

Abstract
The causal role of aneuploidy in cancer initiation remains under debate since mutations of euploidy-controlling genes reduce cell fitness but aneuploidy strongly associates with human cancers. Telomerase activation allows immortal growth by stabilizing telomere length, but its role in aneuploidy survival has not been characterized. Here, we analyze the response of primary human cells and murine hematopoietic stem cells (HSCs) to aneuploidy induction and the role of telomeres and the telomerase in this process. The study shows that aneuploidy induces replication stress at telomeres leading to telomeric DNA damage and p53 activation. This results in p53/Rb-dependent, premature senescence of human fibroblast, and in the depletion of hematopoietic cells in telomerase-deficient mice. Endogenous telomerase expression in HSCs and enforced expression of telomerase in human fibroblasts are sufficient to abrogate aneuploidy-induced replication stress at telomeres and the consequent induction of premature senescence and hematopoietic cell depletion. Together, these results identify telomerase as an aneuploidy survival factor in mammalian cells based on its capacity to alleviate telomere replication stress in response to aneuploidy induction.
Keywords: aneuploidy, replication stress, senescence, telomerase, telomere
Introduction
Although being present in the majority of human cancers, the functional role of aneuploidy in tumorigenesis is still poorly understood. In humans and mice, most autosomal monosomies and trisomies are not compatible with organismal survival (Magnuson et al, 1985; Munne et al, 2004), and it was shown that aneuploidy impairs cellular proliferation, survival, and spontaneous immortalization (Segal & McCoy, 1974; Castedo et al, 2004; Zhivotovsky & Kroemer, 2004; Williams et al, 2008). In contrast to its negative effects on cell viability and immortalization, aneuploidy is a hallmark of cancer cells (Boveri, 2008; Gordon et al, 2012). Recent bioinformatic analysis revealed evidence that the number of genes that drive carcinogenesis is higher than previously thought and aneuploidy appears to be instrumental to shape the complexity of cancer genomes affecting multiple cancer driver genes (Davoli et al, 2013).
Studies on genetic mouse models provided experimental evidence that mutation in genes that control chromosome segregation promotes cancer formation by increasing loss of heterozygosity (Baker & van Deursen, 2010), but the effect on carcinogenesis was context dependent and aneuploidy suppressed tumorigenesis in some mouse strains (Weaver et al, 2007; Schvartzman et al, 2010). Genetic studies in yeast identified genes that influence cell survival in response to aneuploidy (Torres et al, 2010). Aneuploidy survival factors in human cells and its role in tumor formation remain yet to be defined. It is known that proliferation of aneuploid tumor cells is limited by the activation of ATM/p53-dependent DNA damage checkpoints (Li et al, 2010). However, mechanistically, it remains poorly understood how aneuploidy triggers DNA damage responses. Here, we characterized DNA damage response induced by aneuploidy. The study shows that aneuploidy induces telomere replication stress resulting in DNA damage responses, premature senescence, and hematopoietic stem cell depletion in mice. Telomerase is identified as an aneuploidy survival factor that increases cell viability in response to aneuploidy induction by alleviating telomere replication stress.
Results
Telomerase expression rescues aneuploidy-induced senescence
Current data suggest that a variety of cellular stress factors can lead to chromosome mis-segregation (Duijf & Benezra, 2013; Orr & Compton, 2013). Supported by recent iRNA screens on focus set libraries (Conery & Harlow, 2010), the number of aneuploidy-inducing gene mutations is higher than previously thought and involves a variety of cellular signaling pathways. In iRNA screen, we identified a set of gene knockdowns that increase aneuploidy in the human cancer cell line U2OS (Meena, Günes and Rudolph in preparation). Here, we analyzed the cellular responses to the knockdown of the top 3 enriched candidate genes (GJB3, RXFP1—also known as LGR7—and OSBPL3) in two primary human fibroblast cell lines (BJ and IMR90) as well as in murine hematopoietic stem and progenitor cells.
The top 3 candidate genes have not been directly associated with aneuploidy induction, but associations with cancer formation and ploidy-affecting pathways were reported. Specifically, OSBPL3 (ORP3) was described as an R-Ras interaction partner regulating cell adhesion (Lehto et al, 2008). Knockdown of RXFP1 (relaxin family peptide receptor 1) was shown to suppress the proliferation of prostate cancer cells (Feng et al, 2010). In addition, RXFP1 activates a wide spectrum of signaling pathways to generate second messengers, including cAMP and nitric oxide—two known signaling pathways influencing ploidy control (see Bathgate et al, 2013). Mutations in the connexin gene Cx31 (encoding the GJB3 protein) have been associated with deafness and erythrokeratodermia variabilis (EKV), a skin disorder accompanied by hyperproliferation of the skin (Richard et al, 1998). A close homolog of GJB3, the GJB5 (Cx31.1), acts as a tumor suppressor in NSCLC (Zhang et al, 2012), and connexin 32 (Cx32) knockout mice exhibit increased chemical- and radiation-induced liver and lung tumor formation (King & Lampe, 2004a,b).
In addition to these three top candidate genes, two control genes with a known role in ploidy control were included: (i) STARD9, a recently identified regulator of spindle pole assembly (Torres et al, 2011) that was also found in our screen to aggravate aneuploidy of human cancer cells and (ii) MAD2L1 (referred to as MAD2 hereafter), a well-characterized component of the spindle-assembly checkpoint that prevents the onset of anaphase until all chromosomes are properly aligned at the metaphase plate (Li & Benezra, 1996). To exclude off-target effects, two shRNAs were used per gene and the knockdown efficiencies of the shRNAs were verified by qPCR (Supplementary Fig S1A and B). The knockdown of the candidate genes in human primary cells led to a significant increase in chromosomal aberrations (Fig1A, Supplementary Fig S1C and D). Chromosome banding analysis revealed that most of the aberrations were whole chromosome gains and losses (Fig1B and Supplementary Table S1).
Figure 1.

Candidate shRNAs induce aneuploidy in primary human fibroblasts, and telomerase rescues aneuploidy-induced senescence
- The dot plot shows chromosome numbers of metaphase spreads from BJ cells that were infected with shRNAs targeting the indicated candidate genes (n = 20 metaphases per construct, P-values were calculated against the control scramble construct).
- Representative example of Giemsa-banding showing a gain of chromosome 11 in a metaphase of RXFP1 shRNA-infected BJ-hTERT cells.
- Growth curves of BJ and IMR90 fibroblasts that were not infected or infected with shRNAs targeting the indicated genes. The graphs depict mean values ± SEM. P-values were calculated by one-way ANOVA comparing pooled data for non-infected/scrambled shRNA-infected cells with pooled data of cells infected with shRNAs against candidate genes (three independent experiments per construct).
- Western blot analysis of phospho-p38, phospho-p53, p21 and GAPDH in protein lysates from early passage (passage 2) BJ cells infected with shRNAs against the indicated candidate genes.
- Growth curves of IMR90-ST/LT fibroblasts that were not infected or infected with shRNAs targeting the indicated genes. The graphs depict mean values ± SEM. P-values were calculated by one-way ANOVA comparing pooled data for non-infected/scrambled shRNA-infected cells with pooled data of cells infected with shRNAs against candidate genes (two independent experiments per construct).
- Chromosome numbers of metaphase spreads from IMR90-ST/LT cells that were infected with shRNAs targeting the indicated candidate genes (n = 20 metaphases per construct).
- The dot plot shows chromosome numbers of metaphase spreads from BJ-hTERT cells treated and analyzed as described in (A).
- Growth curves of BJ-hTERT and IMR90-hTERT fibroblast treated and analyzed as described in (C).
Source data are available online for this figure.
Aneuploidy induced by candidate gene-targeting shRNAs led to a premature loss of proliferation potential compared to control fibroblasts infected with a scrambled shRNA (Fig1C). Cessation of proliferation of fibroblasts infected with aneuploidy-inducing shRNAs was characterized by senescence morphology, senescence-associated β-galactosidase activity (Supplementary Fig S2B–D and F), phosphorylation of p53 and p38 (Fig1D), and induction of p21 expression (Fig1D and E, Supplementary Fig S2H and J). Combined inactivation of the p53 and Rb pathways by the SV-40 ST/LT antigens abrogated the induction of premature senescence and led to an increase in the number of chromosomal gains and losses in primary human cells that were targeted by aneuploidy-inducing shRNAs (Fig1F and G).
Together, these results indicated that the knockdown of candidate genes from our screen led to aneuploidy in primary human fibroblasts, but this resulted in p53/Rb-dependent checkpoint activation and premature senescence—a phenomenon that was previously described as aneuploidy-induced senescence (AIS) (Humbert et al, 2010; Lentini et al, 2012). Of note, the knockdown of MAD2 induced a stronger phenotype characterized by a more rapid growth arrest (Fig1C) and multinucleated cells (Supplementary Fig S2A). These effects stand in line with previous reports (Lentini et al, 2012). Due to this rapid proliferation arrest, it was not possible to prepare metaphase spreads from MAD2-deficient primary human fibroblasts. Therefore, the subsequent analyses focused on the analysis of aneuploidy-inducing candidate shRNAs from our screen that induced aneuploidy but a delayed growth arrest.
Primary human fibroblasts lack telomerase expression, and telomere shortening limits the proliferative lifespan of human fibroblasts to 50–70 cell divisions. It was shown that telomerase expression is sufficient to immortalize human fibroblasts by stabilizing telomere length, thus preventing the induction of DNA damage responses at dysfunctional telomeres in late passages of human fibroblasts (Bodnar et al, 1998; d’Adda di Fagagna et al, 2003). To test whether telomerase expression would influence the induction of AIS, shRNAs against GJB3, RXFP1, OSBPL3 and STARD9 were infected in BJ and IMR90 fibroblast cell lines that over-expressed telomerase reverse transcriptase (hTERT)—the limiting component for telomerase activity in human cells (Weinrich et al, 1997). In both fibroblast cell lines, TERT expression fully prevented the induction of DNA damage checkpoints and AIS in response to the infection of aneuploidy-inducing shRNAs (Fig1I and Supplementary Fig S2E, G, I and K). Of note, TERT-expressing human fibroblasts developed aberrations in chromosomal copy numbers to a similar extent as TERT-negative HFs in response to the knockdown of the candidate genes (Fig1A versus H). The suppression of AIS in response to the expression of aneuploidy-inducing shRNAs in human cells was dependent on the enzymatic activity of telomerase as a catalytically dead mutant of TERT (Hahn et al, 1999) was not able to rescue growth arrest and senescence in the same setting (Supplementary Fig S2L).
Together, the above result indicated that single-gene mutation can lead to aneuploidy in primary human cells, but aneuploid cells can only survive and continue to proliferate in the presence of telomerase or when p53/Rb-dependent checkpoints are defective.
Telomerase activity suppresses aneuploidy-induced telomere replication stress
The primary function of telomerase is to compensate for the loss of telomeric DNA in response to cell proliferation (Bodnar et al, 1998). It was also shown that telomerase activity prevents the accumulation of DNA damage and the induction of senescence in response to oncogene activation (Suram et al, 2012). To test whether aneuploidy induction would lead to accumulation of DNA damage and whether this would be influenced by telomerase gene status, the activation of DNA damage responses (DDR) was measured in telomerase-positive and telomerase-negative human fibroblasts infected with aneuploidy-inducing shRNAs. In agreement with the AIS phenotype (Fig1C–E, Supplementary Fig S2B–D, F, H and J), telomerase-negative BJ and IMR90 fibroblasts exhibited a strong accumulation of DNA damage (staining positive for both γH2AX and 53BP1) at early passage after shRNA transduction (Fig2A, C and D, Supplementary Fig S3A and B). In contrast, telomerase-positive BJ and IMR90 fibroblasts contained a significantly reduced number of DDR foci in response to the transduction of aneuploidy-inducing shRNAs (Fig2B and C, Supplementary Fig S3C and D).
Figure 2.

Aneuploidy induction in primary human fibroblasts provokes DNA damage responses (DDR) that are suppressed by telomerase
- A–D Analysis of DNA damage foci. (A–C) Quantification of nuclear 53BP1 foci: comparison of scrambled shRNA versus aneuploidy-inducing shRNAs in (A) BJ and (B) BJ-hTERT fibroblasts, and (C) comparison of BJ versus BJ-hTERT fibroblasts transduced with aneuploidy-inducing shRNAs (pooled data from shRNAs against GJB3, RXFP1, OSBPL3, STARD9). (D) Representative images of 53BP1 foci (scale bar: 50 μm).
- E–H Metaphase spreads were stained with a telomere-specific probe. The number of multi-telomeric signals (MTS) was analyzed (n = 15–30 metaphases per sample): comparison of scrambled shRNA versus aneuploidy-inducing shRNAs in (E) BJ and (F) BJ-hTERT fibroblasts. (G) Comparison of BJ versus BJ-hTERT fibroblasts transduced with aneuploidy-inducing shRNAs (pooled data from shRNAs against GJB3, RXFP1, OSBPL3, STARD9). (H) Representative images of metaphases derived from aneuploidy-induced BJ cells (scale bar: 5 μm). The arrows indicate multi-telomeric signals at the chromosome ends.
- I–M Quantification of BLM-coated anaphase bridges (APB). The percentage of Bloom coating was determined in those anaphases showing chromatid bridges (n = 30–50 APBs per sample were analyzed from asynchronously growing cells): comparison of scrambled shRNA versus aneuploidy-inducing shRNAs in (I) BJ and (J) BJ-hTERT fibroblasts. (K) Comparison of BJ versus BJ-hTERT fibroblasts transduced with aneuploidy-inducing shRNAs (pooled data from shRNAs against RXFP1, OSBPL3). (L, M) Representative images of an anaphase bridge (L) and a BLM-coated anaphase bridge (M) (scale bar: 5 μm).
Data information: All histograms depict mean values ± SEM; P-values were calculated by two-tailed t-test.
To explore the involvement of telomeres in the accumulation of DNA damage in human fibroblasts in response to shRNA-mediated aneuploidy induction, telomere length was determined by quantitative fluorescence in situ hybridization (qFISH) and telomere stability was analyzed in metaphase spreads using a telomere-specific FISH probe (fluorescence-labeled TTTAGG3 PNA probe). Telomere length was not affected by aneuploidy-inducing shRNAs in primary fibroblasts (Supplementary Fig S3E). However, increased numbers of fragile telomeres were observed in telomerase-negative BJ and IMR90 fibroblast cell lines in response to the transduction of aneuploidy-inducing shRNAs (Fig2E and H). Specifically, all four selected candidate shRNAs led to an increase in the number of chromosome ends exhibiting multiple telomere signals—a marker for telomere replication stress (van Steensel et al, 1998; Sfeir et al, 2009). This increase in multi-telomeric signals was not observed in telomerase-positive human fibroblasts infected with the same aneuploidy-inducing shRNAs (Fig2F and G).
Anaphase bridges represent a hallmark feature of chromosomal fusions that form in response to: (i) telomere shortening (Rudolph et al, 2001), (ii) mutations in telomeric DNA (Kirk et al, 1997), (iii) the deletion of telomere-binding proteins (van Steensel et al, 1998), or (iv) telomere replication stress (Chan et al, 2009; Sfeir et al, 2009). Coating of anaphase bridges by the Bloom DNA helicase is indicative of DNA replication stress as a cause of chromosomal fusions. These ultrafine Bloom-containing bridges form in the context of replication stress by linking sister chromatids at fragile sites that carry intrinsic difficulties to replicate (Chan et al, 2009; Sfeir et al, 2009; Barefield & Karlseder, 2012). Our analysis revealed a significant increase in anaphase bridges (Supplementary Fig S3F and H) and in Bloom-coated anaphase bridges (Fig2I–M) in telomerase-negative human fibroblasts expressing aneuploidy-inducing shRNAs compared to scrambled shRNA-infected cells. Of note, this phenotype was significantly ameliorated by telomerase expression (Fig2J and K, Supplementary Fig S3G and H).
Phosphorylated RPA2 (S33-pRPA2), a target of ATR kinase, represents a hallmark feature of DNA replication stress (Vassin et al, 2009; Crasta et al, 2012). Immunofluorescence staining depicted a strong increase in nuclear pRPA2 foci in telomerase-negative human fibroblast infected with aneuploidy-inducing shRNAs compared to scrambled shRNA-infected fibroblasts (Fig3A and C, Supplementary Fig S4A). Of note, this phenotype was completely rescued by telomerase expression (Fig3B). The suppression of the pRPA2 foci was dependent on the enzymatic activity of telomerase since the expression of a catalytically inactive TERT mutant (DN-hTERT) did not rescue the induction of pRPA2 foci in HFs expressing aneuploidy-inducing shRNAs (Supplementary Fig S4B).
Figure 3.

Telomerase rescues aneuploidy-induced replication stress
- A–C Analysis of phosphorylated RPA2 (pRPA2) foci by immunofluorescence staining: quantification of nuclear pRPA2 foci in (A) BJ and (B) BJ-hTERT fibroblasts; (C) representative images of pRPA2 foci (scale bar: 50 μm). Note that telomerase expression completely suppresses the induction of pRPA2 foci in BJ fibroblasts infected with aneuploidy-inducing shRNAs (B).
- D Co-staining of pRPA2 foci and telomeric DNA by FISH was used to quantify the number of pRPA2 foci co-localizing to telomeres in cells infected with aneuploidy-inducing shRNAs compared to scrambled shRNA-infected cells.
- E Representative image of immuno-FISH shows co-localization of pRPA2 foci and telomeric DNA. Here, 5 of 6 pRPA2 foci (dashed arrows) co-localize with telomeres (scale bar: 10 μm).
- F–H Chromatin immunoprecipitation (ChIP) was carried out using a pRPA2 antibody on lysates of cells infected with aneuploidy-inducing shRNAs or scrambled shRNA control. qPCR-based quantification of telomeric DNA in the immunoprecipitate of (F) BJ and (G) BJ-hTERT cells. (H) Quantification of telomeric DNA (telomere probe) and non-telomeric DNA (Alu probe) in the immunoprecipitate by dot blot and radioactive labeling. IgG-ChIP was used as a negative control. Note that aneuploidy-inducing shRNAs induced pRPA2 binding at telomeric DNA, which was rescued by telomerase expression. There was no detectable increase in pRPA2 foci at chromosomal localizations outside telomeres.
- I–L Quantification of cell cycle-dependent accumulation of pRPA2 foci formation in BJ cell infected with an aneuploidy-inducing shRNA against RXFP1 or with a scrambled shRNA control. Serum-starved, G1-arrested cells were re-stimulated and labeled at different time points with EdU. (I) Representative 2D cell cycle profile (EdU versus DAPI) at 24 h post-release from serum starvation. (J) Representative pictures of pRPA2 staining at the indicated cell cycle stages (scale bar: 10 μm). Quantification of pRPA2 foci at the indicated cell cycle stages in shRNA RXFP1- and scrambled shRNA-infected (K) BJ and (L) BJ-hTERT cells. Note the significant increase in pRPA2 foci in shRNA RXFP1-infected cells in late S-phase.
Data information: (A, B, D) Mean ± SEM, two-tailed t-test, n ≥ 50 cells per sample. (F, G) Mean ± SEM, two-tailed t-test, n = 3 replicates. (K, L) Mean ± SEM, one-way ANOVA + Tukey’s test, n = 3–7 replicates.
Source data are available online for this figure.
Telomeres are known to represent fragile sites containing tertiary DNA structures that are difficult to replicate, for example, G-quadruplexes and T-loops (Sfeir et al, 2009; Paeschke et al, 2011); for review see Gunes and Rudolph (2013). Since it is known from studies in yeast and ciliates that telomerase itself enables unfolding of telomeric G-quadruplexes (Paeschke et al, 2008) and rescues telomere replication stress (Chang et al, 2009), it was tempting to speculate that aneuploidy of human cells induced replication stress at telomeres, which was rescued by telomerase expression. In agreement with this hypothesis, immuno-FISH staining (combining a telomere-specific FISH probe with a pRPA2 antibody) revealed that > 80% of pRPA2 foci that formed in response to aneuploidy induction in telomerase-negative HFs co-localized with telomeres (Fig3D and E). Furthermore, quantification of telomeric DNA after chromatin immunoprecipitation (ChIP) with a pRPA2 antibody revealed a significant enrichment of telomeric DNA in telomerase-negative human fibroblasts infected with aneuploidy-inducing shRNAs compared to scrambled shRNA-infected cells (Fig3F and H). The enrichment of pRPA2-bound telomeric DNA in human fibroblasts that were infected with aneuploidy-inducing shRNAs was completely suppressed by telomerase expression (Fig3G and H).
Studies in ciliates showed that telomerase assists the opening of telomeric G-quadruplexes (required for telomere replication) by interacting with ciliate POT1/TPP1 complex (Paeschke et al, 2008). Interestingly, POT1 was one of the hits in one of our repeat screens on aneuploidy-inducing shRNAs (data not shown). Using two different shRNAs against POT1 confirmed that POT1 knockdown led to an induction of aneuploidy in primary human fibroblast (Supplementary Fig S5A and B). Similar to the results on the other aneuploidy-inducing shRNAs (see above), POT1 knockdown led to telomeric DNA damage and AIS (Supplementary Fig S5A, C, E, G and I). Telomerase overexpression rescued induction of telomeric DNA damage and AIS in response to POT1 knockdown (Supplementary Fig S5D, F and H). These data suggest that telomerase can rescue telomere replication when POT1 levels become limiting. The data do not exclude the possibility that the remaining 15–30% of POT1 expression in our shRNA experiments (Supplementary Fig S5A) contribute to the telomerase-mediated rescue in telomere integrity by mediating telomerase binding to telomeres. Interestingly, studies on Pot1b null mice revealed that telomerase haploinsufficiency severely impairs the survival of Pot1b-deficient mice by impairing the maintenance of highly proliferating stem and progenitor cell compartments (He et al, 2009). These data suggest that telomerase can rescue telomere replication in a Pot1b-independent manner in mice.
Replication of human telomeres occurs at distinct time points during S-phase with a subset of telomeres replicating in late S-phase (Arnoult et al, 2010). We therefore determined the appearance of pRPA2 foci at early, mid and late S-phase by high-content microscopy analysis of synchronized cells (Fig3I and J). These experiments revealed that pRPA2 foci accumulated in late S in telomerase-negative human fibroblasts infected with RXFP1-shRNA compared to scrambled shRNA-infected cells (Fig3K). Again, pRPA2 foci were suppressed by telomerase expression (Fig3L). Together, these data indicated that telomere replication stress evolves in response to aneuploidy induction in late S-phase in telomerase-negative human fibroblast, but this phenotype is suppressed by telomerase expression.
Transient tetraploidy can be an initiating event leading to the induction of aneuploidy (Andreassen et al, 1996; Fujiwara et al, 2005). To examine whether tetraploidy would also provoke telomere replication stress and a senescence arrest as seen in AIS, primary human fibroblasts were treated with the cytokinesis inhibitor dihydrocytochalasin B (DCB). In accordance with previous studies (Ganem et al, 2014), DCB treatment resulted in rapid induction of tetraploidy (Supplementary Fig S6A–D). DCB treatment induced a rapid proliferation arrest, which, however, was independent of telomerase gene status and not rescued by telomerase overexpression (Supplementary Fig S6E). Moreover, tetraploidy induction by DCB treatment did not result in an accumulation of replication stress as determined by the accumulation of pRPA2-foci (Supplementary Fig S6E). These data indicate that tetraploidy-induced growth arrest involves different mechanisms than AIS. A recent study indicated that the Hippo pathway, specifically the phosphorylation of the transcription co-activator YAP1, contributes to the induction of proliferation arrest in response to tetraploidy (Ganem et al, 2014). In contrast, experiments on the here identified aneuploidy-inducing shRNAs show that aneuploidy induction in primary BJ fibroblasts does not lead to a significant increase in phosphorylated YAP1 (Supplementary Fig S6G).
Together, these data indicate that replication stress at telomeres is induced by aneuploidy but not by tetraploidy. A possible explanation indicates that imbalances in the expression of replication factors could contribute to aneuploidy-induced replication stress, which can be rescued by telomerase expression. Instead, tetraploidy-induced senescence involves a different mechanism (activation of the Hippo pathway) that is not associated with telomere replication stress and is telomerase independent.
Mammalian stem and progenitor cells express endogenous levels of telomerase (Morrison et al, 1996). The finding from this study showing that that telomerase expression ameliorates AIS could therefore be relevant for aneuploidy-induced transformation of stem and progenitor cells. This question is of particular interest given that stem and progenitor cells were identified as the cell type of origin of cancer formation in cancer mouse models and cancer patients (Alcantara Llaguno et al, 2009; Barker et al, 2009; Jan et al, 2012; Welch et al, 2012; Genovese et al, 2014; Jaiswal et al, 2014). To directly test whether endogenous levels of telomerase were required for survival of stem cells in response to aneuploidy induction in an in vivo setting, freshly isolated hematopoietic stem and progenitor cells (HSPCs) from wild-type and first-generation telomerase knockout mice (TERC−/−) were infected with a scrambled shRNA or aneuploidy-inducing shRNAs (targeting GJB3 or OSBPL3, Supplementary Fig S7A) and transplanted into lethally irradiated recipients. These mouse experiments confirmed that the knockdown of candidate genes induced aneuploidy in murine hematopoietic cells (Lin-negative) re-isolated from primary recipients (Fig4A and B). Of note, in response to aneuploidy induction, the in vivo maintenance of hematopoietic cells from mTERC−/− mice was compromised compared to hematopoietic cells from mTERC+/+ mice as indicated by a decrease in peripheral blood cell chimerism 6 weeks after transplantation of targeted cells into lethally irradiated recipient (Fig4C). These results indicated that endogenous telomerase expression improved the long-term survival of aneuploid hematopoietic stem and progenitor cells in vivo.
Figure 4.
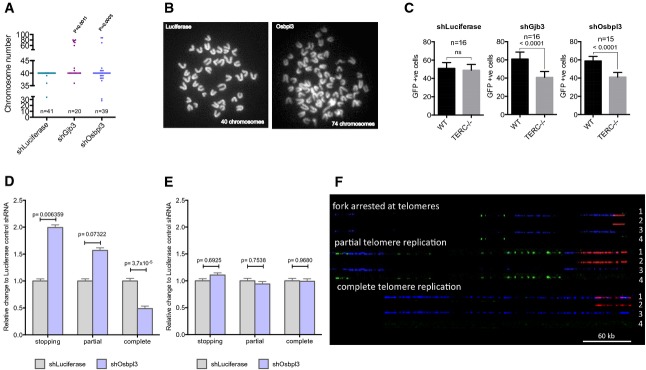
- A, B Knockdown of the indicated genes by mouse-specific shRNAs (shGJB3 and shOSBPL3) induces aneuploidy in mouse hematopoietic stem cells (HSCs). (A) Metaphase countings and (B) representative metaphase pictures from mouse lineage-negative HSPCs derived from the transplanted mice (n: numbers are indicated). Fisher’s exact test was used to calculate significance.
- C In vivo survival of murine HSPCs (telomerase positive or negative) carrying shRNA (GFP-expressing vector) of control luciferase (left), Gjb3 (middle) and Osbpl3 (right), assessed from transplantation assay in lethally irradiated mice. See Materials and Methods for experimental details. Bar diagram shows 6-weeks GFP chimerism of CD45.1 (wild-type, WT) and CD45.2 (TERC−/−) cells in peripheral blood of transplanted mice (mean ± SEM, two-tailed t-test, n = 15–16 mice/group).
- D, E Quantification of DNA replication patterns at telomeres in freshly isolated G1-TERC (D) or wild-type (E) HSPCs with the shOsbpl3 vector or, as a control, shLuciferase vector. See (F) for representative pictures. Counted DNA molecule numbers: n = 80 for G1-Luci, n = 99 for G1 Osbpl3, n = 100 for WT-Luci and n = 79 for WT-Osbpl3. Error bars indicate the standard error of the mean (SEM). P-values were calculated by chi-square test.
- F Representative images of DNA replication patterns occurring at telomeres. A fork arresting at a telomere was defined as an event in which the DNA replication signal stops within 2 kb from the telomeric border; partial telomere replication was defined as an event in which the DNA replication signal is only partially overlapping in length with the telomeric signal; complete telomere replication was defined as an event in which the DNA replication signal completely overlaps with the telomeric signal. Scale bar: 60 kb. 1: merge; 2: PNA-Telomeric Probe (red); 3: IdU (blue); 4: CldU (green). Freshly isolated wild-type (WT) or telomerase knockout (G1-TERC) mouse HSPCs carrying shRNA against Osbpl3 or, as a control, against firefly luciferase were labeled with thymidine analogues following a pulse and chase protocol. DNA was then isolated, combed and processed by immuno-FISH (see Supplementary Fig S7 for details).
To directly test whether endogenous telomerase expression in HSPCs was sufficient and required to suppress telomere replication stress in response to aneuploidy induction, DNA combing experiments were combined with telomere fluorescence in situ hybridization on freshly isolated HSPCs from recipient mice transplanted with mTERC+/+ of mTERC−/− HSPCs that were infected with shRNAs against the OSBPL3 candidate gene or a control shRNA against Luciferase. These experiments demonstrated that aneuploidy induction resulted in impaired telomeric DNA replication in telomerase-negative HSPCs, but endogenous telomerase expression in mTERC+/+ HSPCs was sufficient to suppress this phenotype (Fig4D–F and Supplementary Fig S7B and C).
Discussion
It has previously been noted that aneuploidy induces ATM-dependent activation of DNA damage signals and senescence, also referred to as aneuploidy-induced senescence (AIS) (Humbert et al, 2010; Li et al, 2010). However, the mechanistic basis of the activation of this tumor suppressor checkpoint in response to aneuploidy remained elusive. This study demonstrates that aneuploidy induces replication stress at telomeres, and this represents the main cause of AIS, which can be bypassed by telomerase expression. The relevance of this new concept is supported by in vivo experiments demonstrating that endogenous telomerase expression in murine hematopoietic stem cells is sufficient to abrogate telomere replication stress and the depletion of stem cells in response to aneuploidy induction. Recently, it was reported that genome-wide DNA replication stress by itself can contribute to the generation of structural and numerical chromosome abnormalities in tumor cells (Burrell et al, 2013). However, as shown here, the outcome is different in primary cells where aneuploidy induces telomere replication stress limiting the proliferation of cells that lack telomerase expression. Given the increasing evidences that aneuploidy contributes to the transformation of cells during cancer formation (Davoli et al, 2013), it is tempting to speculate that telomere replication stress contributes to tumor suppression in somatic cells that lack telomerase expression.
It will be of future interest to delineate molecular mechanisms that contribute to the induction of replications stress at telomeres in response to aneuploidy. Studies on DNA replication indicated that some components of the replication machinery represent limiting factors that need to be re-utilized for completion of DNA replication (Douglas & Diffley, 2012). It is conceivable that alteration in chromosome copy numbers results in imbalances in the amount of molecules required for replication and the amount of genomic DNA that needs to be replicated. Such imbalances would eventually lead to replication stress at chromosomal regions that replicate late during S-phase such as certain telomeres (Arnoult et al, 2010). In agreement with this assumption, the current study shows that replication stress in response to aneuploidy induction evolves in late S-phase (Fig3I–L). In addition to the timing of replication, specificities in the tertiary structure of telomeres likely contribute to the concentration of replication stress at telomeres in response to aneuploidy induction. Telomeres form tertiary structures, for example, G-quadruplexes and T-Loops, that are difficult to replicate and can be a source of genomic instability (Sfeir et al, 2009; Paeschke et al, 2011). Experimental evidences indicate that specific DNA-helicases (Ribeyre et al, 2009; Paeschke et al, 2011, 2013), telomere-binding proteins (Sfeir et al, 2009), and telomerase itself (Paeschke et al, 2008) are required for the opening of telomere tertiary structures and for the faithful replication of these structures (for review see Gunes & Rudolph, 2013).
The current study could extend our understanding of the role of telomerase in cancer formation. Studies in yeast provided the first evidence that the telomerase enzyme contributes to telomere replication by enabling the unwinding G-quadruplexes (Paeschke et al, 2008; Chang et al, 2009). Here, described experiments indicate that telomerase has a conserved function in alleviating replication stress at telomeres that occurs in response to aneuploidy induction in mammalian cells. The findings could be of particular importance for the transformation of tissue stem cells that are known to express high levels of telomerase (Morrison et al, 1996; Ritz et al, 2005) and represent an important cell type of origin for the evolution of cancer in both humans and mice (Alcantara Llaguno et al, 2009; Barker et al, 2009; Welch et al, 2012). It is tempting to speculate that telomerase expression although being essential to ensure prolonged proliferation of stem and progenitor cells also renders these cells more vulnerable to undergo transformation in response to aneuploidy-inducing gene mutations by abrogating the efficiency of aneuploidy-induced senescence (Fig5). Of note, the increase in cancer incidence in humans with Down Syndrome predominantly affects stem cell system with intrinsically high levels of telomerase activity, for example, the germline and the hematopoietic system (Satge et al, 1998; Ayed et al, 2012).
Figure 5.
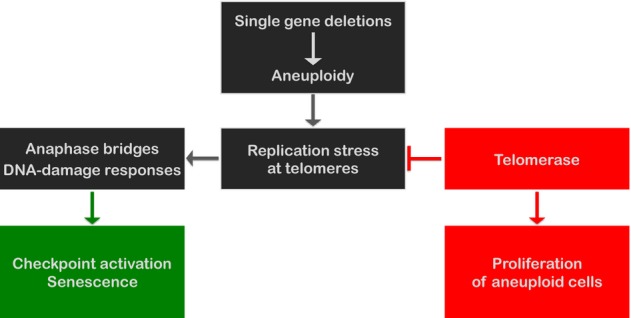
Model for aneuploidy-induced proliferation arrest and telomerase-induced survival of aneuploid cells
Schematic diagram representing how aneuploidy induced by single-gene knockdown could lead to senescence by the induction of replication stress at telomeres and DNA damage. Telomerase activity alleviates telomere replication stress and facilitates continuous proliferation of aneuploidy cells.
Materials and Methods
Plasmid DNA
Lentiviral constructs are pGIPZ-TurboGFP-Puro for all human shRNA constructs and library, and retroviral constructs are pMSCV-Neo-hTERT and pMSCV-Neo-hTERT-DN. IMR90-ST/LT expression vector was generated by subcloning the SV40 small + Large T from pENTR1A Plasmid [SV40 small + Large T in pENTR1A (w611-7) was a gift from Eric Campeau (Addgene plasmid # 22297)] into pBABE-Bla vector.
Cell lines and growth curves
293T, BJ and IMR90 cells were purchased from ATCC. BJ-hTERT and IMR90-hTERT were generated by transduction with the pMSCV-Neo-hTERT retroviral vector. IMR90-ST/LT cells were generated by transduction with the pBABE-Bla-ST/LT vector. All cells were cultured in DMEM high glucose medium (Sigma), penicillin (100 unit/ml), streptomycin (100 μg/ml) (Sigma) and 10% FBS (Lonza) at 37°C and 5% CO2 atmosphere. For growth curves, 5 × 105 cells were seeded in 6-well plates and counted using a hemocytometer and automated cell counter after reaching 70–80% confluency. Population doublings (PD) were calculated using n = (logN-logN0)/log2. N = number of cells after n divisions, N0 = number of cells at time point 0, n = PD difference.
DNA transfection and transduction
All plasmid DNAs were amplified using maxi-prep kit (Promega) in the E. coli DH5α strain. The calcium phosphate method was used for transfection of lentiviral or retroviral construct together with VSVG and GAG/POL (lenti or retro) into 293T cells to produce virus. Virus supernatant was collected at 36 and 48 h after transfection. For transduction, targeted cells were incubated with medium containing virus for 24 h. 8 μg/ml polybrene was added to the medium for enhanced transduction. Twenty-five hours later, appropriate antibiotic selection was started (puromycin 1 μg/ml, neomycin 500 μg/ml, blasticidin 2 μg/ml).
Chimerism analyses by in vivo transplantation of hematopoietic stem and progenitor cells (HSPCs)
Lin-ve HSPCs were isolated from CD45.1 (Ly5.1) wild-type and CD45.2 (Ly5.2) first-generation telomerase knockout (G1-TERC−/−) mice. Cells were cultured (over night) in serum-free stem cells maintaining medium (STEM CELL technologies, 09650) supplemented with cytokines mSCF (50 ng/ml; Peprotech, 250-03) and TPO (50 ng/ml; Peprotech, 315-14). On the next morning, cells were infected for 12 h with lentivirus harboring shRNA against the candidate genes. Twelve hours later, medium was changed with stem cell maintaining medium and kept in culture. Two days post-infection, cells were sorted for GFP-expressing Lin-ve HSC and mixed in equal numbers from both (Ly5.1 and Ly5.2) genotypes. Mixed cells (50% wild-type + 50% TERC−/−) were transplanted in lethally irradiated mice and analyzed 6 weeks after transplantation.
Senescence-associated β-galactosidase assay
Cells were fixed in 4% PFA and washed 3× with PBS and incubated with beta-Gal staining solution (40 mM citric acid sodium phosphate buffer, 1 M NaCl, 5 mM ferrocyanide, 5 mM ferricyanide, 2% DMF, 20 mM MgCl2, X-GAL 1 mg/ml in DMSO) 14–16 h at 37°C. Stained cells were counted under a bright microscope.
Microscopy
Microscopic analysis of bright-field, immunofluorescence and FISH stainings were carried out on Leica and Zeiss systems with the help of appropriate software. Fluorescence microscopy for DNA molecular combing was performed on an UltraVIEW VoX (Perkin-Elmer) spinning disk confocal unit, equipped an EclipseTi inverted microscope (Nikon), a C9100-50 emCCD camera (Hamamatsu) and driven by Volocity software (Improvision, Perkin-Elmer). Images were acquired using a 40×/0.95 NA dry objective. The right focal plane was maintained using the Nikon Perfect Focus system.
Metaphase spreads and telomere qFISH (quantitative fluorescence in situ hybridization)
Cells were treated with 0.035 μg/ml KaryoMAX® Colcemid (Life Technologies; 15212-012) at 70–80% confluency and incubated for 2–6 h at 37°C depending on the proliferation rate of cell lines. Cells were then treated with hypotonic solution (0.075 M KCL) at 37°C and fixed with freshly prepared Carnoy’s fixative (3:1, methanol: acetic acid) on ice and washed subsequently 3× with Carnoy’s fixative and spread on humid slides.
For chromosome counting analysis, metaphases were stained with DAPI mounting medium (VECTASHIELD; H-1200) and chromosomes were counted under a fluorescence microscope. For metaphase qFISH analysis, slides containing metaphase spreads were dried over night at room temperature and rehydrated in PBS, fixed in 4% PFA and washed in PBS, treated with pepsin solution (0.1% pepsin, 0.031% HCl in H2O) at 37°C and dehydrated in ethanol series (70, 95 and 100%). Completely dried slides were hybridized with fluorescently labeled telomere PNA probe (Panagene, TelC-Cy3, F1002) (1 mM Tris pH 7.2, 21.4 μl MgCl2 buffer (25 mM MgCl2/9 mM citric acid/82 mM Na2HPO4), 175 μl deionized formamide, 5 μl PNA probe (25 μg/ml) and 12.5 μl blocking reagent (Roche, 10% solution)) at 80 °C and incubated over night in a humid chamber at 4°C. Slides were washed 2× with 70% formamide, 10 mM Tris pH 7.2, 0.1% BSA solution, 28 ml ddH2O, 3× TBS and 2× PBS. Slides were mounted with DAPI-containing fluorescence medium and analyzed under a fluorescence microscope.
Metaphase countings in the mice experiments were performed with lineage-negative (Lin-) cells. For this experiment, lineage-negative mice HSCs were isolated, kept in culture over night and infected with lentiviral particle harboring the indicated shRNA-encoding vectors. After 48 h, cells were transplanted into lethally irradiated mice. Six weeks post-transplantation, mice were killed, lineage-negative cells were isolated, and metaphases were prepared as described above.
Immunofluorescence and IF-FISH (immunofluorescence–fluorescence in situ hybridization)
Cells were grown on coverslips, fixed with 4% PFA, followed by 2 washes with cold PBS, permeabilized with 0.25% Triton X-100 in PBS, 3× washed with PBS, and blocked with 1% BSA in PBST at room temperature. Coverslips were then incubated with antibody solution in 1% BSA PBST. Antibodies were anti-phospho-H2AX (Ser139) (Millipore; 07-164), anti-53BP1 (Abcam; ab36823), anti-p21 (F-5) (Santa Cruz; sc-6246), anti-BLM (Sigma-Aldrich; HPA005689) and anti-Phospho RPA32 (S33) (Bethyl; A300-246A). Cells were incubated with antibody solution over night at 4°C followed by 3 washes in PBS, 1 h incubation with an appropriate secondary antibody labeled with fluorophore, anti-mouse Cy3 (Sigma, C2181), anti-rabbit Cy3 (Jackson ImmunoResearch, 111-165-006) and anti-rabbit Cy5 (Invitrogen, 81-6116) and 3 washes in PBS. Finally, samples were mounted with DAPI fluorescence mounting medium and analyzed under a fluorescence microscope. For IF-FISH, immunofluorescence stainings were done as described above, and slides were shortly fixed in 2% PFA at room temperature. And then, slides were processed for telomere qFISH as described before.
Analysis of DNA replication dynamics at telomeres by DNA combing and FISH
DNA molecular combing coupled with FISH was performed as described in Suram et al (2012) with minor modifications. HSCs were sequentially labeled with 25 μM IdU for 1 h, followed by 200 μM CldU for one additional hour in the culture medium. Since mouse HSCs were maintained in suspension, no medium changes were performed between the two labelings, and a higher concentration of the second analogue was used in order to compete over the first one during incorporation into the replicating DNA. Cells were then harvested and centrifuged twice and incubated for 3 h in media without IdU/CldU. This procedure was repeated three times. Halogenated nucleotides were detected with specific primary antibodies (IdU: mouse anti-BrdU, Becton Dickinson; CldU: rat anti-BrdU, Abcam) and secondary antibodies (Alexa Fluor 647-conjugated goat anti-mouse, Molecular Probes; Alexa Fluor 488-conjugated goat anti-rat; Molecular Probes). Images were acquired with a spinning disk fluorescence microscope, and labeled DNA molecules were individually manually measured by ImageJ software and analyzed by Excel.
Chromatin immunoprecipitation (ChIP)
To crosslink protein and DNA, 1% formaldehyde (final concentration) was added to cell culture dishes containing medium and neutralized by adding 0.125 M glycine solution. Cells were washed in ice cold PBS and collected by scraping in PBS. Cell membrane was lysed by NP-40 lysis buffer and then treated with micrococcal nuclease at 37°C. Nuclei were lysed using nucleus lysis buffer, and digested chromatin was sonicated to get equal sizes of chromatin fragments (average size about 500 base pairs). Obtained chromatin was incubated with rabbit IgG and anti-phospho RPA32 (S4/S8) (Bethyl; A300-245A) antibody over night at 4°C. Chromatin was then incubated with protein A agarose beads/ssDNA to capture the antibody and washed with high-salt buffer, low-salt buffer, LiCl buffer and TE buffer at 4°C. Antibody/protein/DNA complexes were eluted by incubating the washed samples at RT in IP dilution buffer containing SDS and Triton X-100. To reverse the formaldehyde crosslink of chromatin and DNA, RNase A and NaCl were added and incubated at 65°C for 5 h followed by ethanol precipitation. The pellet was dissolved in nuclease-free H2O and treated with Proteinase K at 45°C for 2 h. Chromatin DNA was extracted with phenol/chloroform/isoamyl alcohol and chloroform/isoamyl alcohol and then precipitated with ethanol in the presence of tRNA and glycogen at −80°C for 1 h. Precipitate was dissolved in nuclease-free water. HSCs were cultured in SFEM medium supplemented with 10 ng/ml IL-3 (Peprotech; 213-13).
RNA extraction and qPCR
RNA was isolated by Trifast (PeqGold, PeqLab) according to the recommendations of the supplier. For RT–PCR, cDNA was prepared from 1 μg total RNA using Go script reverse transcriptase (Promega; A5000). Quantitative PCR (qPCR) was performed using the iQ SYBR Green super mix (Bio-Rad; 170-8880) by Applied Biosystem 7300 real-time PCR system. All samples were normalized to GAPDH.
Sequences of hairpin (from human pGIPZ shRNA library)
GJB3: TGCTGTTGACAGTGAGCGACCACTCTGAGTTCACTAAGTTTAGTGAAGCCACAGATGTAAACTTAGTGAACTCAGAGTGGGTGCCTACTGCCTCGGA
and
TGCTGTTGACAGTGAGCGAAAACAAGCGAATTAACTATCTTAGTGAAGCCACAGATGTAAGATAGTTAATTCGCTTGTTTCTGCCTACTGCCTCGGA
RXFP1: TGCTGTTGACAGTGAGCGCCCACAAGACCATTTAAAGAAATAGTGAAGCCACAGATGTATTTCTTTAAATGGTCTTGTGGTTGCCTACTGCCTCGGA
and
TGCTGTTGACAGTGAGCGCACGAGACTCAATTCCTATTCATAGTGAAGCCACAGATGTATGAATAGGAATTGAGTCTCGTTTGCCTACTGCCTCGGA
OSBPL3: TGCTGTTGACAGTGAGCGCGGCACAAACCTGTGTTTATATTAGTGAAGCCACAGATGTAATATAAACACAGGTTTGTGCCATGCCTACTGCCTCGGA
and
TGCTGTTGACAGTGAGCGCTCCGGTTCTTGGAGAAACATATAGTGAAGCCACAGATGTATATGTTTCTCCAAGAACCGGATTGCCTACTGCCTCGGA
STARD9: TGCTGTTGACAGTGAGCGACGAGTTATGACGAAACTTATTTAGTGAAGCCACAGATGTAAATAAGTTTCGTCATAACTCGCTGCCTACTGCCTCGGA
and
TGCTGTTGACAGTGAGCGACCTCAACAGCTCAGTCAGCAATAGTGAAGCCACAGATGTATTGCTGACTGAGCTGTTGAGGCTGCCTACTGCCTCGGA
MAD2: TGCTGTTGACAGTGAGCGCGGTCCTGAAAGTAACTCATAATAGTGAAGCCACAGATGTATTATGAGTTACTTTCAGGACCTTGCCTACTGCCTCGGA
POT1: TGCTGTTGACAGTGAGCGAGCTATTACAACTTACAACTAATAGTGAAGCCACAGATGTATTAGTTGTAAGTTGTAATAGCGTGCCTACTGCCTCGGA
and
TGCTGTTGACAGTGAGCGCCGAACAAGTTTAATAGTGTAATAGTGAAGCCACAGATGTATTACACTATTAAACTTGTTCGATGCCTACTGCCTCGGA
shRNA hairpin sequences used in mouse transplantation experiments
Gjb3: TGCTGTTGACAGTGAGCGACAAGGTGTTTCACACCCTAATTAGTGAAGCCACAGATGTAATTAGGGTGTGAAACACCTTGCTGCCTACTGCCTCGGA
and
TGCTGTTGACAGTGAGCGATCACCATCTGTGAGATCTGCTTAGTGAAGCCACAGATGTAAGCAGATCTCACAGATGGTGAGTGCCTACTGCCTCGGA
Osbpl3: TGCTGTTGACAGTGAGCGCCAGGAGGGTTCTTGTACTAAATAGTGAAGCCACAGATGTATTTAGTACAAGAACCCTCCTGATGCCTACTGCCTCGGA
and
TGCTGTTGACAGTGAGCGACAAGATGTGAGATGGAAAAATTAGTGAAGCCACAGATGTAATTTTTCCATCTCACATCTTGCTGCCTACTGCCTCGGA
Luciferase: TGCTGTTGACAGTGAGCGCCCGCCTGAAGTCTCTGATTAATAGTGAAGCCACAGATGTATTAATCAGAGACTTCAGGCGGTTGCCTACTGCCTCGGA
Primer sequences for qPCR (all from 5′ to 3′)
Human
GJB3-F: CACTCCAGGCCCTACTGAG; GJB3-R: GCATGTGACGAAGATGAGCTG; RXFP1-F: CAAGTGCTCCCTTGGCTATTT; RXFP1-R: TTGTCCTCATCGGCCTGATTC; OSBPL3-F: GTCATCCGCCCTAGCACAAAA; OSBPL3-R: AGAGACTCGGCATGGATTCTG; STARD9-F: AGGTGGACAATCGACCAGATG; STARD9-R: CCAGTAGCAGTAATCAAAGCCAA; MAD2-F: GGACTCACCTTGCTTGTAACTAC; MAD2-R: GATCACTGAACGGATTTCATCCT
Mouse
Osbpl3-F: TTTTTCTGGCCCTGTGAGACT; Osbpl3-R: TCCCTGGAGTTTTCGGATGAC; Gjb3-F: GCTCCAAGACCTATTGAGTGGC; Gjb3-R: GCCTGGTGTTACAGTCAAAGTC
Telomere amount quantification by qPCR
Telomere amounts were determined essentially as described previously (Gonzalez et al, 2014). In brief, amplification plot of telomeres or HBG was determined for the linear range according to the telomere or HBG standard. Signal in the ChIPed sample was subtracted by telomere signal of IgG control and corrected for DNA amount by HBG. Finally, values obtained with the indicated shRNAs were calculated relative to the values obtained with the scrambled shRNA and plotted.
PCRs for telomere quantification
Telomere: 10 μl Bio-Rad 2× SYBR mix, 25 mM DTT, 1 μl Tel1b (2 μM), 1 μl Tel2b (18 μM), 2 μl sample and 4 μl nuclease-free H2O.
HBG: 10 μl Bio-Rad 2× SYBR mix, 1 μl HBG 3 (6 μM), 1 μl HBG 4 (14 μM), 2 μl sample and 6 μl nuclease-free H2O.
Telomere qPCR primers
Tel1b 5′-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT
Tel2b 5′-GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT
HBG qPCR primers
HBG 3 5′-TGTGCTGGCCCATCACTTTG
HBG 4 5′-ACCAGCCACCACTTTCTGATAGG
Telomere standard
TTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGG
HBG standard
CCACACCAGCCACCACTTTCTGATAGGCAGCCTGCACTGGTGGGGTGAATTCTTTGCCAAAGTGATGGGCCAGCACACAGA
Dot blot
A total of 50 ng ChIPed DNA was denatured incubating for 10 min at 99°C with 0.4 N NaOH, and 10 mM EDTA. Denatured DNA samples were applied to hybond N+ (Amersham; RPN119B) membrane using a dot-blot apparatus connected to a vacuum source and washed with 2× SSC once. DNA was denatured on a membrane using 1.5 M NaCl/0.5 N NaOH for 10 min and then neutralized using 1.5 N NaCl/0.5 M Tris pH 7. DNA was UV crosslinked using a UV Stratagene crosslinker. Membrane was prehybridized by rapid hybridization buffer (Amersham; RPN1635) for 1 h and incubated over night with radiolabeled telomere (TTAGGG)3 or Alu probe (5′-CGGGAAGCAGAGGTTGTAGTGAGCC). Reaction mixture was 1 μl telomere restriction fragment, 13.5 μl H2O, 2 μl T4 PNK buffer, 2.5 μl 32P-γATP and 1 μl T4-polynucleotide kinase (New England Biolabs) at 37°C. The membrane was washed the next day once with 0.1% SDS/2× SSC buffer and twice with 0.1% SDS/1× SSC. Radiolabeled membranes were exposed to a phosphorimager (Fuji), and signal intensities were quantified after 3 days exposure.
SDS–PAGE and immunoblotting
Same numbers of cells were used to extract proteins in 1× passive lysis buffer (Promega; E194A), and protein supernatant was collected after centrifugation at 16,200 g at room temperature. Proteins were denatured in 1× sample buffer (10% glycerol, 0.002% bromophenol blue, 0.075 M Tris, 2% SDS and 5% beta-mercaptoethanol) by boiling at 99°C for 10 min and centrifuged at 16,200 g for 10 min. Western blotting was performed as described (Kumar et al, 2013). Following antibodies were used for immunoblot analyses: anti-phospho p53 (s15) (Cell Signaling; #9284), anti-phospho p38 MAPK (Thr 180/Tyr 182) (Cell Signaling; 4631), anti-phospho-YAP (Ser127) (Cell Signaling; #4911), anti-p21 (H-164) sc-756 (Santa Cruz Biotech) and anti-GAPDH (Bethyl; A300-641A).
Analysis of cell cycle phase-dependent phospho-RPA2 foci
Cells were synchronized by serum starvation for 120 h in 0.1% serum and released in 10% serum-containing medium. 0, 18 and 24 h post-release, cells were pulse labeled with 4 μM EdU for 40 min, followed by fixation, permeabilization and staining with phospho-RPA2 (Bethyl; A300-246A) antibody, click reaction (C10083, Invitrogen) and 1 μg/ml DAPI (D1306, Invitrogen). Images were captured using a “Pathway 435 Bioimager” high-content analysis microscope (Becton Dickinson, Heidelberg, Germany). Confocal images were recorded from the phospho-RPA2 channel. Identification of nuclei and foci as well as signal densitometry was performed by CellProfiler 2.0 software (Kamentsky et al, 2011). Cell cycle scatter plots were generated from EdU signal (total intensity = DNA synthesis) versus DAPI signal (total intensity = DNA content), and G1 (2N, EdU−), S (EdU+) and G2/M (4N, EdU−) phases were gated. S-phase was divided into early, mid and late S-phase based on total DAPI intensity. G0/G1, early and mid S-phase were evaluated at 18 and 24 h post-release; late S-phase was evaluated at 24 h post-release. For each cell cycle phase, the percentage of nuclei with at least three phospho-RPA2 foci was counted in at least three regions of at least two independent samples.
G-Banding and Karyotyping
Slides containing metaphases were treated with SSC at 60°C for 7–14 h and stained with 4% Giemsa staining solution. Slides were analyzed using the scanning program Metafer4, and karyotyping was done using the Ikaros program (Metasystems).
Acknowledgments
We thank Lars Zender and Torsten Wüstefeld for providing the lentiviral shRNA vectors. We also thank Sabrina Eichwald, Amanda Oldani and Dario Parazzoli for technical support. This work was supported by the Erich und Gertrud Roggenbuck Stiftung and Deutsche Krebshilfe (C. Günes, Grant 10-2086-Gü 2), FP7 PEOPLE 2012 ITN (CodeAge, GA 316354), the European Research Council advanced grant (323136) and the Austrian Science Fund (M.R. Speicher, Grant P23284). Work in F.d’A.d.F. laboratory is supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) Grant Number 8866; Human Frontier Science Program (HFSP), contract number: RGP 0014/2012; Association for International Cancer Research (AICR), 14–1331; Telethon (GGP12059); PRIN 2010–2011, the Italian Ministry of Education, Universities, and Research (MIUR, 2010M4NEFY_006); EPIGEN Project; and European Research Council advanced Grant (322726). A.C. was supported by FIRC Fellowship.
Author contributions
JKM, AC, CB, MK, CB, YM and CG performed the experiments. JMK, HAK, Fd’AdF, Z-QW and MRS analyzed the data. KLR and CG planned the experiments, analyzed the data and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Table S1
Supplementary Information
Source Data for Supplementary Figure S6G
Review Process File
Source Data for Figure 1D
Source Data for Figure 3H
References
- d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, Alvarez-Buylla A, Parada LF. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15:45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen PR, Martineau SN, Margolis RL. Chemical induction of mitotic checkpoint override in mammalian cells results in aneuploidy following a transient tetraploid state. Mutat Res. 1996;372:181–194. doi: 10.1016/s0027-5107(96)00138-8. [DOI] [PubMed] [Google Scholar]
- Arnoult N, Schluth-Bolard C, Letessier A, Drascovic I, Bouarich-Bourimi R, Campisi J, Kim SH, Boussouar A, Ottaviani A, Magdinier F, Gilson E, Londono-Vallejo A. Replication timing of human telomeres is chromosome arm-specific, influenced by subtelomeric structures and connected to nuclear localization. PLoS Genet. 2010;6:e1000920. doi: 10.1371/journal.pgen.1000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayed W, Gouas L, Penault-Llorca F, Amouri A, Tchirkov A, Vago P. [Trisomy 21 and cancers] Morphologie. 2012;96:57–66. doi: 10.1016/j.morpho.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Baker DJ, van Deursen JM. Chromosome missegregation causes colon cancer by APC loss of heterozygosity. Cell Cycle. 2010;9:1711–1716. doi: 10.4161/cc.9.9.11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barefield C, Karlseder J. The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res. 2012;40:7358–7367. doi: 10.1093/nar/gks407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- Bathgate RA, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ. Relaxin family peptides and their receptors. Physiol Rev. 2013;93:405–480. doi: 10.1152/physrev.00001.2012. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci. 2008;121(Suppl. 1):1–84. doi: 10.1242/jcs.025742. [DOI] [PubMed] [Google Scholar]
- Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, Chew SK, Rowan AJ, Schenk A, Sheffer M, Howell M, Kschischo M, Behrens A, Helleday T, Bartek J, Tomlinson IP, et al. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–496. doi: 10.1038/nature11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Roumier T, Valent A, Raslova H, Yakushijin K, Horne D, Feunteun J, Lenoir G, Medema R, Vainchenker W, Kroemer G. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene. 2004;23:4362–4370. doi: 10.1038/sj.onc.1207572. [DOI] [PubMed] [Google Scholar]
- Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11:753–760. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- Chang M, Luke B, Kraft C, Li Z, Peter M, Lingner J, Rothstein R. Telomerase is essential to alleviate pif1-induced replication stress at telomeres. Genetics. 2009;183:779–791. doi: 10.1534/genetics.109.107631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conery AR, Harlow E. High-throughput screens in diploid cells identify factors that contribute to the acquisition of chromosomal instability. Proc Natl Acad Sci USA. 2010;107:15455–15460. doi: 10.1073/pnas.1010627107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–58. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–962. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas ME, Diffley JF. Replication timing: the early bird catches the worm. Curr Biol. 2012;22:R81–R82. doi: 10.1016/j.cub.2011.12.028. [DOI] [PubMed] [Google Scholar]
- Duijf PH, Benezra R. The cancer biology of whole-chromosome instability. Oncogene. 2013;32:4727–4736. doi: 10.1038/onc.2012.616. [DOI] [PubMed] [Google Scholar]
- Feng S, Agoulnik IU, Truong A, Li Z, Creighton CJ, Kaftanovskaya EM, Pereira R, Han HD, Lopez-Berestein G, Klonisch T, Ittmann MM, Sood AK, Agoulnik AI. Suppression of relaxin receptor RXFP1 decreases prostate cancer growth and metastasis. Endocr Relat Cancer. 2010;17:1021–1033. doi: 10.1677/ERC-10-0073. [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–1047. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Cornils H, Chiu SY, O’Rourke KP, Arnaud J, Yimlamai D, Thery M, Camargo FD, Pellman D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell. 2014;158:833–848. doi: 10.1016/j.cell.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, Purcell SM, Svantesson O, Landen M, Hoglund M, Lehmann S, Gabriel SB, Moran JL, Lander ES, Sullivan PF, Sklar P, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez OG, Assfalg R, Koch S, Schelling A, Meena JK, Kraus J, Lechel A, Katz SF, Benes V, Scharffetter-Kochanek K, Kestler HA, Gunes C, Iben S. Telomerase stimulates ribosomal DNA transcription under hyperproliferative conditions. Nat Commun. 2014;5:4599. doi: 10.1038/ncomms5599. [DOI] [PubMed] [Google Scholar]
- Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet. 2012;13:189–203. doi: 10.1038/nrg3123. [DOI] [PubMed] [Google Scholar]
- Gunes C, Rudolph KL. The role of telomeres in stem cells and cancer. Cell. 2013;152:390–393. doi: 10.1016/j.cell.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, Beijersbergen RL, Knoll JH, Meyerson M, Weinberg RA. Inhibition of telomerase limits the growth of human cancer cells. Nat Med. 1999;5:1164–1170. doi: 10.1038/13495. [DOI] [PubMed] [Google Scholar]
- He H, Wang Y, Guo X, Ramchandani S, Ma J, Shen MF, Garcia DA, Deng Y, Multani AS, You MJ, Chang S. Pot1b deletion and telomerase haploinsufficiency in mice initiate an ATR-dependent DNA damage response and elicit phenotypes resembling dyskeratosis congenita. Mol Cell Biol. 2009;29:229–240. doi: 10.1128/MCB.01400-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert N, Navaratnam N, Augert A, Da Costa M, Martien S, Wang J, Martinez D, Abbadie C, Carling D, de Launoit Y, Gil J, Bernard D. Regulation of ploidy and senescence by the AMPK-related kinase NUAK1. EMBO J. 2010;29:376–386. doi: 10.1038/emboj.2009.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G, Correa A, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, Majeti R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4:149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics. 2011;27:1179–1180. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King TJ, Lampe PD. The gap junction protein connexin 32 is a mouse lung tumor suppressor. Cancer Res. 2004a;64:7191–7196. doi: 10.1158/0008-5472.CAN-04-0624. [DOI] [PubMed] [Google Scholar]
- King TJ, Lampe PD. Mice deficient for the gap junction protein Connexin 32 exhibit increased radiation-induced tumorigenesis associated with elevated mitogen-activated protein kinase (p44/Erk1, p42/Erk2) activation. Carcinogenesis. 2004b;25:669–680. doi: 10.1093/carcin/bgh071. [DOI] [PubMed] [Google Scholar]
- Kirk KE, Harmon BP, Reichardt IK, Sedat JW, Blackburn EH. Block in anaphase chromosome separation caused by a telomerase template mutation. Science. 1997;275:1478–1481. doi: 10.1126/science.275.5305.1478. [DOI] [PubMed] [Google Scholar]
- Kumar M, Witt B, Knippschild U, Koch S, Meena JK, Heinlein C, Weise JM, Krepulat F, Kuchenbauer F, Iben S, Rudolph KL, Deppert W, Gunes C. CEBP factors regulate telomerase reverse transcriptase promoter activity in whey acidic protein-T mice during mammary carcinogenesis. Int J Cancer. 2013;132:2032–2043. doi: 10.1002/ijc.27880. [DOI] [PubMed] [Google Scholar]
- Lehto M, Mayranpaa MI, Pellinen T, Ihalmo P, Lehtonen S, Kovanen PT, Groop PH, Ivaska J, Olkkonen VM. The R-Ras interaction partner ORP3 regulates cell adhesion. J Cell Sci. 2008;121:695–705. doi: 10.1242/jcs.016964. [DOI] [PubMed] [Google Scholar]
- Lentini L, Barra V, Schillaci T, Di Leonardo A. MAD2 depletion triggers premature cellular senescence in human primary fibroblasts by activating a p53 pathway preventing aneuploid cells propagation. J Cell Physiol. 2012;227:3324–3332. doi: 10.1002/jcp.24030. [DOI] [PubMed] [Google Scholar]
- Li Y, Benezra R. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 1996;274:246–248. doi: 10.1126/science.274.5285.246. [DOI] [PubMed] [Google Scholar]
- Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, Han S, van Deursen JM, Zhang P. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci USA. 2010;107:14188–14193. doi: 10.1073/pnas.1005960107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson T, Debrot S, Dimpfl J, Zweig A, Zamora T, Epstein CJ. The early lethality of autosomal monosomy in the mouse. J Exp Zool. 1985;236:353–360. doi: 10.1002/jez.1402360313. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Prowse KR, Ho P, Weissman IL. Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity. 1996;5:207–216. doi: 10.1016/s1074-7613(00)80316-7. [DOI] [PubMed] [Google Scholar]
- Munne S, Bahce M, Sandalinas M, Escudero T, Marquez C, Velilla E, Colls P, Oter M, Alikani M, Cohen J. Differences in chromosome susceptibility to aneuploidy and survival to first trimester. Reprod Biomed Online. 2004;8:81–90. doi: 10.1016/s1472-6483(10)60501-9. [DOI] [PubMed] [Google Scholar]
- Orr B, Compton DA. A double-edged sword: how oncogenes and tumor suppressor genes can contribute to chromosomal instability. Front Oncol. 2013;3:164. doi: 10.3389/fonc.2013.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paeschke K, Juranek S, Simonsson T, Hempel A, Rhodes D, Lipps HJ. Telomerase recruitment by the telomere end binding protein-beta facilitates G-quadruplex DNA unfolding in ciliates. Nat Struct Mol Biol. 2008;15:598–604. doi: 10.1038/nsmb.1422. [DOI] [PubMed] [Google Scholar]
- Paeschke K, Capra JA, Zakian VA. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell. 2011;145:678–691. doi: 10.1016/j.cell.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paeschke K, Bochman ML, Garcia PD, Cejka P, Friedman KL, Kowalczykowski SC, Zakian VA. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature. 2013;497:458–462. doi: 10.1038/nature12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeyre C, Lopes J, Boule JB, Piazza A, Guedin A, Zakian VA, Mergny JL, Nicolas A. The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo. PLoS Genet. 2009;5:e1000475. doi: 10.1371/journal.pgen.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard G, Smith LE, Bailey RA, Itin P, Hohl D, Epstein EH, Jr, DiGiovanna JJ, Compton JG, Bale SJ. Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis. Nat Genet. 1998;20:366–369. doi: 10.1038/3840. [DOI] [PubMed] [Google Scholar]
- Ritz JM, Kuhle O, Riethdorf S, Sipos B, Deppert W, Englert C, Gunes C. A novel transgenic mouse model reveals humanlike regulation of an 8-kbp human TERT gene promoter fragment in normal and tumor tissues. Cancer Res. 2005;65:1187–1196. doi: 10.1158/0008-5472.CAN-04-3046. [DOI] [PubMed] [Google Scholar]
- Rudolph KL, Millard M, Bosenberg MW, DePinho RA. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet. 2001;28:155–159. doi: 10.1038/88871. [DOI] [PubMed] [Google Scholar]
- Satge D, Sommelet D, Geneix A, Nishi M, Malet P, Vekemans M. A tumor profile in Down syndrome. Am J Med Genet. 1998;78:207–216. [PubMed] [Google Scholar]
- Schvartzman JM, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer. 2010;10:102–115. doi: 10.1038/nrc2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal DJ, McCoy EE. Studies on Down’s syndrome in tissue culture. I. Growth rates and protein contents of fibroblast cultures. J Cell Physiol. 1974;83:85–90. doi: 10.1002/jcp.1040830112. [DOI] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- Suram A, Kaplunov J, Patel PL, Ruan H, Cerutti A, Boccardi V, Fumagalli M, Di Micco R, Mirani N, Gurung RL, Hande MP, d’Adda di Fagagna F, Herbig U. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. 2012;31:2839–2851. doi: 10.1038/emboj.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, Dunham MJ, Amon A. Identification of aneuploidy-tolerating mutations. Cell. 2010;143:71–83. doi: 10.1016/j.cell.2010.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres JZ, Summers MK, Peterson D, Brauer MJ, Lee J, Senese S, Gholkar AA, Lo YC, Lei X, Jung K, Anderson DC, Davis DP, Belmont L, Jackson PK. The STARD9/Kif16a kinesin associates with mitotic microtubules and regulates spindle pole assembly. Cell. 2011;147:1309–1323. doi: 10.1016/j.cell.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassin VM, Anantha RW, Sokolova E, Kanner S, Borowiec JA. Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J Cell Sci. 2009;122:4070–4080. doi: 10.1242/jcs.053702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Weinrich SL, Pruzan R, Ma L, Ouellette M, Tesmer VM, Holt SE, Bodnar AG, Lichtsteiner S, Kim NW, Trager JB, Taylor RD, Carlos R, Andrews WH, Wright WE, Shay JW, Harley CB, Morin GB. Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat Genet. 1997;17:498–502. doi: 10.1038/ng1297-498. [DOI] [PubMed] [Google Scholar]
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, Kandoth C, Fulton RS, McLellan MD, Dooling DJ, Wallis JW, Chen K, Harris CC, Schmidt HK, Kalicki-Veizer JM, Lu C, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–709. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Chen C, Li Y, Fu X, Xie Y, Li Y, Huang Y. Cx31.1 acts as a tumour suppressor in non-small cell lung cancer (NSCLC) cell lines through inhibition of cell proliferation and metastasis. J Cell Mol Med. 2012;16:1047–1059. doi: 10.1111/j.1582-4934.2011.01389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivotovsky B, Kroemer G. Apoptosis and genomic instability. Nat Rev Mol Cell Biol. 2004;5:752–762. doi: 10.1038/nrm1443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Table S1
Supplementary Information
Source Data for Supplementary Figure S6G
Review Process File
Source Data for Figure 1D
Source Data for Figure 3H