

Abstract
Ubiquitin and ubiquitin-like proteins (UBLs) function in a wide array of cellular processes. UBL5 is an atypical UBL that does not form covalent conjugates with cellular proteins and which has a known role in modulating pre-mRNA splicing. Here, we report an unexpected involvement of human UBL5 in promoting the function of the Fanconi anemia (FA) pathway for repair of DNA interstrand crosslinks (ICLs), mediated by a specific interaction with the central FA pathway component FANCI. UBL5-deficient cells display spliceosome-independent reduction of FANCI protein stability, defective FANCI function in response to DNA damage and hypersensitivity to ICLs. By mapping the sequence determinants underlying UBL5–FANCI binding, we generated separation-of-function mutants to demonstrate that key aspects of FA pathway function, including FANCI–FANCD2 heterodimerization, FANCD2 and FANCI monoubiquitylation and maintenance of chromosome stability after ICLs, are compromised when the UBL5–FANCI interaction is selectively inhibited by mutations in either protein. Together, our findings establish UBL5 as a factor that promotes the functionality of the FA DNA repair pathway.
Keywords: DNA damage response, FANCI, Fanconi anemia, protein stability, UBL5
Introduction
Post-translational modifications (PTMs) of proteins by ubiquitin and ubiquitin-like modifiers (UBLs) control a wide variety of cellular processes including protein degradation, DNA repair, transcriptional regulation and endocytosis (Hochstrasser, 2009; van der Veen & Ploegh, 2012). The covalent attachment of a C-terminal glycine residue in ubiquitin and UBLs such as SUMO, NEDD8 and ISG15 to target proteins is catalyzed by an enzymatic cascade involving the sequential actions of E1 activating enzymes, E2 conjugating enzymes and E3 ligases (van der Veen & Ploegh, 2012). Ubiquitin-like protein 5 (UBL5), known as Hub1 in yeast, is an evolutionarily conserved member of the UBL family that shares a low degree of sequence similarity with ubiquitin, yet closely resembles it structurally (McNally et al, 2003; Ramelot et al, 2003). However, unlike other UBLs, UBL5/Hub1 lacks a C-terminal di-glycine motif and is thus unable to form covalent conjugates with other proteins (Luders et al, 2003; Yashiroda & Tanaka, 2004). Previous studies showed that yeast Hub1 interacts non-covalently with the spliceosomal protein Snu66 (also termed SART1 in mammals), and this interaction is critical for alternative splicing of SRC1, the only gene known to undergo this type of modification in budding yeast (Mishra et al, 2011). The non-covalent binding of Hub1 to Snu66 is mediated through a salt bridge formed between D22 in Hub1 and a Hub1 interaction domain (HIND) in Snu66 (Mishra et al, 2011). Recent studies demonstrated that the involvement of Hub1 in pre-mRNA splicing is conserved in higher eukaryotes, and that RNAi-mediated knockdown of UBL5 in human cells compromises global pre-mRNA splicing efficiency, leading to intron retention (Ammon et al, 2014; Oka et al, 2014). Among other phenotypes, the splicing defects arising from depletion of UBL5 or other pre-mRNA splicing factors manifest as complete loss of mitotic sister chromatid cohesion, due to mis-splicing of the sister chromatid cohesion protection factor Sororin (van der Lelij et al, 2014; Oka et al, 2014; Sundaramoorthy et al, 2014; Watrin et al, 2014). This defect can be fully relieved by depletion of the cohesion dissociation factor WAPL. Despite its established role in pre-mRNA splicing in both yeast and human cells, the cellular function of UBL5 may not be limited to this process. For instance, a genome-wide RNAi-based screen in the nematode C. elegans showed that UBL5 is required for signaling in the mitochondrial unfolded protein response, through interaction with the transcription factor DVE-1 and upregulation of chaperone genes in response to mitochondrial stress (Haynes et al, 2007).
Fanconi anemia (FA) is a genetic disorder characterized by bone marrow failure, predisposition to cancer and congenital abnormalities (Moldovan & D’Andrea, 2009). Cells derived from FA patients display chromosomal instability and hypersensitivity to DNA interstrand crosslink (ICL)-inducing agents (Kottemann & Smogorzewska, 2013). There are at least 16 FA genes, the products of which function in ICL repair and stabilization of stalled replication forks (Kottemann & Smogorzewska, 2013; Zhang & Walter, 2014). In response to ICLs or replication stress, FANCD2 is monoubiquitylated by the FA core complex, which is composed of 8 FA proteins (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL and FANCM) and 5 FA-related proteins (FAAP20, FAAP24, FAAP100, MHF1 and MHF2) (Kottemann & Smogorzewska, 2013). Monoubiquitylated FANCD2 is relocalized to genotoxic stress sites, where it coordinates ICL repair in conjunction with a paralogous protein, FANCI, with which FANCD2 forms a heterodimer (Kim & D’Andrea, 2012). FANCI is also monoubiquitylated by the FA core complex following DNA damage, in a manner dependent on its ATR-dependent phosphorylation and FANCD2 (Sims et al, 2007; Smogorzewska et al, 2007; Ishiai et al, 2008). This cascade of PTMs is required for the recruitment of FANCI to nuclear foci upon DNA damage. The monoubiquitylation of FANCI and FANCD2 by the FA core complex is mutually interdependent in human cells (Sims et al, 2007; Smogorzewska et al, 2007). However, while FANCD2 monoubiquitylation has a pivotal role in the FA pathway, mutation of the corresponding monoubiquitylation site in FANCI only confers a mild ICL repair defect (Smogorzewska et al, 2007; Ishiai et al, 2008). FANCI binds directly to the USP1–UAF1 complex, regulating FANCD2 deubiquitylation (Yang et al, 2011) and conferring site specificity to FANCD2 monoubiquitylation in vitro (Alpi et al, 2008; Sato et al, 2012; Rajendra et al, 2014).
While it is clear that the FANCD2–FANCI complex has a central role in ICL repair, the assembly, functions and regulation of this complex remain only partially understood. Here, we discovered an unexpected role of UBL5 in supporting the functionality of the FA pathway, distinct from its canonical function in pre-mRNA splicing. We show that UBL5 directly binds to and stabilizes FANCI, promoting the integrity of the FANCI-FANCD2 complex to protect cells from the cytotoxicity of ICLs.
Results
UBL5 interacts with FANCI
Our recent proteomic survey of UBL5-associated proteins revealed that UBL5 binds to a number of spliceosomal proteins including SART1, MFAP1 and EFTUD2 in human cells (Oka et al, 2014). In addition to pre-mRNA splicing components, we identified FANCI as a major non-spliceosomal UBL5-associated protein (Supplementary Fig S1A), and we set out to investigate this link in more detail. First, we validated the UBL5–FANCI interaction in co-immunoprecipitation experiments, in which endogenous FANCI could be co-purified with Strep-HA-UBL5 and endogenous UBL5 co-precipitated with GFP-FANCI (Fig1A and B). Importantly, a complex between the endogenous UBL5 and FANCI proteins could also be detected in cells (Fig1C). We did not observe interaction between FANCI and SART1 (Supplementary Fig S1B), suggesting that the association between UBL5 and FANCI does not occur in the context of the spliceosome. Moreover, unlike the effect of knocking down SART1 or UBL5 (Oka et al, 2014), depletion of FANCI did not have a marked impact on global transcriptome profiles (data not shown). The interaction between UBL5 and FANCI appeared to be direct as it could be readily observed with purified components in vitro (Fig1D; Supplementary Fig S4B), and occurred independently of FANCD2 in vivo (Fig1E). We also assessed whether the UBL5–FANCI interaction was regulated during the cell cycle and in response to DNA damage. To this end, we performed pull-down experiments using cells inducibly expressing Strep-HA-tagged UBL5 and synchronized by a double thymidine block and release protocol. This revealed that the UBL5–FANCI interaction was constitutive during the S and G2 phases of the cell cycle and that the ubiquitylation state of FANCI did not appreciably influence its association with UBL5 (Fig1F). Similarly, the UBL5–FANCI interaction was unaffected by treatment with the ICL-inducing agent mitomycin C (MMC) (Fig1C; Supplementary Fig S1C), indicating that it was not regulated in a DNA damage-dependent manner. Together, these data suggest that FANCI is a direct cellular binding partner of UBL5 outside the context of the pre-mRNA splicing machinery.
Figure 1.
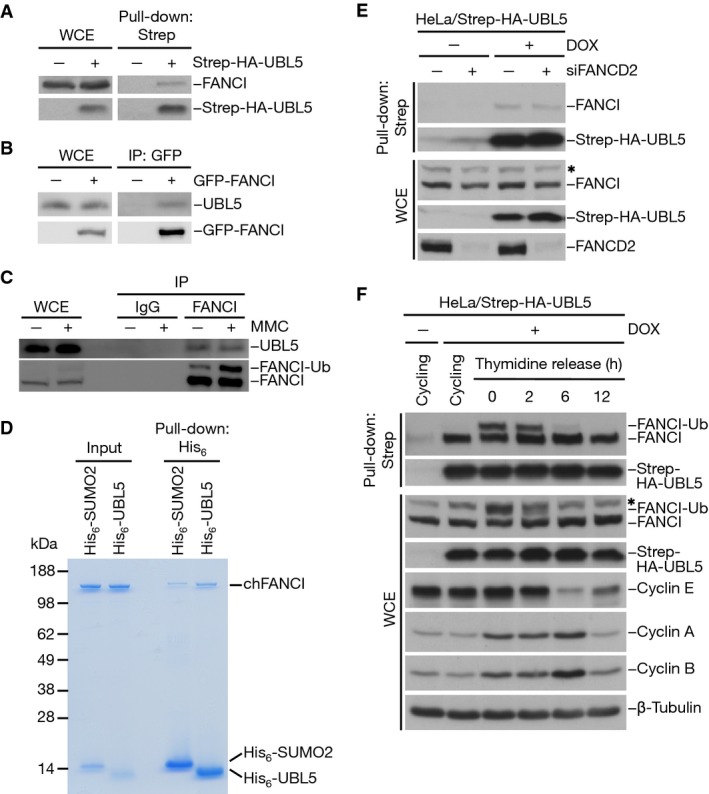
- Whole-cell extracts (WCE) of HeLa cells transfected with Strep-HA-UBL5 plasmid were subjected to Strep-Tactin pull-down followed by immunoblotting with FANCI and HA antibodies.
- Whole-cell extracts (WCE) of HEK293T cells transfected with cDNA encoding GFP-FANCI were subjected to GFP immunoprecipitation followed by immunoblotting with UBL5 and GFP antibodies.
- Whole-cell extracts (WCE) of HeLa cells treated or not with 0.5 μM mitomycin C (MMC) for 24 h were subjected to immunoprecipitation with FANCI antibody or pre-immune serum (IgG) followed by immunoblotting with UBL5 and FANCI antibodies.
- Recombinant chicken FANCI (chFANCI) was incubated with His6-SUMO2 or His6-UBL5. Bound proteins were resolved by SDS–PAGE and visualized by colloidal blue staining.
- HeLa/Strep-HA-UBL5 cells induced or not with doxycycline (DOX) were transfected with non-targeting or FANCD2 siRNAs for 48 h. Whole-cell extracts (WCE) were subjected to Strep-Tactin pull-down followed by immunoblotting with the indicated antibodies. * non-specific band.
- HeLa/Strep-HA-UBL5 cells induced or not with doxycycline (DOX) were left untreated or synchronized by double thymidine block and released for the indicated times. Whole-cell extracts (WCE) were subjected to Strep-Tactin pull-down followed by immunoblotting with the indicated antibodies. * non-specific band.
UBL5 promotes the integrity of the FA pathway
The emerging link between UBL5 and FANCI prompted us to ask whether UBL5 has a role in the FA pathway. Interestingly, knockdown of UBL5 by independent siRNAs was accompanied by strongly reduced expression levels of FANCI and FANCD2, but not several FA core complex components, including FANCC and FANCE (Fig2A; Supplementary Fig S2A). This effect could be rescued by reintroduction of siRNA-resistant UBL5 (Fig2B and C), demonstrating that it was a specific consequence of UBL5 depletion. In contrast, levels of UBL5 were not affected by knockdown of FANCI (Supplementary Fig S2B). It has previously been shown that FANCI is required for FANCD2 stability, but not vice versa (Dorsman et al, 2007; Sims et al, 2007; Smogorzewska et al, 2007), suggesting that the observed destabilization of FANCD2 in UBL5-depleted cells may be due to the lower levels of FANCI expression resulting from UBL5 knockdown. Consistent with the reduced abundance of FANCI and FANCD2, no detectable accumulation of FANCI at ICLs induced by UVA laser-activated psoralen or MMC was evident in UBL5-depleted cells, and FANCD2 monoubiquitylation in response to MMC treatment was impaired (Fig2D and E; Supplementary Fig S2C). Based on these observations, we tested whether knockdown of UBL5 confers cellular sensitivity to ICL-inducing agents in clonogenic survival assays (Fig2F). We have recently shown that knockdown of UBL5 leads to strong induction of precocious sister chromatid separation in metaphase (Oka et al, 2014). This phenotype can be fully rescued by co-depletion of the sister chromatid cohesion release factor WAPL, which is prerequisite for the continued proliferation and colony formation of UBL5-depleted cells. Assessing DNA damage sensitivity of cells lacking UBL5 therefore necessitates concomitant knockdown of WAPL, which did not in itself confer MMC sensitivity (Fig2F; Supplementary Fig S2D). We found that co-depletion of UBL5 and WAPL significantly sensitized cells to MMC, although to a lesser extent than did knockdown of FANCI or FANCD2 (Fig2F; Supplementary Fig S2D), consistent with the substantial reduction, but not complete loss, of FANCI and FANCD2 expressions in cells lacking UBL5. Importantly, the enhanced MMC sensitivity resulting from depletion of endogenous UBL5 could be fully restored by ectopic expression of UBL5 in these cells (Fig2G). We conclude from these results that UBL5 is important for the functionality of the FA pathway for ICL repair.
Figure 2.
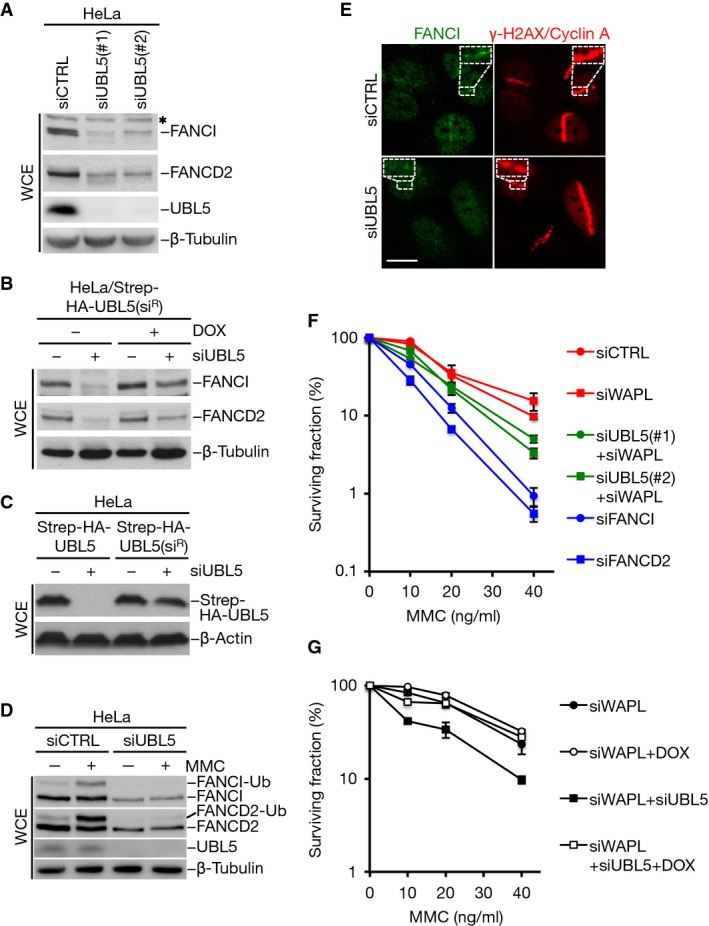
- Extracts of HeLa cells transfected with non-targeting control (CTRL) or UBL5 siRNAs for 48 h were analyzed by immunoblotting with the indicated antibodies. * non-specific band.
- HeLa cells stably expressing a doxycycline-inducible siRNA-resistant form (siR) of Strep-HA-UBL5 were transfected with non-targeting control (CTRL) or UBL5 siRNAs for 48 h. Cell extracts were analyzed by immunoblotting with FANCI, FANCD2 and β-tubulin antibodies.
- HeLa cells transfected with non-targeting or UBL5 siRNAs were transfected with Strep-HA-UBL5 or siRNA-resistant form (siR) of Strep-HA-UBL5. Cell extracts were analyzed by immunoblotting with HA and β-actin antibodies.
- HeLa cells transfected with non-targeting control (CTRL) or UBL5 siRNAs and then treated or not with MMC for 15 h were lysed and processed for immunoblotting with FANCI, FANCD2, UBL5 and β-tubulin antibodies.
- U2OS cells transfected with non-targeting control (CTRL) or UBL5 siRNAs were subjected to laser microirradiation in the presence of trioxsalen, fixed 2 h later and co-immunostained with FANCI, γ-H2AX and cyclin A antibodies. Representative images are shown. Scale bar, 10 μm.
- Clonogenic survival of U2OS cells transfected with indicated siRNAs and exposed to various doses of MMC. Results from at least three independent experiments (mean ± SEM) are shown.
- Clonogenic survival of U2OS/Strep-HA-UBL5(siR) cells treated or not with doxycycline (DOX) to induce expression of the transgenes, transfected with the indicated siRNAs and then exposed to various doses of MMC. Results from three independent experiments (mean ± SEM) are shown.
The role of UBL5 in the FA pathway can be uncoupled from its pre-mRNA splicing function
Given the established role of UBL5 in modulating pre-mRNA splicing (Ammon et al, 2014; Oka et al, 2014), we next sought to clarify to which extent UBL5 supports FANCI expression through direct interaction and indirectly via its involvement in spliceosomal processes. To this end, we compared FANCI expression levels after depletion of UBL5 or spliceosomal factors with which it interacts, including SART1 and EFTUD2. While knockdown of each of these factors reduced the expression level of FANCI, UBL5 depletion consistently had the most pronounced effect (Fig3A), despite that treatment with UBL5 and SART1 siRNAs gave rise to a similar decrease in FANCI mRNA levels as measured by quantitative RT–PCR (Supplementary Fig S3A). This suggests that UBL5 has an additional, spliceosome-unrelated role in maintaining FANCI expression. In agreement with this idea, we found that the expression of an intron-less, and therefore splicing-insensitive, FANCI construct was impaired in cells depleted of UBL5, but not SART1 or EFTUD2 (Fig3B). Treating cells with a proteasome inhibitor largely corrected the FANCI expression defect in UBL5-depleted cells (Supplementary Fig S3B). These data suggest that in addition to maintaining FANCI mRNA levels via its previously described role in pre-mRNA splicing, UBL5 binds and stabilizes FANCI through direct protein–protein interaction.
Figure 3.
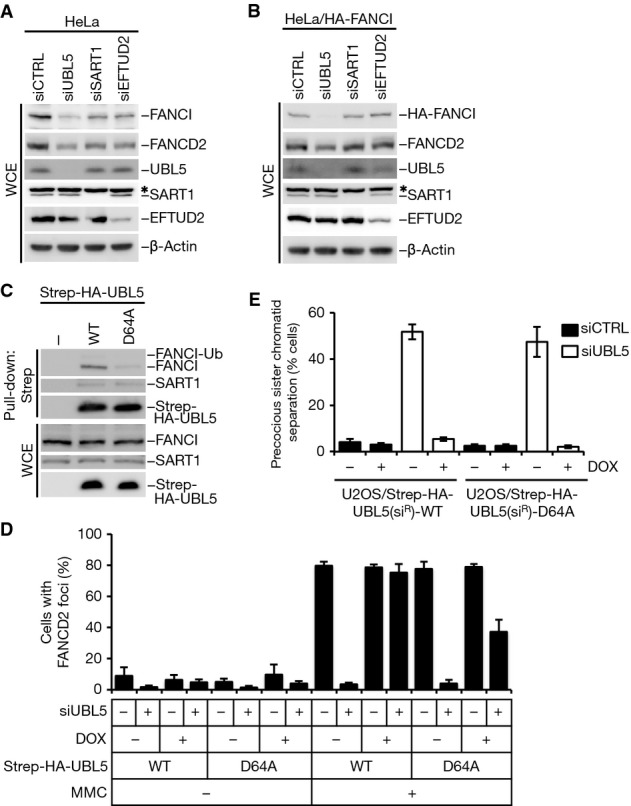
- HeLa cells were transfected with the indicated siRNAs for 48 h. Cell extracts were analyzed by immunoblotting with the indicated antibodies. * non-specific band.
- HeLa cells stably expressing HA-tagged FANCI were transfected with the indicated siRNAs for 48 h. Cell extracts were analyzed by immunoblotting with the indicated antibodies. * non-specific band.
- Whole-cell extracts (WCE) of HeLa cells transfected with the indicated Strep-HA-UBL5 constructs were subjected to Strep-Tactin pull-down followed by immunoblotting with FANCI, SART1 and HA antibodies.
- U2OS cells stably expressing siRNA-resistant (siR) forms of indicated Strep-HA-UBL5 alleles were induced or not with doxycycline, transfected with non-targeting control (CTRL) or UBL5 siRNAs and treated with MMC for 24 h. Cells were then fixed and immunostained with FANCD2 antibody. FANCD2 foci formation in cells was enumerated, and cells containing more than five foci were defined as foci positive. More than 200 cells were counted for each condition. Results from three independent experiments (mean ± SD) are shown.
- Quantification of precocious sister chromatid separation in U2OS cells stably expressing doxycycline-inducible Strep-HA-UBL5(siR) constructs after knockdown of endogenous UBL5. At least 200 metaphase spreads were counted in each experiment. Mean values (± SD) from three independent experiments are shown.
To better understand the potential dual contributions of UBL5 in maintaining FANCI protein levels, we sought to generate a separation-of-function UBL5 mutant that would disrupt its interaction with FANCI while leaving its spliceosomal function intact. To achieve this, we adopted a systematic mutagenesis approach, in which the effect of introducing alanine substitutions of highly conserved amino acids in UBL5 on binding to FANCI and SART1 was evaluated. We noted that a D64A mutation in UBL5 reduced the interaction with FANCI but not SART1 (Fig3C; Supplementary Fig S3C), suggesting that it might selectively compromise UBL5 function in the FA pathway but not in pre-mRNA splicing. To test this, we generated stable cell lines inducibly expressing siRNA-resistant forms of WT and mutant UBL5. In these cell lines, the D64A mutant was less efficient than UBL5 WT in restoring the reduced levels of FANCI and FANCD2 and their MMC-induced monoubiquitylation resulting from depletion of endogenous UBL5 (Supplementary Fig S3D). Consistently, while ectopic expression of UBL5 WT fully restored the formation of MMC-induced FANCD2 foci in cells depleted of endogenous UBL5, the D64A mutant only showed a partial rescue effect (Fig3D). We have previously shown that siRNA-mediated knockdown of UBL5 impairs spliceosome functionality, leading to loss of Sororin and defective sister chromatid cohesion in metaphase cells (Oka et al, 2014). To test whether the UBL5 D64A mutant supports pre-mRNA splicing, we assayed for cohesion defects under conditions where endogenous UBL5 was replaced by ectopically expressed UBL5 alleles. We found that expression of UBL5 WT and D64A both rescued mitotic sister chromatid cohesion (Fig3E), implying that the UBL5 D64A mutant is fully proficient in promoting the spliceosome-related functions of UBL5. Taken together, our data suggest that the direct interaction between UBL5 and FANCI promotes FANCI stability and FA pathway integrity independently of the known role of UBL5 in pre-mRNA splicing. Unlike FANCI, however, we did not observe detectable accumulation of UBL5 at damaged DNA (data not shown); hence, UBL5 is likely to mainly facilitate FANCI function prior to its direct engagement at DNA lesions.
Mapping the UBL5-interacting region in FANCI
To pinpoint the region of FANCI responsible for its interaction with UBL5, we generated a series of overlapping GFP-tagged FANCI fragments (Fig4A) and tested their ability to bind ectopically expressed UBL5. From these interaction studies, we first narrowed down the UBL5-interacting region in FANCI to the N-terminal 300 amino acids (aa) (Fig4A and B; Supplementary Fig S4). A fragment spanning residues 1–200 did not interact with UBL5 (Fig4C), further localizing the UBL5-binding determinant in FANCI to the region encompassing aa 200–300. Subsequently, using a series of additional FANCI fragments containing small, 20-aa internal deletions within this region (Fig4A), we found that only a fragment lacking residues 261–280 was defective for interaction with UBL5 (Fig4D), suggesting that this sequence harbors the key UBL5-binding site in FANCI. While this region is well conserved among vertebrate FANCI orthologues (Fig4E), we could not detect a similar motif in the paralogous FANCD2 protein. We introduced point mutations into the most highly conserved residues within the aa261–280 region in the context of full-length FANCI and assessed their impact on UBL5 interaction. We found that a double point mutant containing alanine substitutions of the highly conserved E268 and H274 residues (hereafter referred to as ∆UBL5) displayed markedly attenuated binding to UBL5 when expressed in cells (Fig4E and F). Likewise, pull-down assays using FANCI purified from HEK293T cells and bacterially expressed recombinant UBL5 showed that the FANCI ∆UBL5 mutant was deficient for interaction with UBL5 in vitro (Fig4G). Hence, it is possible to obstruct the UBL5-binding ability of FANCI by means of subtle point mutations in the full-length protein, and we set out to test the impact of expressing such a UBL5-binding-deficient FANCI mutant on the functionality of the FA pathway.
Figure 4.
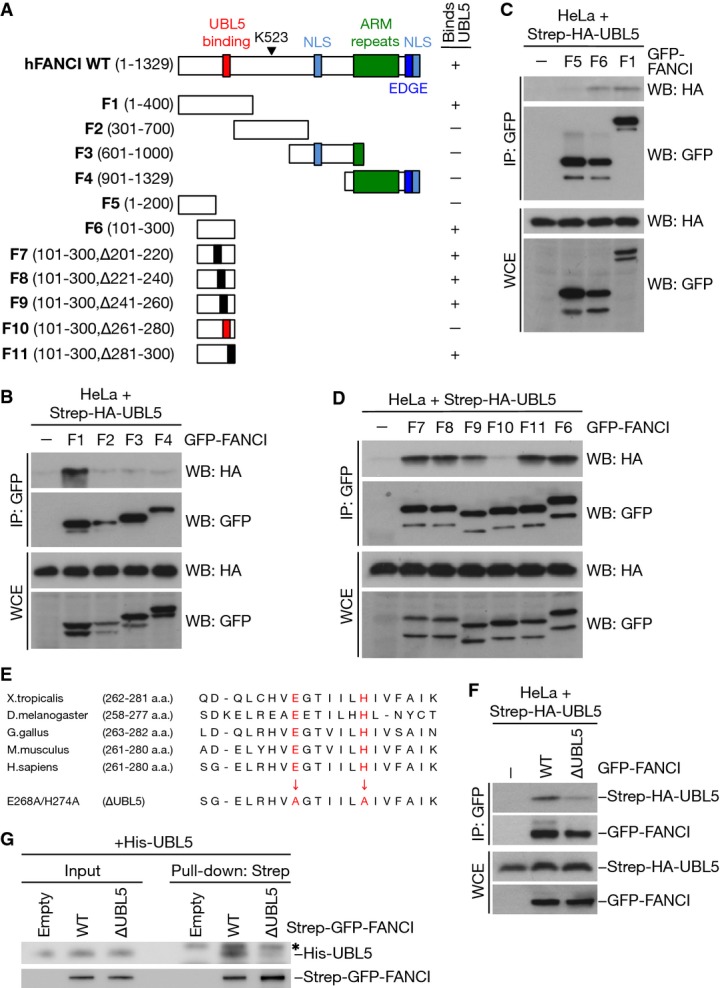
- A Diagram of FANCI fragments used to test interaction with UBL5. The UBL5-binding region is highlighted in red.
- B Whole-cell extracts (WCE) of HeLa cells co-transfected with Strep-HA-UBL5 and the indicated GFP-FANCI fragments were subjected to GFP immunoprecipitation followed by immunoblotting with HA and GFP antibodies.
- C, D As in (B), using the indicated GFP-FANCI constructs.
- E Sequence alignment of amino acids in the UBL5-interacting region in FANCI proteins from different organisms. Amino acid substitutions of the highly conserved E268 and H274 residues are indicated in red.
- F Whole-cell extracts (WCE) of HeLa cells co-transfected with Strep-HA-UBL5 and the indicated GFP-tagged FANCI constructs were subjected to GFP immunoprecipitation followed by immunoblotting with HA and GFP antibodies.
- G Strep-GFP-FANCI WT or ∆UBL5 purified from HEK293T cells under stringent conditions was used to pull down recombinant His6-UBL5. Input and pull-down samples were subjected to immunoblotting with His6 and GFP antibodies. * non-specific band.
UBL5 promotes FANCI–FANCD2 complex formation in cells
Based on our observations that loss of UBL5 negatively affects the expression level of FANCI in both a spliceosome-dependent and spliceosome-independent manner (Fig3A and B; Supplementary Fig S3A and B), we next utilized the FANCI ∆UBL5 mutant to more directly address the impact of UBL5 binding on FANCI protein stability and function. To this end, we first compared the half-lives of ectopically expressed, intron-less FANCI WT and ∆UBL5 alleles in cells treated with the protein synthesis inhibitor cycloheximide. Whereas FANCI WT remained stable over the course of several hours, levels of the FANCI ∆UBL5 mutant dropped rapidly within the same time frame (Fig5A and B), consistent with its compromised ability to bind UBL5. In contrast, in line with previous reports (Dorsman et al, 2007; Sims et al, 2007; Smogorzewska et al, 2007), depletion of FANCD2 did not induce similar destabilization of FANCI (Supplementary Fig S5A and B). These findings highlight UBL5 as a factor that is crucial for the stability of FANCI, unlike other known FANCI binding partners.
Figure 5.
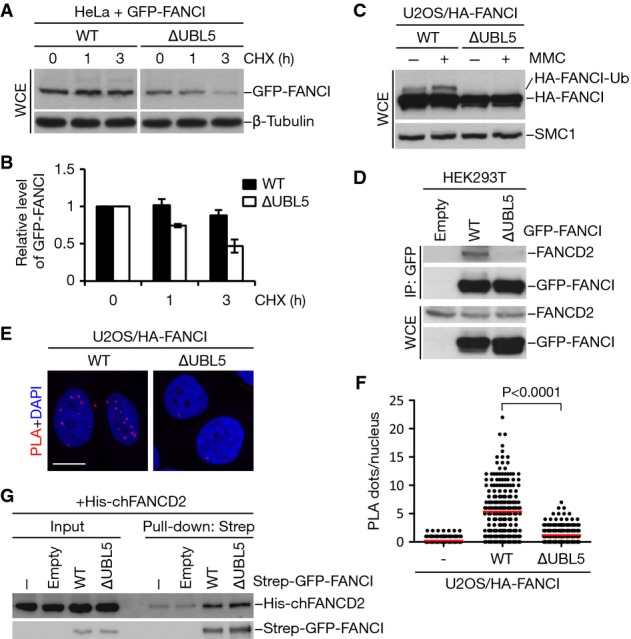
- HeLa cells transfected with GFP-tagged FANCI WT or ∆UBL5 were harvested at the indicated times following addition of cycloheximide (CHX). Cell lysates were immunoblotted with GFP and β-tubulin antibodies.
- Quantification of GFP-FANCI levels in (A) by image analysis, normalized to β-tubulin levels. Mean values (± SD) from three independent experiments are shown.
- U2OS cells stably expressing HA-tagged FANCI WT or ∆UBL5 were treated or not with 1 μM mitomycin C (MMC) for 16 h. Cell extracts were analyzed by immunoblotting with HA and SMC1 antibodies.
- Extracts of HEK293T cells transfected with the indicated GFP-FANCI constructs were subjected to GFP immunoprecipitation followed by immunoblotting with FANCD2 and GFP antibodies.
- U2OS cells stably expressing HA-FANCI WT or ∆UBL5 were fixed and incubated with antibodies against HA and FANCD2. The interaction between HA-FANCI and FANCD2 was visualized using PLA. Nuclei were stained with DAPI. Representative images are shown. Scale bar, 10 μm.
- Quantification of data in (E). Graph shows the quantification of PLA dots per nucleus. More than 200 cells were counted for each condition. Red lines indicate the mean of the data plotted. P-value was calculated by the Mann–Whitney U-test.
- Strep-GFP-FANCI WT or ∆UBL5 was purified from HEK293T cells and incubated with bacterially produced, recombinant His6-FANCD2. Complexes were immobilized on Strep-Tactin Sepharose and washed extensively. Bound material and input samples were then subjected to immunoblotting with His6 and GFP antibodies.
To address the importance of the UBL5–FANCI interaction for the functionality of the FA pathway, we generated cell lines stably expressing WT and mutant (∆UBL5) forms of FANCI. To circumvent issues pertaining to the reduced stability of the FANCI ∆UBL5 mutant, we carefully selected clones with near-identical levels of ectopic FANCI expression (Fig5C). We noticed that only WT, but not mutant, FANCI was competent to undergo monoubiquitylation by the FA core complex following MMC treatment (Fig5C). Both FANCD2 and FANCI can be deubiquitylated by the USP1–UAF1 complex (Nijman et al, 2005; Cohn et al, 2007; Smogorzewska et al, 2007; Yang et al, 2011). However, while treatment with ML323, a specific inhibitor of USP1–UAF1 (Liang et al, 2014), increased monoubiquitylation of FANCI WT in undamaged cells, it did not induce monoubiquitylation of FANCI ∆UBL5 (Supplementary Fig S5C), indicating that this UBL5-binding mutant is refractory to monoubiquitylation under both basal and genotoxic stress conditions. Because FANCI monoubiquitylation is fully dependent on its association with FANCD2 (Sims et al, 2007; Smogorzewska et al, 2007), we reasoned that the FANCI ∆UBL5 mutant might be deficient for binding to FANCD2. Indeed, when we probed for interaction between FANCD2 and FANCI using co-immunoprecipitation analysis, we found that mutation of the UBL5-binding region in FANCI abolished complex formation between these factors (Fig5D). To assess FANCI–FANCD2 interaction by a complementary approach, we performed proximity ligation assays (PLA) (Soderberg et al, 2006). Again, whereas interaction between FANCI WT and FANCD2 in situ could be readily observed, FANCI ∆UBL5 showed markedly reduced FANCD2 binding also under these experimental conditions (Fig5E and F). FANCI and FANCD2 are paralogous proteins that interact with each other through extensive homologous binding surfaces (Smogorzewska et al, 2007; Joo et al, 2011). Importantly, binding experiments using recombinant FANCD2 and ectopic FANCI proteins purified from cells under stringent conditions revealed that the FANCI point mutations introduced to disrupt UBL5 binding did not impair FANCI–FANCD2 heterodimer formation per se, as the FANCI ∆UBL5 mutant was fully competent to interact with FANCD2 in vitro (Fig5G). We conclude from these findings that UBL5 facilitates FANCI heterodimerization with FANCD2 in cells.
FANCI forms UBL5-dependent homomeric complexes
In keeping with the notion that FANCI appears to be more abundant in cells than FANCD2 (Beck et al, 2011), we hypothesized that FANCI might also be capable of forming homodimers independently of FANCD2. In support of this idea, we found that ectopically expressed GFP-FANCI recovered substantial amounts of co-expressed HA-tagged FANCI and vice versa, indicating that FANCI homodimers or oligomers can indeed be observed in cells (Fig6A; Supplementary Fig S6A). The formation of such FANCI complexes was unaffected by knockdown of FANCD2 (Fig6A). To test whether UBL5 has a role in this process similar to its ability to promote FANCI–FANCD2 interaction in cells, we assessed the ability of the ∆UBL5 mutant to engage in FANCI homodimer formation. Unlike FANCI WT, the ∆UBL5 mutant was deficient for homodimerization (Fig6B), suggesting that the formation of FANCI homodimers in cells is also stimulated by UBL5.
Figure 6.
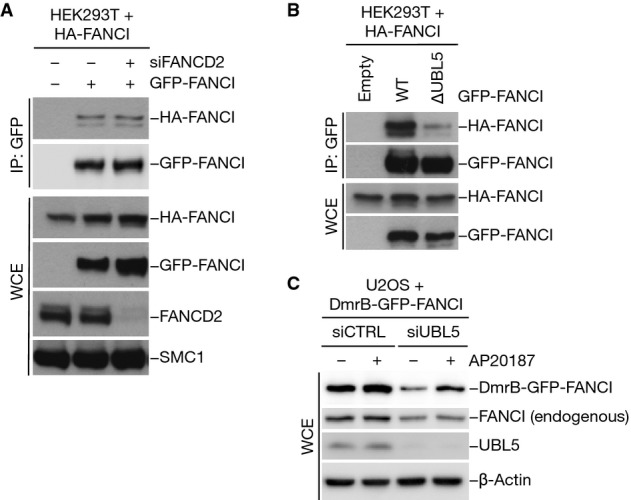
- HEK293T cells transfected with non-targeting control or FANCD2 siRNA were co-transfected with HA-FANCI and GFP-FANCI constructs as indicated. Whole-cell extracts (WCE) were subjected to GFP immunoprecipitation followed by immunoblotting with the indicated antibodies.
- Extracts of HEK293T cells co-transfected with indicated combinations of HA-FANCI and GFP-FANCI constructs were subjected to GFP immunoprecipitation followed by immunoblotting with HA and GFP antibodies.
- U2OS cells were transfected with non-targeting control (CTRL) or UBL5 siRNA, subsequently transfected with a construct encoding DmrB-GFP-FANCI and then treated with 100 nM AP20187 (B/B homodimerizer) or left untreated. Cell extracts were analyzed by immunoblotting with the indicated antibodies.
To further study the properties of FANCI homodimers, we took advantage of a previously published system to enforce protein dimerization (Clackson et al, 1998; Feng et al, 2001). Briefly, we fused a mutant form of the FK506 protein domain DmrB to FANCI, which undergoes dimerization upon the addition of the rapamycin derivative “B/B homodimerizer” (also known as AP20187) (Supplementary Fig S6B). To test whether FANCI homodimers are more stable than monomeric FANCI and whether UBL5 underpins FANCI stability by promoting formation of such complexes, we monitored the expression levels of the above constructs in cells containing or lacking UBL5. We found that the reduced abundance of DmrB-GFP-FANCI resulting from UBL5 knockdown could be reversed by treatment with AP20187 (Fig6C). In contrast, the destabilization of endogenous FANCI resulting from UBL5 depletion was not rescued by AP20187 (Fig6C). The reduced stability of the FANCI ∆UBL5 mutant compared to FANCI WT could also be counteracted by forced homodimerization of this mutant (Supplementary Fig S6C). Collectively, these findings suggest that FANCD2-independent FANCI homodimers exist in cells and that their formation is stimulated by UBL5. Moreover, while FANCI stability is not affected by loss of FANCD2 but appears to be enhanced by homodimer formation, the ability of UBL5 to facilitate the latter process may, at least in part, explain its role in maintaining FANCI protein levels.
UBL5–FANCI interaction is required for FA pathway function after DNA damage
The FANCI ∆UBL5 mutant allowed us to test directly whether UBL5 binding to FANCI is needed for FA pathway function in response to DNA damage. In agreement with its inability to bind FANCD2 in cells, FANCI ∆UBL5 failed to form foci after MMC treatment (Fig7A and B). Consistent with the lack of binding to FANCD2, we did not observe dominant-negative effects of overexpressing FANCI ∆UBL5 on FANCD2 foci formation under these conditions (Fig7A), unlike what has been reported for monoubiquitylation- and phosphorylation-deficient FANCI mutants (Smogorzewska et al, 2007; Ishiai et al, 2008). To further investigate the importance of UBL5–FANCI interaction in the FA pathway, we expressed FANCI WT or ∆UBL5 in cells depleted of endogenous FANCI, using siRNAs targeting the 5′-UTR region of the FANCI gene. While complementation with WT FANCI largely restored MMC-induced FANCD2 foci formation and suppressed chromosome aberrations in cells depleted of endogenous FANCI, the ∆UBL5 mutant was unable to promote such rescue (Fig7C and D). Together, these results suggest that UBL5–FANCI interaction is required for the functionality of the FA pathway, highlighting UBL5 as an important factor in ICL repair.
Figure 7.
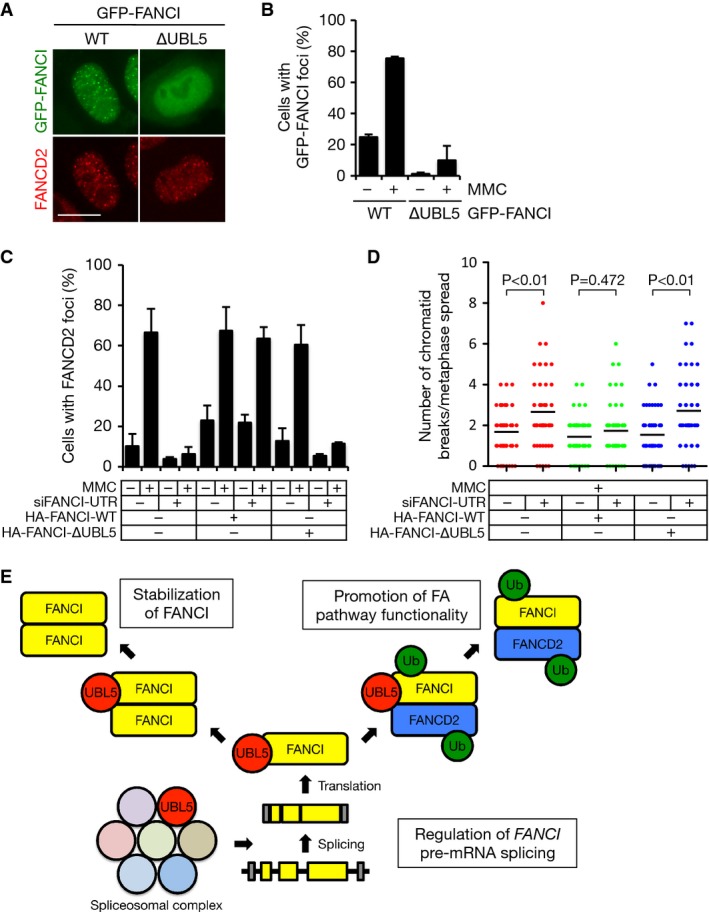
- Representative images of HeLa cells transfected with GFP-tagged FANCI WT or ∆UBL5 and treated with 0.2 μM MMC for 12 h. Scale bar, 10 μm.
- Quantification of data in (A). Results from three independent experiments (mean ± SD) are shown. More than 200 cells were counted for each condition.
- Quantification of cells containing FANCD2 foci. U2OS cells stably expressing HA-FANCI WT or ∆UBL5 transfected with siRNA targeting the 5′-UTR region of FANCI were treated with 0.2 μM MMC for 16 h, fixed and analyzed for FANCD2 foci formation by immunostaining with FANCD2 antibody. Cells containing more than five foci were scored as foci positive. Results from three independent experiments (mean ± SD) are shown. More than 300 cells were counted for each condition.
- Quantification of chromosomal aberrations. U2OS cells stably expressing HA-FANCI WT or ∆UBL5 transfected with siRNA targeting the 5′-UTR region of FANCI were treated with 120 nM MMC for 24 h, then treated with nocodazole for additional 2 h and collected. Metaphase spreads were prepared and chromosomal aberrations were quantified. Black lines indicate the mean of the data plotted. P-values were calculated using Mann–Whitney U-test (n = 50).
- Model of UBL5 function in the FA pathway (see main text for details). Ub, ubiquitin.
Discussion
The repair of ICLs is orchestrated by the Fanconi anemia (FA) pathway in an exquisitely complex reaction that requires a large number of proteins and is regulated by several PTMs, including ubiquitylation, phosphorylation and SUMOylation (Kottemann & Smogorzewska, 2013; Walden & Deans, 2014; Gibbs-Seymour et al, 2015). Here, we discovered a parallel, dual role of the ubiquitin-like protein UBL5 in ensuring the proper functioning of the FA pathway (Fig7E). Unlike other UBLs, UBL5 does not form covalent conjugates with cellular proteins but instead exerts its function in the FA pathway by acting as a direct binding partner of FANCI that promotes its stability and functionality.
Stabilization of proteins through interactions with dedicated binding partners is a common theme in cell biology. As an example, in S. cerevisiae, the formation of heterodimers between MATα2 and MATa1 masks their ubiquitin-dependent degradation signals, increasing the stability of both proteins (Johnson et al, 1998). In the context of the FA pathway, such proteostatic relationships have also been described. For instance, FAAP100, a component of the FA core complex, is crucial for the stabilization of its binding partners FANCB and FANCL (Ling et al, 2007). Moreover, FAAP20 physically interacts with FANCA, and these proteins are mutually dependent on each other for stability (Ali et al, 2012; Kim et al, 2012; Leung et al, 2012; Yan et al, 2012). In addition to the FA core complex, it is well established that FANCD2 is largely absent in patient cell lines containing FANCI mutations and FANCI siRNA-treated cells (Dorsman et al, 2007; Sims et al, 2007; Smogorzewska et al, 2007). FANCD2 thus appears to rely strongly on heterodimerization with FANCI for its own stability. Interestingly, however, the stability of FANCI is not affected by loss of FANCD2. Our results suggest that UBL5 stabilizes FANCI through direct protein–protein interaction, as protection of FANCI from proteasomal degradation is weakened in cells expressing mutated forms of UBL5 or FANCI that impair their association. This may, at least in part, account for the observed absence of FANCI destabilization in cells lacking FANCD2. In addition to this direct mechanism of FANCI stabilization, UBL5 indirectly regulates the levels of FANCI via its canonical involvement in modulating pre-mRNA splicing. Importantly, these dual mechanisms by which UBL5 maintains FANCI expression levels can be uncoupled by a specific separation-of-function mutation that reduces UBL5–FANCI interaction but leaves the spliceosomal function of UBL5 intact. As the FA pathway is compromised in cells expressing such a mutant, our studies suggest that UBL5 is directly involved in ensuring the functionality of the FA pathway and thus plays a significant role in ICL repair in human cells. Despite extensive efforts to identify novel FA genes, there are still FA patients for which the underlying genetic defects have not been established. Given the strong cell proliferation defect associated with complete elimination of UBL5 function (Oka et al, 2014), biallelic inactivating mutations in UBL5 are unlikely to be compatible with life. However, the notion that a separation-of-function UBL5 mutant selectively compromising FA pathway functionality can be created formally renders possible that more subtle alterations of the UBL5 gene could manifest as an FA-related phenotype.
In addition to our finding that UBL5 stabilizes FANCI, our data also suggest that UBL5 is important for FANCI–FANCD2 heterodimerization and, consequently, for their ICL-induced monoubiquitylation. X-ray crystallography studies of FANCD2 and FANCI have shown that the binding surfaces in these proteins are composed of four α-solenoid and two helical segments (Joo et al, 2011). Based on a 3D structure of a partial FANCI–FANCD2 heterodimer, the UBL5-interacting region that we mapped resides in Solenoid 1 of FANCI, which is in close proximity to Solenoid 2 of FANCD2. Interestingly, while the FANCI ∆UBL5 mutant showed strongly impaired binding to FANCD2 in cells, it remained competent to interact with FANCD2 in vitro. It is therefore possible that UBL5 may engage FANCI in an intermediary binding reaction that facilitates its ensuing heterodimerization with FANCD2 in cells. The absence of detectable UBL5 accumulation at sites of DNA damage suggests that this chaperone-like role of UBL5 in supporting FANCI functionality is mainly exerted outside the context of DNA repair processes involving FANCI.
In the present study, we also observed an ability of FANCI to form FANCD2-independent homodimers and/or oligomers. The formation of such higher-order FANCI complexes also requires the UBL5-binding site and confers stabilization of FANCI. Thus, UBL5 may promote FANCI stability through direct binding, by stimulation of FANCI homomer formation, or both. Interestingly, previous studies suggested that recombinant FANCI can form homodimers in vitro (Takahashi et al, 2014) and that also a small fraction of FANCD2 molecules exist as homo-oligomers in vivo (Zhi et al, 2010). Recent measurements of absolute copy numbers of proteins in U2OS cells estimated that FANCI is considerably more abundant than FANCD2 (Beck et al, 2011). Our data suggest that UBL5-mediated FANCI homodimerization might be a mechanism to protect this excess pool of FANCI molecules from degradation. As FANCD2 is highly dependent on binding to FANCI for its own stability, such a mechanism, perhaps actively regulated by UBL5, could help to accurately buffer the cellular levels of FANCD2 in cells. It is also possible that FANCI homomers might have a distinct functional role in the DNA damage response independently of FANCD2. To this end, it has been shown that FANCI possesses DNA binding activity in the absence of FANCD2 (Longerich et al, 2009, 2014; Yuan et al, 2009; Joo et al, 2011; Sato et al, 2012) and that unmodified FANCI binds DNA with similar affinity as phosphomimetic or ubiquitylated forms (Sato et al, 2012; Longerich et al, 2014). Thus, FANCI may have as-yet unappreciated, FANCD2-independent roles in preserving genome integrity, and UBL5 may be involved in maintaining this pool of non-FANCD2-associated FANCI molecules.
In conclusion, we have identified UBL5 as a FANCI-binding protein and demonstrated an important role of this interplay in promoting FANCI stability, FANCI hetero- and homodimerization and thus the overall functionality of the FA pathway.
Materials and Methods
Plasmids and siRNA
Full-length human UBL5 cDNA was amplified by PCR and inserted into pcDNA4/TO (Invitrogen) containing an N-terminal Strep-HA-tag. UBL5 cDNAs were rendered insensitive to UBL5 siRNA #1 by introducing the underlined silent mutations (5′-CCTAGAACTGTACTACCAG-3′). A plasmid expressing HA-tagged FANCI WT was a kind gift from Dr. Tony T. Huang (New York University, USA). Plasmid encoding DmrB-GFP-FANCI was generated by insertion of a synthetically produced FK506-binding protein derivative DmrB cDNA into the NheI and AgeI sites between the CMV promoter and GFP coding region. Cloning and site-directed mutagenesis were performed using PrimeSTAR Max Polymerase (Clontech) and KOD Hot Start Polymerase (Novagen). All constructs were verified by sequencing. Plasmid DNA and siRNA transfections were performed using GeneJuice (Novagene) and Lipofectamine RNAiMAX (Invitrogen), respectively, according to the manufacturer’s instructions. siRNA target sequences used in this study were as follows: Control (CTRL) (5′-GGGAUACCUAGACGUUCUA-3′); UBL5(#1) (5′-CCUGGAGCUUUAUUAUCAA-3′); UBL5(#2) (5′-CGAUUUUUAAGGACCACGU-3′); FANCI(#1) (5′-GCAGAAAGAAAUAGCGUCU-3′); FANCI(5′-UTR) (5′-GGAAGUUUGUGGCGGAGUU-3′); FANCI(3′-UTR) (5′-GCGCUUCACCUGAAAGAUA-3′); FANCD2 (5′-CAACAUACCUCGACUCAUU-3′); SART1 (5′-GCAUCGAGGAGACUAACAA-3′); EFTUD2 (5′-GCAUGUAUUCCACUGAUGA-3′); and WAPL (5′-CGGACUACCCUUAGCACAA-3′).
Cell culture, cell synchronization and colony survival assays
Human U2OS, HeLa and HEK293T cells were cultured in DMEM containing 10% fetal bovine serum. Cell lines stably expressing HA-FANCI were obtained by selecting U2OS or HeLa cells transfected with pIRESpuro3-HA-FANCI plasmids in medium containing 1 μg/ml puromycin (Sigma-Aldrich). To generate cell lines stably expressing Strep-HA-UBL5, U2OS or HeLa cells were co-transfected with pcDNA4/TO-Strep-HA-UBL5 and pcDNA6/TR (Invitrogen) plasmids, and positive clones were selected in medium containing 5 μg/ml blasticidin S and 200 μg/ml zeocin (both from Invitrogen). All cell lines were regularly tested for mycoplasma infection. Other chemicals used included mitomycin C, doxycycline and cycloheximide (all from Sigma-Aldrich), B/B homodimerizer (AP20187, Clontech) and ML323 (MedChemExpress). To synchronize cells in S phase, cells were grown in the presence of 2 mM thymidine for 17 h, washed with PBS and grown in fresh medium for 9 h. Subsequently, cells were cultured in medium with thymidine for additional 21 h. Samples were harvested at various time points after release from the second thymidine block. For clonogenic survival assays, U2OS cells transfected with siRNAs were treated with MMC for 24 h. Cells were trypsinized and seeded in 6-well plates and allowed to grow for 14 days. Colonies were fixed with methanol and stained with crystal violet, and colonies with more than 50 cells were counted.
Chromosome metaphase spreads
To prepare chromosome spreads, cells treated with nocodazole for 2 h were collected and incubated in 0.075 M KCl solution for 20 min at 37°C. Cells were then fixed in methanol/acetic acid (3:1 ratio), dropped onto glass slides and stained with 5.8% Giemsa solution.
Immunochemical methods
Immunoblotting, immunoprecipitation and Strep-Tactin pull-down were done as described (Poulsen et al, 2012). To detect interaction between endogenous FANCI and UBL5, cells were incubated with 125 U/ml benzonase nuclease (Sigma) in modified RIPA buffer (50 mM Tris, pH 7.5; 150 mM NaCl; 1% NP-40; 0.1% sodium deoxycholate; 1.5 mM MgCl2) supplemented with protease inhibitor cocktail (Roche) for 15 min at 37°C. The extract was then incubated with anti-FANCI antibody (Atlas antibodies) and Dynabeads Protein G (Life Technologies). After five washes in lysis buffer, the immunoprecipitates were eluted with Laemmli sample buffer. Antibodies used in this study included the following: rabbit polyclonals to FANCI, SMC1, WAPL and EFTUD2 (Bethyl), FANCD2 and β-tubulin (Abcam), FANCI and SART1 (Atlas antibodies), FANCD2 (Novus Biologicals), ORC2 (BD Biosciences) and cyclin A (Santa Cruz); mouse monoclonals to HA and GFP (Santa Cruz), cyclin A (BD Biosciences), cyclin B1 (Abcam), γ-H2AX and β-actin (Millipore), 6xHis (Clontech) and FLAG (Sigma); goat polyclonals to MCM6, FANCC and FANCE (Santa Cruz); and rat monoclonal to HA (Roche). Rabbit polyclonal S. pombe Hub1 antibody used for immunoblotting of human UBL5 was a kind gift from Dr. Hideki Yashiroda (University of Tokyo, Japan).
Purification of FANCI from HEK293T cells
After transfection of siRNA against FANCD2, HEK293T cells were transiently transfected with plasmid encoding Strep-GFP-FANCI and collected 24 h later. Cells were lysed in high-salt buffer (50 mM Tris, pH 7.4; 500 mM NaCl; 1% NP-40; 5 mM EDTA; 1 mM DTT) containing protease inhibitor cocktail (Roche). Lysates were sonicated and passed through a 0.45-μm filter and then subjected to pull-down with Strep-Tactin Sepharose (IBA). After washing with high-salt buffer, the bound protein was eluted with D-Desthiobiotin elution buffer (IBA).
In vitro binding assays
For detection of interaction between chicken FANCI and human UBL5, bacterially expressed and purified chicken FANCI, a kind gift from Dr. Hitoshi Kurumizaka (Waseda University, Japan), was mixed with either His6-SUMO2 or His6-UBL5 (Boston Biochem or Enzo Life Sciences, respectively) in binding buffer (20 mM Tris–HCl, pH 7.4; 0.15% Triton X-100; 150 mM NaCl; 10 mM imidazole). The samples were incubated with Ni-NTA agarose beads (Qiagen) under continuous end-over-end rotation at 4°C for 2 h and then washed five times in binding buffer. Bound His6-tagged proteins were eluted with elution buffer (20 mM Tris–HCl, pH 7.4; 0.15% Triton X-100; 150 mM NaCl; 500 mM imidazole). Alternatively, 6 pmol recombinant His6-UBL5 (Enzo Life Sciences) was mixed with either 6 pmol purified Strep-GFP-FANCI WT or ∆UBL5 in binding buffer (20 mM HEPES-KOH, pH 8.0; 0.25% NP-40; 150 mM NaCl; 0.1% BSA). After incubation for 15 min at room temperature, Strep-Tactin Sepharose (IBA) was added, and the mixture was incubated for 1 h at room temperature. After six washes in binding buffer, bound proteins were eluted with Laemmli sample buffer. For detection of interaction between FANCI and FANCD2 in vitro, 2 pmol recombinant chicken His6-FANCD2 (kind gift from Eeson Rajendra and Lori A. Passmore, MRC Laboratory of Molecular Biology, Cambridge, UK) was mixed with 2 pmol Strep-GFP-FANCI WT or ∆UBL5 purified from HEK293T cells in binding buffer (20 mM HEPES-KOH, pH 7.4; 0.05% NP-40; 100 mM NaCl; 1 mM DTT; 0.1% BSA). After incubation for 15 min at room temperature, Strep-Tactin Sepharose (IBA) was added, and the mixture was incubated for 1 h at room temperature. After six washes in binding buffer, bound proteins were eluted with Laemmli sample buffer.
Immunofluorescence staining and laser microirradiation
For immunofluorescence staining, cells were fixed in 4% formaldehyde for 10 min, permeabilized with PBS containing 0.2% Triton X-100 for 5 min and incubated with primary antibodies diluted in DMEM for 1 h at room temperature. Following staining with secondary antibodies (Alexa Fluor 488 and 568; Life Technologies) for 30 min, coverslips were mounted in Vectashield mounting medium (Vector Laboratories) containing DAPI. Images were acquired with an LSM780 confocal microscope (Carl Zeiss Microimaging Inc.) mounted on Zeiss-Axiovert 100M equipped with Plan-Apochromat 40×/1.3 oil immersion objective, using standard settings. Image acquisition and analysis was carried out with ZEN2010 software. To generate laser-induced localized ICLs, cells grown on coverslips were incubated with trioxsalen (6 μM) for 10 min prior to laser irradiation. The coverslips were transferred to a LabTek chamber and irradiated using PALM MicroBeam equipped with an UV-A pulsed laser (Zeiss). Localized irradiation was performed through a 40× objective with a cut speed of 10% and power output of 25%. Operation was assisted by the PALMRobo-Software supplied by the manufacturer.
In situ proximity ligation assay
In situ proximity ligation assay was performed using Duolink II Detection Kit with anti-mouse MINUS and anti-rabbit PLUS PLA Probes, according to the manufacturer’s instructions (Olink Bioscience).
Quantitative RT–PCR
Total RNA was extracted using RNeasy Mini kit (Qiagen), and cDNA synthesis was performed using SuperScript III Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions. Quantitative PCR was performed using Brilliant III Ultra-Fast SYBR Green QPCR Master (Agilent Technologies). Primer sequences for these reactions were as follows: FANCI (forward): 5′-CACCACACTTACAGCCCTTG-3′; FANCI (reverse): 5′-ATTCCTCCGGAGCTCTGAC-3′; β-Actin (forward): 5′-TCACCCACATGTGCCCATCTACGA-3′; and β-Actin (reverse): 5′-CAGCGGAACCGCTCATTGCCAATGG-3′. Experiments were repeated three times and each quantitative PCR experiment was performed in duplicate.
Acknowledgments
We thank Drs. Hideki Yashiroda, Tony T. Huang, Hitoshi Kurumizaka, Eeson Rajendra and Lori A. Passmore for providing reagents. This work was supported by grants from The Novo Nordisk Foundation (Grant no. NNF14CC0001), Danish Council for Independent Research, Danish Cancer Society and The Lundbeck Foundation.
Author contributions
YO performed all experiments, analyzed data and wrote the manuscript. SB-J supervised the project, analyzed data and wrote the manuscript. NM supervised the project, analyzed data and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figures S1–S6
Review Process File
References
- Ali AM, Pradhan A, Singh TR, Du C, Li J, Wahengbam K, Grassman E, Auerbach AD, Pang Q, Meetei AR. FAAP20: a novel ubiquitin-binding FA nuclear core-complex protein required for functional integrity of the FA-BRCA DNA repair pathway. Blood. 2012;119:3285–3294. doi: 10.1182/blood-2011-10-385963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpi AF, Pace PE, Babu MM, Patel KJ. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol Cell. 2008;32:767–777. doi: 10.1016/j.molcel.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Ammon T, Mishra SK, Kowalska K, Popowicz GM, Holak TA, Jentsch S. The conserved ubiquitin-like protein Hub1 plays a critical role in splicing in human cells. J Mol Cell Biol. 2014;6:312–323. doi: 10.1093/jmcb/mju026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, Aebersold R. The quantitative proteome of a human cell line. Mol Syst Biol. 2011;7:549. doi: 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clackson T, Yang W, Rozamus LW, Hatada M, Amara JF, Rollins CT, Stevenson LF, Magari SR, Wood SA, Courage NL, Lu X, Cerasoli F, Jr, Gilman M, Holt DA. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc Natl Acad Sci USA. 1998;95:10437–10442. doi: 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn MA, Kowal P, Yang K, Haas W, Huang TT, Gygi SP, D’Andrea AD. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol Cell. 2007;28:786–797. doi: 10.1016/j.molcel.2007.09.031. [DOI] [PubMed] [Google Scholar]
- Dorsman JC, Levitus M, Rockx D, Rooimans MA, Oostra AB, Haitjema A, Bakker ST, Steltenpool J, Schuler D, Mohan S, Schindler D, Arwert F, Pals G, Mathew CG, Waisfisz Q, de Winter JP, Joenje H. Identification of the Fanconi anemia complementation group I gene, FANCI. Cell Oncol. 2007;29:211–218. doi: 10.1155/2007/151968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Zeng Y, Whitesell L, Katsanis E. Stressed apoptotic tumor cells express heat shock proteins and elicit tumor-specific immunity. Blood. 2001;97:3505–3512. doi: 10.1182/blood.v97.11.3505. [DOI] [PubMed] [Google Scholar]
- Gibbs-Seymour I, Oka Y, Rajendra E, Weinert BT, Passmore LA, Patel KJ, Olsen JV, Choudhary C, Bekker-Jensen S, Mailand N. Ubiquitin-SUMO circuitry controls activated Fanconi anemia ID complex dosage in response to DNA damage. Mol Cell. 2015;57:150–164. doi: 10.1016/j.molcel.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13:467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature. 2009;458:422–429. doi: 10.1038/nature07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, Saberi A, Kinoshita E, Kinoshita-Kikuta E, Koike T, Tashiro S, Elledge SJ, Takata M. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat Struct Mol Biol. 2008;15:1138–1146. doi: 10.1038/nsmb.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PR, Swanson R, Rakhilina L, Hochstrasser M. Degradation signal masking by heterodimerization of MATalpha2 and MATa1 blocks their mutual destruction by the ubiquitin-proteasome pathway. Cell. 1998;94:217–227. doi: 10.1016/s0092-8674(00)81421-x. [DOI] [PubMed] [Google Scholar]
- Joo W, Xu G, Persky NS, Smogorzewska A, Rudge DG, Buzovetsky O, Elledge SJ, Pavletich NP. Structure of the FANCI-FANCD2 complex: insights into the Fanconi anemia DNA repair pathway. Science. 2011;333:312–316. doi: 10.1126/science.1205805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, D’Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–1408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Yang K, Dejsuphong D, D’Andrea AD. Regulation of Rev1 by the Fanconi anemia core complex. Nat Struct Mol Biol. 2012;19:164–170. doi: 10.1038/nsmb.2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lelij P, Stocsits RR, Ladurner R, Petzold G, Kreidl E, Koch B, Schmitz J, Neumann B, Ellenberg J, Peters JM. SNW1 enables sister chromatid cohesion by mediating the splicing of sororin and APC2 pre-mRNAs. EMBO J. 2014;33:2643–2658. doi: 10.15252/embj.201488202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung JW, Wang Y, Fong KW, Huen MS, Li L, Chen J. Fanconi anemia (FA) binding protein FAAP20 stabilizes FA complementation group A (FANCA) and participates in interstrand cross-link repair. Proc Natl Acad Sci USA. 2012;109:4491–4496. doi: 10.1073/pnas.1118720109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Dexheimer TS, Zhang P, Rosenthal AS, Villamil MA, You C, Zhang Q, Chen J, Ott CA, Sun H, Luci DK, Yuan B, Simeonov A, Jadhav A, Xiao H, Wang Y, Maloney DJ, Zhuang Z. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat Chem Biol. 2014;10:298–304. doi: 10.1038/nchembio.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling C, Ishiai M, Ali AM, Medhurst AL, Neveling K, Kalb R, Yan Z, Xue Y, Oostra AB, Auerbach AD, Hoatlin ME, Schindler D, Joenje H, de Winter JP, Takata M, Meetei AR, Wang W. FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. EMBO J. 2007;26:2104–2114. doi: 10.1038/sj.emboj.7601666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longerich S, San Filippo J, Liu D, Sung P. FANCI binds branched DNA and is monoubiquitinated by UBE2T-FANCL. J Biol Chem. 2009;284:23182–23186. doi: 10.1074/jbc.C109.038075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longerich S, Kwon Y, Tsai MS, Hlaing AS, Kupfer GM, Sung P. Regulation of FANCD2 and FANCI monoubiquitination by their interaction and by DNA. Nucleic Acids Res. 2014;42:5657–5670. doi: 10.1093/nar/gku198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luders J, Pyrowolakis G, Jentsch S. The ubiquitin-like protein HUB1 forms SDS-resistant complexes with cellular proteins in the absence of ATP. EMBO Rep. 2003;4:1169–1174. doi: 10.1038/sj.embor.7400025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally T, Huang Q, Janis RS, Liu Z, Olejniczak ET, Reilly RM. Structural analysis of UBL5, a novel ubiquitin-like modifier. Protein Sci. 2003;12:1562–1566. doi: 10.1110/ps.0382803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra SK, Ammon T, Popowicz GM, Krajewski M, Nagel RJ, Ares M, Jr, Holak TA, Jentsch S. Role of the ubiquitin-like protein Hub1 in splice-site usage and alternative splicing. Nature. 2011;474:173–178. doi: 10.1038/nature10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan GL, D’Andrea AD. How the Fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–249. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D’Andrea AD, Bernards R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005;17:331–339. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Oka Y, Varmark H, Vitting-Seerup K, Beli P, Waage J, Hakobyan A, Mistrik M, Choudhary C, Rohde M, Bekker-Jensen S, Mailand N. UBL5 is essential for pre-mRNA splicing and sister chromatid cohesion in human cells. EMBO Rep. 2014;15:956–964. doi: 10.15252/embr.201438679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen M, Lukas C, Lukas J, Bekker-Jensen S, Mailand N. Human RNF169 is a negative regulator of the ubiquitin-dependent response to DNA double-strand breaks. J Cell Biol. 2012;197:189–199. doi: 10.1083/jcb.201109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendra E, Oestergaard VH, Langevin F, Wang M, Dornan GL, Patel KJ, Passmore LA. The genetic and biochemical basis of FANCD2 monoubiquitination. Mol Cell. 2014;54:858–869. doi: 10.1016/j.molcel.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramelot TA, Cort JR, Yee AA, Semesi A, Edwards AM, Arrowsmith CH, Kennedy MA. Solution structure of the yeast ubiquitin-like modifier protein Hub1. J Struct Funct Genomics. 2003;4:25–30. doi: 10.1023/a:1024674220425. [DOI] [PubMed] [Google Scholar]
- Sato K, Toda K, Ishiai M, Takata M, Kurumizaka H. DNA robustly stimulates FANCD2 monoubiquitylation in the complex with FANCI. Nucleic Acids Res. 2012;40:4553–4561. doi: 10.1093/nar/gks053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims AE, Spiteri E, Sims RJ, III, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, Auerbach AD, Huang TT. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol. 2007;14:564–567. doi: 10.1038/nsmb1252. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, III, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, Elledge SJ. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Sundaramoorthy S, Vazquez-Novelle MD, Lekomtsev S, Howell M, Petronczki M. Functional genomics identifies a requirement of pre-mRNA splicing factors for sister chromatid cohesion. EMBO J. 2014;33:2623–2642. doi: 10.15252/embj.201488244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi D, Sato K, Shimomuki M, Takata M, Kurumizaka H. Expression and purification of human FANCI and FANCD2 using Escherichia coli cells. Protein Expr Purif. 2014;103:8–15. doi: 10.1016/j.pep.2014.08.012. [DOI] [PubMed] [Google Scholar]
- van der Veen AG, Ploegh HL. Ubiquitin-like proteins. Annu Rev Biochem. 2012;81:323–357. doi: 10.1146/annurev-biochem-093010-153308. [DOI] [PubMed] [Google Scholar]
- Walden H, Deans AJ. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys. 2014;43:257–278. doi: 10.1146/annurev-biophys-051013-022737. [DOI] [PubMed] [Google Scholar]
- Watrin E, Demidova M, Watrin T, Hu Z, Prigent C. Sororin pre-mRNA splicing is required for proper sister chromatid cohesion in human cells. EMBO Rep. 2014;15:948–955. doi: 10.15252/embr.201438640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Guo R, Paramasivam M, Shen W, Ling C, Fox D, III, Wang Y, Oostra AB, Kuehl J, Lee DY, Takata M, Hoatlin ME, Schindler D, Joenje H, de Winter JP, Li L, Seidman MM, Wang W. A ubiquitin-binding protein, FAAP20, links RNF8-mediated ubiquitination to the Fanconi anemia DNA repair network. Mol Cell. 2012;47:61–75. doi: 10.1016/j.molcel.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Moldovan GL, Vinciguerra P, Murai J, Takeda S, D’Andrea AD. Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev. 2011;25:1847–1858. doi: 10.1101/gad.17020911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiroda H, Tanaka K. Hub1 is an essential ubiquitin-like protein without functioning as a typical modifier in fission yeast. Genes Cells. 2004;9:1189–1197. doi: 10.1111/j.1365-2443.2004.00807.x. [DOI] [PubMed] [Google Scholar]
- Yuan F, El Hokayem J, Zhou W, Zhang Y. FANCI protein binds to DNA and interacts with FANCD2 to recognize branched structures. J Biol Chem. 2009;284:24443–24452. doi: 10.1074/jbc.M109.016006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Walter JC. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair. 2014;19:135–142. doi: 10.1016/j.dnarep.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi G, Chen X, Newcomb W, Brown J, Semmes OJ, Kupfer GM. Purification of FANCD2 sub-complexes. Br J Haematol. 2010;150:88–92. doi: 10.1111/j.1365-2141.2010.08217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1–S6
Review Process File