR | Vandetanib | NCT00732745 (I) |
| Sorafenib | NCT00917462 (II) | |
| VEGFA | Bevacizumab | NCT01212822 (II) |
| PD-L1 | MEDI4736 | NCT01938612 (I) |
| Bcl-2 mRNA | Oblimersen | NCT00003103 (I/II) |
| CDK9 | Alvocidib | NCT00006245 (II) |
| CRM1 | Selinexor | NCT02213133 (II) |
| FGFR | AZD4547 | NCT01795768 (II) |
| KIF11 | Litronesib | NCT01059643 (II) |
| TACSTD2 | IMMU-132 | NCT01631552 (I/II) |
1"Esophageal squamous cell carcinoma" was searched at the website (www.clinicaltrials.gov). Targeted therapy has been or is being tried in 62/204 studies. Some of these agents target multiple molecules, for example, lapatinib (EGFR and Erbb2), rigosertib (PI3K and PLK), vandetanib (VEGFR, EGFR, and RET), and sorafenib (VEGFR, PDGFR and RAF).
If the target is not druggable, its regulatory proteins or functional pathway may be targeted. For example, cyclin D1 amplification is commonly seen in human ESCC. As cyclin D1 mainly functions through CDK activation, CDK4 and CDK6 can be targeted instead of cyclin D1[44]. TP53, which encodes p53, is the most commonly mutated gene in human ESCC. Instead of targeting TP53, many strategies have been tested to restore the functions of p53 by delivering wildtype TP53, targeting the MDM2-p53 interaction, restoring the functions of mutant p53, targeting p53 family proteins, or eliminating the mutation in p53[45,46].
In addition to selecting drugs for targeted therapy, analysis of drug-metabolism genes in germ-line DNA can also optimize dosing and identify drug toxicity risk[47,48]. With the help of a database, such as Pharmacogenetics and Pharmacogenomics Knowledge Base, genetic variations can be associated with drug response[49].
ISSUES IN TARGETED THERAPY
Cancer heterogeneity
Various combinations of drivers and pathways result in intratumoral, intermetastatic, intrametastatic, or interpatient heterogeneities. It may explain why the same treatment brings about either a favorable response or resistance in different patients, and why a patient that responds well initially can develop resistance over time. Intratumoral heterogeneity has been validated using single-cell RNA-seq of primary glioblastomas[50]. As the majority of cancer gene mutations appear in multiple regions of the same tumor, single-region sequencing may be adequate to identify the majority of cancer gene mutations[51]. It can be predicted that most cancer cells in the same tumor may share the major alterations. If this is proven true in ESCC, it will make treatment more predictable.
Intermetastatic and intrametastatic heterogeneity may not be a great concern. Despite many years of research, we have failed to identify a group of so-called metastasis genes. Metastasis is probably stochastic depending on the environment in the metastatic site[52]. Therefore, if we can understand genetic and epigenetic alterations in the primary tumor well, all cancer cells left at the primary site or metastatic sites would be expected to behave in the same way. Nevertheless, the prevalence of different patterns of tumor heterogeneity needs to be more robustly assessed in large patient cohorts, and new patterns will probably be identified as the wealth of genomic data of ESCC is analyzed[53].
Drug resistance
If carcinogenesis is regarded as an evolutionary process with successive new mutations driven by natural selection, chemotherapy, radiotherapy, and target therapy may all provide a potent source of artificial selection to alter clonal dynamics. Consequently, the antitumor therapy may lead to resistance[54]. Indeed, targeted therapy is associated with a high rate of resistance at the very beginning when vemurafenib, a BRAFV600E inhibitor, was clinically used for melanoma. Combination of a BRAFV600E inhibitor (dabrafenib) and a MEK inhibitor (trametinib) resulted in better response, yet did not prevent resistance from occurring. Distinct mechanisms include mutations in the target, reactivation of the targeted pathway, hyperactivation of alternative pathways, and cross-talk with the microenvironment[55]. Resistant cells may undergo a process called phenotype switching under the selection of targeted therapy[56]. Understanding these mechanisms has led to additional efforts in finding new therapies targeting the same target, the same pathway, or alternative pathways[57-59].
Three strategies are feasible measures in the handling of drug resistance. Before treatment, both bioinformatics and experimental modeling can provide information concerning heterogeneity[60-62]. There is a need to develop clinically useful measures of heterogeneity[63]. Secondly, during treatment, limited success can be achieved with a single agent. The combination strategy may be the best way to refrain from the inevitable development of resistance to single drug-targeted therapies[31]. Thirdly, longitudinal tumor sampling will be essential to decipher the impact of tumor heterogeneity on cancer evolution, and developing minimally invasive methods to profile heterogeneous tumor genomes will play a major part in following clonal dynamics in real time[61]. For ESCC, repeated biopsy, circulating tumor DNA analysis[64,65], and exfoliative cells[66,67] are all valid options for this purpose.
Exceptional responders
As opposed to drug resistance, exceptional responders are patients who have a unique response to treatments that are not effective for most other patients. The National Cancer Institute has embarked on the Exceptional Responders Initiative to understand the molecular underpinnings of exceptional responses to treatment in cancer patients. In the past, exceptional responders led to clinical breakthroughs in treatments of certain types of cancer, and understanding of novel molecular mechanisms of carcinogenesis[68]. It is foreseeable that careful characterization and follow-up of these exceptional responders will be of great value in the future practice of personalized and targeted therapy of ESCC.
Side effects
As compared with traditional chemotherapy, targeted therapy is better tolerated. However, it does produce toxicities based on several major mechanisms, including on-target, off-target, hypersensitivity-related, and metabolite-induced toxicities. Vascular endothelial growth factor receptor inhibitors cause hypertension, and epidermal growth factor receptor inhibitors cause toxicities in tissues where they normally play an important functional role in tissue maintenance (e.g., skin and gastrointestinal epithelia). Some of these on-target toxicities may serve as surrogate biomarkers for clinical response[69-73]. Considering these potential side effects, clinical oncologists should be prepared to educate the patients and undertake respective preventive and therapeutic measures.
RESEARCH APPROACHES FOR TARGETED THERAPY
For genomics-guided research, cell line-based platforms have become an indispensable tool[74,75]. Clarification of genetic and epigenetic alterations of established ESCC cell lines would be great tools for preclinical drug development[76,77], in particular, the KYSE series of ESCC cell lines that have been sequenced[7-9]. Patient-derived ESCC cells can be used for selection of potential individualized therapy[78,79]. These cells are particularly useful in identifying effective drug combinations for acquired resistance[57].
Several models have been put into preclinical research and even clinical applications. A patient-derived xenograft model of ESCC is created when cancerous tissue from a patient’s primary tumor is implanted directly into immunodeficient mice. This model provides solutions to the translational challenges that researchers and clinicians face in cancer drug research and selection[80,81]. Carcinogen-induced models, for example, the N-nitrosomethylbenzylamine-induced model, represent classical models for ESCC research. They mimic human ESCC in not only etiology and histopathology, but also in molecular alterations (e.g., TP53 mutations[82,83]). However, exactly how well this model can mimic human ESCC at the genomics level has not been well studied. Whole exome sequencing has already shown that carcinogen-induced and genetically engineered models lead to carcinogenesis through different routes. A carcinogen-induced model is particularly important in understanding the complex mutation spectra seen in human cancers[84]. It is encouraging that genomic alterations in 4-nitroquinoline 1-oxide-induced mouse tongue cancer are well preserved[83].
Genetically engineered mouse models of human cancers have proven essential to dissect the molecular mechanisms behind carcinogenesis[85] and provide robust preclinical platforms for investigating drug efficacy[86] and resistance[87-89]. As an example, transgenic overexpression of Sox2, an amplified oncogene in ESCC[90], drives the complete process of carcinogenesis in mice[91]. This model can readily be used for preclinical drug development for SOX2-overexpressing ESCC. Although it may be difficult to target SOX2 itself, its downstream genes or pathways, such as the Akt/mTOR pathway, can be targeted[79]. Biochemical outcomes may be used for assessment of the efficacy of a Sox2-targeting therapy even when it does not reduce tumor incidence or size in mice. Genome engineering with CRISPR-Cas9 in vivo is an extremely promising technique in identifying cancer-driver genes and testing drug targets[92]. It may ultimately be used for human gene therapy in the future[93].
As a hallmark of human cancer and a crucial determinant of variable response to treatment[75], genomic heterogeneity calls for revision of clinical trial design currently in use in order to implement personalized therapy[94]. The majority of traditional prospective clinical trials are disease or histopathology based. Genomics-driven trials, for example, mutation-, pathway-, and subtype-based trials, will be more widely used in drug development[95]. Two genomics-based study designs are currently being utilized to develop targeted therapies, and for exploratory and multi-agent sequential design[96]. ESCC fits both study designs very well because the esophagus can be biopsied before and after treatment.
FUTURE PERSPECTIVES
The biggest challenge in ESCC treatment is the translation of genomic discoveries into personalized therapies based on strategies sketched from patients’ individual profiles[94]. The evasiveness of cancer cells has been a frustrating observation of clinical oncologists. Vogelstein et al[16] proposed that “there is order in cancer,” pointing to the need to tackle ESCC as a disease status with its own homeostatic mechanisms. From the perspective of ten hallmarks of human cancer[97], Hanahan[98] proposed three strategically distinct “battlespace-guided plans” for cancer treatment: disruption of the enemy’s many capabilities, defense against cancer’s armed forces, and integration of the geographies of the battlefields. It is clear that combination therapy targeting multiple mechanisms would be the only option in the future. Using immunotherapy as an example, tremelimumab (anti-CTLA4) has been tested as a second-line therapy for esophageal cancer. Although the clinical response was not impressive, its biologic effect on T-cell activation seemed to be associated with clinical response[99]. Recent development of immunotherapy based on ERBB2IP mutation-specific CD4+ T cells[100] and programmed-death ligand 1 (PD-L1) suppression is also quite promising. For patients in which pre-existing immunity is suppressed by PD-L1, blocking PD-L1 enhanced anti-cancer immunity (including one case of esophageal cancer)[101]. A realistic option in the near future can be a combination of target drugs and traditional chemoradiotherapy for ESCC. Target drugs are expected to kill cancer cells with specific genomic alterations, while traditional therapy acts in a much broader manner.
Technical issues continue to represent large hurdles for next-generation sequencing and bioinformatics, and they prevent us from gaining full insights into the mechanisms of carcinogenesis and metastasis of ESCC. Nonetheless, whole genome sequencing correlates with incomplete coverage of inherited disease genes, low reproducibility of genetic variation with the highest potential clinical effects, and uncertainty about clinically reportable sequencing findings[102]. Whole exome sequencing is particularly prone to errors, as only 61% of the mutated genes in ESCC are transcribed[8]. This is similar to what has been observed in pancreatic cancer: only 63% of the expected 251 driver-gene mutations were identified, suggesting a 37% false-negative rate. Marked discrepancies in the detection of missense mutations in identical cell lines (57.38%) have been reported due to inadequate sequencing of GC-rich areas of the exome[103]. The protein-coding genes account for only about 1.5% of the total genome. Although the vast majority of the alterations in noncoding regions are presumably passengers, some of these may be drivers, for example, mutations in the Tert promoter[104,105].
New computational and bioinformatics tools still need to be developed and improved due to low concordance of multiple variant-calling pipelines[106,107]. Directly comparing genome sequence reads may improve data quality as compared with initial alignment of reads to a reference genome[108].
Apart from the logistic challenges, financial, social and ethical challenges are also posed by personalized and targeted therapy[39]. In addition to viewing a patient’s cancer as a biologic phenomenon waiting for medical attention alone, personalized therapy emphasizes biopsychosocial care by including communication and information giving, psychologic and emotional well-being, enhancement of function, addressing financial and spiritual concerns, and providing symptom control and social support[109]. If we look at one specific patient’s ESCC from all these perspectives, a tumor board should involve not only medical staff but also supporting staff (Figure 1).
Figure 1.
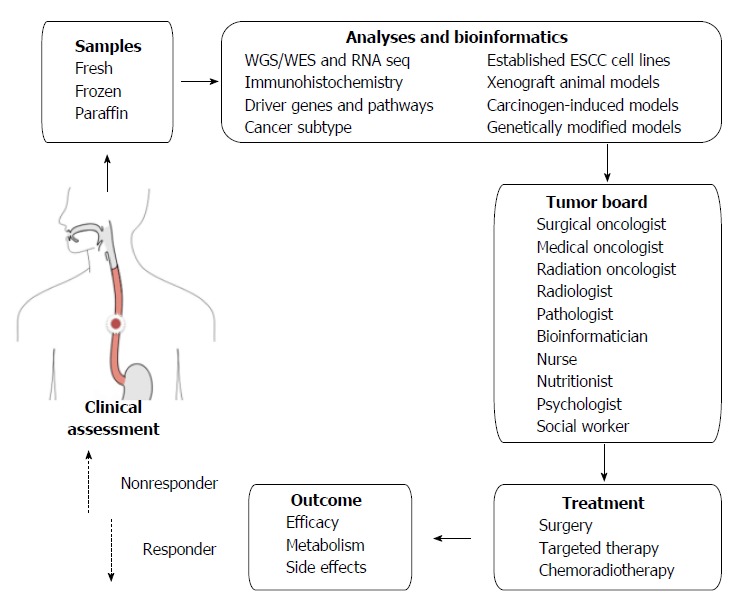
Personalized and targeted therapy for esophageal squamous cell carcinoma. The strategy is based on the concept that a patient’s genetic makeup should guide his/her treatment. After a series of molecular analyses on tumor samples, bioinformatics is expected to identify driver genes, pathways, cancer subtype, and target drugs. A tumor board will synthesize all information and generate a personalized treatment plan. Nonresponders may be analyzed in a similar manner during subsequent surveillance and further treated. ESCC: Esophageal squamous cell carcinoma; WES: Whole exome sequencing; WGS: Whole genome sequencing.
Footnotes
Supported by Grants from Beijing Academic Leaders Program, NO. 2009-2-17; Beijing Natural Science Foundation, No. 7102029; Capital Medical Developed Research Fund, No. 2007-1023; New Scholar Star Program of Ministry of Education; National Basic Research Program of China, No. 2011CB504300; Specialized Research Fund for the Doctoral Program of Higher Education, No. 20130001110108; National Natural Science Foundation for Distinguished Young Scholars, No. 81301748; Science Fund for Creative Research Groups of the National Natural Science Foundation of China, No. IRT13003 and No. NIH/NCI U54 CA156735.
Conflict-of-interest statement: Dr. D’Amico TA serves as a consultant for Scanlan; other authors have no potential conflicts of interest relevant to this article to disclose.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: February 4, 2015
First decision: March 10, 2015
Article in press: April 28, 2015
P- Reviewer: Hsu PK, Sato Y S- Editor: Yu J L- Editor: AmEditor E- Editor: Wang CH
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama H, Tsurumaru M, Udagawa H, Kajiyama Y. Radical lymph node dissection for cancer of the thoracic esophagus. Ann Surg. 1994;220:364–372; discussion 372-373. doi: 10.1097/00000658-199409000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schweigert M, Dubecz A, Stein HJ. Oesophageal cancer--an overview. Nat Rev Gastroenterol Hepatol. 2013;10:230–244. doi: 10.1038/nrgastro.2012.236. [DOI] [PubMed] [Google Scholar]
- 4.Grünberger B, Raderer M, Schmidinger M, Hejna M. Palliative chemotherapy for recurrent and metastatic esophageal cancer. Anticancer Res. 2007;27:2705–2714. [PubMed] [Google Scholar]
- 5.Rustgi AK, El-Serag HB. Esophageal carcinoma. N Engl J Med. 2014;371:2499–2509. doi: 10.1056/NEJMra1314530. [DOI] [PubMed] [Google Scholar]
- 6.Ajani JA, Barthel JS, Bentrem DJ, D’Amico TA, Das P, Denlinger CS, Fuchs CS, Gerdes H, Glasgow RE, Hayman JA, et al. Esophageal and esophagogastric junction cancers. J Natl Compr Canc Netw. 2011;9:830–887. doi: 10.6004/jnccn.2011.0072. [DOI] [PubMed] [Google Scholar]
- 7.Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM, Zhang F, Zhao ZR, Li ZT, Liu ZY, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46:1097–1102. doi: 10.1038/ng.3076. [DOI] [PubMed] [Google Scholar]
- 8.Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, Meng X, Sato Y, Okuno Y, Varela AM, Ding LW, et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet. 2014;46:467–473. doi: 10.1038/ng.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng H, Zheng R, Guo Y, Zhang S, Zou X, Wang N, Zhang L, Tang J, Chen J, Wei K, et al. Cancer survival in China, 2003-2005: a population-based study. Int J Cancer. 2015;136:1921–1930. doi: 10.1002/ijc.29227. [DOI] [PubMed] [Google Scholar]
- 10.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agrawal N, Jiao Y, Bettegowda C, Hutfless SM, Wang Y, David S, Cheng Y, Twaddell WS, Latt NL, Shin EJ, et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012;2:899–905. doi: 10.1158/2159-8290.CD-12-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nones K, Waddell N, Wayte N, Patch AM, Bailey P, Newell F, Holmes O, Fink JL, Quinn MC, Tang YH, et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat Commun. 2014;5:5224. doi: 10.1038/ncomms6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15:585–598. doi: 10.1038/nrg3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torkamani A, Schork NJ. Identification of rare cancer driver mutations by network reconstruction. Genome Res. 2009;19:1570–1578. doi: 10.1101/gr.092833.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komatsu M, Sasaki H. DNA methylation is a key factor in understanding differentiation phenotype in esophageal squamous cell carcinoma. Epigenomics. 2014;6:567–569. doi: 10.2217/epi.14.56. [DOI] [PubMed] [Google Scholar]
- 19.Cheng CP, Kuo IY, Alakus H, Frazer KA, Harismendy O, Wang YC, Tseng VS. Network-based analysis identifies epigenetic biomarkers of esophageal squamous cell carcinoma progression. Bioinformatics. 2014;30:3054–3061. doi: 10.1093/bioinformatics/btu433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamborero D, Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Kandoth C, Reimand J, Lawrence MS, Getz G, Bader GD, Ding L, et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci Rep. 2013;3:2650. doi: 10.1038/srep02650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.An O, Pendino V, D’Antonio M, Ratti E, Gentilini M, Ciccarelli FD. NCG 4.0: the network of cancer genes in the era of massive mutational screenings of cancer genomes. Database (Oxford) 2014;2014:bau015. doi: 10.1093/database/bau015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abbott KL, Nyre ET, Abrahante J, Ho YY, Isaksson Vogel R, Starr TK. The Candidate Cancer Gene Database: a database of cancer driver genes from forward genetic screens in mice. Nucleic Acids Res. 2015;43:D844–D848. doi: 10.1093/nar/gku770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alamo P, Gallardo A, Di Nicolantonio F, Pavón MA, Casanova I, Trias M, Mangues MA, Lopez-Pousa A, Villaverde A, Vázquez E, et al. Higher metastatic efficiency of KRas G12V than KRas G13D in a colorectal cancer model. FASEB J. 2015;29:464–476. doi: 10.1096/fj.14-262303. [DOI] [PubMed] [Google Scholar]
- 24.Park JT, Johnson N, Liu S, Levesque M, Wang YJ, Ho H, Huso D, Maitra A, Parsons MJ, Prescott JD, et al. Differential in vivo tumorigenicity of diverse KRAS mutations in vertebrate pancreas: A comprehensive survey. Oncogene. 2015;34:2801–2806. doi: 10.1038/onc.2014.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Ye Y, Sun H, Shi G. Association between KRAS codon 13 mutations and clinical response to anti-EGFR treatment in patients with metastatic colorectal cancer: results from a meta-analysis. Cancer Chemother Pharmacol. 2013;71:265–272. doi: 10.1007/s00280-012-2005-9. [DOI] [PubMed] [Google Scholar]
- 26.Hou JP, Ma J. DawnRank: discovering personalized driver genes in cancer. Genome Med. 2014;6:56. doi: 10.1186/s13073-014-0056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nevins JR. Pathway-based classification of lung cancer: a strategy to guide therapeutic selection. Proc Am Thorac Soc. 2011;8:180–182. doi: 10.1513/pats.201006-040MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gatza ML, Lucas JE, Barry WT, Kim JW, Wang Q, Crawford MD, Datto MB, Kelley M, Mathey-Prevot B, Potti A, et al. A pathway-based classification of human breast cancer. Proc Natl Acad Sci USA. 2010;107:6994–6999. doi: 10.1073/pnas.0912708107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez-Perez A, Mustonen V, Reva B, Ritchie GR, Creixell P, Karchin R, Vazquez M, Fink JL, Kassahn KS, Pearson JV, et al. Computational approaches to identify functional genetic variants in cancer genomes. Nat Methods. 2013;10:723–729. doi: 10.1038/nmeth.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877–1888. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 32.Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012;22:398–406. doi: 10.1101/gr.125567.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shang L, Liu HJ, Hao JJ, Jiang YY, Shi F, Zhang Y, Cai Y, Xu X, Jia XM, Zhan QM, et al. A panel of overexpressed proteins for prognosis in esophageal squamous cell carcinoma. PLoS One. 2014;9:e111045. doi: 10.1371/journal.pone.0111045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe H, Ma Q, Peng S, Adelmant G, Swain D, Song W, Fox C, Francis JM, Pedamallu CS, DeLuca DS, et al. SOX2 and p63 colocalize at genetic loci in squamous cell carcinomas. J Clin Invest. 2014;124:1636–1645. doi: 10.1172/JCI71545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verhaegh W, van Ooijen H, Inda MA, Hatzis P, Versteeg R, Smid M, Martens J, Foekens J, van de Wiel P, Clevers H, et al. Selection of personalized patient therapy through the use of knowledge-based computational models that identify tumor-driving signal transduction pathways. Cancer Res. 2014;74:2936–2945. doi: 10.1158/0008-5472.CAN-13-2515. [DOI] [PubMed] [Google Scholar]
- 37.Hofree M, Shen JP, Carter H, Gross A, Ideker T. Network-based stratification of tumor mutations. Nat Methods. 2013;10:1108–1115. doi: 10.1038/nmeth.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, Ostos LC, Lannon WA, Grotzinger C, Del Rio M, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, Kalyana-Sundaram S, Sam L, Balbin OA, Quist MJ, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011;3:111ra121. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dutton SJ, Ferry DR, Blazeby JM, Abbas H, Dahle-Smith A, Mansoor W, Thompson J, Harrison M, Chatterjee A, Falk S, et al. Gefitinib for oesophageal cancer progressing after chemotherapy (COG): a phase 3, multicentre, double-blind, placebo-controlled randomised trial. Lancet Oncol. 2014;15:894–904. doi: 10.1016/S1470-2045(14)70024-5. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y, McGee J, Chen X, Doman TN, Gong X, Zhang Y, Hamm N, Ma X, Higgs RE, Bhagwat SV, et al. Identification of druggable cancer driver genes amplified across TCGA datasets. PLoS One. 2014;9:e98293. doi: 10.1371/journal.pone.0098293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin C, Zhang C, Zhu F, Xu F, Chen SY, Zhang P, Li YH, Yang SY, Wei YQ, Tao L, et al. Therapeutic target database update 2014: a resource for targeted therapeutics. Nucleic Acids Res. 2014;42:D1118–D1123. doi: 10.1093/nar/gkt1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 45.Hong B, van den Heuvel AP, Prabhu VV, Zhang S, El-Deiry WS. Targeting tumor suppressor p53 for cancer therapy: strategies, challenges and opportunities. Curr Drug Targets. 2014;15:80–89. doi: 10.2174/1389450114666140106101412. [DOI] [PubMed] [Google Scholar]
- 46.Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13:217–236. doi: 10.1038/nrd4236. [DOI] [PubMed] [Google Scholar]
- 47.McLeod HL. Cancer pharmacogenomics: early promise, but concerted effort needed. Science. 2013;339:1563–1566. doi: 10.1126/science.1234139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harper AR, Topol EJ. Pharmacogenomics in clinical practice and drug development. Nat Biotechnol. 2012;30:1117–1124. doi: 10.1038/nbt.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thorn CF, Klein TE, Altman RB. PharmGKB: the pharmacogenetics and pharmacogenomics knowledge base. Methods Mol Biol. 2005;311:179–191. doi: 10.1385/1-59259-957-5:179. [DOI] [PubMed] [Google Scholar]
- 50.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, Seth S, Chow CW, Cao Y, Gumbs C, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346:256–259. doi: 10.1126/science.1256930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Komori J, Boone L, DeWard A, Hoppo T, Lagasse E. The mouse lymph node as an ectopic transplantation site for multiple tissues. Nat Biotechnol. 2012;30:976–983. doi: 10.1038/nbt.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramos P, Bentires-Alj M. Mechanism-based cancer therapy: resistance to therapy, therapy for resistance. Oncogene. 2014:Epub ahead of print. doi: 10.1038/onc.2014.314. [DOI] [PubMed] [Google Scholar]
- 56.Kemper K, de Goeje PL, Peeper DS, van Amerongen R. Phenotype switching: tumor cell plasticity as a resistance mechanism and target for therapy. Cancer Res. 2014;74:5937–5941. doi: 10.1158/0008-5472.CAN-14-1174. [DOI] [PubMed] [Google Scholar]
- 57.Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger P, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346:1480–1486. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, Ebbesen SH, Ainscough BJ, Ramu A, Iyer G, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature. 2015;518:240–244. doi: 10.1038/nature13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martz CA, Ottina KA, Singleton KR, Jasper JS, Wardell SE, Peraza-Penton A, Anderson GR, Winter PS, Wang T, Alley HM, et al. Systematic identification of signaling pathways with potential to confer anticancer drug resistance. Sci Signal. 2014;7:ra121. doi: 10.1126/scisignal.aaa1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Diaz LA, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kreso A, O’Brien CA, van Galen P, Gan OI, Notta F, Brown AM, Ng K, Ma J, Wienholds E, Dunant C, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339:543–548. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 64.Spellman PT, Gray JW. Detecting cancer by monitoring circulating tumor DNA. Nat Med. 2014;20:474–475. doi: 10.1038/nm.3564. [DOI] [PubMed] [Google Scholar]
- 65.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kadri S, Lao-Sirieix P, Fitzgerald RC. Developing a nonendoscopic screening test for Barrett’s esophagus. Biomark Med. 2011;5:397–404. doi: 10.2217/bmm.11.40. [DOI] [PubMed] [Google Scholar]
- 67.Sepehr A, Razavi P, Saidi F, Salehian P, Rahmani M, Shamshiri A. Esophageal exfoliative cytology samplers. A comparison of three types. Acta Cytol. 2000;44:797–804. doi: 10.1159/000328564. [DOI] [PubMed] [Google Scholar]
- 68.Subbiah IM, Subbiah V. Exceptional responders: in search of the science behind the miracle cancer cures. Future Oncol. 2015;11:1–4. doi: 10.2217/fon.14.204. [DOI] [PubMed] [Google Scholar]
- 69.Liu S, Kurzrock R. Toxicity of targeted therapy: Implications for response and impact of genetic polymorphisms. Cancer Treat Rev. 2014;40:883–891. doi: 10.1016/j.ctrv.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 70.Pessi MA, Zilembo N, Haspinger ER, Molino L, Di Cosimo S, Garassino M, Ripamonti CI. Targeted therapy-induced diarrhea: A review of the literature. Crit Rev Oncol Hematol. 2014;90:165–179. doi: 10.1016/j.critrevonc.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 71.Jensen SB, Peterson DE. Oral mucosal injury caused by cancer therapies: current management and new frontiers in research. J Oral Pathol Med. 2014;43:81–90. doi: 10.1111/jop.12135. [DOI] [PubMed] [Google Scholar]
- 72.Macdonald JB, Macdonald B, Golitz LE, LoRusso P, Sekulic A. Cutaneous adverse effects of targeted therapies: Part II: Inhibitors of intracellular molecular signaling pathways. J Am Acad Dermatol. 2015;72:221–236; quiz 237-238. doi: 10.1016/j.jaad.2014.07.033. [DOI] [PubMed] [Google Scholar]
- 73.Macdonald JB, Macdonald B, Golitz LE, LoRusso P, Sekulic A. Cutaneous adverse effects of targeted therapies: Part I: Inhibitors of the cellular membrane. J Am Acad Dermatol. 2015;72:203–218; quiz 219-220. doi: 10.1016/j.jaad.2014.07.032. [DOI] [PubMed] [Google Scholar]
- 74.Abaan OD, Polley EC, Davis SR, Zhu YJ, Bilke S, Walker RL, Pineda M, Gindin Y, Jiang Y, Reinhold WC, et al. The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res. 2013;73:4372–4382. doi: 10.1158/0008-5472.CAN-12-3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sharma SV, Haber DA, Settleman J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat Rev Cancer. 2010;10:241–253. doi: 10.1038/nrc2820. [DOI] [PubMed] [Google Scholar]
- 76.Ahmed D, Eide PW, Eilertsen IA, Danielsen SA, Eknæs M, Hektoen M, Lind GE, Lothe RA. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis. 2013;2:e71. doi: 10.1038/oncsis.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martin D, Abba MC, Molinolo AA, Vitale-Cross L, Wang Z, Zaida M, Delic NC, Samuels Y, Lyons JG, Gutkind JS. The head and neck cancer cell oncogenome: a platform for the development of precision molecular therapies. Oncotarget. 2014;5:8906–8923. doi: 10.18632/oncotarget.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shimada Y, Maeda M, Watanabe G, Yamasaki S, Komoto I, Kaganoi J, Kan T, Hashimoto Y, Imoto I, Inazawa J, et al. Cell culture in esophageal squamous cell carcinoma and the association with molecular markers. Clin Cancer Res. 2003;9:243–249. [PubMed] [Google Scholar]
- 79.Gen Y, Yasui K, Nishikawa T, Yoshikawa T. SOX2 promotes tumor growth of esophageal squamous cell carcinoma through the AKT/mammalian target of rapamycin complex 1 signaling pathway. Cancer Sci. 2013;104:810–816. doi: 10.1111/cas.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu X, Zhang J, Zhen R, Lv J, Zheng L, Su X, Zhu G, Gavine PR, Xu S, Lu S, et al. Trastuzumab anti-tumor efficacy in patient-derived esophageal squamous cell carcinoma xenograft (PDECX) mouse models. J Transl Med. 2012;10:180. doi: 10.1186/1479-5876-10-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang J, Jiang D, Li X, Lv J, Xie L, Zheng L, Gavine PR, Hu Q, Shi Y, Tan L, et al. Establishment and characterization of esophageal squamous cell carcinoma patient-derived xenograft mouse models for preclinical drug discovery. Lab Invest. 2014;94:917–926. doi: 10.1038/labinvest.2014.77. [DOI] [PubMed] [Google Scholar]
- 82.Wang D, Weghorst CM, Calvert RJ, Stoner GD. Mutation in the p53 tumor suppressor gene in rat esophageal papillomas induced by N-nitrosomethylbenzylamine. Carcinogenesis. 1996;17:625–630. doi: 10.1093/carcin/17.4.625. [DOI] [PubMed] [Google Scholar]
- 83.Onken MD, Winkler AE, Kanchi KL, Chalivendra V, Law JH, Rickert CG, Kallogjeri D, Judd NP, Dunn GP, Piccirillo JF, et al. A surprising cross-species conservation in the genomic landscape of mouse and human oral cancer identifies a transcriptional signature predicting metastatic disease. Clin Cancer Res. 2014;20:2873–2884. doi: 10.1158/1078-0432.CCR-14-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Westcott PM, Halliwill KD, To MD, Rashid M, Rust AG, Keane TM, Delrosario R, Jen KY, Gurley KE, Kemp CJ, et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature. 2015;517:489–492. doi: 10.1038/nature13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tuveson DA, Jacks T. Technologically advanced cancer modeling in mice. Curr Opin Genet Dev. 2002;12:105–110. doi: 10.1016/s0959-437x(01)00272-6. [DOI] [PubMed] [Google Scholar]
- 86.Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov. 2006;5:741–754. doi: 10.1038/nrd2110. [DOI] [PubMed] [Google Scholar]
- 87.Pirazzoli V, Nebhan C, Song X, Wurtz A, Walther Z, Cai G, Zhao Z, Jia P, de Stanchina E, Shapiro EM, et al. Acquired resistance of EGFR-mutant lung adenocarcinomas to afatinib plus cetuximab is associated with activation of mTORC1. Cell Rep. 2014;7:999–1008. doi: 10.1016/j.celrep.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rottenberg S, Nygren AO, Pajic M, van Leeuwen FW, van der Heijden I, van de Wetering K, Liu X, de Visser KE, Gilhuijs KG, van Tellingen O, et al. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc Natl Acad Sci USA. 2007;104:12117–12122. doi: 10.1073/pnas.0702955104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41:1238–1242. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu K, Jiang M, Lu Y, Chen H, Sun J, Wu S, Ku WY, Nakagawa H, Kita Y, Natsugoe S, et al. Sox2 cooperates with inflammation-mediated Stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell. 2013;12:304–315. doi: 10.1016/j.stem.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sánchez-Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A, Joshi NS, Subbaraj L, Bronson RT, Xue W, et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature. 2014;516:428–431. doi: 10.1038/nature13906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 94.Wheeler DA, Wang L. From human genome to cancer genome: the first decade. Genome Res. 2013;23:1054–1062. doi: 10.1101/gr.157602.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roychowdhury S, Chinnaiyan AM. Translating genomics for precision cancer medicine. Annu Rev Genomics Hum Genet. 2014;15:395–415. doi: 10.1146/annurev-genom-090413-025552. [DOI] [PubMed] [Google Scholar]
- 96.Simon R, Roychowdhury S. Implementing personalized cancer genomics in clinical trials. Nat Rev Drug Discov. 2013;12:358–369. doi: 10.1038/nrd3979. [DOI] [PubMed] [Google Scholar]
- 97.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 98.Hanahan D. Rethinking the war on cancer. Lancet. 2014;383:558–563. doi: 10.1016/S0140-6736(13)62226-6. [DOI] [PubMed] [Google Scholar]
- 99.Ralph C, Elkord E, Burt DJ, O’Dwyer JF, Austin EB, Stern PL, Hawkins RE, Thistlethwaite FC. Modulation of lymphocyte regulation for cancer therapy: a phase II trial of tremelimumab in advanced gastric and esophageal adenocarcinoma. Clin Cancer Res. 2010;16:1662–1672. doi: 10.1158/1078-0432.CCR-09-2870. [DOI] [PubMed] [Google Scholar]
- 100.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344:641–645. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dewey FE, Grove ME, Pan C, Goldstein BA, Bernstein JA, Chaib H, Merker JD, Goldfeder RL, Enns GM, David SP, et al. Clinical interpretation and implications of whole-genome sequencing. JAMA. 2014;311:1035–1045. doi: 10.1001/jama.2014.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hudson AM, Yates T, Li Y, Trotter EW, Fawdar S, Chapman P, Lorigan P, Biankin A, Miller CJ, Brognard J. Discrepancies in cancer genomic sequencing highlight opportunities for driver mutation discovery. Cancer Res. 2014;74:6390–6396. doi: 10.1158/0008-5472.CAN-14-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhao Y, Gao Y, Chen Z, Hu X, Zhou F, He J. Low frequency of TERT promoter somatic mutation in 313 sporadic esophageal squamous cell carcinomas. Int J Cancer. 2014;134:493–494. doi: 10.1002/ijc.28360. [DOI] [PubMed] [Google Scholar]
- 105.Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA, Friedman AH, Friedman H, Gallia GL, Giovanella BC, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. 2013;110:6021–6026. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ding L, Wendl MC, McMichael JF, Raphael BJ. Expanding the computational toolbox for mining cancer genomes. Nat Rev Genet. 2014;15:556–570. doi: 10.1038/nrg3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.O’Rawe J, Jiang T, Sun G, Wu Y, Wang W, Hu J, Bodily P, Tian L, Hakonarson H, Johnson WE, et al. Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome Med. 2013;5:28. doi: 10.1186/gm432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Moncunill V, Gonzalez S, Beà S, Andrieux LO, Salaverria I, Royo C, Martinez L, Puiggròs M, Segura-Wang M, Stütz AM, et al. Comprehensive characterization of complex structural variations in cancer by directly comparing genome sequence reads. Nat Biotechnol. 2014;32:1106–1112. doi: 10.1038/nbt.3027. [DOI] [PubMed] [Google Scholar]
- 109.Cherny NI, de Vries EG, Emanuel L, Fallowfield L, Francis PA, Gabizon A, Piccart MJ, Sidransky D, Soussan-Gutman L, Tziraki C. Words matter: distinguishing “personalized medicine” and “biologically personalized therapeutics”. J Natl Cancer Inst. 2014;106:dju321. doi: 10.1093/jnci/dju321. [DOI] [PMC free article] [PubMed] [Google Scholar]