

Summary
Angiogenesis is an important ‘hallmark’ of cancer. We reviewed the various pathways controlling angiogenesis, summarized the possible role of specific environmental chemicals disrupting these pathways and listed assays for assessing the effects of low-dose exposures to chemicals in promoting tumor angiogenesis.
Abstract
One of the important ‘hallmarks’ of cancer is angiogenesis, which is the process of formation of new blood vessels that are necessary for tumor expansion, invasion and metastasis. Under normal physiological conditions, angiogenesis is well balanced and controlled by endogenous proangiogenic factors and antiangiogenic factors. However, factors produced by cancer cells, cancer stem cells and other cell types in the tumor stroma can disrupt the balance so that the tumor microenvironment favors tumor angiogenesis. These factors include vascular endothelial growth factor, endothelial tissue factor and other membrane bound receptors that mediate multiple intracellular signaling pathways that contribute to tumor angiogenesis. Though environmental exposures to certain chemicals have been found to initiate and promote tumor development, the role of these exposures (particularly to low doses of multiple substances), is largely unknown in relation to tumor angiogenesis. This review summarizes the evidence of the role of environmental chemical bioactivity and exposure in tumor angiogenesis and carcinogenesis. We identify a number of ubiquitous (prototypical) chemicals with disruptive potential that may warrant further investigation given their selectivity for high-throughput screening assay targets associated with proangiogenic pathways. We also consider the cross-hallmark relationships of a number of important angiogenic pathway targets with other cancer hallmarks and we make recommendations for future research. Understanding of the role of low-dose exposure of chemicals with disruptive potential could help us refine our approach to cancer risk assessment, and may ultimately aid in preventing cancer by reducing or eliminating exposures to synergistic mixtures of chemicals with carcinogenic potential.
Introduction
Angiogenesis, the formation of new blood vessels from existing blood vessels, was identified as one of the ‘hallmarks of cancer’ by Hanahan and Weinberg (1,2) due to the recognition that this process is of crucial importance during the transition from benign hyperplastic nodules to malignant lesions (3). This review article focused on angiogenesis constitutes an integral component of the 2013 Halifax Project on ‘Assessing the Carcinogenic Potential of Low-Dose Exposures to Chemical Mixtures in the Environment’ (see Capstone Article for details). Tumor expansion is dependent on the ability of the tumor to induce the growth of new blood vessels, which provide nutrients and oxygen to the growing tumor mass and simultaneously serve as a conduit for tumor cells to metastasize to distant organs (4,5). Tumor angiogenesis is integral not only in solid tumor progression but also in leukemia (6). Recent cancer treatments target tumor angiogenesis using antiangiogenesis inhibitors (7,8), which prevent new vessel formation, or by using vascular-disrupting/damaging agents (9–11) and neovascular-targeting immunoconjugates (12–14). However, angiogenesis is also necessary for normal organ function, tissue growth and regeneration (e.g. wound healing, female menstruation, ovulation and pregnancy), necessitating a fine balance to avoid complications due to antiangiogenic therapy (15–17).
Though human exposures to environmental chemicals, which often occur due to the leaching of plastics into food and water (18), have been found to promote tumorigenesis of multiple cancers through various mechanisms (19–24), less attention has been focused on their role in tumor angiogenesis. With increases in our knowledge of endocrine disruptors (25), new concerns have arisen about potential exposures to low doses of environmental chemicals that are generally regarded as non-carcinogens, but may be acting as proangiogenic agents. Here, we consider the possibility that certain chemical disruptors, which are prevalent in the environment (e.g. as pesticides and industrial surfactants) (26), may have a role to play in environmental carcinogenesis by stimulating proangiogenic pathways, providing an environment conducive to tumor growth and metastasis.
In this review, we discuss emerging data on specific environmental chemicals that may act as proangiogenic agents, and identify key angiogenesis pathways and corresponding molecular components as prioritized targets for future study. We briefly summarize in vitro and in vivo angiogenesis model systems with an emphasis on high-throughput screening (HTS) assays. We also consider the cross-hallmark relationships that a number of important angiogenic pathway targets have with other hallmarks of the disease and we make recommendations for future research.
Identifying VEGFR- and TF-mediated signaling as two key tumor angiogenesis pathways and corresponding molecular components as prioritized targets for assessing the carcinogenic potential of low-dose exposures to chemical mixtures in the environment
Tumor growth and metastasis require angiogenesis to provide a circuit for increased blood supply and dissemination of tumor cells (27). Angiogenesis is tightly controlled by diverse subsets of ligands and receptors. Enrichment of ligands, including growth factors, chemokines and cytokines or a decrease in the production of endogenous angiogenesis inhibitors, has been extensively observed in tumors during vascularization. The biology and mechanisms of tumor angiogenesis have been elegantly summarized elsewhere (4,28–33). Here, we will only briefly review some of the key angiogenic pathways [vascular endothelial growth receptor (VEGFR) and tissue factor (TF)-mediated signaling] (Figure 1A) and pathway-associated molecular components (Figure 1B) to provide a framework for our review and discussion of potential chemical disruptors (Figure 1B).
Figure 1.
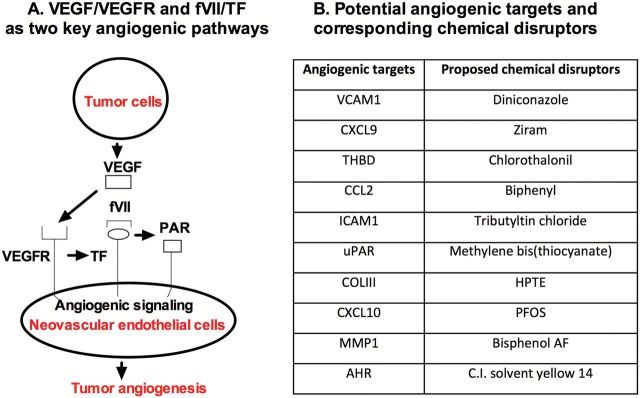
VEGF and TF-signaling pathways as prioritized tumor angiogenic pathways (A) and proposed angiogenic molecular targets and their corresponding chemical disruptors (B). (A) The diagram shows VEGF produced by tumor cells binds to VEGFR on vascular endothelial cells to activate VEGF signaling pathways in tumor angiogenesis. In addition, VEGF binding to endothelial cells can induce TF expression, an angiogenic endothelial receptor in pathological neovasculature. After its ligand fVII binds, TF could contribute to tumor angiogenesis via proteolysis-dependent pathways through PARs or proteolysis-independent pathway through its cytoplasmic domain. (B) Proposed list of specific angiogenesis molecular targets and corresponding chemical disruptors.
The vascular endothelial growth factor (VEGF) pathway is crucial for cancer angiogenesis. As a tumor enlarges, the tissue becomes hypoxic and deprived of nutrients leading to increased expression of factors involved in both fighting against and adapting to these stressful conditions (34). Such factors will activate the growth of new blood vessels to increase the oxygen and nutrients supply but also lower the oxygen-dependent metabolism by causing a shift to glycolytic metabolism in the tumor cells (35). A well-studied example of hypoxia-induced tumor angiogenesis is the stabilization of hypoxia-inducible factor 1 alpha (HIF-1α) in hypoxic tumor tissues which lead to production of VEGF-A and nitric oxide synthase (NOS) that act as drivers of neovascularization and dilation of the existing blood vessels, respectively (36). In addition to VEGF-A, other growth factors including angiopoeitin-2 (Ang-2), fibroblast growth factors (FGFs), platelet-derived growth factors, insulin-like growth factor and transforming growth factor-beta (TGF-β) are also produced at high levels by hypoxic tumor or tumor stromal cells and lead to disruption of the tumor vessels (37). The tumor milieu, which has been compared to that of a healing wound (38), also leads to massive recruitment and activation of inflammatory cells types, including macrophages, neutrophils and lymphocytes, which are producing proangiogenic cytokines including tumor necrosis factor-alpha, interleukin 1 beta (IL-1β) and interleukin 6 (IL-6). In addition, carcinoma-associated fibroblasts are also rich sources of a wide range of angiogenic factors, further complicating the proangiogenic phenotype of solid tumors (39,40). In addition to angiogenic factors, deregulated vessel sprouting and path finding through disruptions in, for instance Notch-activation by delta-like ligand 4 (Dll4) and Jagged1 ligands (41), are involved in disrupting the tumor vascular functions further contributing to the pathological phenotypes of the tumor blood vessels. Activated endothelial cells and tumor-associated macrophages produce matrix metalloproteinases (MMPs) including a disintegrin and metalloproteases, MMP-2 and MMP-9, which cleave extracellular matrix (ECM) to release more ECM-bound angiogenic factors and further reduce the integrity of the vasculature, leading to a vicious circle driving pathologic progression in cancer (42,43). In addition to expressed proteins, angiogenesis-modulating miRNAs, so called angiomiRs, directly repress gene expression of several angiogenic or antiangiogenic factors by binding to the 3′-untranslated regions (3′-UTR) of target mRNAs (44). For instance, miR-21 promotes cancer progression and angiogenesis through Akt and ERK pathways (45).
As a consequence of such untamed and exaggerated angiogenic signaling by the broad palette of proangiogenic factors existing at high levels in the tumor, the vasculature become highly chaotic, immature and of very low quality (in terms of the stability and barrier function of the vascular wall) and functionality (in terms of supporting efficient perfusion through the tumor) (46). As such, tumor blood vessels exhibit excessive leakage, causing highly elevated interstitial fluid pressure and inhibited delivery of blood, paradoxically further contributing to tumor hypoxia and decreasing delivery of drugs injected to the blood stream. At the same time, such deregulated tumor blood vessels pose little opposition against tumor-cell intravasation and metastatic dissemination. As such the pathological vasculature can be considered a main cause of resistance to therapy and progression of the cancer to metastatic disease (47).
While tumor angiogenic vascular endothelial cells (VECs) may express VEGFR at higher levels (48), VEGFRs are not specific for angiogenic endothelial cells, but are constitutively expressed also in the quiescent vasculature in normal organs (49,50). In contrast, TF may be a promising target, which is specifically expressed by angiogenic vessels, making it more specific for the tumor vasculature than VEGF receptors. Under physiological conditions, TF is only expressed on some cells outside of vessels, but is not expressed by quiescent endothelial cells of blood vessels in normal organs (51). Accumulating evidence suggests that TF also contribute directly and indirectly to tumor angiogenesis (52–56). TF is a transmembrane protein receptor (57–60), which is composed of 263 amino acid (aa) residues with an extracellular domain (1–219 aa), a transmembrane domain (220–242 aa) and a short cytoplasmic domain (243–263 aa). As a type I membrane bound receptor, TF forms an exceptionally strong and specific complex with its natural ligand coagulation factor VII (61,62), the initial step of the coagulation pathway (63). In tumor angiogenesis, it is found that TF expression is only detected on angiogenic tumor VECs (13,64–66), a downstream product induced by VEGF that can be secreted by cancer cells (67,68) and cancer stem cells (69). More importantly, TF is selectively expressed in vivo in the tumor neovasculature (12,13,64,65,70) and in vitro by VEGF-stimulated VECs, thus the latter could serve as an in vitro model of angiogenic endothelial cells (71–73). Other angiogenic factors and inflammatory chemokines (such as bFGF, IL-1β, tumor necrosis factor-alpha, bacterial lipopolysaccharide (LPS)) can also induce TF expression on VECs under pathological conditions (54). Thus TF can be regarded as an angiogenic-specific endothelial receptor (65,72,73). We believe that this unique feature makes TF a promising therapeutic target for neovascular-targeted therapy (73) and an interesting angiogenic receptor for discussion in this review and for future studies of chemical angiogenesis.
After induction by VEGF and other factors, vascular endothelial TF contributes to tumor angiogenesis via proteolysis-dependent and -independent signaling pathways (Figure 1A). More details on TF signaling in tumor angiogenesis were previously described and reviewed (52,74–77). Briefly, coagulation factor VII/TF complex can initiate the proteolysis-dependent pathway by activating protease-activated receptors (PARs), which is modulated by thrombomodulin (THBD), the endothelial-specific type I membrane receptor that binds thrombin to reduce thrombin generation, and ultimately results in the transcription of genes such as early growth response-1, adhesion molecules [intercellular adhesion molecule 1 (ICAM1), vascular cell-adhesion molecule 1 (VCAM1), P- and E-selectin], growth factors and cytokines (IL-6, IL-8, chemokines), whereas the cytoplasmic serine residues can be phosphorylated and ultimately influences endothelial cell migration. Note that many of these angiogenic components involved in VEGFR- and TF-mediated signaling are chosen as potential angiogenic targets for selected chemical disruptors (Figure 1B).
To review the role of low-dose exposures to environmental chemical disruptors in tumor angiogenesis, our angiogenesis team as part of the Halifax Project was asked to identify 10 angiogenesis molecular targets and 10 corresponding potential chemical disruptors for these angiogenic targets. We choose the following angiogenic components involved in VEGFR- and TF-signaling pathways as prioritized VCAM1, C-X-C motif chemokine ligands 9 and 10 (CXCL9 and CXCL10), THBD, monocyte-like chemoattractant protein (CCL2), ICAM1, urokinase-type plasminogen activator receptor (uPAR), collagen III, MMP1 and aryl-hydrocarbon receptor (AHR). These targets were chosen based on their relevance to the signaling pathways reviewed above, and, importantly, based on previous work that examined a large database of animal toxicity studies (ToxRefDB; http://actor.epa.gov/toxrefdb/) and the concordance between tumor incidence in vivo and chemical activity profiles in vitro. The 10 molecular targets in Figure 1B were angiogenic signaling molecules that showed statistically significant associations with mammalian carcinogenicity (78).
This list of target sites was not intended to be comprehensive. Other targets exist, including well-known molecules such as collagen IV, CXCL4, thrombospondin, MMP9, etc., But we selected these targets because each of them are actively involved in tumor angiogenesis and all of them have been shown to be of considerable importance. For example, suppression of the angiostatic molecules CXCL9 and CXCL10 and upregulation of the proangiogenic chemokine CCL2 would provide a local environment of proliferative and migratory signals to endothelial cells forming new vessels to feed a tumor (79,80). Decreased THBD expression was highly correlated with tumor invasiveness, metastasis and lower survival rates (81,82). CCL2 is complementary to angiogenesis, through p53 regulation of CCL2 gene expression (83,84). ICAM1 is also complementary to angiogenesis through NF-κB-independent role for p53 in ICAM1 regulation that may link p53 to ICAM1 function in various physiological and pathological settings (85). CXCL10 is complementary to angiogenesis through activation of p53 and p53-responsive genes. Over expression of IP10 upregulated p53 and resulted in altered expression of p53-responsive genes such as the p21Cip1, p27kip1, NF-κB, Bax and PUMA genes and the mitochondrial translocation of Bax (86). The AHR is complementary to angiogenesis through its role in cell cycle regulation. AHR modulates angiogenesis through a mechanism requiring VEGF activation in the endothelium and TGF-β inactivation in the stroma. Activation of AHR by its various ligands disrupts contact inhibition and induces cell proliferation depending on the tissue and cell type involved (87–93). THBD is contrary to angiogenesis due to over expression of p53 suppressed THBD expression (94,95). It is also complementary to genetic instability (96,97) and resistance to cell death (98). uPAR is contrary to angiogenesis (wild type p53 downregulates uPAR expression). P53 acts as an uPAR mRNA binding protein that downregulates uPAR mRNA stability and decreases cellular uPAR expression. Codepletion of Cathepsin B and uPAR reduced the expression of cyclin D1, cyclin D2, p-Rb and cyclin E while the expression of Cdk2 was unaffected. The MMP1 is contrary when cross validated with evasion of antigrowth signaling hallmark (99–102). Inactivation of Rb leads to increased expression of MMP1 and dysfunction of p53. p53 inhibits basal and UV-induced MMP-1 expression in human dermal fibroblasts and p53 dysfunction caused by XPC defects in lung cancers may enhance tumor metastasis via increased MMP1 expression (99–101,103).
To examine the role of these angiogenic pathways and prioritized targets in chemical angiogenesis, we also identify 10 corresponding chemical disruptors (Figure 1B) as novel environmental chemicals in tumor angiogenesis, which are discussed below in the Sections of ‘Environmental Carcinogens Affecting Angiogenic Pathways’ and ‘Identifying Novel Environmental Chemical Disruptors’.
Environmental carcinogens affecting angiogenic pathways
Here, we review the evidence for proangiogenic actions of cigarette smoke, nicotine and arsenic as case study compounds that provide supporting evidence for the subsequent selection of environmental chemicals that disrupt angiogenic signaling targets and potentially contribute to cancer.
Cigarette smoke
Cigarette smoke is one of the oldest environmental exposures linked to cancer (104) and contains numerous carcinogenic compounds, such as nicotine and its derivatives, described elsewhere (105,106). Cigarette and second hand smoke have both been shown to induce or be associated with angiogenesis by a variety of mechanisms, although separating angiogenic effects from other carcinogenic activities is a challenge. Mouse models of chronic colitis were found to have dose-dependent increases in blood vessel formation and VEGF protein expression following exposure to CS (107). Tumor growth, capillary density, plasma VEGF levels and circulating endothelial progenitor cells were significantly increased in mice subcutaneously injected with Lewis lung cancer cells after a 17-day exposure to second hand smoke compared to mice exposed to clean room air. These results were attenuated with mecamylamine, an inhibitor of nicotine cholinergic receptors (108).
A hospital-based case-control study consisting of 730 urothelial carcinoma cases, 470 bladder cancers, 260 upper urinary tract urothelial carcinomas and 850 age-matched controls found significant correlations between bladder and upper urinary tract urothelial carcinomas (UUTUC) and both cigarette smoking and arsenic exposure (109). The risk for both bladder cancer (6.6; 95% CI, 3.1–13.9) and UUTUC (9.9; 95% CI, 4–24.5) were increased with the presence of VEGF polymorphisms associated with increased cancer risk.
Nicotine
Nicotine, one of the main carcinogenic components of cigarettes, has been found to influence angiogenesis. Several in vitro studies have linked nicotine to proangiogenic effects in cancer. The ERK/COX-2 pathway was suggested to play a role in nicotine-induced VEGF expression in gastric cancer cells after VEGF levels were decreased by inhibitors of MEK (U0126) and COX-2 (SC-236) (110). Nicotine was further shown to influence angiogenesis in lung cancer (111). Nicotine significantly stimulated HIF-1α protein and VEGF expression in human non-small cell lung cancer (NSCLC) cells. Increased capillary and tubule formation was shown in human umbilical VECs (HUVECs) following treatment with conditioned medium containing nicotine. The possible mechanism of nicotine-induced VEGF expression was investigated with human dermal microvascular endothelial cells, which showed that the nicotine acetylcholine receptor was needed for pro-migratory effects of VEGF and bFGF in culture (111). In addition, cultured HUVECs were observed to have increased cell proliferation, migration and tube formation following exposure to nicotine at concentrations similar to those found in smokers (112).
Although in vitro studies provide some evidence that nicotine has proangiogenic properties, animal studies further bolster nicotine as a promoter of neovascularization, as well as provide possible biological mechanisms. An increase in lesion growth and lesion vascularity was seen in lung cancer and atherosclerosis mouse models following nicotine exposure (113). In addition, in a mouse model of hind-limb ischemia systemically administered nicotine (100 μg/ml in drinking water) resulted in an increase of capillary density in the hind limb from 0.38 to 0.71 (95% CI 0.55–1.01) capillaries/myocyte compared to control. Later it was shown that nude mice injected subcutaneously with HT-29 cells, a colon cancer cell line, exhibited significant increases in both blood vessels and microvessel densities after drinking water containing 200 μg/ml nicotine for 25 days. VEGF expression correlated with microvessel density. B1 and b2-selective antagonists reversed nicotine-induced tumor growth; suggesting b-adrenoceptors may be involved in nicotine-induced angiogenesis in colon cancer (114). The growth rate of breast, colon and lung cancer tumor cells implanted in a chorioallantoic membrane model exhibited significant increases following 1 week of exposure to nicotine (115). This study further showed that nicotinic receptor antagonists and integrin avb3 antagonists abrogated nicotine-mediated angiogenesis, suggesting molecular and cellular mechanisms of nicotine-mediated angiogenesis (116).
Arsenic
Another carcinogen that shows angiogenic properties is arsenic, an environmental contaminant that humans may be exposed to via environmental, medical and occupational sources (117). An in vitro study using HUVECs revealed that low concentrations (≤ 1 μM) of sodium arsenite increased cell growth and vascular tubular formation compared to higher concentrations (> 5 μM) that induced cytotoxicity and inhibited angiogenesis (117). Low concentrations of arsenic also induced transcript expression of VEGF and von Willebrand Factor, an early detector of endothelial activation, in tumor metastasis. Several subsequent in vitro studies focused on the proangiogenic properties of arsenic in human microvascular endothelial (HMVEC) cells. Klei et al. (118) investigated signaling interactions between arsenic and alcohols using non-cytotoxic concentrations of arsenite (1–5mM) with or without the presence of 0.1% ethanol. Data in this study showed that both agents together, but not ethanol alone, increased phosphorylation of the regulator of vascular integrin signaling PYK2 and VEGF gene expression as well as endothelial tube formation (118). Another study revealed that the sphingsine-1-phosphate type 1 receptor is important for arsenic-stimulated signaling for angiogenic effects (119) and that heme oxygenase-1 plays a role in arsenic-induced angiogenesis (120). Moreover, environmentally relevant levels of arsenic were shown to promote angiogenesis, neovascularization and inflammatory cell infiltration in Matrigel plugs implanted in C57BL/6 mice following 5 weeks exposures (drinking water) to concentrations ranging from 5 to 500 ppb (121).
These examples from the literature on known carcinogens indicate that environmental exposures to cigarette smoke, nicotine and arsenic can result in the increase of angiogenic activity through several pathways. There is a diversity of techniques available for investigating angiogenic activity, though there are challenges to separating effects that are specific to angiogenic pathways from other hallmark pathways.
Other environmental chemicals with proangiogenic properties
In addition to cigarette smoke, nicotine and arsenic, other potentially carcinogenic compounds have been identified that induce proangiogenic effects. Whole diesel exhaust has been shown to enhance angiogenesis in mice with either subcutaneous scaffold implantation or hindlimb ischemia (122). Increased CD31 expression, vessel volume and VEGF and HIF-1 gene expression was observed in these models. Bisphenol A has been intensively studied over the past few years due to its detrimental effects on developmental processes and metabolic effects and has recently been shown to influence angiogenesis (123). Increased gene expression of VEGFR-2, VEGF-A, eNOS and Cx43 and production of nitric oxide was found after HUVECs were exposed to 1M bisphenol A for 6h (123). Furthermore, manganese induced hypoxia-associated transcript expression of proangiogenic genes in mice (124) and both dioxin (125) and trimethyltin chloride (109) were found to influence angiogenesis and vascularization during early development in rat and zebra fish models.
Identifying novel environmental chemical disruptors
As discussed above, tumor angiogenesis is critical for carcinogenesis, and despite the evidence that several known carcinogens are targeting proangiogenic pathways the role of most environmental chemicals in tumor angiogenesis is largely unknown. In this project, we were tasked to identify ‘prototypical’ environmental chemicals with disruptive potential that met the following criteria: chemicals that are ubiquitous in the environment; chemicals that have been shown to disrupt specific mechanisms/pathways for angiogenesis; and chemical exposures that are not related to ‘lifestyle’ choices (i.e. chemicals that are not already known or designated to be human carcinogens). Our intent was to explore the possible synergies of disruptive environmental chemicals with proangiogenic capabilities that could potentially contribute to carcinogenesis (especially when combined, or when acting with other chemicals that are known to perturb other cancer hallmark pathways).
Thousands of untested chemicals in the environment lack hazard characterization of their carcinogenic potential. The Tox21 partnership of regulatory and scientific federal agencies, including USA. EPA, the National Toxicology Program (NTP), the National Center for Advancing Translational Science (NCATS) and USA. FDA, are addressing this data gap using in vitro HTS and computational modeling to predict hazard and prioritize compounds for targeted testing (126,127). The EPA’s ToxCast research project (127), part of Tox21, has tested over a thousand chemicals with known and unknown toxicities in hundreds of assays for human gene and protein targets in pathways linked to cancer disease processes (128). This effort is concurrent with the creation of the Toxicity Reference Database (ToxRefDB) containing >40 years’ worth of in vivo animal toxicity data, such as 2-year chronic cancer studies, broken down into a computable and searchable ontology structure (129). A model was recently published that used the ToxCast Phase I HTS data to predict in vivo rodent carcinogenicity endpoints from ToxRefDB (78). This work employed an unsupervised statistical approach to identify significant correlations between in vitro assay activity and preneoplastic and neoplastic lesions in a variety of tissue types, across a training set of 232 compounds with both in vitro and in vivo data. The model was able to accurately predict carcinogenicity classifications from the EPA’s Office of Pesticide Programs for an external test set of compounds, based solely on their in vitro HTS data. Interestingly, the majority of HTS assays that were strongly associated with particular types of rodent cancers were linked to genes, pathways and hallmark processes documented to be involved in tumor biology and cancer progression, including stimulation of angiogenesis.
Most of the chemical carcinogens in the model training set were food-use pesticides, meaning they are non-genotoxic and instead act as tumor promoters (130). In addition to broad activity across assays that were mapped to other hallmark processes (i.e. apoptosis, proliferative signaling, evading growth suppression, enabling replicative immortality, metastasis, avoiding immune destruction, tumor-promoting inflammation and deregulating cellular energetics) some of these compounds were linked to targets in angiogenic pathways (1,2). Thus, a subset of these chemicals may have the potential to act as tumor promoters primarily via induction of angiogenesis, based on specific patterns of bioactivity against in vitro targets associated with vascular development. Many of these targets were from enzyme-linked immunosorbent assay-based chemokine expression assays in human primary cell cocultures. Statistically significant associations were observed between pesticide exposures causing rodent liver, thyroid, spleen and kidney tumors and differential regulation of inflammatory chemokines as well as cellular adhesion molecules, and elements of the plasminogen activating system. Many of these targets, shown in Figure 1, belong to signaling pathways reviewed above. Therefore, the results from the in vitro screens of these mammalian carcinogens were in all cases consistent with a proangiogenic and thus a protumorigenic program.
Analysis of bioactivity patterns of over a thousand chemicals across hundreds of in vitro assays revealed that other carcinogens were preferentially affecting targets in chemokine signaling, vascular cellular adhesion molecules and ECM interactions controlling vascular growth factor release (78). These results strongly support the concept that at some point in cancer progression, the angiogenic switch is turned ‘ON’, facilitating tumor growth, and that carcinogenic environmental chemicals may participate in this process by regulating cellular signaling in a proangiogenic fashion.
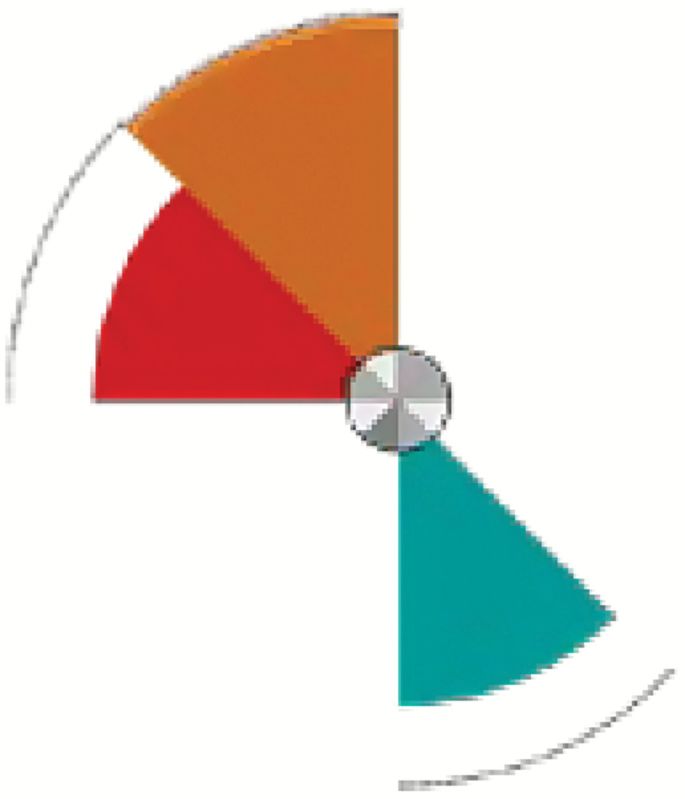
A number of environmental chemicals tested in the ToxCast program were identified as potential tumor promoters through their ability to interact with the angiogenic signaling molecules in vitro that had been shown to be significantly associated with in vivo tumorigenesis. Many of these compounds had associated in vivo data and evidence in the literature confirming their carcinogenic effects (78), while others are candidates for further study. In the ToxCast Phase I study, there were 27 chemicals tested in the in vitro assays for which there was no corresponding in vivo guideline data or EPA carcinogenicity classification (examples shown in Table 1). As shown in Figure 1B, we identify several of these Phase I compounds that may be acting via proangiogenic mechanisms, their cancer hallmark score and the specific angiogenic targets affected. All of these compounds are present in the environment, are predicted to be selectively disruptive, are not ‘lifestyle’-related, and not known to be ‘Carcinogen to Humans’ (i.e. IARC Group 1). The Toxicological Priority Index (130) (ToxPi, key shown in Figure 2) displays the activity of each chemical against the angiogenic in vitro assay targets that were previously identified as significantly associated with tumor endpoints in vivo. The size of the slice is determined by the potency of the compound in the assay, based on the half-maximal activity concentration (AC50). The chosen angiogenic prototypical disruptors are Bisphenol AF, Methoxychlor, perfluorooctane sulfonate (PFOS), Diniconazole, Ziram, Chlorothalonil, Biphenyl, Tributyltin Chloride, 2,2-bis-(p-hydroxyphenyl)-1,1,1-trichloroethane (HPTE) and C.I. Solvent Yellow 14 (Figure 1B). For several of these compounds, there is literature evidence that supports their potential angiogenic activity. For example, Bisphenol AF may induce angiogenesis via inactivation of the p53 axis and underlying deregulation of proliferation kinetics and cell death in human epithelial cells, as well as through its effect on Estrogen Receptor (ERα) (131). Bisphenol AF also affected a number of vascular targets in the ToxCast assay portfolio, including uPAR, THBD and ICAM1, as well as downregulating the antiangiogenic chemokines CXCL9 and CXCL10. Methoxychlor (the parent compound to HPTE) was shown to induce increases in histological expression of angiogenic factors such as VEGF, VEGFR2 and ANG1 in rat pituitary and uterus (132). The angiogenic HTS targets of HPTE include CXCL10, CXCL9, MMP1, uPAR, THBD, ICAM1 and VCAM1. Exposure to PFOS induced actin filament remodeling and endothelial permeability changes as well as ROS production in human microvascular endothelial cells (133). PFOS could also overwhelm homeostasis of antioxidative systems, boost ROS generation, impact the mitochondria and affect protein expression of apoptotic regulators in endothelial cells (134). Diniconazole (a pesticide) is predicted to be carcinogenic and shown to target certain angiogenic molecules CXCL10, uPAR and VCAM1 in vitro. Ziram may induce angiogenesis through activation of mitogen-activated protein kinases (MAPK) and decreases cytolytic protein levels in human natural killer cells (135,136).
Table 1.
Examples of ToxCast Phase I chemicals predicted to be carcinogens and shown to target certain angiogenic molecules in vitro, but lacking in vivo data or EPA carcinogenicity classifications
| Chemical name | Chemical use class | Cancer hazard model score (#cancer hallmark assays hit) | Angiogenic targets | Proangiogenic ToxPi |
|---|---|---|---|---|
| Diniconazole | Pesticide | 18 | CXCL10, uPAR, VCAM1 |
|
| HPTE | Pesticide metabolite | 17 | CXCL10, CXCL9, MMP1, uPAR, ICAM1, THBD, VCAM1 | 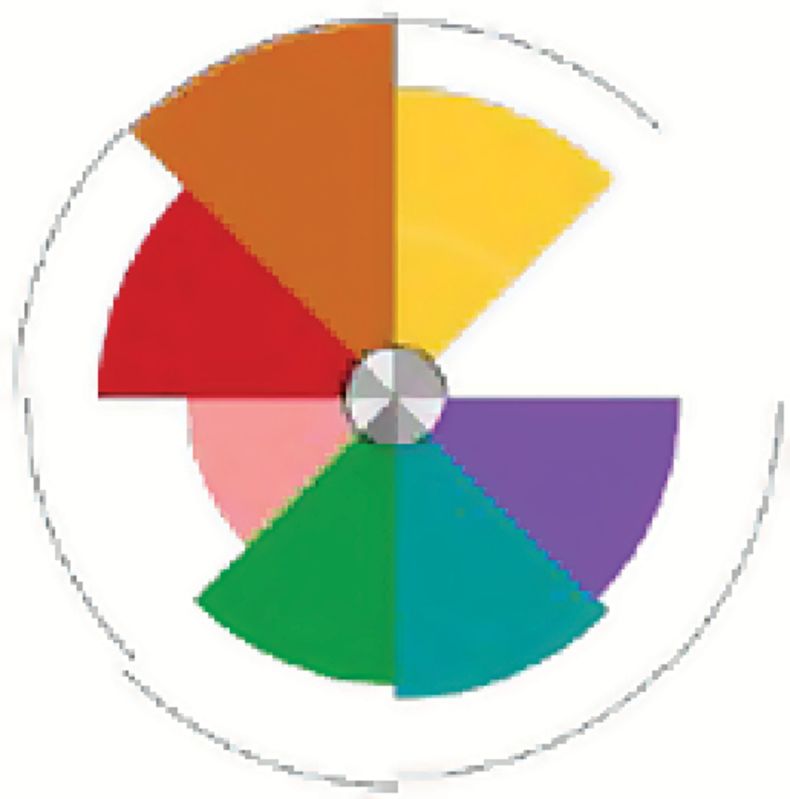 |
| Methylene bis(thiocyanate) | Pesticide | 16 | CXCL10, CXCL9, MMP1, uPAR, ICAM1, THBD, VCAM1 | 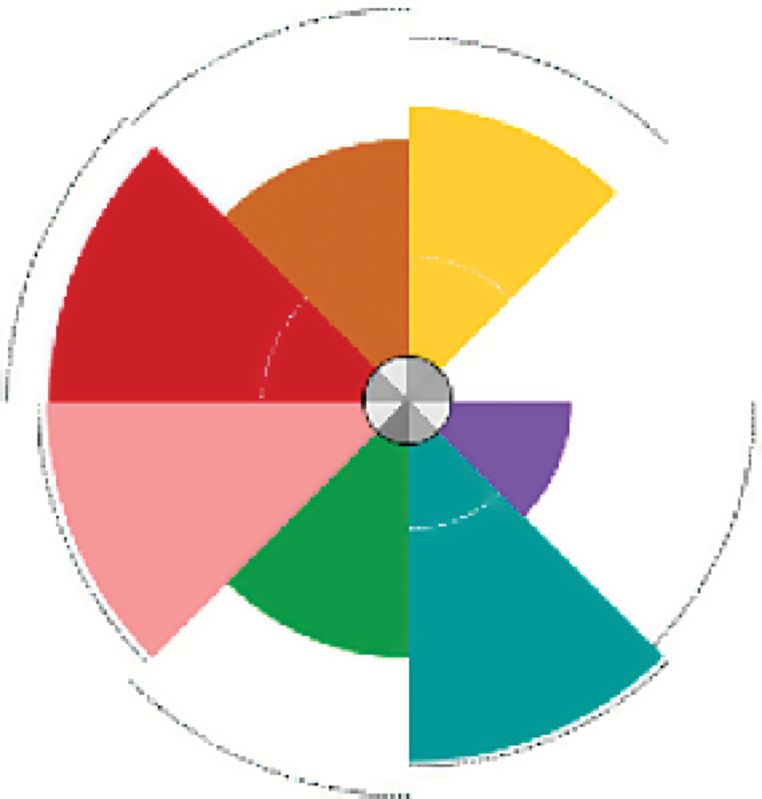 |
| PFOS | Industrial surfactant | 7 | CXCL10, MMP1, uPAR, VCAM1 |  |
These compounds were identified in an analysis linking rodent chemical carcinogenesis to HTS assay targets in cancer hallmark pathways (78). All of these compounds are ubiquitous in the environment, are predicted to be selectively disruptive, are not ‘lifestyle’ related and not known to be ‘Carcinogen to Humans’ (i.e. IARC Group 1). The Toxicological Priority Index (ToxPi) key mapping assays to slices is shown in Figure 2. CXCL9 and 10, C-X-C motif chemokine ligands 9 and 10.
Figure 2.
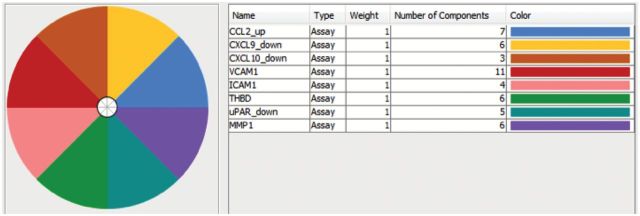
The ToxPi key for proangiogenic in vitro assay targets that were previously identified as being statistically significantly associated with tumor endpoints in vitro. The number of components represents the number of ToxCast assays for that target. Results for certain ToxCast Phase I test chemicals are shown in Figure 1B.
Phase II of the ToxCast program expanded the chemical library beyond pesticides to over a thousand compounds, many of which lack cancer data but appear to be targeting angiogenic signaling and may also be candidates for future examination. A number of organotin compounds, including tributyltin chloride, tributyltin methacrylate and triphenyltin hydroxide, caused a decrease in expression of THBD in vascular smooth muscle cells as well as other proangiogenic activity in the ToxCast assays. As in the case of dioxin, AHR ligands may be potential tumor promoters via angiogenic pathways, and it has been hypothesized that AHR signaling may suppress VEGF-A expression by competing with HIF-1α for their common dimerization partner ARNT (137). Compounds such as C.I. solvent yellow 14, Benzo(b) fluoranthene and 7,12 dimethyl(benz)anthracene are active in the AHR assay in addition to multiple other angiogenic targets, however their downstream effects on VEGF expression and angiogenesis will be dependent on their agonist vs. antagonist activity and are not yet known. Other chemicals exhibited specific activity on cytokine signaling, such as acrylamide and biphenyl, both of which caused increased expression of the proangiogenic chemokine CCL2 in vascular smooth muscle cells. The release of the full ToxCast Phase II dataset in late 2013 (http://www.epa.gov/ncct) is assisting in further identification of key assay targets and prioritization of potential chemical modulators of tumor angiogenesis. There are also a number of compounds that emerged from this analysis that have been tested in animals and assigned positive carcinogenicity classifications, but whose effects have not been well characterized histologically. If some of these were studied in more depth, they could also potentially serve as proangiogenic reference compounds.
In vitro and in vivo angiogenesis assays including HTS assay for assessing the effects of environmental chemicals in tumor angiogenesis
To screen the effects of environmental chemicals in tumor angiogenesis, there are many well developed in vitro and in vivo angiogenesis model systems that can be used or adapted (138–151). Each model has distinct advantages and disadvantages. Microvascular endothelial cells or well-characterized immortalized microvascular endothelial cell lines are generally considered superior to HUVEC in tumor angiogenesis studies, since tumor blood vessels are presumably microvessels. In vitro assays are usually designed to examine endothelial cell proliferation, migration and ability to form tube-like structures in coculture, matrigel or other matrix-containing environments. In vivo assays include, but are not limited to, the chorioallantoic membrane assay (chicken embryos), mesenteric window assay (small gut of rats and mice), corneal angiogenesis assay (rabbit, rat or mouse eyes), matrigel plug assay (mice and rats), sponge implant assay (rats) and alternate animal models such as hamster and zebrafish.
With technological advancement and the development of HTS, several in vitro angiogenesis assays have been used to screen and profile large numbers of chemical compounds that can be assayed in 96-well to 1536-well microplates. Because cancer cells can survive through compensation pathways, a battery of angiogenesis assays in HTS formats are needed to rapidly profile thousands of environmental chemicals and to build better predictive toxicology models. These assays are grouped into biochemical and cell-based categories and summarized in Table 2.
Table 2.
HTS assays for assessing the role of environmental chemicals in tumor angiogenesis
| Assay technology | Target | Assay principle | HTS format | Reference |
|---|---|---|---|---|
| Biochemical HTS assays | ||||
| Fluorescence intensity | Integrin | Binding to dye-labeled fibronectin | Microarray | (163) |
| FP | VEGF, | Competitive binding of dye-labeled proteins or ligands | 384 well, microfluidics | (164) |
| TRF | HIF-1α | Protein–ligand binding interactions | 96 well | (176) |
| AlphaScreen | VEGFR | Protein–ligand binding interactions | 1536 well | (165) |
| TR-FRET | TGF, VEGFR | Product formation catalyzed by active enzymes | 96 well, 384 well | (175,166) |
| Cell-based HTS assays | ||||
| Phenotype | Tube formation | Total tube length measured by dsTomato fluorescent protein, nuclear stains or cell permeable dyes | 96 well, 384 well, 1536 well, microfluidics | (179,167,168, 169, 170,172) |
| Wound closures | Scratch assays or stopper assays, with some measured by cell permeable dyes | 96 well, 384 well, microfluidics | (173,174, 357, 358,359) | |
| Chemifluorescence | IL-1α/β, IL-6, IL-10 | Detection of endogenous target proteins | 96 well | (360) |
| β-lactamase reporter | IL-6, HIF-1α, NFκB, | Target-driven β-lactamase reporter gene system and β-lactamase- cleavable FRET substrates | 384 well, 1536 well | (181,182,361,184) |
| GFP reporter | NFκB, VEGF, IL-8 | Target-driven GFP reporter gene system | 96 well, 384 well | (362–364) |
| Luciferase reporter | NFκB, HIF-1/2, VEGFR, IL-8, TGF- β | Target-driven luciferase reporter gene system | 96 well, 384 well, 1536 well | (184,365,366,180, 185) |
| HIF-1α | Degradation of a luciferase-fused HIF-1α reporter | 384 well | (183) | |
| TRF | E-selectin, ICAM-1, VCAM-1 | Detection of endogenous targets | 96 well | (367) |
| RT-PCR | VEGFR | mRNA levels of ICAM-1 and tissue factor | 96 well | (171) |
FP, fluorescence polarization; GFP, green fluorescent protein; HIF-1, HIF-2, HIF-1α, hypoxia-inducible factor 1, 2 and 1 alpha; ICAM-1, intracellular cell adhesion molecule 1; IL-1α, IL-1β, IL-6, IL-8 and IL-10, Interleukin 1 alpha, 1 beta, 6, 8 and 10; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; RT-PCR, real-time polymerase chain reaction; TRF, time-resolved fluorescence; TR-FRET, time resolved fluorescence resonance energy transfer.
Biochemical HTS assays directly measure the effects of test chemicals on target protein or peptide samples. These methods are particularly useful for well-validated angiogenic signaling components. Several biochemical assays have been implemented in large scale screens for VEGFR (166), TF (171), TGF-β (175), HIF (176) and integrins (177). Particularly, Yauch et al. (171) described a HUVEC-based HTS assay for the VEGF signaling pathway followed by quantitative real-time PCR for measuring downstream gene products TF and ICAM1 as transcriptional readouts. This HTS/real-time qPCR assay could be improved, e.g. using microvascular endothelial cells as discussed above, for future study of assessing chemical disruptors in tumor angiogenesis, as we propose in this review.
Cell-based HTS assays can be used to assess phenotypic changes or specific pathway activation/inhibition caused by exposure to test chemicals in cells or tissues. Active compounds identified from biochemical screens do not always exhibit similar activities in physiological conditions, thus cell-based assays, especially human primary cells, are useful to identify chemicals that exert adverse effects in the natural environment. Angiogenesis-associated phenotypic changes such as proliferation, apoptosis, motility and tube formation are routinely quantified in endothelial cells by a wide selection of commercially available assay kits and instruments (178,179). Chemicals that alter gene expression or protein–protein interaction can be detected by immunofluorescence or intracellular reporter gene assays. A battery of such assays have been applied to screen and identify chemicals targeting cellular signal pathways including HIFs (180,182,183,361), NF-κB (184), IL-6 (181), IL-8 (185) and TGFs (186,187).
The environmental chemicals can be assessed and profiled using the aforementioned assays in a quantitative HTS platform in which each test chemical is assayed at multiple concentrations covering at least four-log concentrations (188). The quantitative HTS-generated concentration response curves greatly reduce rates of false positives and false negatives, facilitating chemical prioritization for follow-up in-depth studies. For example, a cell-based hypoxia-response element-β-lactamase reporter assay has been optimized and miniaturized into a 1536-well format, and utilized to identify inhibitors and activators of the HIF-1 signaling pathway from 73 000 compounds from the Molecular Libraries Screening Centers Networks (MLSCN) (361) and 1408 environmental chemicals from the collection of the National Toxicology Program (182). Three environmental chemicals—iodochlorohydroxyquinoline, cobalt sulfate and O-phenanthroline were identified as chemical inducers of hypoxia signaling pathway. These quantitative HTS assays combined with a robotic system will greatly increase screening throughput for future assessment of environmental chemicals that may be affecting angiogenesis and other cancer hallmarks (189).
Discussion
When tumor vasculature was first successfully targeted in cancer to prevent growth and dispersion of malignant cells, it appeared that not only the blood vessels but the entire microenvironment within the tumor was participating in tumor growth, progression and resistance to treatment (152). A new concept providing additional relevant factors in this already complex multifaceted pathology was emerging to explain why current therapies are not fully or only transiently efficient (153). It is not only that ‘normal cells’ could turn into ‘conscripted or subverted cells’ to establish a cancer but some other normal cells would be triggered by the mutant cancer cells to help them proliferate and survive. These include normal host cells such as endothelial cells, fibroblasts, monocytes/macrophages, mesenchymal cells and cells of hematopoietic origin, at sites distant from and local to the site at which malignant transformation occurs (154). In addition, host and cancer cell interactions are occurring within a network that governs and influences both cancer and host cell properties. This ECM is now recognized as a crucial regulator of cancer evolution (152). Thus, several cell types in a complex and dynamic non-cellular environment collaborate to stimulate angiogenesis. One would therefore predict that chemical mixtures potentially modifying the tumor environment would therefore affect angiogenesis for the benefit of the cancer cells. On the other hand, tumor angiogenesis is also closely tied to hypoxia and thus deregulated metabolism, tumor-promoting inflammation, accelerated tumor growth, invasion and metastasis.
The carcinogenicity of low-dose exposures to chemical mixtures in any given tissue will probably depend upon simultaneous activation of several important tumor promotion mechanisms and the disruption of several important defense mechanisms. The potential synergies of combinations of chemicals will ultimately be involved in several mechanisms of disruptive actions that are known to be relevant in cancer biology. We undertook a thorough cross validation activity to illustrate the importance of the prioritized target sites for disruption (i.e. across multiple aspects of cancer’s biology) and to illustrate the extent to which the prototypical chemical disruptors that were identified disrupt other mechanisms that are also relevant to carcinogenesis. Since tumor angiogenesis is not only an early and central event in the development of a tumor, but also critical and essential for tumor growth, invasion and metastasis. In addition, it is closely tied to hypoxia and deregulated metabolism. Therefore, we cross validate their potential participation of these angiogenic targets in other cancer hallmarks (Table 3) and their potential effects of chemical disruptors of angiogenesis in other cancer hallmarks (Table 4).
Table 3.
Cross-validation of angiogenic target pathways
| Angiogenesis priority targets |
Deregulated metabolism | Evasion of antigrowth signaling | Genetic instability | Immune system evasion | Resistance to cell death | Replicative immortality | Sustained proliferative signaling | Tissue invasion and metastasis | Tumor-promoting inflammation | Tumor microenvironment |
|---|---|---|---|---|---|---|---|---|---|---|
| VCAM1 | + (190) |
0 | 0 | 0 | + (191) |
0 | + (192) |
+ (193–196) |
+ (197) |
+ (198) |
| CXCL9 | 0 | 0 | 0 | − (199,200) |
0 | 0 | + (201) |
− (202–204) |
+ (201,205,206) |
+ (207) |
| THBD | 0 | − (94,95) |
+ (96,97) |
0 | + (98) |
0 | 0 | − (81,82,208,209) |
− (209–211) |
0 |
| CCL2 | + (212) |
+ (83,213) |
− (214) |
+ (215,216) |
+ (217) |
0 | + (218) |
+ (219–221) |
+ (222) |
+ (223) |
| ICAM1 | + (190,224) |
+ (85) |
− (225) |
− (226,227) |
− (228) |
0 | + (229) |
+ (191,229–232) |
+ (233,234) |
+ (198,235) |
| uPAR | + (236) |
− (103,237–239) |
0 | 0 | + (239,240) |
+ (241) |
+ (242) |
+ (243–248) |
+ (249) |
− (250) |
| COLIII | + (251) |
0 | 0 | 0 | 0 | 0 | 0 | + (252–254) |
+ (255) |
0 |
| CXCL10 | 0 | + (86) |
0 | − (199,256–259) |
0 | 0 | + (260) |
+/− (261–265) |
+ (266,267) |
+ (199,207) |
| MMP1 | + (268) |
− (99–102) | + (269) |
0 | 0 | 0 | + (270) |
+ (271–276) |
+ (277) |
+ (250,278) |
| AHR | + (279) |
+ (88–93,280) |
+ (281) |
0 | +/− (87,282,283) |
+ (284–286) |
+ (287,288) |
+/− (289) |
+ (290) |
+ (291) |
Complementary effect (+): Targets and chemicals that were not only relevant for angiogenesis, but also relevant for other areas of cancer biology (i.e. procarcinogenic). Contrary effects (−): Targets and chemicals that were found to have opposing actions (i.e. anticarcinogenic). Both (+/−): Instances where reports on relevant actions in other aspects of cancer biology were mixed (i.e. reports showing both procarcinogenic potential and anticarcinogenic potential). Not known (0): Instances where no literature support was found to document the relevance of a target site or chemical in a particular aspect of cancer biology. VCAM1, vascular cell adhesion molecule 1; CXCL9 and 10, C-X-C motif chemokine ligands 9 and 10; CCL2, monocyte-like chemoattractant protein; ICAM1, intercellular adhesion molecule 1.
Table 4.
Cross-validation of disruptors in other cancer hallmarks
| Angiogenesis prototypical disruptors | Deregulated metabolism | Evasion of antigrowth signaling | Genetic instability | Immune system evasion | Resistance to cell death | Replicative immortality | Sustained proliferative signaling | Tissue invasion and metastasis | Tumor promoting inflammation | Tumor microenvironment |
|---|---|---|---|---|---|---|---|---|---|---|
| Diniconazole | + (292) |
0 | 0 | 0 | 0 | 0 | 0 | 0 | + (293) |
0 |
| Ziram | +/− (294,295) |
+ (296) |
+ (297) |
0 | − (298) |
0 | 0 | 0 | + (298,299) |
0 |
| Chlorothalonil | + (300,301) |
+ (302) |
+ (303) |
0 | − (304) |
0 | + (305) |
0 | + (306,307) |
0 |
| Biphenyl | 0 | Do not alter the levels of p53 (135) |
+ (307) |
0 | − (308,309) |
0 | +/− (310,311) |
− (312–314) |
+ (315) |
0 |
| Tributyltin chloride | + (316–319) |
0 | + (320) |
0 | − (321–324) |
0 | 0 | 0 | + (321,325) |
0 |
| Methylene bis(thiocyanate) | 0 | 0 | + (326) |
0 | 0 | 0 | 0 | − (327–329) | + (330) |
0 |
| HPTE | 0 | 0 | + (331,332) |
0 | + (333) |
0 | + (334) |
0 | + (335) |
0 |
| PFOS | 0 | + (134,336) |
+ (337) |
0 | − (338,339) |
0 | + (340) |
0 | + (341,342) |
0 |
| Bisphenol AF | + (343,344) |
+ (131) |
0 | 0 | − (345) | 0 | + (346) |
+ (347–349) |
+ (350,351) |
0 |
| C.I. solvent yellow 14 | + (352,353) |
+ (352,354) |
+ (355) |
0 | 0 | 0 | 0 | 0 | + (356) |
0 |
Complementary effect (+): Targets and chemicals that were not only relevant for angiogenesis, but also relevant for other areas of cancer biology (i.e. procarcinogenic). Contrary effects (−): Targets and chemicals that that were found to have opposing actions (i.e. anticarcinogenic). Both (+/−): Instances where reports on relevant actions in other aspects of cancer biology were mixed (i.e. reports showing both procarcinogenic potential and anticarcinogenic potential). Not known (0): Instances where no literature support was found to document the relevance of a target site or chemical in a particular aspect of cancer’s biology. HPTE,2,2-bis-(p-hydroxyphenyl)-1,1,1-trichloroethane; PFOS, perfluorooctane sulfonate.
When studying the role of chemical disruptors in tumor angiogenesis, it is also important to keep in mind that inflammation and angiogenesis are closely linked (126,155–157). Many of the angiogenic molecule targets that are selected as important targets in this review are also involved in inflammation pathways. However, the critical role of VEGFR and TF pathways in chemical angiogenesis can be examined in vitro with HTS systems where individual chemical disruptors can be added to the assay wells to explore their role in angiogenesis, followed by a variety of assay techniques as reviewed and summarized above and in Table 2 for measuring the changes of these angiogenic priority targets (CCL2, ICAM1, CXCL9, CXCL10, AHR, THBD, uPAR, MMP1, VCAM1 and collagen III) that we choose as potential targets for chemical disruptors (Bisphenol AF, Methoxychlor, PFOS, Diniconazole, Ziram, Chlorothalonil, Biphenyl, Tributyltin Chloride, HPTE and C.I. Solvent Yellow 14).
It is worth noting that many common drugs and some dietary compounds can prevent cancer by inhibiting tumor angiogenesis. For example, aspirin and metformin are two cases where epidemiological evidence indicates cancer prevention (158,159), and experimental evidence suggested that inhibition of angiogenesis plays a part in this role (160,161). As well, there is substantial experimental evidence for phytochemicals, in particular dietary phytochemicals, preventing angiogenesis (162). So simultaneous exposures to both antiangiogenic and proangiogenic substances may represent two competing forces that could influence the process of environmental carcinogenesis. However, it is beyond the scope of this review to simultaneously consider these antiangiogenic exposures as well. Primarily, we believe that proangiogenic environmental exposures have not been considered in detail elsewhere, so they are the focus of this review. However, we do recognize that the combined effects of these constituents with other chemicals warrant careful consideration.
Conclusions
In conclusion, we propose to study the role of environmental chemicals on angiogenesis, particularly at low doses of selective chemical disruptors. We believe there is a great need for future research that explores the potentially carcinogenic synergies produced by low-dose exposures to a wide range of chemicals with disruptive potential. Those with proangiogenic potential may be non-carcinogenic, but combinations of those chemicals may warrant further research and how they might combine with other chemicals that act on other hallmarks may help us better understand whether or not these types of combination exposures have a role to play in environmental carcinogenesis. In this regard, we identify prioritized vascular signaling targets, identify various environmental chemicals as novel, potential selectively disruptive agents in tumor angiogenesis, consider the cross-hallmark relationships within tumor angiogenesis pathways and targets as well as with other cancer hallmarks and make suggestions for assessing environmental chemicals in tumor angiogenesis for future studies. Understanding of the role of low-dose exposure of chemicals with disruptive potential could help us to refine our approach to cancer risk assessment, and may ultimately aid in preventing cancer by reducing or eliminating exposures to synergistic mixtures of chemicals with carcinogenic potential.
Funding
The Ohio State University College of Medicine, The OSU James Comprehensive Cancer Center (OSUCCC), OSUCCC Translational Therapeutics Program (to Z. H.); Ministry of Science and Technology of Taiwan (NSC93-2314-B-320-006 and NSC94-2314-B-320-002 to H.-Y. H.); Taipei Medical University (TMU101-AE3-Y19 to L.-T. L.); INSERM and University of Strasbourg, France (to T. M.); Fondazione Cariplo (2011-0370 to C.M.); Kuwait Institute for the Advancement of Sciences (2011-1302-06 to F. al-M.); Grant University Scheme (RUGs) Ministry Of Education Malaysia (04-02-12-2099RU to R.A.H.); Italian Ministry of University and Research (2009FZZ4XM_002 to A.A); the University of Florence (ex60%2012 to A.A.); US Public Health Service Grants (RO1 CA92306, RO1 CA92306-S1, RO1 CA113447 to R.R.); Department of Science and Technology, Government of India (SR/FT/LS-063/2008 to N.S.); NIEHS contracts (N01-ES 35504 and HHSN27320140003C to N.K.).
Acknowledgements
All authors contributed to writing/revising the review article. Specifically, Z.H. for overall, Prioritized Targets, TF Signaling Pathways and In vitro and In vitro Angiogenesis Assays including Figure 1, Introduction, Discussion and Conclusion; N.K. for overall, Topic Area Overview and Selective Chemical Disruptors including Figures 1 and 2 and Table 1; T.M. and V.D. for Tumor environment and angiogenesis, Establishing molecular identity, and Drug development for antiangiogenesis that target the tumor environment; M.X. and C.-W. H. for VEGF Signaling Pathways and HTS assays including Table 2; W.K.R. and S.B. for Selective Chemical Disruptors and Introduction and Discussion/Conclusion; all other authors for Tables 3 and 4; H.K.S. and W.H.B. for the section of ‘Cross-talk between angiogenesis and the other hallmarks of cancer’ involving all Prioritized Targets and Selective Chemical Disruptors. We are thankful for Dr Michael Gilbertson’s critical reading, thoughtful edits and comments on the review article. Particularly, we thank Mr. Leroy Lowe for initiating and organizing the Halifax Project in the past 3 years, his assistance in structuring this article, and his critical reading of the manuscripts and his inputs as well. We are also thankful for National Institute of Environmental Health Sciences of USA for travel grant support for team members to attend the Halifax Workshop in August 2013.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- AHR
aryl-hydrocarbon receptor
- CXCL9 and 10
C-X-C motif chemokine ligands 9 and 10
- CCL2
monocyte-like chemoattractant protein
- COLIII
collagen III
- ECM
extracellular matrix
- FGF
fibroblast growth factor
- HIF-1α
hypoxia-inducible factor 1 alpha
- HUVEC
human umbilical vein endothelial cells
- HPTE
2,2-bis-(p-hydroxyphenyl)-1,1,1-trichloroethane
- HTS
high-throughput screening
- IL
interleukin
- ICAM1
intercellular adhesion molecule 1
- MMP1
matrix metalloproteinase-1
- PAR
protease-activated receptors
- PFOS
perfluorooctane sulfonate
- THBD
thrombomodulin
- TF
tissue factor
- TGF-β
transforming growth factor-beta
- uPAR
urokinase-type plasminogen activator receptor
- VCAM1
vascular cell-adhesion molecule 1
- VEGF/VEGFR
vascular endothelial growth factor/receptor
- VEC
vascular endothelial cells
References
- 1. Hanahan D., et al. (2000) The hallmarks of cancer. Cell, 100, 57–70. [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D., et al. (2011) Hallmarks of cancer: the next generation. Cell, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 3. Folkman J. (1971) Tumor angiogenesis: therapeutic implications. N. Engl. J. Med., 285, 1182–1186. [DOI] [PubMed] [Google Scholar]
- 4. Carmeliet P., et al. (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature, 473, 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ferrara N., et al. (2005) Angiogenesis as a therapeutic target. Nature, 438, 967–974. [DOI] [PubMed] [Google Scholar]
- 6. Folkman J. (2001) Angiogenesis-dependent diseases. Semin. Oncol., 28, 536–542. [DOI] [PubMed] [Google Scholar]
- 7. Folkman J. (2003) Angiogenesis inhibitors: a new class of drugs. Cancer Biol. Ther., 2(4 suppl. 1), S127–S133. [PubMed] [Google Scholar]
- 8. Cao Y., et al. (2011) Forty-year journey of angiogenesis translational research. Sci. Transl. Med., 3, 114rv3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Siemann D.W., et al. (2005) Differentiation and definition of vascular-targeted therapies. Clin. Cancer Res., 11, 416–420. [PubMed] [Google Scholar]
- 10. Thorpe P.E. (2004) Vascular targeting agents as cancer therapeutics. Clin. Cancer Res., 10, 415–427. [DOI] [PubMed] [Google Scholar]
- 11. Patterson D.M., et al. (2007) Vascular damaging agents. Clin. Oncol. (R. Coll. Radiol)., 19, 443–456. [DOI] [PubMed] [Google Scholar]
- 12. Hu Z., et al. (1999) Targeting tumor vasculature endothelial cells and tumor cells for immunotherapy of human melanoma in a mouse xenograft model. Proc. Natl. Acad. Sci. USA, 96, 8161–8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu Z., et al. (2001) Targeting tissue factor on tumor vascular endothelial cells and tumor cells for immunotherapy in mouse models of prostatic cancer. Proc. Natl. Acad. Sci. USA, 98, 12180–12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu Z., et al. (2010) Natural killer cells are crucial for the efficacy of Icon (factor VII/human IgG1 Fc) immunotherapy in human tongue cancer. BMC Immunol., 11, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Heeckeren W.J., et al. (2007) Complications from vascular disrupting agents and angiogenesis inhibitors: aberrant control of hemostasis and thrombosis. Curr. Opin. Hematol., 14, 468–480. [DOI] [PubMed] [Google Scholar]
- 16. Bair S.M., et al. (2013) Cardiovascular complications associated with novel angiogenesis inhibitors: emerging evidence and evolving perspectives. Trends Cardiovasc. Med., 23, 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ellis L.M., et al. (2008) VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat. Rev. Cancer, 8, 579–591. [DOI] [PubMed] [Google Scholar]
- 18. Muncke J. (2011) Release of chemicals from plastics: lessons from food contact with plastics. Integr. Environ. Assess. Manag., 7, 688–690. [DOI] [PubMed] [Google Scholar]
- 19. Hecht S.S. (2012) Lung carcinogenesis by tobacco smoke. Int. J. Cancer, 131, 2724–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Letašiová S., et al. (2012) Bladder cancer, a review of the environmental risk factors. Environ. Health, 11 (suppl. 1), S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Loeb L.A., et al. (1984) Smoking and lung cancer: an overview. Cancer Res., 44(12 Pt 1), 5940–5958. [PubMed] [Google Scholar]
- 22. Pogribny I.P., et al. (2013) Environmental toxicants, epigenetics, and cancer. Adv. Exp. Med. Biol., 754, 215–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sankpal U.T., et al. (2012) Environmental factors in causing human cancers: emphasis on tumorigenesis. Tumour Biol., 33, 1265–1274. [DOI] [PubMed] [Google Scholar]
- 24. Torti S.V., et al. (2013) Iron and cancer: more ore to be mined. Nat. Rev. Cancer, 13, 342–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vandenberg L.N. (2014) Low-dose effects of hormones and endocrine disruptors. Vitam. Horm., 94, 129–165. [DOI] [PubMed] [Google Scholar]
- 26. Collotta M., et al. (2013) Epigenetics and pesticides. Toxicology, 307, 35–41. [DOI] [PubMed] [Google Scholar]
- 27. Cao Y. (2005) Opinion: emerging mechanisms of tumour lymphangiogenesis and lymphatic metastasis. Nat. Rev. Cancer, 5, 735–743. [DOI] [PubMed] [Google Scholar]
- 28. Risau W. (1997) Mechanisms of angiogenesis. Nature, 386, 671–674. [DOI] [PubMed] [Google Scholar]
- 29. Chung A.S., et al. (2010) Targeting the tumour vasculature: insights from physiological angiogenesis. Nat. Rev. Cancer, 10, 505–514. [DOI] [PubMed] [Google Scholar]
- 30. Potente M., et al. (2011) Basic and therapeutic aspects of angiogenesis. Cell, 146, 873–887. [DOI] [PubMed] [Google Scholar]
- 31. Weis S.M., et al. (2011) Tumor angiogenesis: molecular pathways and therapeutic targets. Nat. Med., 17, 1359–1370. [DOI] [PubMed] [Google Scholar]
- 32. Jain R.K., et al. (2012) SnapShot: Tumor angiogenesis. Cell, 149, 1408–1408.e1. [DOI] [PubMed] [Google Scholar]
- 33. Welti J., et al. (2013) Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J. Clin. Invest., 123, 3190–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaelin W.G., Jr (2008) The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat. Rev. Cancer, 8, 865–873. [DOI] [PubMed] [Google Scholar]
- 35. Semenza G.L. (2009) Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin. Cancer Biol., 19, 12–16. [DOI] [PubMed] [Google Scholar]
- 36. Harris A.L. (2002) Hypoxia–a key regulatory factor in tumour growth. Nat. Rev. Cancer, 2, 38–47. [DOI] [PubMed] [Google Scholar]
- 37. Lewis C.E., et al. (2007) Tie2-expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2. Cancer Res., 67, 8429–8432. [DOI] [PubMed] [Google Scholar]
- 38. Dvorak H.F. (1986) Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med., 315, 1650–1659. [DOI] [PubMed] [Google Scholar]
- 39. Xue Y., et al. (2012) PDGF-BB modulates hematopoiesis and tumor angiogenesis by inducing erythropoietin production in stromal cells. Nat. Med., 18, 100–110. [DOI] [PubMed] [Google Scholar]
- 40. Avraamides C.J., et al. (2008) Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer, 8, 604–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weis S.M., et al. (2011) Tumor angiogenesis: molecular pathways and therapeutic targets. Nat. Med., 17, 1359–1370. [DOI] [PubMed] [Google Scholar]
- 42. Singh S., et al. (2007) Chemokines in tumor angiogenesis and metastasis. Cancer Metastasis Rev., 26, 453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Hinsbergh V.W., et al. (2006) Pericellular proteases in angiogenesis and vasculogenesis. Arterioscler. Thromb. Vasc. Biol., 26, 716–728. [DOI] [PubMed] [Google Scholar]
- 44. Wang S.S., et al. (2009) AngiomiRs-Key regulators of angiogenesis. Curr. Opin. Genet. Dev., 19, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bao L., et al. (2013) MicroRNA-21 suppresses PTEN and hSulf-1 expression and promotes hepatocellular carcinoma progression through AKT/ERK pathways. 2013 337(2):226–36. [DOI] [PubMed] [Google Scholar]
- 46. Nagy J.A., et al. (2007) VEGF-A and the induction of pathological angiogenesis. Annu. Rev. Pathol., 2, 251–275. [DOI] [PubMed] [Google Scholar]
- 47. Jain R.K. (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science, 307, 58–62. [DOI] [PubMed] [Google Scholar]
- 48. Smith N.R., et al. (2010) Vascular endothelial growth factor receptors VEGFR-2 and VEGFR-3 are localized primarily to the vasculature in human primary solid cancers. Clin. Cancer Res., 16, 3548–3561. [DOI] [PubMed] [Google Scholar]
- 49. Christofori G., et al. (1995) Vascular endothelial growth factor and its receptors, flt-1 and flk-1, are expressed in normal pancreatic islets and throughout islet cell tumorigenesis. Mol. Endocrinol., 9, 1760–1770. [DOI] [PubMed] [Google Scholar]
- 50. Witmer A.N., et al. (2002) Expression of vascular endothelial growth factor receptors 1, 2, and 3 in quiescent endothelia. J. Histochem. Cytochem., 50, 767–777. [DOI] [PubMed] [Google Scholar]
- 51. Semeraro N., et al. (1997) Tissue factor in health and disease. Thromb. Haemost., 78, 759–764. [PubMed] [Google Scholar]
- 52. Folkman J. (1996) Tumor angiogenesis and tissue factor. Nat. Med., 2, 167–168. [DOI] [PubMed] [Google Scholar]
- 53. Rickles F.R., et al. (2001) The role of the hemostatic system in tumor growth, metastasis, and angiogenesis: tissue factor is a bifunctional molecule capable of inducing both fibrin deposition and angiogenesis in cancer. Int. J. Hematol., 73, 145–150. [DOI] [PubMed] [Google Scholar]
- 54. Fernandez P.M., et al. (2002) Tissue factor and angiogenesis in cancer. Curr. Opin. Hematol., 9, 401–406. [DOI] [PubMed] [Google Scholar]
- 55. Pawlinski R., et al. (2004) Role of tissue factor in haemostasis, thrombosis, angiogenesis and inflammation: lessons from low tissue factor mice. Thromb. Haemost., 92, 444–450. [DOI] [PubMed] [Google Scholar]
- 56. Rak J., et al. (2006) Tissue factor in cancer and angiogenesis: the molecular link between genetic tumor progression, tumor neovascularization, and cancer coagulopathy. Semin. Thromb. Hemost., 32, 54–70. [DOI] [PubMed] [Google Scholar]
- 57. Fisher K.L., et al. (1987) Cloning and expression of human tissue factor cDNA. Thromb. Res., 48, 89–99. [DOI] [PubMed] [Google Scholar]
- 58. Morrissey J.H., et al. (1987) Molecular cloning of the cDNA for tissue factor, the cellular receptor for the initiation of the coagulation protease cascade. Cell, 50, 129–135. [DOI] [PubMed] [Google Scholar]
- 59. Spicer E.K., et al. (1987) Isolation of cDNA clones coding for human tissue factor: primary structure of the protein and cDNA. Proc. Natl. Acad. Sci. USA, 84, 5148–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Konigsberg W.H., et al. (1988) Molecular cloning of the cDNA for human tissue factor. Cell, 52, 639–640. [DOI] [PubMed] [Google Scholar]
- 61. O’Hara P.J., et al. (1987) Nucleotide sequence of the gene coding for human factor VII, a vitamin K-dependent protein participating in blood coagulation. Proc. Natl. Acad. Sci. USA, 84, 5158–5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Idusogie E., et al. (1996) Characterization of a cDNA encoding murine coagulation factor VII. Thromb. Haemost., 75, 481–487. [PubMed] [Google Scholar]
- 63. Nemerson Y. (1983) Regulation of the initiation of coagulation by factor VII. Haemostasis, 13, 150–155. [DOI] [PubMed] [Google Scholar]
- 64. Contrino J., et al. (1996) In situ detection of tissue factor in vascular endothelial cells: correlation with the malignant phenotype of human breast disease. Nat. Med., 2, 209–215. [DOI] [PubMed] [Google Scholar]
- 65. Duanmu J., et al. (2011) Effective treatment of chemoresistant breast cancer in vitro and in vivo by a factor VII-targeted photodynamic therapy. Br. J. Cancer, 104, 1401–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shoji M., et al. (1998) Activation of coagulation and angiogenesis in cancer: immunohistochemical localization in situ of clotting proteins and vascular endothelial growth factor in human cancer. Am. J. Pathol., 152, 399–411. [PMC free article] [PubMed] [Google Scholar]
- 67. Gabrilovich D.I., et al. (1996) Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med., 2, 1096–1103. [DOI] [PubMed] [Google Scholar]
- 68. Goel H.L., et al. (2013) VEGF targets the tumour cell. Nat. Rev. Cancer, 13, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yao X.H., et al. (2008) Glioblastoma stem cells produce vascular endothelial growth factor by activation of a G-protein coupled formylpeptide receptor FPR. J. Pathol., 215, 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Abdulkadir S.A., et al. (2000) Tissue factor expression and angiogenesis in human prostate carcinoma. Hum. Pathol., 31, 443–447. [DOI] [PubMed] [Google Scholar]
- 71. Zucker S., et al. (1998) Vascular endothelial growth factor induces tissue factor and matrix metalloproteinase production in endothelial cells: conversion of prothrombin to thrombin results in progelatinase A activation and cell proliferation. Int. J. Cancer, 75, 780–786. [DOI] [PubMed] [Google Scholar]
- 72. Hu Z., et al. (2010) Targeting tissue factor on tumour cells and angiogenic vascular endothelial cells by factor VII-targeted verteporfin photodynamic therapy for breast cancer in vitro and in vivo in mice. BMC Cancer, 10, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hu Z. (2011) Factor VII-targeted photodynamic therapy for breast cancer and its therapeutic potential for other solid cancers and leukemia. Breast Cancer - Current and Alternative Therapeutic Modalities, Prof. Esra Gunduz (Ed.), pp.175–196. ISBN: 978-953-307-776-5, InTech, doi:10.5772/20398. [Google Scholar]
- 74. Ruf W., et al. (2010) Tissue factor in cancer progression and angiogenesis. Thromb. Res., 125 (suppl. 2), S36–S38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Belting M., et al. (2004) Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat. Med., 10, 502–509. [DOI] [PubMed] [Google Scholar]
- 76. Fernandez P.M., et al. (2004) Tissue factor and fibrin in tumor angiogenesis. Semin. Thromb. Hemost., 30, 31–44. [DOI] [PubMed] [Google Scholar]
- 77. Versteeg H.H., et al. (2003) Tissue factor signal transduction in angiogenesis. Carcinogenesis, 24, 1009–1013. [DOI] [PubMed] [Google Scholar]
- 78. Kleinstreuer N.C., et al. (2013) In vitro perturbations of targets in cancer hallmark processes predict rodent chemical carcinogenesis. Toxicol. Sci., 131, 40–55. [DOI] [PubMed] [Google Scholar]
- 79. Vicari A.P., et al. (2002) Chemokines in cancer. Cytokine Growth Factor Rev., 13, 143–154. [DOI] [PubMed] [Google Scholar]
- 80. Romagnani P., et al. (2004) CXC chemokines: the regulatory link between inflammation and angiogenesis. Trends Immunol., 25, 201–209. [DOI] [PubMed] [Google Scholar]
- 81. Horowitz N.A., et al. (2011) Thrombomodulin is a determinant of metastasis through a mechanism linked to the thrombin binding domain but not the lectin-like domain. Blood, 118, 2889–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liu P.L., et al. (2010) Decreased expression of thrombomodulin is correlated with tumor cell invasiveness and poor prognosis in nonsmall cell lung cancer. Mol. Carcinog., 49, 874–881. [DOI] [PubMed] [Google Scholar]
- 83. Tang X., et al. (2012) p53 is an important regulator of CCL2 gene expression. Curr. Mol. Med., 12, 929–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lin T.H., et al. (2013) CCL2 increases alphavbeta3 integrin expression and subsequently promotes prostate cancer migration. Biochim. Biophys. Acta, 1830, 4917–4927. [DOI] [PubMed] [Google Scholar]
- 85. Gorgoulis V.G., et al. (2003) p53 activates ICAM-1 (CD54) expression in an NF-kappaB-independent manner. EMBO J., 22, 1567–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhang H.M., et al. (2005) Gamma interferon-inducible protein 10 induces HeLa cell apoptosis through a p53-dependent pathway initiated by suppression of human papillomavirus type 18 E6 and E7 expression. Mol. Cell. Biol., 25, 6247–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. O’Donnell E.F., et al. (2014) The aryl hydrocarbon receptor mediates raloxifene-induced apoptosis in estrogen receptor-negative hepatoma and breast cancer cells. Cell Death Dis., 5, e1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kung T., et al. (2009) The aryl hydrocarbon receptor (AhR) pathway as a regulatory pathway for cell adhesion and matrix metabolism. Biochem. Pharmacol., 177, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Dietrich C., et al. (2010) The aryl hydrocarbon receptor (AhR) in the regulation of cell-cell contact and tumor growth. Carcinogenesis, 31, 1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Andrysík Z., et al. (2007) The aryl hydrocarbon receptor-dependent deregulation of cell cycle control induced by polycyclic aromatic hydrocarbons in rat liver epithelial cells. Mutat. Res., 615, 87–97. [DOI] [PubMed] [Google Scholar]
- 91. Weiss C., et al. (2008) TCDD deregulates contact inhibition in rat liver oval cells via Ah receptor, JunD and cyclin A. Oncogene, 27, 2198–2207. [DOI] [PubMed] [Google Scholar]
- 92. Roman A.C., et al. (2009) Dioxin receptor deficiency impairs angiogenesis by a mechanism involving VEGF-A depletion in the endothelium and transforming growth factor-beta overexpression in the stroma. J. Biol. Chem., 284, 25135–25148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Feng S., et al. (2013) Role of aryl hydrocarbon receptor in cancer. Biochim Biophys Acta, 1836, 14. [DOI] [PubMed] [Google Scholar]
- 94. Huang H.C., et al. (2013) UVB irradiation regulates ERK1/2- and p53-dependent thrombomodulin expression in human keratinocytes. PLoS One, 8, e67632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kumar A., et al. (2011) p53 impairs endothelial function by transcriptionally repressing Kruppel-like factor 2. Arterioscler. Thromb. Vasc. Biol., 31, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Maekita T., et al. (2006) High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin. Cancer Res., 12(3 Pt 1), 989–995. [DOI] [PubMed] [Google Scholar]
- 97. Lengauer C., et al. (1997) DNA methylation and genetic instability in colorectal cancer cells. Proc. Natl. Acad. Sci. USA, 94, 2545–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ikezoe T., et al. (2012) Thrombomodulin protects endothelial cells from a calcineurin inhibitor-induced cytotoxicity by upregulation of extracellular signal-regulated kinase/myeloid leukemia cell-1 signaling. Arterioscler. Thromb. Vasc. Biol., 32, 2259–2270. [DOI] [PubMed] [Google Scholar]
- 99. Pickard A., et al. (2012) Inactivation of Rb in stromal fibroblasts promotes epithelial cell invasion. EMBO J., 31, 3092–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kim S., et al. (2008) Basal and UV-induced MMP-1 expression are inhibited by p53 in human dermal fibroblasts. Exp. Dermatol., 17, 939–945. [DOI] [PubMed] [Google Scholar]
- 101. Wu Y.H., et al. (2010) p53 dysfunction by xeroderma pigmentosum group C defects enhance lung adenocarcinoma metastasis via increased MMP1 expression. Cancer Res., 70, 10422–10432. [DOI] [PubMed] [Google Scholar]
- 102. Singh P.K., et al. (2008) Phosphorylation of MUC1 by Met modulates interaction with p53 and MMP1 expression. J. Biol. Chem., 283, 26985–26995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Gopinath S., et al. (2010) Co-depletion of cathepsin B and uPAR induces G0/G1 arrest in glioma via FOXO3a mediated p27 upregulation. PLoS One, 5, e11668. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 104. Doll R. (1986) Tobacco: an overview of health effects. IARC Scientific Publications, pp. 11–22. [PubMed] [Google Scholar]
- 105. Costa F., et al. (2009) Nicotine: a pro-angiogenic factor. Life Sci., 84, 785–790. [DOI] [PubMed] [Google Scholar]
- 106. Hecht S.S., et al. (1975) Chemical studies on tobacco smoke. XXXIII. N’ -nitrosonornicotine in tobacco: analysis of possible contributing factors and biologic implications. J. Natl. Cancer Inst., 54, 1237–1244. [DOI] [PubMed] [Google Scholar]
- 107. Liu E.S., et al. (2003) Cigarette smoke exposure increases ulcerative colitis-associated colonic adenoma formation in mice. Carcinogenesis, 24, 1407–1413. [DOI] [PubMed] [Google Scholar]
- 108. Zhu B.Q., et al. (2003) Second hand smoke stimulates tumor angiogenesis and growth. Cancer Cell, 4, 191–196. [DOI] [PubMed] [Google Scholar]
- 109. Wang Y.H., et al. (2013) Comparing the joint effect of arsenic exposure, cigarette smoking and risk genotypes of vascular endothelial growth factor on upper urinary tract urothelial carcinoma and bladder cancer. J. Hazard. Mater 262:1139–46. [DOI] [PubMed] [Google Scholar]
- 110. Shin V.Y., et al. (2004) Nicotine promotes gastric tumor growth and neovascularization by activating extracellular signal-regulated kinase and cyclooxygenase-2. Carcinogenesis, 25, 2487–2495. [DOI] [PubMed] [Google Scholar]
- 111. Zhang Q., et al. (2007) Nicotine induces hypoxia-inducible factor-1alpha expression in human lung cancer cells via nicotinic acetylcholine receptor-mediated signaling pathways. Clin. Cancer Res., 13, 4686–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Park Y.J., et al. (2008) Effect of nicotine on human umbilical vein endothelial cells (HUVECs) migration and angiogenesis. Vascul. Pharmacol., 49, 32–36. [DOI] [PubMed] [Google Scholar]
- 113. Heeschen C., et al. (2001) Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat. Med., 7, 833–839. [DOI] [PubMed] [Google Scholar]
- 114. Wong H.P., et al. (2007) Nicotine promotes colon tumor growth and angiogenesis through beta-adrenergic activation. Toxicol. Sci., 97, 279–287. [DOI] [PubMed] [Google Scholar]
- 115. Mousa S., et al. (2006) Cellular and molecular mechanisms of nicotine’s pro-angiogenesis activity and its potential impact on cancer. J. Cell. Biochem., 97, 1370–1378. [DOI] [PubMed] [Google Scholar]
- 116. Martin J.W., et al. (2009) The multiple faces of nicotine and its implications in tissue and wound repair. Exp. Dermatol., 18, 497–505. [DOI] [PubMed] [Google Scholar]
- 117. Kao Y.H., et al. (2003) Low concentrations of arsenic induce vascular endothelial growth factor and nitric oxide release and stimulate angiogenesis in vitro . Chem. Res. Toxicol., 16, 460–468. [DOI] [PubMed] [Google Scholar]
- 118. Klei L.R., et al. (2008) Positive signaling interactions between arsenic and ethanol for angiogenic gene induction in human microvascular endothelial cells. Toxicol. Sci., 102, 319–327. [DOI] [PubMed] [Google Scholar]
- 119. Straub A.C., et al. (2009) Arsenic requires sphingosine-1-phosphate type 1 receptors to induce angiogenic genes and endothelial cell remodeling. Am. J. Pathol., 174, 1949–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Meng D., et al. (2010) Arsenic promotes angiogenesis in vitro via a heme oxygenase-1-dependent mechanism. Toxicol. Appl. Pharmacol., 244, 291–299. [DOI] [PubMed] [Google Scholar]
- 121. Straub A.C., et al. (2007) Low level arsenic promotes progressive inflammatory angiogenesis and liver blood vessel remodeling in mice. Toxicol. Appl. Pharmacol., 222, 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Xu X., et al. (2009) Diesel exhaust exposure induces angiogenesis. Toxicol. Lett., 191, 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Andersson H., et al. (2012) Proangiogenic effects of environmentally relevant levels of bisphenol A in human primary endothelial cells. Arch. Toxicol., 86, 465–474. [DOI] [PubMed] [Google Scholar]
- 124. Bredow S., et al. (2007) Subchronic inhalation of soluble manganese induces expression of hypoxia-associated angiogenic genes in adult mouse lungs. Toxicol. Appl. Pharmacol., 221, 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ishimura R., et al. (2009) Dioxin-induced toxicity on vascular remodeling of the placenta. Biochem. Pharmacol., 77, 660–669. [DOI] [PubMed] [Google Scholar]
- 126. Halin C., et al. (2008) Chapter 1. Inflammation, angiogenesis, and lymphangiogenesis. Methods Enzymol., 445, 1–25. [DOI] [PubMed] [Google Scholar]
- 127. Tice R.R., et al. (2013) Improving the human hazard characterization of chemicals: a Tox21 update. Environ. Health Perspect., 121, 756–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kavlock R., et al. (2012) Update on EPA’s ToxCast program: providing high throughput decision support tools for chemical risk management. Chem. Res. Toxicol., 25, 1287–1302. [DOI] [PubMed] [Google Scholar]
- 129. Martin M.T., et al. (2009) Profiling the reproductive toxicity of chemicals from multigeneration studies in the toxicity reference database. Toxicol. Sci., 110, 181–190. [DOI] [PubMed] [Google Scholar]
- 130. Judson R.S., et al. (2010) In vitro screening of environmental chemicals for targeted testing prioritization: the ToxCast project. Environ. Health Perspect., 118, 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Dairkee S.H., et al. (2013) Bisphenol-A-induced inactivation of the p53 axis underlying deregulation of proliferation kinetics, and cell death in non-malignant human breast epithelial cells. Carcinogenesis, 34, 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Goldman J.M., et al. (2004) Methoxychlor-induced alterations in the histological expression of angiogenic factors in pituitary and uterus. J. Mol. Histol., 35, 363–375. [DOI] [PubMed] [Google Scholar]
- 133. Qian Y., et al. (2010) Perfluorooctane sulfonate (PFOS) induces reactive oxygen species (ROS) production in human microvascular endothelial cells: role in endothelial permeability. J. Toxicol. Environ. Health. A, 73, 819–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zhang Y.H., et al. (2013) Mechanism of perfluorooctanesulfonate (PFOS)-induced apoptosis in the immunocyte. J. Immunotoxicol., 10, 49–58. [DOI] [PubMed] [Google Scholar]
- 135. Jessen-Eller K., et al. (2002) A new invertebrate member of the p53 gene family is developmentally expressed and responds to polychlorinated biphenyls. Environ. Health Perspect., 110, 377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Taylor T.R., et al. (2011) Ziram activates mitogen-activated protein kinases and decreases cytolytic protein levels in human natural killer cells. Toxicol. Mech. Methods, 21, 577–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Wu P.Y., et al. (2013) Aromatic hydrocarbon receptor inhibits lysophosphatidic acid-induced vascular endothelial growth factor-A expression in PC-3 prostate cancer cells. Biochem. Biophys. Res. Commun., 437, 440–445. [DOI] [PubMed] [Google Scholar]
- 138. Madri J.A., et al. (1986) Endothelial cell-matrix interactions: in vitro models of angiogenesis. J. Histochem. Cytochem., 34, 85–91. [DOI] [PubMed] [Google Scholar]
- 139. Carmeliet P., et al. (1998) Mouse models of angiogenesis, arterial stenosis, atherosclerosis and hemostasis. Cardiovasc. Res., 39, 8–33. [DOI] [PubMed] [Google Scholar]
- 140. Vailhé B., et al. (2001) In vitro models of vasculogenesis and angiogenesis. Lab. Invest., 81, 439–452. [DOI] [PubMed] [Google Scholar]
- 141. Bernardini G., et al. (2004) In vitro and in vivo models to study chemokine regulation of angiogenesis. Methods Mol. Biol., 239, 223–232. [DOI] [PubMed] [Google Scholar]
- 142. Radovanovic I., et al. (2004) Angiogenesis in transgenic models of multistep angiogenesis. Cancer Treat. Res., 117, 97–114. [DOI] [PubMed] [Google Scholar]
- 143. Hoang M.V., et al. (2005) In vivo and in vitro models of Mammalian angiogenesis. Methods Mol. Biol., 294, 269–285. [DOI] [PubMed] [Google Scholar]
- 144. Norrby K. (2006) In vivo models of angiogenesis. J. Cell. Mol. Med., 10, 588–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Ucuzian A.A., et al. (2007) In vitro models of angiogenesis. World J. Surg., 31, 654–663. [DOI] [PubMed] [Google Scholar]
- 146. Deryugina E.I., et al. (2008) Chapter 2. Chick embryo chorioallantoic membrane models to quantify angiogenesis induced by inflammatory and tumor cells or purified effector molecules. Methods Enzymol., 444, 21–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Shiba Y., et al. (2008) Models for the study of angiogenesis. Curr. Pharm. Des., 14, 371–377. [DOI] [PubMed] [Google Scholar]
- 148. Jensen L.D., et al. (2009) In vivo angiogenesis and lymphangiogenesis models. Curr. Mol. Med., 9, 982–991. [DOI] [PubMed] [Google Scholar]
- 149. Qutub A.A., et al. (2009) Multiscale models of angiogenesis. IEEE Eng. Med. Biol. Mag., 28, 14–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Eklund L., et al. (2013) Mouse models for studying angiogenesis and lymphangiogenesis in cancer. Mol. Oncol., 7, 259–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Morin K.T., et al. (2013) In vitro models of angiogenesis and vasculogenesis in fibrin gel. Exp Cell Res 319(16):2409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Swartz M.A., et al. (2012) Tumor microenvironment complexity: emerging roles in cancer therapy. Cancer Res., 72, 2473–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Sounni N.E., et al. Targeting the tumor microenvironment for cancer therapy. Clin. Chem., 59, 85–93. [DOI] [PubMed] [Google Scholar]
- 154. Shang B., et al. (2012) Deciphering the key features of malignant tumor microenvironment for anti-cancer therapy. Cancer Microenviron., 5, 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Kwee B.J., et al. (2014) Manipulating the intersection of angiogenesis and inflammation. Ann. Biomed. Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Schweighofer B., et al. (2009) The VEGF-induced transcriptional response comprises gene clusters at the crossroad of angiogenesis and inflammation. Thromb. Haemost., 102, 544–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Kobayashi H., et al. (2009) Angiogenesis links chronic inflammation with cancer. Methods Mol. Biol., 511, 185–191. [DOI] [PubMed] [Google Scholar]
- 158. Alfonso L., et al. (2014) Molecular targets of aspirin and cancer prevention. Br. J. Cancer, 111, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Gronich N., et al. (2013) Beyond aspirin-cancer prevention with statins, metformin and bisphosphonates. Nat. Rev. Clin. Oncol., 10, 625–642. [DOI] [PubMed] [Google Scholar]
- 160. Borthwick G.M., et al. (2006) Therapeutic levels of aspirin and salicylate directly inhibit a model of angiogenesis through a Cox-independent mechanism. FASEB J., 20, 2009–2016. [DOI] [PubMed] [Google Scholar]
- 161. Etulain J., et al. (2013) Platelet-mediated angiogenesis is independent of VEGF and fully inhibited by aspirin. Br. J. Pharmacol., 170, 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Singh A.V., et al. (2006) Soy phytochemicals prevent orthotopic growth and metastasis of bladder cancer in mice by alterations of cancer cell proliferation and apoptosis and tumor angiogenesis. Cancer Res., 66, 1851–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Kim E.Y., et al. (2008) A novel integrin alpha5beta1 antagonistic peptide, A5-1, screened by Protein Chip system as a potent angiogenesis inhibitor. Biochem. Biophys. Res. Commun., 377, 1288–1293. [DOI] [PubMed] [Google Scholar]
- 164. Peterson K.J., et al. (2008) A fluorescence polarization assay for identifying ligands that bind to vascular endothelial growth factor. Anal. Biochem., 378, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Wang K., et al. (2011) Development of an AlphaScreen-based high-throughput screening assay for inhibitors of human vascular endothelial growth factor receptor-3. J. Immunoassay Immunochem., 32(3):219–232. [DOI] [PubMed] [Google Scholar]
- 166. Moshinsky D.J., et al. (2003) A widely applicable, high-throughput TR-FRET assay for the measurement of kinase autophosphorylation: VEGFR-2 as a prototype. J. Biomol. Screen., 8, 447–452. [DOI] [PubMed] [Google Scholar]
- 167. Santos A.F., et al. (2008) Angiogenesis: an improved in vitro biological system and automated image-based workflow to aid identification and characterization of angiogenesis and angiogenic modulators. Assay Drug Dev. Technol., 6, 693–710. [DOI] [PubMed] [Google Scholar]
- 168. Wang H.S., et al. (2004) A simple quantitative method for evaluation of angiogenesis activity. Assay Drug Dev. Technol., 2(1):31–38. [DOI] [PubMed] [Google Scholar]
- 169. Reed M.J., et al. (2007) Culture of murine aortic explants in 3-dimensional extracellular matrix: a novel, miniaturized assay of angiogenesis in vitro . Microvasc. Res., 73, 248–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Sanz L., et al. (2002) Development of a computer-assisted high-throughput screening platform for anti-angiogenic testing. Microvasc. Res., 63, 335–339. [DOI] [PubMed] [Google Scholar]
- 171. Yauch R.L., et al. (2004) Transcriptional-based screens for pathway-specific, high-throughput target discovery in endothelial cells. J. Biomol. Screen., 9, 704–711. [DOI] [PubMed] [Google Scholar]
- 172. Bischel L.L., et al. (2013) Tubeless microfluidic angiogenesis assay with three-dimensional endothelial-lined microvessels. Biomaterials, 34(5):1471–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Yarrow J.C., et al. (2004) A high-throughput cell migration assay using scratch wound healing, a comparison of image-based readout methods. BMC Biotechnol., 4, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Kumar N., et al. (2006) A high-throughput migration assay reveals HER2-mediated cell migration arising from increased directional persistence. Biophys. J., 91, L32–L34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Huynh Q.K., et al. (2011) Screening and identification of a novel class of TGF-beta type 1 receptor kinase inhibitor. J. Biomol. Screen., 16, 724–33. [DOI] [PubMed] [Google Scholar]
- 176. Kung A.L., et al. (2004) Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell, 6, 33–43. [DOI] [PubMed] [Google Scholar]
- 177. Lee Y., et al. (2004) High-throughput screening of novel peptide inhibitors of an integrin receptor from the hexapeptide library by using a protein microarray chip. J. Biomol. Screen., 9, 687–694. [DOI] [PubMed] [Google Scholar]
- 178. Staton C.A., et al. (2009) A critical analysis of current in vitro and in vivo angiogenesis assays. Int. J. Exp. Pathol., 90, 195–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Evensen L., et al. (2010) A novel imaging-based high-throughput screening approach to anti-angiogenic drug discovery. Cytometry. A, 77, 41–51. [DOI] [PubMed] [Google Scholar]
- 180. Ji D.B., et al. (2008) Establishment of a cell-based assay to screen regulators of the hypoxia-inducible factor-1-dependent vascular endothelial growth factor promoter. Biol. Pharm. Bull., 31, 2255–2259. [DOI] [PubMed] [Google Scholar]
- 181. Johnson R.L., et al. (2009) A quantitative high-throughput screen for modulators of IL-6 signaling: a model for interrogating biological networks using chemical libraries. Mol. Biosyst., 5, 1039–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Xia M., et al. (2009) Identification of chemical compounds that induce HIF-1alpha activity. Toxicol. Sci., 112, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Smirnova N.A., et al. (2010) Utilization of an in vivo reporter for high throughput identification of branched small molecule regulators of hypoxic adaptation. Chem. Biol., 17, 380–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Miller S.C., et al. (2010) Identification of known drugs that act as inhibitors of NF-kappaB signaling and their mechanism of action. Biochem. Pharmacol., 79, 1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Takahashi T., et al. (2011) An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol. Sci., 124, 359–369. [DOI] [PubMed] [Google Scholar]
- 186. Cash J.N., et al. (2013) Development of a small-molecule screening method for inhibitors of cellular response to myostatin and activin A. J. Biomol. Screen. [DOI] [PubMed] [Google Scholar]
- 187. Li X., et al. (2009) Identification of upregulators of BMP2 expression via high-throughput screening of a synthetic and natural compound library. J. Biomol. Screen., 14, 1251–1256. [DOI] [PubMed] [Google Scholar]
- 188. Shukla S.J., et al. (2010) The future of toxicity testing: a focus on in vitro methods using a quantitative high-throughput screening platform. Drug Discov. Today, 15, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Attene-Ramos M.S., et al. (2013) The Tox21 robotic platform for the assessment of environmental chemicals - from vision to reality. Drug Discov. Today. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Dejardin E. (2006) The alternative NF-kappaB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem. Pharmacol., 72, 1161–1179. [DOI] [PubMed] [Google Scholar]
- 191. Chen L.M., et al. (2011) RANKL increases migration of human lung cancer cells through intercellular adhesion molecule-1 up-regulation. J. Cell. Biochem., 112, 933–941. [DOI] [PubMed] [Google Scholar]
- 192. Liu S.Q., et al. (2013) Sphingosine kinase 1 promotes tumor progression and confers malignancy phenotypes of colon cancer by regulating the focal adhesion kinase pathway and adhesion molecules. Int. J. Oncol., 42, 617–626. [DOI] [PubMed] [Google Scholar]
- 193. Madhavan M., et al. (2002) Down regulation of endothelial adhesion molecules in node positive breast cancer: possible failure of host defence mechanism. Pathol. Oncol. Res., 8, 125–128. [DOI] [PubMed] [Google Scholar]
- 194. Ding Y.B., et al. (2003) Association of VCAM-1 overexpression with oncogenesis, tumor angiogenesis and metastasis of gastric carcinoma. World J. Gastroenterol., 9, 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Heidemann J., et al. (2006) Expression of vascular cell adhesion molecule-1 (CD 106) in normal and neoplastic human esophageal squamous epithelium. Int. J. Oncol., 28, 77–85. [PubMed] [Google Scholar]
- 196. Slack-Davis J.K., et al. (2009) Vascular cell adhesion molecule-1 is a regulator of ovarian cancer peritoneal metastasis. Cancer Res., 69, 1469–1476. [DOI] [PubMed] [Google Scholar]
- 197. Yepuri G., et al. (2012) Positive crosstalk between arginase-II and S6K1 in vascular endothelial inflammation and aging. Aging Cell, 11, 1005–1016. [DOI] [PubMed] [Google Scholar]
- 198. Bose A., et al. (2013) Tumor-derived vascular pericytes anergize Th cells. J. Immunol., 191, 971–981. [DOI] [PubMed] [Google Scholar]
- 199. Petro M., et al. (2013) Cutaneous tumors cease CXCL9/Mig production as a result of IFN-γ-mediated immunoediting. J. Immunol., 190, 832–841. [DOI] [PubMed] [Google Scholar]
- 200. Kryczek I., et al. (2009) Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood, 114, 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Chang K.P., et al. (2013) Serum levels of chemokine (C-X-C motif) ligand 9 (CXCL9) are associated with tumor progression and treatment outcome in patients with oral cavity squamous cell carcinoma. Oral Oncol., 49, 802–807. [DOI] [PubMed] [Google Scholar]
- 202. Walser T.C., et al. (2007) Immune-mediated modulation of breast cancer growth and metastasis by the chemokine Mig (CXCL9) in a murine model. J. Immunother., 30, 490–498. [DOI] [PubMed] [Google Scholar]
- 203. Reckamp K.L., et al. (2007) Expression of CXCR3 on mononuclear cells and CXCR3 ligands in patients with metastatic renal cell carcinoma in response to systemic IL-2 therapy. J. Immunother., 30, 417–424. [DOI] [PubMed] [Google Scholar]
- 204. Bronger H., et al. (2012) Modulation of CXCR3 ligand secretion by prostaglandin E2 and cyclooxygenase inhibitors in human breast cancer. Breast Cancer Res., 14, R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. O’Garra A., et al. (2012) Editorial overview. Curr. Opin. Immunol., 24, 361–363. [DOI] [PubMed] [Google Scholar]
- 206. Musah S., et al. (2012) Tumor necrosis factor-α mediates interactions between macrophages and epithelial cells underlying proinflammatory gene expression induced by particulate matter. Toxicology, 299, 125–132. [DOI] [PubMed] [Google Scholar]
- 207. Wong J.L., et al. (2013) IL-18-primed helper NK cells collaborate with dendritic cells to promote recruitment of effector CD8+ T cells to the tumor microenvironment. Cancer Res., 73, 4653–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Hanly A.M., et al. (2006) Thrombomodulin expression in colorectal carcinoma is protective and correlates with survival. Br. J. Cancer, 94, 1320–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Chen L.M., et al. (2013) Thrombomodulin mediates the progression of epithelial ovarian cancer cells. Tumour Biol., 34, 3743–3751. [DOI] [PubMed] [Google Scholar]
- 210. Tai C.J., et al. (2014) Thrombomodulin mediates the migration of cervical cancer cells through the regulation of epithelial-mesenchymal transition biomarkers. Tumour Biol., 35, 47–54. [DOI] [PubMed] [Google Scholar]
- 211. Rajashekhar G., et al. (2012) Soluble thrombomodulin reduces inflammation and prevents microalbuminuria induced by chronic endothelial activation in transgenic mice. Am. J. Physiol. Renal Physiol., 302, F703–F712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Joven J., et al. (2013) Multifunctional targets of dietary polyphenols in disease: a case for the chemokine network and energy metabolism. Food Chem. Toxicol., 51, 267–279. [DOI] [PubMed] [Google Scholar]
- 213. Hacke K., et al. (2010) Regulation of MCP-1 chemokine transcription by p53. Mol. Cancer, 9, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Redon C.E., et al. (2010) Tumors induce complex DNA damage in distant proliferative tissues in vivo . Proc. Natl. Acad. Sci. USA, 107, 17992–17997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Kudo-Saito C., et al. (2013) CCL2 is critical for immunosuppression to promote cancer metastasis. Clin. Exp. Metastasis, 30, 393–405. [DOI] [PubMed] [Google Scholar]
- 216. Huang B., et al. (2007) CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett., 252, 86–92. [DOI] [PubMed] [Google Scholar]
- 217. Eugenin E.A., et al. (2003) MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J. Neurochem., 85, 1299–1311. [DOI] [PubMed] [Google Scholar]
- 218. Hu W.T., et al. (2014) IL-33 enhances proliferation and invasiveness of decidual stromal cells by up-regulation of CCL2/CCR2 via NF-κB and ERK1/2 signaling. Mol. Hum. Reprod., 20, 358–372. [DOI] [PubMed] [Google Scholar]
- 219. Soria G., et al. (2011) Inflammatory mediators in breast cancer: coordinated expression of TNFalpha & IL-1beta with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer, 11, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Li M.Q., et al. (2011) CD82 gene suppression in endometrial stromal cells leads to increase of the cell invasiveness in the endometriotic milieu. J. Mol. Endocrinol., 47, 195–208. [DOI] [PubMed] [Google Scholar]
- 221. Izumi K., et al. (2013) Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol. Med., 5, 1383–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Karasawa F., et al. (2012) Essential role of gastric gland mucin in preventing gastric cancer in mice. J. Clin. Invest., 122, 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Borsig L., et al. (2014) Inflammatory chemokines and metastasis–tracing the accessory. Oncogene, 33, 3217–3224. [DOI] [PubMed] [Google Scholar]
- 224. Fang W., et al. (2005) The effects of polygoni multiflori total glycosides on the experimentally atherosclerotic formation in apoE-deficient mice. Zhongguo Zhong Yao Za Zhi, 30, 1542–1545. [PubMed] [Google Scholar]
- 225. Krutmann J., et al. (1994) Evidence that DNA damage is a mediate in ultraviolet B radiation-induced inhibition of human gene expression: ultraviolet B radiation effects on intercellular adhesion molecule-1 (ICAM-1) expression. J. Invest. Dermatol., 102, 428–432. [DOI] [PubMed] [Google Scholar]
- 226. Shirai A., et al. (2003) Expression of intercellular adhesion molecule (ICAM)-1 in adenoid cystic carcinoma of the head and neck. Laryngoscope, 113, 1955–1960. [DOI] [PubMed] [Google Scholar]
- 227. Gallaher A.M., et al. (2013) Proteomic screening of human targets of viral microRNAs reveals functions associated with immune evasion and angiogenesis. PLoS Pathog., 9, e1003584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Li X., et al. (2010) The helix-loop-helix transcription factor TWIST is dysregulated in myelodysplastic syndromes. Blood, 116, 2304–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Usami Y., et al. (2013) Intercellular adhesion molecule-1 (ICAM-1) expression correlates with oral cancer progression and induces macrophage/cancer cell adhesion. Int. J. Cancer, 133, 568–578. [DOI] [PubMed] [Google Scholar]
- 230. Schröder C., et al. (2011) Prognostic value of intercellular adhesion molecule (ICAM)-1 expression in breast cancer. J. Cancer Res. Clin. Oncol., 137, 1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Buitrago D., et al. (2012) Intercellular adhesion molecule-1 (ICAM-1) is upregulated in aggressive papillary thyroid carcinoma. Ann. Surg. Oncol., 19, 973–980. [DOI] [PubMed] [Google Scholar]
- 232. Ramer R., et al. (2012) Cannabidiol inhibits lung cancer cell invasion and metastasis via intercellular adhesion molecule-1. FASEB J., 26, 1535–1548. [DOI] [PubMed] [Google Scholar]
- 233. Rafat M., et al. (2012) Engineered endothelial cell adhesion via VCAM1 and E-selectin antibody-presenting alginate hydrogels. Acta Biomater., 8, 2697–2703. [DOI] [PubMed] [Google Scholar]
- 234. Oberyszyn T.M., et al. (1998) Beta2 integrin/ICAM-1 adhesion molecule interactions in cutaneous inflammation and tumor promotion. Carcinogenesis, 19, 445–455. [DOI] [PubMed] [Google Scholar]
- 235. Chen Q., et al. (2012) Molecular pathways: VCAM-1 as a potential therapeutic target in metastasis. Clin. Cancer Res., 18, 5520–5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Grismayer B., et al. (2012) Overexpression of the urokinase receptor splice variant uPAR-del4/5 in breast cancer cells affects cell adhesion and invasion in a dose-dependent manner and modulates transcription of tumor-associated genes. Biol. Chem., 393, 1449–1455. [DOI] [PubMed] [Google Scholar]
- 237. Shetty S., et al. (2007) Regulation of urokinase receptor expression by p53: novel role in stabilization of uPAR mRNA. Mol. Cell. Biol., 27, 5607–5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Shetty P., et al. (2008) Urokinase expression by tumor suppressor protein p53: a novel role in mRNA turnover. Am. J. Respir. Cell Mol. Biol., 39, 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Gupta R., et al. (2011) Oncogenic role of p53 is suppressed by si-RNA bicistronic construct of uPA, uPAR and cathepsin-B in meningiomas both in vitro and in vivo . Int. J. Oncol., 38, 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 240. Wang F., et al. (2011) Virtual screening targeting the urokinase receptor, biochemical and cell-based studies, synthesis, pharmacokinetic characterization, and effect on breast tumor metastasis. J. Med. Chem., 54, 7193–7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Hodjat M., et al. (2013) Urokinase receptor mediates doxorubicin-induced vascular smooth muscle cell senescence via proteasomal degradation of TRF2. J. Vasc. Res., 50, 109–123. [DOI] [PubMed] [Google Scholar]
- 242. Bhandary Y.P., et al. (2013) Regulation of lung injury and fibrosis by p53-mediated changes in urokinase and plasminogen activator inhibitor-1. Am. J. Pathol., 183, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Testa J.E., et al. (1990) The role of urokinase-type plasminogen activator in aggressive tumor cell behavior. Cancer Metastasis Rev., 9, 353–367. [DOI] [PubMed] [Google Scholar]
- 244. Stahl A., et al. (1994) Binding of urokinase to its receptor promotes migration and invasion of human melanoma cells in vitro . Cancer Res., 54, 3066–3071. [PubMed] [Google Scholar]
- 245. Cantero D., et al. (1997) Enhanced expression of urokinase plasminogen activator and its receptor in pancreatic carcinoma. Br. J. Cancer, 75, 388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Reuning U., et al. (1998) Multifunctional potential of the plasminogen activation system in tumor invasion and metastasis (review). Int. J. Oncol., 13, 893–906. [DOI] [PubMed] [Google Scholar]
- 247. Lund I.K., et al. (2011) uPAR as anti-cancer target: evaluation of biomarker potential, histological localization, and antibody-based therapy. Curr. Drug Targets, 12, 1744–1760. [DOI] [PubMed] [Google Scholar]
- 248. Noh H., et al. (2013) Role of urokinase receptor in tumor progression and development. Theranostics, 3, 487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Thunø M., et al. (2009) suPAR: the molecular crystal ball. Dis. Markers, 27, 157–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Zhuang T., et al. (2013) Involvement of nitric oxide synthase in matrix metalloproteinase-9- and/or urokinase plasminogen activator receptor-mediated glioma cell migration. BMC Cancer, 13, 590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Marcus H.J., et al. (2010) In vivo assessment of high-grade glioma biochemistry using microdialysis: a study of energy-related molecules, growth factors and cytokines. J. Neurooncol., 97, 11–23. [DOI] [PubMed] [Google Scholar]
- 252. Wapnir I.L., et al. (1996) Collagen gene expression in the neomatrix of carcinoma of the breast. Invas. Metastasis, 16, 308–316. [PubMed] [Google Scholar]
- 253. Menke A., et al. (2001) Down-regulation of E-cadherin gene expression by collagen type I and type III in pancreatic cancer cell lines. Cancer Res., 61, 3508–3517. [PubMed] [Google Scholar]
- 254. Imamichi Y., et al. (2007) Signaling pathways involved in collagen-induced disruption of the E-cadherin complex during epithelial-mesenchymal transition. Cells. Tissues. Organs, 185, 180–190. [DOI] [PubMed] [Google Scholar]
- 255. Zhang X., et al. (2013) Histone deacetylase inhibition downregulates collagen 3A1 in fibrotic lung fibroblasts. Int. J. Mol. Sci., 14, 19605–19617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Boyd A., et al. (2013) Quiescent innate response to infective filariae by human Langerhans cells suggests a strategy of immune evasion. Infect. Immun., 81, 1420–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Srivastava M.K., et al. (2012) Myeloid suppressor cell depletion augments antitumor activity in lung cancer. PLoS One, 7, e40677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Thakur A., et al. (2013) Immunotherapy and immune evasion in prostate cancer. Cancers (Basel)., 5, 569–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Kryczek I., et al. (2009) Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood, 114, 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Lee Y., et al. (2012) Protumoral role of monocytes in human B-cell precursor acute lymphoblastic leukemia: involvement of the chemokine CXCL10. Blood, 119, 227–237. [DOI] [PubMed] [Google Scholar]
- 261. Martins V.L., et al. (2009) Increased invasive behaviour in cutaneous squamous cell carcinoma with loss of basement-membrane type VII collagen. J. Cell Sci., 122(Pt 11), 1788–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Jiang Z., et al. (2010) CXCL10 expression and prognostic significance in stage II and III colorectal cancer. Mol. Biol. Rep., 37, 3029–3036. [DOI] [PubMed] [Google Scholar]
- 263. Kawada K., et al. (2004) Pivotal role of CXCR3 in melanoma cell metastasis to lymph nodes. Cancer Res., 64, 4010–4017. [DOI] [PubMed] [Google Scholar]
- 264. Tannenbaum C.S., et al. (1998) The CXC chemokines IP-10 and Mig are necessary for IL-12-mediated regression of the mouse RENCA tumor. J. Immunol., 161, 927–932. [PubMed] [Google Scholar]
- 265. Walser T.C., et al. (2006) Antagonism of CXCR3 inhibits lung metastasis in a murine model of metastatic breast cancer. Cancer Res., 66, 7. [DOI] [PubMed] [Google Scholar]
- 266. Ling C.C., et al. (2014) Post-transplant endothelial progenitor cell mobilization via CXCL10/CXCR3 signaling promotes liver tumor growth. J. Hepatol., 60, 103–109. [DOI] [PubMed] [Google Scholar]
- 267. Billottet C., et al. (2013) CXCR3, a double-edged sword in tumor progression and angiogenesis. Biochim. Biophys. Acta, 1836, 287–295. [DOI] [PubMed] [Google Scholar]
- 268. Nakayama K. (2013) cAMP-response element-binding protein (CREB) and NF-κB transcription factors are activated during prolonged hypoxia and cooperatively regulate the induction of matrix metalloproteinase MMP1. J. Biol. Chem., 288, 22584–22595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Radisky D.C., et al. (2006) Matrix metalloproteinase-induced genomic instability. Curr. Opin. Genet. Dev., 16, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Urtasun R., et al. (2009) Reactive nitrogen species switch on early extracellular matrix remodeling via induction of MMP1 and TNFalpha. Gastroenterology, 136, 1410–22, e1. [DOI] [PubMed] [Google Scholar]
- 271. Nutt J.E., et al. (1998) Matrix metalloproteinase-1 is induced by epidermal growth factor in human bladder tumour cell lines and is detectable in urine of patients with bladder tumours. Br. J. Cancer, 78, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Pritchard S.C., et al. (2001) Expression of matrix metalloproteinases 1, 2, 9 and their tissue inhibitors in stage II non-small cell lung cancer: implications for MMP inhibition therapy. Oncol. Rep., 8, 421–424. [PubMed] [Google Scholar]
- 273. Lu X., et al. (2009) ADAMTS1 and MMP1 proteolytically engage EGF-like ligands in an osteolytic signaling cascade for bone metastasis. Genes Dev., 23, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Bao W., et al. (2010) HER2-mediated upregulation of MMP-1 is involved in gastric cancer cell invasion. Arch. Biochem. Biophys., 499, 49–55. [DOI] [PubMed] [Google Scholar]
- 275. Garamszegi N., et al. (2012) Matrix metalloproteinase-1 contribution to sarcoma cell invasion. J. Cell. Mol. Med., 16, 1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Liao M., et al. (2012) Prognostic value of matrix metalloproteinase-1/ proteinase-activated receptor-1 signaling axis in hepatocellular carcinoma. Pathol. Oncol. Res., 18, 7. [DOI] [PubMed] [Google Scholar]
- 277. Fanjul-Fernández M., et al. (2013) Matrix metalloproteinase Mmp-1a is dispensable for normal growth and fertility in mice and promotes lung cancer progression by modulating inflammatory responses. J. Biol. Chem., 288, 14647–14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Al-Hassan N.N., et al. (2012) Differential roles of uPAR in peritoneal ovarian carcinomatosis. Neoplasia, 14, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Reyes-Hernández O.D., et al. (2009) Aromatic hydrocarbons upregulate glyceraldehyde-3-phosphate dehydrogenase and induce changes in actin cytoskeleton. Role of the aryl hydrocarbon receptor (AhR). Toxicology, 266, 30–37. [DOI] [PubMed] [Google Scholar]
- 280. Marlowe J.L., et al. (2005) Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J. Cell. Biochem., 96, 1174–1184. [DOI] [PubMed] [Google Scholar]
- 281. Korkalainen M., et al. (2012) induces genomic instability in mouse embryonic fibroblasts. PLoS One, 7, e37895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Caruso J.A., et al. (2006) Aryl hydrocarbon receptor modulation of tumor necrosis factor-alpha-induced apoptosis and lysosomal disruption in a hepatoma model that is caspase-8-independent. J. Biol. Chem., 281, 10954–10967. [DOI] [PubMed] [Google Scholar]
- 283. Park K.T., et al. (2005) The aryl hydrocarbon receptor predisposes hepatocytes to Fas-mediated apoptosis. Mol. Pharmacol., 67, 612–622. [DOI] [PubMed] [Google Scholar]
- 284. Shervington A., et al. (2007) Identification of a novel co-transcription of P450/1A1 with telomerase in A549. Genes Dev., 388, 7. [DOI] [PubMed] [Google Scholar]
- 285. Sarkar P., et al. (2006) Activation of telomerase in BeWo cells by estrogen and 2,3,7,8-tetrachlorodibenzo-p-dioxin in co-operation with c-Myc. I. Int. J. Oncol., 28, 9. [PubMed] [Google Scholar]
- 286. Ray S., et al. (2009) Activation of the aryl hydrocarbon receptor by TCDD inhibits senescence: a tumor promoting event. Biochem. Pharmacol., 77, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Lahoti T.S., et al. (2014) Aryl hydrocarbon receptor antagonism attenuates growth factor expression, proliferation, and migration in fibroblast-like synoviocytes from patients with rheumatoid arthritis. J. Pharmacol. Exp. Ther., 348, 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Budinsky R.A., et al. (2014) Mode of action and dose-response framework analysis for receptor-mediated toxicity: The aryl hydrocarbon receptor as a case study. Crit. Rev. Toxicol., 44, 83–119. [DOI] [PubMed] [Google Scholar]
- 289. Goode G.D., et al. (2013) Knockdown of aberrantly upregulated aryl hydrocarbon receptor reduces tumor growth and metastasis of MDA-MB-231 human breast cancer cell line. Int. J. Cancer, 133, 2769–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Pierre S., et al. (2014) Aryl hydrocarbon receptor-dependent induction of liver fibrosis by dioxin. Toxicol. Sci., 137, 11. [DOI] [PubMed] [Google Scholar]
- 291. Opitz C.A., et al. (2011) An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature, 478, 197–203. [DOI] [PubMed] [Google Scholar]
- 292. Hosokawa S., et al. (1993) Hormonal disregulation mechanism in the rat thyroid tumor induced by diniconazole. J. Toxicol. Sci., 18, 11. [DOI] [PubMed] [Google Scholar]
- 293. Koç H., et al. (2012) Severe hepatotoxicity and acute renal failure caused by diniconazole. Clin. Res. Hepatol. Gastroenterol., 36, e104–e105. [DOI] [PubMed] [Google Scholar]
- 294. Wang X.F., et al. (2006) Inhibitory effects of pesticides on proteasome activity: implication in Parkinson’s disease. Neurobiol. Dis., 23, 198–205. [DOI] [PubMed] [Google Scholar]
- 295. Gopinath S., et al. (2011) Mechanism of p27 upregulation induced by downregulation of cathepsin B and uPAR in glioma. Mol. Oncol., 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Taylor T.R., et al. (2011) Ziram activates mitogen-activated protein kinases and decreases cytolytic protein levels in human natural killer cells. Toxicol. Mech. Methods., 21, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Scarabelli L., et al. (1993) Relationship between poly(ADP-ribose) polymerase activity and DNA damage induced by zinc dithiocarbamates in mouse and rat liver. Mutat. Res., 302, 1–6. [DOI] [PubMed] [Google Scholar]
- 298. Li Q., et al. (2012) Mechanism of ziram-induced apoptosis in human natural killer cells. Int. J. Immunopathol. Pharmacol., 25, 883–891. [DOI] [PubMed] [Google Scholar]
- 299. Coussens L.M., et al. (2013) Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science, 339, 286–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Blog D.N. (2012) Take action! New farm bill amendments attack your health and environment; beyond pesticides. Daily News Blog. [Google Scholar]
- 301. Cima F., et al. (2008) Toxic effects of new antifouling compounds on tunicate haemocytes I. Sea-nine 211 and chlorothalonil. Aquat. Toxicol., 86, 299–312. [DOI] [PubMed] [Google Scholar]
- 302. Pariseau J., et al. (2011) Effects of pesticide compounds (chlorothalonil and mancozeb) and benzo[a]pyrene mixture on aryl hydrocarbon receptor, p53 and ubiquitin gene expression levels in haemocytes of soft-shell clams (Mya arenaria). Ecotoxicology, 20, 1765–1772. [DOI] [PubMed] [Google Scholar]
- 303. Lebailly P., et al. (1997) Assessment of DNA damage induced in vitro by etoposide and two fungicides (carbendazim and chlorothalonil) in human lymphocytes with the comet assay. Mutat. Res., 375, 205–217. [DOI] [PubMed] [Google Scholar]
- 304. Greenlee A.R., et al. (2004) Low-dose agrochemicals and lawn-care pesticides induce developmental toxicity in murine preimplantation embryos. Environ. Health Perspect., 112, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Tessier D.M., et al. (2001) Increased ErbB-2 tyrosine kinase activity, MAPK phosphorylation, and cell proliferation in the prostate cancer cell line LNCaP following treatment by select pesticides. Toxicol. Sci., 60, 6. [DOI] [PubMed] [Google Scholar]
- 306. Wilkinson C.F., et al. (1996) A mechanistic interpretation of the oncogenicity of chlorothalonil in rodents and an assessment of human relevance. Regul. Toxicol. Pharmacol., 24(1 Pt 1), 69–84. [DOI] [PubMed] [Google Scholar]
- 307. Rencüzoğullari E., et al. (2008) The effects of food protector biphenyl on sister chromatid exchange, chromosome aberrations, and micronucleus in human lymphocytes. Drug Chem. Toxicol., 31, 263–274. [DOI] [PubMed] [Google Scholar]
- 308. Park J.B., et al. (2012) Magnolol-induced apoptosis in HCT-116 colon cancer cells is associated with the AMP-activated protein kinase signaling pathway. Biol. Pharm. Bull., 35, 1614–1620. [DOI] [PubMed] [Google Scholar]
- 309. Pisano M., et al. (2007) Antiproliferative and pro-apoptotic activity of eugenol-related biphenyls on malignant melanoma cells. Mol. Cancer Ther., 6, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Holland J.P., et al. (2013) Synthesis and evaluation of biphenyl compounds as kinesin spindle protein inhibitors. Chem. Biodivers., 10, 18. [DOI] [PubMed] [Google Scholar]
- 311. Brown J.F., Jr, et al. (2007) Polychlorinated biphenyls modulated tumorigenesis in Sprague Dawley rats: correlation with mixed function oxidase activities and superoxide (O2*) formation potentials and implied mode of action. Toxicol. Sci., 98(2):375–394. [DOI] [PubMed] [Google Scholar]
- 312. Nozaki S., et al. (2003) Activity of biphenyl matrix metalloproteinase inhibitor BAY 12–9566 in a human breast cancer orthotopic model. Clin. Exp. Metastasis., 20, 6. [DOI] [PubMed] [Google Scholar]
- 313. Shin K.D., et al. (2005) Blocking tumor cell migration and invasion with biphenyl isoxazole derivative KRIBB3, a synthetic molecule that inhibits Hsp27 phosphorylation. J. Biol. Chem., 280, 10. [DOI] [PubMed] [Google Scholar]
- 314. Hwang E.S., et al. (2010) Magnolol suppresses metastasis via inhibition of invasion, migration, and matrix metalloproteinase-2/-9 activities in PC-3 human prostate carcinoma cells. Biosci. Biotechnol. Biochem., 74, 961–967. [DOI] [PubMed] [Google Scholar]
- 315. Bunaciu R.P., et al. (2007) The effect of dietary glycine on the hepatic tumor promoting activity of polychlorinated biphenyls (PCBs) in rats. Toxicology, 239, 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Yamada S., et al. (2013) AMP-activated protein kinase-mediated glucose transport as a novel target of tributyltin in human embryonic carcinoma cells. Metallomics, 5, 484–491. [DOI] [PubMed] [Google Scholar]
- 317. Isomura M., et al. (2013) Tributyltin-induced endoplasmic reticulum stress and its Ca(2+)-mediated mechanism. Toxicol. Appl. Pharmacol., 272, 10. [DOI] [PubMed] [Google Scholar]
- 318. Kirchner S., et al. (2010) Prenatal exposure to the environmental obesogen tributyltin predisposes multipotent stem cells to become adipocytes. Mol. Endocrinol., 24, 526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Tsunoda M., et al. (2006) Subacute administration of tributyltin chloride modulates neurotransmitters and their metabolites in discrete brain regions of maternal mice and their F1 offspring. Toxicol. Ind. Health, 22, 15–25. [DOI] [PubMed] [Google Scholar]
- 320. Tiano L., et al. (2001) DNA damage induced by organotins on trout-nucleated erythrocytes. Appl. Organometal. Chem., 15, 6. [Google Scholar]
- 321. Mitra S., et al. (2013) Tributyltin chloride induced testicular toxicity by JNK and p38 activation, redox imbalance and cell death in sertoli-germ cell co-culture. Toxicology, 314, 12. [DOI] [PubMed] [Google Scholar]
- 322. Zhang J., et al. (2011) Tributyltin chloride results in dorsal curvature in embryo development of Sebastiscus marmoratus via apoptosis pathway. Chemosphere, 82, 437–442. [DOI] [PubMed] [Google Scholar]
- 323. Nakatsu Y., et al. (2007) Concentration dependence of the mechanisms of tributyltin-induced apoptosis. Toxicol. Sci., 97, 438–447. [DOI] [PubMed] [Google Scholar]
- 324. Tiano L., et al. (2003) Effect of tributyltin on trout blood cells: changes in mitochondrial morphology and functionality. Biochim. Biophys. Acta., 1640, 8. [DOI] [PubMed] [Google Scholar]
- 325. Ishihara Y., et al. (2012) Tributyltin induces oxidative stress and neuronal injury by inhibiting glutathione S-transferase in rat organotypic hippocampal slice cultures. Neurochem. Int., 60, 782–790. [DOI] [PubMed] [Google Scholar]
- 326. Singh T., et al. (2006) Microscopic, biochemical and physiological assessment of the effect of methylene bisthiocyanate on the sapstain fungus ophiostoma floccosum. Eur. J. Plant Pathol., 114, 12. [Google Scholar]
- 327. Thejass P., et al. (2006) Antimetastatic activity of sulforaphane. Life Sci., 78, 8. [DOI] [PubMed] [Google Scholar]
- 328. Jee H.G., et al. (2011) Sulforaphane inhibits oral carcinoma cell migration and invasion in vitro. Phytother. Res., 25, 6. [DOI] [PubMed] [Google Scholar]
- 329. Xu T., et al. (2012) Dual roles of sulforaphane in cancer treatment. Anticancer Agents Med. Chem., 12, 12. [DOI] [PubMed] [Google Scholar]
- 330. Burka L.T. (1993) NTP technical report on the toxicity studies of Methylene Bis(thiocyanate) (CAS No. 6317-18-6) Administered by Gavage to F344/N Rats and B6C3F1 Mice. Toxic. Rep. Ser., 32, 1–E7. [PubMed] [Google Scholar]
- 331. Bulger W.H., et al. (1978) Interactions of methoxychlor, methoxychlor base-soluble contaminant, and 2,2-bis(p-hydroxyphenyl)-1,1,1-trichloroethane with rat uterine estrogen receptor. J. Toxicol. Environ. Health, 4, 881–893. [DOI] [PubMed] [Google Scholar]
- 332. Skinner M.K., et al. (2011) Epigenetic transgenerational actions of endocrine disruptors. Reprod. Toxicol., 31, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Symonds D.A., et al. (2005) Methoxychlor induces proliferation of the mouse ovarian surface epithelium. Toxicol. Sci., 83, 8. [DOI] [PubMed] [Google Scholar]
- 334. Hall J.M., et al. (2013) Endocrine disrupting chemicals promote the growth of ovarian cancer cells via the ER-CXCL12-CXCR4 signaling axis. Mol. Carcinog., 52, 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Shishodia S., et al. (2005) N-(4-hydroxyphenyl)retinamide inhibits invasion, suppresses osteoclastogenesis, and potentiates apoptosis through down-regulation of I(kappa)B(alpha) kinase and nuclear factor-kappaB-regulated gene products. Cancer Res., 65, 9555–9565. [DOI] [PubMed] [Google Scholar]
- 336. Dong G.H., et al. (2012) Induction of p53-mediated apoptosis in splenocytes and thymocytes of C57BL/6 mice exposed to perfluorooctane sulfonate (PFOS). Toxicol. Appl. Pharmacol., 264, 292–299. [DOI] [PubMed] [Google Scholar]
- 337. Eriksen K.T., et al. (2010) Genotoxic potential of the perfluorinated chemicals PFOA, PFOS, PFBS, PFNA and PFHxA in human HepG2 cells. Mutat. Res., 700, 39–43. [DOI] [PubMed] [Google Scholar]
- 338. Wang X., et al. (2013) PFOS-induced apoptosis through mitochondrion-dependent pathway in human-hamster hybrid cells. Mutat. Res., 754, 51–57. [DOI] [PubMed] [Google Scholar]
- 339. Lee Y.J., et al. (2013) Perfluorooctane sulfonate-induced apoptosis of cerebellar granule cells is mediated by ERK ½ pathway. Chemosphere, 90, 1597–1602. [DOI] [PubMed] [Google Scholar]
- 340. Elcombe C.R., et al. (2012) Hepatocellular hypertrophy and cell proliferation in Sprague-Dawley rats from dietary exposure to potassium perfluorooctanesulfonate results from increased expression of xenosensor nuclear receptors PPARα and CAR/PXR. Toxicology, 293, 16–29. [DOI] [PubMed] [Google Scholar]
- 341. Fair P.A., et al. (2011) Effects of environmentally-relevant levels of perfluorooctane sulfonate on clinical parameters and immunological functions in B6C3F1 mice. J. Immunotoxicol., 8, 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Liao Y., et al. (2012) Evaluation of cellular response to perfluorooctane sulfonate in human umbilical vein endothelial cells. Toxicol. In Vitro, 26, 421–428. [DOI] [PubMed] [Google Scholar]
- 343. Betancourt A.M., et al. (2012) Altered carcinogenesis and proteome in mammary glands of rats after prepubertal exposures to the hormonally active chemicals bisphenol a and genistein. J. Nutr., 142, 1382S–1388S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Fillon M. (2012) Getting it right: BPA and the difficulty proving environmental cancer risks. J. Natl. Cancer Inst., 104, 652–655. [DOI] [PubMed] [Google Scholar]
- 345. Lee S., et al. (2013) Neurotoxic effects of bisphenol AF on calcium-induced ROS and MAPKs. Neurotox. Res., 23, 249–259. [DOI] [PubMed] [Google Scholar]
- 346. Pupo M., et al. (2012) Bisphenol A induces gene expression changes and proliferative effects through GPER in breast cancer cells and cancer-associated fibroblasts. Environ. Health Perspect., 120, 1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Zhu H., et al. (2010) Environmental endocrine disruptors promote invasion and metastasis of SK-N-SH human neuroblastoma cells. Oncol. Rep., 23, 129–139. [PubMed] [Google Scholar]
- 348. Jenkins S., et al. (2011) Chronic oral exposure to bisphenol A results in a nonmonotonic dose response in mammary carcinogenesis and metastasis in MMTV-erbB2 mice. Environ. Health Perspect., 119, 1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Derouiche S., et al. (2013) Bisphenol A stimulates human prostate cancer cell migration via remodelling of calcium signalling. Springerplus, 2, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Feng Y., et al. (2012) Bisphenol AF may cause testosterone reduction by directly affecting testis function in adult male rats. Toxicol. Lett., 211, 201–209. [DOI] [PubMed] [Google Scholar]
- 351. Pitot H.C., et al. (1989) Regulation of the expression of some genes for enzymes of glutathione metabolism in hepatotoxicity and hepatocarcinogenesis. Toxicol. Appl. Pharmacol., 97, 23–34. [DOI] [PubMed] [Google Scholar]
- 352. Stiborová M., et al. (2002) Sudan I is a potential carcinogen for humans: evidence for its metabolic activation and detoxication by human recombinant cytochrome P450 1A1 and liver microsomes. Cancer Res., 62, 5678–5684. [PubMed] [Google Scholar]
- 353. Stiborová M., et al. (2009) Oxidation of the carcinogenic non-aminoazo dye 1-phenylazo-2-hydroxy-naphthalene (Sudan I) by cytochromes P450 and peroxidases: a comparative study. Interdiscip. Toxicol., 2, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Ohno M., et al. (2012) Sudan III dye strongly induces CYP1A1 mRNA expression in HepG2 cells. J. Biochem. Mol. Toxicol., 26, 7. [DOI] [PubMed] [Google Scholar]
- 355. Elliott B.M., et al. (1997) CI solvent yellow 14 shows activity in the bone marrow micronucleus assay in both the rat and mouse. Mutagenesis, 12, 255–258. [DOI] [PubMed] [Google Scholar]
- 356. Pitot H.C., et al. (1989) Critical parameters in the quantitation of the stages of initiation, promotion, and progression in one model of hepatocarcinogenesis in the rat. Toxicol. Pathol., 17(4 Pt 1), 594–611; discussion 611. [DOI] [PubMed] [Google Scholar]
- 357. Gough W., et al. (2011) A quantitative, facile, and high-throughput image-based cell migration method is a robust alternative to the scratch assay. J. Biomol. Screen., 16, 155–63. [DOI] [PubMed] [Google Scholar]
- 358. Wang L., et al. (2008) An automatic and quantitative on-chip cell migration assay using self-assembled monolayers combined with real-time cellular impedance sensing. Lab Chip, 8, 872–878. [DOI] [PubMed] [Google Scholar]
- 359. Nie F.Q., et al. (2007) On-chip cell migration assay using microfluidic channels. Biomaterials, 28, 4017–4022. [DOI] [PubMed] [Google Scholar]
- 360. Moody M.D., et al. (2001) Array-based ELISAs for high-throughput analysis of human cytokines. Biotechniques, 31, 186–190, 192. [DOI] [PubMed] [Google Scholar]
- 361. Xia M., et al. (2009) Identification of small molecule compounds that inhibit the HIF-1 signaling pathway. Mol. Cancer, 8, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Hellweg C.E., et al. (2003) Generation of stably transfected mammalian cell lines as fluorescent screening assay for NF-kappaB activation-dependent gene expression. J. Biomol. Screen., 8, 511–521. [DOI] [PubMed] [Google Scholar]
- 363. Huang H., et al. (2012) High-throughput screening for bioactive molecules using primary cell culture of transgenic zebrafish embryos. Cell Rep., 2, 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Kiss-Toth E., et al. (2000) A novel mammalian expression screen exploiting green fluorescent protein-based transcription detection in single cells. J. Immunol. Methods, 239, 125–135. [DOI] [PubMed] [Google Scholar]
- 365. Rapisarda A., et al. (2002) Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res., 62, 4316–4324. [PubMed] [Google Scholar]
- 366. Woldemichael G.M., et al. (2006) Development of a cell-based reporter assay for screening of inhibitors of hypoxia-inducible factor 2-induced gene expression. J. Biomol. Screen., 11, 678–687. [DOI] [PubMed] [Google Scholar]
- 367. Zerwes H.G., et al. (2002) A multiparameter screening assay to assess the cytokine-induced expression of endothelial cell adhesion molecules. Anal. Biochem., 304, 166–173. [DOI] [PubMed] [Google Scholar]