

Summary
Exposure to common environmental chemicals at low doses and in mixtures that act as immune disruptors and destabilize the balance of pro- and anti-inflammatory responses in favor of inflammation may increase cancer risk in exposed individuals. This risk and the potential for transgenerational inheritance through epigenetic effects to offspring are understudied in the cancer field and overlooked by current toxicant screening platforms for carcinogenesis.
Abstract
An emerging area in environmental toxicology is the role that chemicals and chemical mixtures have on the cells of the human immune system. This is an important area of research that has been most widely pursued in relation to autoimmune diseases and allergy/asthma as opposed to cancer causation. This is despite the well-recognized role that innate and adaptive immunity play as essential factors in tumorigenesis. Here, we review the role that the innate immune cells of inflammatory responses play in tumorigenesis. Focus is placed on the molecules and pathways that have been mechanistically linked with tumor-associated inflammation. Within the context of chemically induced disturbances in immune function as co-factors in carcinogenesis, the evidence linking environmental toxicant exposures with perturbation in the balance between pro- and anti-inflammatory responses is reviewed. Reported effects of bisphenol A, atrazine, phthalates and other common toxicants on molecular and cellular targets involved in tumor-associated inflammation (e.g. cyclooxygenase/prostaglandin E2, nuclear factor kappa B, nitric oxide synthesis, cytokines and chemokines) are presented as example chemically mediated target molecule perturbations relevant to cancer. Commentary on areas of additional research including the need for innovation and integration of systems biology approaches to the study of environmental exposures and cancer causation are presented.
Introduction
The assessment of the cancer potential of chemicals has historically relied on in vitro genotoxicity assays and evaluation of tumor formation in rodents. This approach emphasizes the ‘tumor initiation’ properties of individual compounds and a one-at-a-time testing paradigm. This strategy, while experimentally robust, is highly reductionist and does not consider the complex and permutable pathogenesis of tumorigenesis. The complex pathogenesis of cancer has been synthesized into discrete aspects or hallmark features by Hanahan et al. (1) as the ‘cancer hallmarks’. These cancer hallmarks are the features of carcinogenesis that encompass the multiple perturbations of the host and tissue anti-tumor defense mechanisms. Integrating this complex etiology into environmental cancer causation studies is an imposing challenge to the field. Over the past few decades, there has been a rapid expansion of chemicals in the human environment with ever-increasing exposure of humans to low-dose, mixtures of man-made chemicals. This is occurring in the absence of much needed attention and resources to innovate within the field of chemical carcinogenesis including expanding beyond genotoxicity and single agent research to the study of mixtures in biological systems as targets of chemicals in carcinogenesis.
As reviewed in refs. (2–4), effective tumor immunity is provided through the pleiotropy or duality (polarity) of the immune system via the self-terminating and protective properties of acute inflammation or maintenance of balance in tumoricidal (yin) and tumorigenic (yang) properties of immune surveillance (Figure 1). Tissue exposure to foreign elements induces specific and non-specific local and/or systemic signals as a host defense response to protect the host. These immune ‘perturbagens’ are numerous and include pathogens, biological, chemical or environmental hazards (e.g. pollen, dust, prescription and over-the-counter drugs, asbestos, paints, detergents, hair sprays, cosmetics, food additives, pesticides), oxidized metabolites of chemical mixtures, as well as defective cells (e.g. senescent and cancerous cells). Whereas humans have evolved controlled responses to foreign pathogens, altered self and other naturally occurring plant exposures, it is less understood how man-made environmental chemicals impact the immune system. Emerging evidence suggests that chronic and mixed exposures to specific chemicals may act to disrupt or perturb the balance of highly evolved regulatory mechanisms of the immune system to deal with xenobiotics, altered-self and other exposures. While increasingly recognized as potentially important in disorders of the immune and nervous system, little attention has been given to the role of environmental chemicals as carcinogens that act through indirect effects on inflammatory response and resolution mechanisms.
Figure 1.

Graphic representation of ‘yin’ and ‘yang’ arms of acute inflammation. The scheme depicts two, tightly controlled and biologically opposing arms of self-terminating acute inflammatory responses. Stimuli induce activation of innate and/or adaptive immune cells by expression of appropriate ‘death factors’ in yin (apoptosis, growth-arrest) processes to destroy foreign elements and injured tissue; while yang simultaneously produces ‘growth factors’ (wound healing, growth-promote) to terminate and resolve inflammation. Yin and yang processes are intimately facilitated by activation of a vasculature response and expression of apoptotic and wound-healing mediators. Reproduced with permission (3) [Exp. Opin. Biol. Ther. 2008, All Rights Reserved.]
Overview of inflammation and cancer
Inflammation enables tumor development
Inflammation is mediated by immune cells as an immediate defense in response to infection or injury by noxious stimuli. Innate immune cells such as neutrophils, mast cells, and macrophages possess receptors that signal the activation and production of an array of biologically active proteins and defense molecules in response to foreign substances as well as to damaged or altered self-molecules (2). The infiltration of immune cells into sites of solid tumors, observed first by Rudolf Virchow in 1863, has for many years been pursued as a failed effort of the immune system to resist tumor development. Though this latter is true and the basis of tumor escape from immune surveillance, Virchow’s idea that the immune cells associated with tumors reflected a role for these cells in the origination of cancer was the first to suggest that the immune cells ‘themselves’ were active participants in tumor development.
It is now well recognized that the presence of inflammatory cells commonly precedes tumor development (5). Demonstration that inflammation plays a causal role in tobacco-related carcinogenesis, viral carcinogenesis and asbestos-associated carcinogenesis, highlights the significance of inflammation in tumorigenesis. Substantial evidence from both experimental models and human studies have demonstrated that inflammation fosters the development of tumors by acting on or with the cancer hallmarks identified by Hanahan et al. (1). This includes effects on evasion of apoptosis, uncontrolled growth and dissemination, as well as altering/deregulating tumor immune surveillance. In fact, Colotta et al. (5) suggested that inflammation be considered a separate cancer hallmark, an idea supported in the update to the cancer hallmarks, where because of the broad acting role of inflammatory cells in tumor development, Hanahan et al. (6) conceptualized the role of inflammation as one of ‘enabling’ tumorigenesis.
As discussed by Khatami (3), some of the earliest evidence for a direct association between inflammation and tumorigenesis were obtained in experimental models of acute and chronic inflammatory ocular diseases. Analyses of a series of these studies led to one of the first reports on time-course kinetics of inflammation-induced ‘phases’ of immune dysfunction. These and the studies of others have led to the identification of at least three distinct inflammation response phases. During the acute phase, there is an initial response to an irritant or infectious organism that mimics the healing response to a wound or during an infectious process. This phase is often followed by an intermediate response phase that, in a healthy state, serves to down-regulate or dampen the acute response to resolve inflammation. Finally, there is a chronic response phase that, if unresolved, can have potent pathologic properties. As a consequence of persistence, a ‘pro-inflammatory’ state sustains the release of cytokines and chemokines with the capability of causing progressive alterations in the cellular and molecular composition of the microenvironment. This leads to elevated levels of promutagenic reactive oxygen (ROS) and reactive nitrogen species (RNS), alterations in the vasculature (e.g. vascular hyperpermeability, neovascularization, and angiogenesis), disturbances in mitochondrial function, and, importantly, the disruption of normal cell-cell signaling/cross-talk such as recruitment of macrophages with suppressive function to disable T cell-mediated tumor immunity. It is this chronically inflamed state or ‘failed wound healing’ response or localized ‘system’ response that has been identified as a common feature in tumor development and metastasis.
Acute versus chronic inflammation and carcinogenesis
Acute inflammation possesses two balanced and biologically opposing effector arms represented in a ‘yin’ (pro-apoptotic or tumoricidal) and ‘yang’ (wound healing or pro-tumorigenic) relationship model, where immune cells participate with the non-immune cells in the local environment (e.g. epithelial, vasculature and neuronal) (3). Local or systemic adaptive immune responses (cell-mediated and humoral immunity) are mobilized by selective signaling between the activated innate immune effector cells (e.g. macrophages and mast cells) and their counterparts in the adaptive immune system (e.g. T and B lymphocytes). In acute inflammation, immune cells possess shared and specialized properties that function in the recognition and elimination of intrinsic or extrinsic foreign elements and that injure or damage host tissue (acute phase/’yin’ response]. In the intermediate or resolution (‘yang’) phase response, the immune cells function to resolve inflammation and repair the damaged tissue.
Unresolved and persistent inflammation has been described as the loss of or deregulation in the balance between the ‘yin’ and ‘yang’ responses. The role of persistent inflammation as a contributing factor in tumorigenesis is well accepted and, in many cancers, thought to be a necessary component. Examples include a causal relationship between inflammation and infectious agent-associated cancers [e.g. hepatitis B and C virus (liver), human papilloma virus (e.g. cervix, anal) and the bacterium Helicobacter pylori (stomach)]. The relationship between cancer and inflammation is also supported by the elevated risk of cancer in chronic inflammatory conditions, such as colitis-associated colorectal cancer. Importantly, the cause-effect relationship between inflammation and cancer is a challenging concept as it implies that inflammation precedes the processes. However, current evidence widely suggests that in the case of cancer, which is a multi-step and complex process, inflammation is an integral component of the overall pathogenesis of disease at the microenvironment level that not only contributes in a causal way but also supports a permissive state for tumors to grow (6). As such, it is important to recognize that tumor-associated inflammation (TAI) in solid tumors is itself a complex pathologic process, with contributions from classic immune cells as well as poorly characterized, cancer-associated fibroblasts and the epithelial tumor cell compartment.
Cellular mechanisms of inflammation and tumorigenesis
Over the past two decades, our understanding of inflammation in tumorigenesis has led to the identification of a number of molecules that are strongly linked to the development of human cancers (5,7,8). Like tumorigenesis, tumor-promoting inflammation and TAI are the phenotypic product of a complex set of cellular and molecular interactions that result in an imbalance in local microenvironment cross-talk that is most analogous to an unresolved ‘wound-healing’ response (8). The cellular and molecular composition of TAI has been the subject of a number of extensive recent reviews (5,8) including work from co-author Khatami (2–4), which are abbreviated below and illustrated in Figure 1.
A number of the cellular and molecular mechanisms involved in inflammation-induced tumor initiation, promotion, and progression are now well described (see examples in Box 1). Essential to these inflammation-induced changes at the cellular and tissue level is the diverse array of immune cell-derived effector molecules (Figure 1). Among the best characterized are the pro-inflammatory ROS and RNS, cytokines, chemokines and lipid-derived products of the inducible COX-2 in arachidonic acid metabolism, including the highly potent PGE2 molecule.
Box 1: Examples of molecular, cellular and tissue alterations observed with chronic inflammation and tumor promoting consequence
• Genomic instability, chromosome remodeling, epigenetic changes and altered gene and miRNA expression
• Altered post-translational modification, activity and localization of cell proteins
• Altered cell metabolism
• Induction of cell growth and anti-apoptotic signals→ uncontrolled cell growth and retention of cells with damaged genomes
• Vasodilation, leakage of the vasculature and infiltration of leukocytes → disrupted tissue integrity and altered microenvironment and immuno-suppression and recruitment of myeloid suppressor cells
• Altered cell polarity → disturbance in stroma/epithelial tissue matrix and loss of differentiation signals
• Tissue necrosis → neovascularization and hypoxia
• Induction of matrix metalloproteinases → invasiveness and spread
Nitric oxide and ROS
At physiological levels, both ROS and RNS are important cell signaling molecules (9). However, at high levels or with aberrant production, ROS and RNS are capable of causing considerable cellular damage resulting in cell injury, DNA damage and prompting an inflammatory response (10,11). During tumorigenesis, ROS and RNS have been characterized for their ability to induce a plethora of effects on cells and on the local environment that include DNA damage, adduct of cellular protein and lipids, and, in the absence of apoptosis at high levels, promotion of abnormal cell proliferation and transformation (8). Considerable levels of ROS and RNS are produced by the innate immune system in response to tissue injury or damage. Thus, ROS and RNS produced in response to cell-damage by inflammatory cells, that unresolved have the potential to set up a vicious cycle leading to chronic and aberrantly high levels of ROS and RNS. These high levels and chronic exposure of cells to reactive species in tissue microenvironments from macrophages and mast cells are linked to a range of tissue pathologies, including neurodegenerative and autoimmune diseases, along with the propagation of mast cells that are thought to promote myeloid-suppressor cell expansion that inhibit tumor immunosurveillance as well as acting to enable the ‘maintenance’ of a tumor promoting microenvironment (8,12,13). As such, uncontrolled or deregulated ROS or RNS production have been, and continue to be, investigated as biological indicators of exogenous and endogenous insults with cancer-causing potential, independent of their DNA-damaging potential.
Mitochondria are the primary source of intracellular ROS (8,10). A number of known carcinogens (e.g. benzene, halocarbons, nitrosamines, etc.) exert adverse human health effects by promoting inflammatory states as a consequence of ROS production (14). Individuals exposed to chemicals that promote ROS, including asbestos, coal, arsenic, vinyl chloride, mustard gas, auto fumes, diesel soot, crystalline silica, inorganic dust and agricultural dusts, have a higher risk of lung and other cancers (15,16). A number of these chemicals are International Agency for Research on Cancer (IARC) group 1 carcinogens, primarily associated with their DNA-damaging or genotoxic effects. However, it is clear that DNA damage alone is not sufficient for the development of metastatic cancers (1,6) and that environmental chemicals do not exist in isolation. As such, it is increasingly clear that, in addition to or independent of their genotoxic effects, the activity of a chemical or complex mixture to perturb ROS or RNS balance, should be considered when evaluating its carcinogenic capacity.
A well-studied example of chemical mixtures in the environment that are capable of acting as ROS inducers is vehicle exhaust. It is through work on diesel exhaust particulates, a mixture of polycyclic aromatic hydrocarbons and metals, in animal model and cell culture that we have a reasonable mechanistic understanding of the relationship between ROS production and inflammation following exposure to diesel exhaust particulates (17–20). Interesting and important work by Zhao et al. (21), aimed at teasing apart mitochondrial and cytosolic nitric oxide stress responses with diesel exhaust particulates exposure, led to the observation that alveolar macrophages activate ROS and nitric oxide (NO) in response to diesel exhaust particulates, but the two have distinct effects. Using an inducible nitric oxide synthase (iNOS) mutant and wildtype mouse model system, this group demonstrated that intracellular ROS production and related mitochondrial dysfunction occurred independently from NO production. In this model, NO production was associated with a pro-inflammatory response and was required to maintain an inflamed state. This pro-inflammatory response was hypothesized by the authors as a counterbalance to a ROS-induced adaptive stress response that promotes an anti-inflammatory response that increases sensitivity to bacterial infections in individuals exposed to diesel exhaust particulates (21). Importantly, knockout of iNOS resulted in a dramatic reduction in lung tumor multiplicity (80% reduction) compared with wild-type animals demonstrating the important role of the NO induced pro-inflammatory response in tumor development (22). The Zhao study is highlighted here to emphasize a few recurrent themes that are relevant across exposures: (i) the dynamic interplay among cells of the immune response and the local microenvironment in determining the ultimate fate of local systems response following toxic exposure and (ii) the importance of developing a better systems level mechanistic understanding of the tissue level response to a toxicant in developing biological indicators of a chemical’s potential to promote a pro-tumor or tumor favorable environment.
Cyclooxygenase, prostaglandins and their receptors
The cyclooxygenase (COX) enzymes were among the first identified molecular targets of interest in TAI. Before the identification of COX-2 as a major enzyme mediator of TAI, a handful of epidemiological studies had reported lower cancer rates in regular users of aspirin and other non-steroidal anti-inflammatory agents that are now explained by the inhibitory activity of these drugs on the pro-inflammatory/pro-tumorigenic effects of PGE2 (23,24). There are three COX isoforms: COX-1 or prostaglandin G/H synthase 1 (PTGS1), which is constitutively expressed; COX-2 (PTGS2), the inducible form of the COX enzymes; and COX-3, an alternative splice variant of COX-1. COX enzymes catalyze the formation of lipid mediators, including prostanoids, prostacyclins and thromboxanes. Of the three, COX-2 is over-expressed in acute and chronic inflammation as well as in tumors. Extensive research efforts over the past three decades have established a strong link between COX-2 expression, inflammation, and cancer, including demonstration that COX-2 suppression prevents neoplasia in numerous rodent models of cancer as well as in human clinical trials (6). COX-2 can be induced by a number of factors including cytokines, chemokines, ROS and environmental chemicals (see later). Induction of COX-2 activates mPGES-1, the inducible enzyme that catalyzes the COX-2-derived lipid intermediate PGH2 to PGE2, the biological mediator of the tumorigenic effects of COX-2. PGE2 is the most abundant prostaglandin (PG) in solid tumors and has been shown to influence tumor cell growth, migration and invasiveness. The tumorigenic actions of PGE2 are numerous and include the induction of angiogenesis, transactivation of the epidermal growth factor receptor, inhibition of apoptosis and immunosuppression (25).
The physiological and pathological effects of PGE2 are mediated through interactions with specific PG receptor subtypes present on an array of cell types, including most immune cells and epithelial cells. PGE2 shows the highest affinity for the EP receptor subtypes 1–4 (PTGER1-4 or EP1-4). Through the recent use of receptor subtype specific inhibitors, antibodies and engineered mouse models, the multiple PGE2/EP signaling pathways associated with human health and disease have become clearer. All four of the EP receptors are present on the majority of cells involved in immune responses (26,27). Under normal physiological conditions, PGE2 attenuates the activity of macrophages and dendritic cells by inhibiting the production of tumor necrosis factor (TNF)-α and interleukin (IL)-10. The EP2 and EP4 receptors mediate these activities as well as regulate the proliferation and differentiation of T and B cells. And while it is clear that the biologically diverse activity of PGE2 is determined by the nature and distribution of the EP receptors, very little is known about the EP receptor subtype/PGE2 interactions, interaction with environmental chemicals and potential contribution of toxicants in the evolution and progression of TAI. This represents an important area for active research in environmental toxicology.
Within the intent of this review, it is important to recognize that COX-2 expression is regulated by a number of transcription factors that themselves can become deregulated leading to the sustained induction of COX-2 as a co-factor in TAI. These include the hypoxia inducible factors (HIF-1α and HIF-2α), NF-κB, CREB and members of the signal transducer and activator of transcription family (STAT) (28,29).
STAT family proteins regulate cytokine-dependent inflammation and immunity. STAT protein family members, including STAT 1–6, are overexpressed in a number of human cancers. The role the STATs in TAI has recently been well characterized in prostate cancer where chronic inflammation is believed to play a major role in tumor development (30). STAT3 has been mechanistically linked to the induction and maintenance of an inflammatory microenvironment in the prostate and to the malignant transformation and progression due to the maintenance of a pro-inflammatory state. The pro-inflammatory cytokine IL-6 is a potent inducer STAT3 where binding to the IL6R induces activation of the Janus tyrosine family kinase (JAK)-signal transducer leading to a phosphorylation dependent activation STAT3. This promotes the dimerization of STAT3 monomers via their SH2 domain and promotes their active transport to the nucleus where the active dimer binds to cytokine-inducible promoter regions of genes containing gamma-activated site motif (31). In normal tissues, this robust response is countered by a SHP phosphatase and the suppressor of cytokine signaling molecule (SOCS3). This negative feedback loop insures resolution of the signaling and restoration to homeostasis. In prostate and other cancers such as breast, STAT3 becomes constitutively activated; a phenotype thought to reflect the influence of the local microenvironment and in particular TAI. Because STAT3 activation induces a number of transcriptional factors that include oncogenes involved in cell survival, proliferation, inflammation and angiogenic factors (32), its constitutive activation is associated with a number of the cancer hallmarks and nicely illustrates the molecular aspects of TAI that enable tumorigenesis (31).
STATs, like other transcription factors, have a dual and self-perpetuating role in inflammation and, like other similar molecules, is considered to be both a friend and a foe in tumorigenesis (33). They can be induced by inflammation and can, in turn, induce inflammation by activating NFκB and IL-6 pathways. If unchecked this leads to an uncontrolled pro-inflammatory/pro-tumorigenesis state (Figure 2). For example in the liver the resident myeloid cells or Kupffer cells, in response to an environmental or endogenous stimuli produce pro-inflammatory cytokines as a result of activation of the IKKβ/NFκB complex. The activation of IKKβ/NFκB is potent stimuli for IL-6 and thus activation of the STAT3 protein. Inflammation is an established risk factor for hepatocellular cancer (HCC) from viral infection and other environmental or drug insults. STAT3 is overexpressed in the majority of HCC in human with high levels correlated with IL-6 levels in the local tumor environment (34); findings that support a role of IL-6 and STAT3 as a TAI phenomenon in HCC in humans. Given the role of STATs in inflammation and evidence as an important signaling molecule in TAI, the STAT transcription factors represent an important and unexplored family of molecules as putative mediators of TAI in the presence of environmental chemicals and other toxicants.
Figure 2.
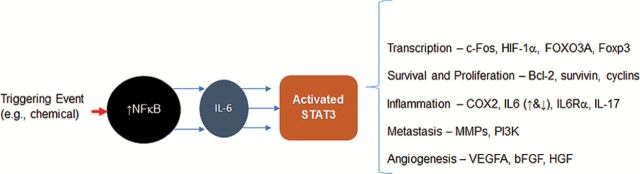
The activation of NFκB is a potent stimuli for IL-6 and IL-6 activates the STAT3 protein. Cancer cells and surrounding inflammatory immune cells have been shown to produce excessive and continuous amounts of IL-6 and other cytokines promoting chronic stimulation of STAT3. If unchecked, this leads to an uncontrolled pro-inflammatory/pro-tumorigenesis state mediated by the effects of STAT3 on gene transcription that promote proliferation, resistance to apoptosis, angiogenesis, immune evasion, invasion and metastasis; all hallmarks of cancer.
Cytokines as immune effector molecules
Cytokines are a large group of small proteins (5–20 kD) that act as pleiotropic paracrine and autocrine messengers with a wide spectrum of biological functions across numerous tissue and cell types. Collectively, the cytokines include chemokines, interferons, interleukins, lymphokines and TNF. Cytokines are produced by cells of the immune system (e.g. B and T lymphocytes, macrophages and mast cells), stromal cells (endothelial cells and fibroblasts) as well as tumor cells. Cytokines exhibit paracrine, and autocrine effects on a wide range of tissues and cells. The cytokine most consistently associated with tumor cell killing is TNFα. Upon engagement of TNFα with its receptor, a subsequent chain of cellular events leads to the activation of the transcription factor nuclear factor (NF)κB and subsequent production of IL-1β, IL-6, IL-8 and IL-17. In the simplest mechanistic model, these pro-inflammatory molecules are coupled to each other via TNFα binding to its receptor (TNFR), which activates the NFκB pathway in the acute phase response. This results in the upregulation of a group of pro-inflammatory cytokines as a programmed response to wounding or infection. It is this response that is triggered in the initial response to injury or infection (35) that, when unresolved or chronic, is widely believed to promote tumorigenesis and contribute or enable tumor progression.
Under homeostatic conditions, two membrane receptors, TNFR1 and TNFR2, mediate the actions of the TNF family of molecules (36). While initially described as an anti-tumor molecule, the role of TNFα as pro-tumorigenic is now well characterized. Tumor and inflammatory cells within the tumor microenvironment constitutively produce TNFα, supporting tumorigenesis and metastasis by promoting: genomic instability through the production of ROS and RNS, cell survival by deregulating apoptotic pathways, promoting invasion through induction of matrix metalloproteinases (MMPs), and angiogenesis via the induction of pro-angiogenic factors. Part of this response may be due to the presence of TNFR1 on tumor, stromal and immune cells, thereby allowing TNFα to exert its activity both directly on the tumor and indirectly within the tumor microenvironment to sustain local inflammation and recruitment of cells with inhibitory effects (i.e. myeloid suppressor cells) on tumor immunity. The effects of TNFα as a pro-tumor molecule have been clearly demonstrated in TNFR1-deficient mice, which are resistant to tumorigenesis. The best-characterized mechanism of the tumor-promoting effects of TNFα are those related to the tumor cell itself and molecular alterations (i.e. mutation, deletion and amplification) in key regulatory genes that lead to the constitutive activation and deregulated activation of NFκB. More recently, the role of non-genetic factors in the localized overproduction of TNFα is recognized. These include previously underappreciated effects of the local microenvironment and the cancer-associated fibroblasts and immune cells that fail to produce or recognize the wound resolving cues. In the presence of active NFκB signaling, TNFα and NFκB interact to induce cytokines (e.g. IL-1, IL-6), COX-2, adhesion proteins and MMPs. In turn, high levels of inflammatory cytokines trigger uncontrolled NFκB expression and activation, ultimately preventing the resolution of the response (5,7). Failed resolution of TAI resulting in a localized mileau of chronic cytokine activation is believed to shift the balance away from cell death toward survival and tumor cell invasion (5–7,36). Thus, independent of direct genotoxicity, this adaptation to the local microenvironment stressors is thought to place a selective pressure on tumor cells that promotes angiogenesis and ultimately escape of tumor cells from the toxic environment; two critical cancer hallmarks of metastasis.
Along with TNF-α, IL-6 is among the most commonly over-expressed cytokine in human tumors (37). Similar to other aspects of inflammation, IL-6 can act as a double-edged sword. Induced in response to injury or infection, IL-6 can induce COX-2 expression and PGE2 synthesis as well as function in the resolution phase of an acute response by inhibiting TNFα and IL-1 and by inducing other anti-inflammatory or resolution cytokines such as IL-10. Thus, IL-6 exhibits both anti- and pro-inflammatory actions at the site of a wound. In the tumor microenvironment, IL-6 has been shown to negatively regulate apoptotic processes, making cells more resistant to cell death in an inflamed, highly reactive microenvironment. Two types of receptors, membrane-bound and soluble, bind IL-6 (38). The membrane bound IL-6 receptor is predominantly expressed in hepatocytes, lymphocytes, neutrophils, monocyte/macrophages and epithelial cells. After binding to IL-6, the receptor associates with the signal-transducing protein gp130 to initiate its signaling cascade. The interaction with gp130 promotes a negative feedback loop responsible for the anti-inflammatory effect of IL-6. The soluble IL6 receptor (IL-6R) is present in body fluids and is linked to the inflammatory action of IL-6 in cells not expressing IL-6R. In this case, the IL-6/IL-6R complex can bind to gp130, which is expressed in all cell types, thus explaining the broad spectrum and systemic action associated with IL-6 in inflammation.
The diverse functions of IL-6 are mechanistically linked to interactions across distinct signaling pathways, including the MAP/STAT pathway and the AKT/PI3K signaling cascade, which negatively regulates apoptosis and promotes cellular proliferation. Recently, IL-6 has been shown to play a key role in maintaining the balance between the regulatory subclass of T cells (Treg) and Th17, an effector T cells that produces IL-17, IL-6, TNFα and other pro-inflammatory chemokines (39). This function, of pivotal importance in immunity and immune pathology, is linked to inflammation which, when chronically maintained, promotes the onset of malignancies in different organs and that acts to suppress tumor immune surveillance and tumor killing through the recruitment of immunosuppressive myeloid suppressor cells (40).
Along with IL-6, a number of other cytokines that participate in inflammation and present in TAI, have been mechanistically implicated in tumor metastasis. In the case of IL-8 and IL-17 (41), these two pro-inflammatory cytokines have received considerable attention for their ability to induce neovascularization and to enhance the activity of the matrix-degrading enzymes MMP-2 and MMP-9 (42). IL-8, which is also known as CXCL8, has received considerable attention as a potential therapeutic target for a number of inflammatory diseases given its critical role in innate immune responses and as a chemoattractant for neutrophils. The activity of IL-8 is mediated by binding of monomeric or dimeric forms of CXCL8 to one of its two receptors CXCR1 and CXCR2. Expressed normally on the surface of leukocytes, these receptors have also been shown to be upregulated on both tumor and tumor-associated stromal cells in a variety of cancers including lung, prostate and colorectal. Via CXCR1/2, IL-8 activates several important signaling pathways that are overactive in tumors (MAPK, PI3K, PKC, FAK and Src) and which function in tumor cell proliferation and migration. IL-8 pathway signaling is induced by a number of factors including inflammatory cytokines (e.g. TNF-α, IL-1), ROS, and steroid hormones. There is now convincing evidence that IL-8 and CXCR1/2 signaling are major drivers in conditions of chronic inflammation including TAI. As such, the IL-8/CRCR receptor interactions receptors are the focus of intensive drug development for use in cancer and other inflammatory disease states (42).
Like IL-8, the IL-17 molecule is a recently recognized potent, pro-inflammatory cytokine that is produced by the Th17 subpopulation of T lymphocytes and is thought to be involved in tumorigenesis (41). After binding to its receptor, IL-17RA, IL-17A then activate the MAPKs ERK1/2 and p38, PI3K/Akt and NFκB pathways, leading to the production and secretion of IL-1β, IL-6, TNFα and IL-8, as well as CXCL1 and CXCL6, which attract neutrophils. Although reported in some other cancers, IL-17 has been strongly linked with tumor development in the colorectum in animal models (43). Here, leakage of bacterial products with tumor development and endotoxin exposure appears to mobilize cells producing IL-17. The presence of IL-17-producing macrophages in these models has been linked directly to suppressive effects on both local and systemic anti-tumor T cell responses. The importance of IL-17 in tumor development is supported by observations that inhibition of IL-17 in animal models of colorectal carcinogenesis prevents tumor formation, an effect that both prevents the pro-inflammatory response and the ‘poisoning’ effect of the pro-inflammatory response on tumor specific immunity.
Lipoxygenases and lipoxins
The lipoxygenases/lipoxin products of polyunsaturated fatty acid metabolism represent a more recently recognized set of bioactive metabolites in inflammation both in its induction and resolution for which there has been little work with regard to environmental exposures and modulation. These are briefly mentioned here. For example 5-lipoxygenase (5-LOX) has been implicated in inflammation-related neoplasia. 5-LOX is a non-heme iron dioxygenase that synthesizes leukotrienes, lipoxins, resolvins, and protectins from different substrates belonging to the polyunsaturated fatty acids (44). The 5-LOX is located in the cytoplasm or nucleus and is activated in the nuclear envelope, where it translocates to interact with 5-lipoxygenase activating protein to mediate the transfer of arachidonic acid from the membrane to 5-LOX. Besides its well-known role in inflammation, the over-expression of 5-LOX occurs in a number of tumor tissues and cell lines (45). Consistent with overexpression, the end products of 5-LOX, such as 5-hydroxyeicosatetraenoic acid and leukotrienes A4 and B4 (LTA4 and LTB4) contribute to cell survival and growth. The inhibition of 5-LOX enzymatic activity or the silencing of 5-LOX and leukotriene receptor expression attenuates the metastatic phenotype in colon cancer cells (46). As with the COXs, there are anti-proliferative effects with 5-LOX inhibitors such as AA-861, zileuton, nordihydroguaiaretic acid and 5-lipoxygenase activating protein inhibitors such as MK 886, MK 591. These molecules induce apoptosis in breast (47), leukemia (48) and pancreatic (49) cell lines. As such, much like the interest in COX2 and PGE2, the LOX pathway is emerging as an important mediator of tumorigenesis with direct effects on tumor-associated and tumor-promoting inflammation.
Environmental chemicals as selective disruptors of inflammation and prioritized targets of activity
Human studies on environmental chemicals, inflammation and cancer
Given the importance of inflammation as an enabling factor in carcinogenesis, we consider the paucity of research on chemicals as pro-inflammatory molecules and carcinogenesis significant. Our ability to study chemically associated cancer-specific outcomes in humans has largely been limited to comparing cancer burden among exposed and unexposed individuals in observational epidemiologic studies. This approach is important and has successfully linked cancer etiology in humans to a number of important carcinogens (e.g. tobacco exposure, asbestos and tumor viruses). However, in the absence of strong and reliable estimates of an exposure (e.g. viral antigens, asbestos fibers or numbers of cigarettes smoked), the protracted and multi-factorial nature of tumor development makes it incredibly difficult to causally link chemical exposures in the environment to cancer risk. This is particularly true when the carcinogenic potential of an exposure is dependent on often unmeasured factors such as the dose/duration of the exposure, timing of the exposure (i.e. when in life), biomarker of adverse effect after exposure and presence in the population of heterogeneity with regard to sensitivity (genetic or other such as sex or diet). And while there are some large-scale, bio-banked cohort studies (particularly in the in the absence of testable hypotheses to relate exposures with cancer outcomes. As a result, there is a need to integrate the knowledge that has been gained about the etiopathogenesis of cancer in the study of environmental chemical effects including effects on specific cellular and molecular processes important in carcinogenesis.
To example a strategy for inflammation and cancer, we focused on chemicals thought to act on immune cells and molecular targets mechanistically linked to TAI. Thus, we undertook a process to identify candidate chemicals in the environment [i.e. Bisphenol A (BPA), polybrominated diphenyl ether (PBDE), vinclozolin, nonylphenol (NP), phthalates and atrazine] shown to on specific target molecules (i.e. ER, iNOS, NFκB, IL-6, COX-2 and TNFα, respectively) that have been identified in the cancer biology field as relevant in TAI. The chemicals that we focused on were prioritized for their ubiquitous nature in the environment and the relative level of evidence that their disruption may promote disturbances in immune and non-immune cells favoring inflammation (Summarized in Table 1). These chemicals are not currently classified as carcinogens and themselves are not considered genotoxic. While a number of these are recognized as toxicants, our goal is to highlight the potential role of these chemicals from the perspective of their ability to disrupt immunomodulatory molecules related to inflammation and to challenge thinking on how these chemicals, alone or in combination with other exposures, influence cancer risk in humans.
Table 1.
Six environmental chemicals and their putative immune disrupting activity on primary mediators of inflammation and tumor-associated inflammation
| Chemicals (uses) | Modulate nuclear receptors (ER, AR, PPARs) and the AhR | iNOS in immune cells/tumor | NFκB | Anti-inflammatory cytokines (IL-10, IL-4) | COX-2/PGE2 | Pro-inflammatory cytokines (IL-6, IL-8, IL-17, TNFα) |
|---|---|---|---|---|---|---|
| BPA (synthesis of polycarbonates, epoxy resins…) | + | ↓ (50) | + (51) | ↑ (52) | ↑ (53) | ↑ (54) and ↓ |
| PBDEs (flame retardants) | + (55) | − | + (56) | + | ? (57) | ↑ (56,57) |
| Vinclozolin (fungicide) | + (58) | − | + (59) | − | − | + (59) |
| 4-NP (degradation of surfactant in household products) | + | ↓ (50) and ↑ (60) | ↓(61) and ↑(62) | ↑(62,63) | +(64) | + |
| Phthalates (plastics) | + (65–67) | ↑ (68) | ↑ (69) | ↑ (70) and ↓ | ? | ↑ (67) and ↓ |
| Atrazine (herbicide) | + (71, 72) | ↑ | No effect(73) and ↑(72) | ↑IL-4 (74) | ↑ (72) | ↓ |
‘+’ indicates evidence that the chemical is probably acting through pathway; ‘−’ indicates no evidence the chemical is acting through this pathway; “?” unclear; ↑ indicates induces; ↓ indicates inhibits.
Bisphenol A
Perhaps the most abundant (>3 million tons/year produced) and best studied environmental endrocrine disruptor is the synthetic xenoestrogen BPA. While the role of BPA as an endocrine disruptor with ligand activity for the estrogen and aryl hydrocarbon receptors (AhRs) has been extensively reviewed elsewhere, the impact of BPA on the immune system and as an immune disruptor is less recognized (75). BPA is present in the environment as a result of its widespread use in the synthesis of polycarbonates, epoxy resins and thermal paper (76), resulting in everyday exposures from food packaging, plastic bottles, water-pipes, electronic equipment, paper and toys (77,78). The physio-chemical properties of BPA, reproductive organ toxicity, activity on the hormone and AhRs, and toxic effects, along with levels and sources of exposure in humans, have recently been reviewed by Michałowicz (75). Notably, this review highlights the evidence for both immune-activating and immune-inhibiting consequences of exposure to BPA and suggests that the inconsistency in reported effects reflect a more generalized disruption in innate immune balance as opposed to more easily defined and specific effects on antigen-driven immune or adaptive immune responses. Most relevant to carcinogenesis are the findings from rodent studies linking BPA exposure to histological changes in the prostate gland. In rats, the Prins laboratory (79) have shown that early life exposure to BPA mimics estrogen-induced prostate intraepithelial neoplasia (a prostate cancer precursor lesion), which includes BPA-dependent epigenetic reprogramming of DNA along with the development of lateral prostate inflammation in the adult animal, reported earlier to reflect BPA effects on prolactin levels (80). Because inflammation of the prostate is ‘insufficient’ for the development of prostate cancer in animal models and since the role of inflammation in human prostate cancer unclear, it has been argued that the effects of BPA in rodents may not be relevant to humans. An alternative explanation is that in the presence of genotoxic or other co-factors, the immune deregulating effects of BPA on the prostate act to enhance or accelerate tumor development in the rat and while not sufficient are necessary exposures for carcinogenesis.
In addition to the work in prostate, evidence for an effect of BPA on the immune system is present from studies of BPA effects on immune cell components, particularly the T cell compartment. BPA appears to largely act on the immune system by promoting ‘immune’ cell proliferation (81), though the exact nature of the effect on specific cells of the immune system and, thus, the consequences are complex and poorly delineated. An example is the effect of BPA on T lymphocytes. CD4+ T lymphocytes, for example comprise the Th1 and Th17 helper T cells that produce pro-inflammatory cytokines whereas the Th2 or Treg cells produce anti-inflammatory or regulatory cytokines. A number of studies have been conducted on BPA effects on CD4+ T cell polarization toward one or the other subtype with highly mixed results. There are results indicating BPA activation of Th1 and Th2, often with dominance of one type over the other, effects which vary depending on the dose, duration and timing (adult or early life) of the exposure, and no reported effects on Th17 cell differentiation. Interesting work from Yan et al. (82), found that prenatal BPA exposure had a much more dramatic inhibitory effect on the anti-inflammatory, Treg cells than that seen in the rodent prostate studies, but the exact mechanisms and a role of BPA in susceptibility to TAI has not been investigated. Currently, it is unclear why BPA-exposed CD4+ cells polarize to either a pro- or anti-inflammatory state, but there is sufficient evidence to support an effect of BPA on CD4+ T cells at exposure levels comparable to those in humans. Much like the BPA-exposed T cells, results from studies on macrophages and B cells are also conflicting (81).
As noted from the prostate studies in rodents, the immunomodulatory effect of BPA on cells has been linked with BPA activity as a ligand for ER (83). CD4+ T cells in humans express ERα and, to a lesser extent, ERβ. Though studied under different model conditions, low estradiol levels have been associated with Th1 T cell development, whereas high estradiol during pregnancy, for example has been shown to promote Th2 polarization; results that may explain the immune effects of BPA through its recognized endocrine disrupting function.
In addition to putative immune effects of BPA mediated through ER, the ability of BPA to bind to the AhR and the reports of BPA activity on the peroxisome proliferator-activated receptor (PPAR), a family of nuclear receptors implicated in inflammatory disease states (84), should be considered. For example the endocrine disrupting potential of BPA has been partially correlated to weak AhR modulation (85). BPA in this study was shown to weakly suppress AhR activation in mouse cells whereas more recent studies proved that BPA toxicity is only partially regulated by AhR pathways suggesting that further studies are needed to clarify the nature of the BPA/AhR interaction. In breast cancer cells ARNT2, a heterodimeric partner for the activated AhR, decreases with BPA exposure in an ERα-dependent fashion (86). This finding contrasts with 2,3,7,8-tetrachlorobenzo-p-dioxin (TCDD), a wide-spread anthropogenic chemical and prototype agonist of the AhR, which acts as an immunosuppressive compound across model systems (87–89). To date, there is no evidence of direct binding of BPA to the AhR PASB domain (the domain TCDD binds to).
In addition to the AhR, there is growing interest on the effects of BPA and BPA analogs on members of the PPAR nuclear receptor family members α, β/δ and γ. Various studies implicate a role for PPARs in the pathogenesis of inflammatory diseases. For example, haploinsufficieny for PPARγ resulted in exacerbated experimental arthritis in mice compared with wildtypes (90). PPARγ is present on macrophages (91), dendritic cells (92), T cells (93) and B cells (90). For BPA exposure, the PPARγ isoform is of particular interest given the findings (94) that bisphenol-A diglycigyl ether, an analog of BPA present in some food containers (95) and in waste waters (81), antagonizes PPARγ. In addition, the role of PPARs as BPA targets is further suggested by observations that other BPA analogs (e.g. tetrabromobisphenol A, a brominated BPA found in flame retardants) antagonize PPARs in direct relation to the bulkiness of the brominated BPA analogs. Bulkier brominated BPA analogs were found to have greater activity as partial agonists of PPARγ and weaker estrogenic activity that could potentially disrupt or deregulate PPAR-dependent anti-inflammatory effects (96).
Polybrominated diphenyl ethers
As flame retardants, PBDEs are ubiquitous in the environment in a number of consumer products from textiles to electronic components. Leaching of PBDEs from treated products results in air, food, water and soil contamination, where exposure through ingestion and inhalation is associated with an estimated half-life of the common congeners in human adipose tissues of 1–3 years (97). Body burdens of PBDE have increased over the past few decades raising concerns on long-term health effects. The need to understand the bioactivity and/or toxicity of PBDEs is made more relevant by the demonstration of increased concentrations of PBDEs in breast milk (98), placenta (99), amniotic fluid and umbilical cord blood (100), with additional evidence that PBDEs cross the placental barrier, accumulating in the cotyledons (100). For women living near electronic waste sites, the placental burden of PBDEs is nearly 20-fold higher than for women residing in a referent site (101). These results support very early life exposures for which the long term health effects are unknown, including risk of cancer. There is currently little experimental evidence that the PBDEs act as direct mutagens. The activity and chemical structure of PBDEs are similar to TCDD. While limited to a handful of studies, recent work on PBDE effects on inflammatory cytokines in placental explant models is notable for its potential implications for other health outcomes, including cancers, where microbes are implicated. Pro- and anti-inflammatory factors play a critical role in the placenta during fetal development and at parturition, wherein the pro-inflammatory cytokines induce PGs that promote uterine contraction and cervical ripening. Thus, during pregnancy, potent anti-inflammatory cytokines, in particular IL-10, are elevated as a defense against preterm birth induced by bacterial infections. Peltier et al. recently found that placental explants treated with a mixture of the cogeners BDE-47, BDE-99 and BDE-100 and then exposed to Escherichia coli were ‘reprogrammed’ toward a pro-inflammatory response (increased IL-1β and TNFα) and away from the expected anti-inflammatory response (decreased IL-10) compared with untreated placenta. The switch from an anti- to pro-inflammatory response was not detectable in the absence of the E.coli stimuli. Interestingly, basal PGE2 levels were increased in the absence of E.coli, suggesting an effect of PBDE on the basal PG pathway that predisposed the treated cells toward a pro-inflammatory response when exposed to E.coli, compared with the untreated cells that exhibited a potent IL-10 induction. An important conclusion drawn by these authors is that chronic PBDE exposure may ‘lower the threshold for bacteria to stimulate a pro-inflammatory response’. The potential relevance of this conclusion to other health outcomes is intriguing. This study is noted here given the established link between bacteria and cancers, such as H.pylori and gastric cancer, where tumor development is dependent on inflammation. Emerging evidence also shows that many other human cancers may have a bacterial component, with cancers of the gastrointestinal tract (esophagus, liver, stomach, pancreas, colon and rectum) strongly believed to involve a disturbance in the interaction between normal flora and the immune system that promotes chronic, low-grade inflammation (i.e. dysbiosis). To our knowledge, there has been no consideration of the role of environmental immune disruptors, such as PBDEs, as contributors to these cancers, where incidence rates have increased in parallel to industrialization.
Vinclozolin
Introduced in the mid-1970s in Germany, the non-systemic, dicarboximide fungicide vinclozolin is classified by the World Health Organization (WHO) as ‘unlikely to present acute hazard in normal use’ due to its extremely low toxicity in rats. This opinion contrasts with a review by the EPA concluding that vinclozolin or a breakdown product of the compound, 3,5-dichloroaniline moiety, induces testicular tumors in rats and tumors of the kidneys and prostate glands in dogs, with species sensitivity identified as a factor for tumor development. As a result, the EPA has classified vinclozolin as a possible human carcinogen, although vinclozolin is not listed as a carcinogen by IARC or the United States NTP Carcinogens program.
More convincing than potential effects on cancer risk is the evidence demonstrating endocrine-disrupting activity of Vinclozolin, anti-androgenic effects on lipid metabolism and storage, deleterious effects on sperm count, reduced prostate weight and delayed puberty in animals (102). Despite toxicity concerns and declining use, vinclozolin remains a common fungicide for use on specific crops in the USA and Europe. There have been efforts to minimize exposure using safe handling practices (protective equipment and clothing), different application methods (to reduce exposure through inhalation or absorption) and reductions in recommended uses (i.e. specific crops to minimize ingestion such as fruit with inedible, thick peel).
Vinclozolin is of particular interest as an environmental chemical, where transient early-life exposures in utero have been linked to both adult-onset disease and transgenerational disease that involves inflammation (103,104). For example, transient vinclozolin exposure in utero has been shown to promote inflammation in the prostate (prostatitis) of postpubertal rats coupled with a down-regulation of the androgen receptor (AR) and increase in nuclear NFκB. The late or delayed effect of exposure is hypothesized to reflect a mechanism whereby vincozolin exposure during a critical development window imprints an irreversible alteration in DNA methyltransferase activity, leading to reprogramming of the AR gene(s), which manifest as inflammation in early adult life with adverse effects on spermatid number. Evidence for early life exposure leading to epigenome alterations that manifest later as disease in the adult is supported by the work of others and raised as a concern in cancer risk (103). Transient vinclozolin exposure during gestation in the F0 generation manifests as adult onset spermatogenic cell defects in the F3 generation, suggesting that, at least in some cases, changes to the methylation status of specific genes are heritable and that the exposure effect acts transgenerationally.
This work on viclozolin is noted for the reader as it demonstrates the inflammation-related changes in the prostate with in utero exposure and raises intriguing possibilities about environmental causes of cancer, where single-generation experimental models may be inadequate to fully detect carcinogenic activity of a given chemical. This is a grossly understudied molecular mechanism by which environmental chemicals may impact human health, including risk of cancer, and represents an important area for future studies.
4-Nonylphenol
A ubiquitous environmental chemical implicated recently in inflammation is 4-nonylphenol (4-NP). Human exposure to 4-NP occurs through ingestion of contaminated food and water from liquid detergents, cosmetics, paints, pesticides and other common products, where NP ethoxylates are used as nonionic surfactants (105). Of special note, 4-NP is present at higher concentrations in treated waste water than at the inlet source as a result of microbial biodegradation of the parent compound NP ethoxylate (106). As an endocrine disruptor, 4-NP is recognized for its potent reproductive effects. More recently, however, 4-NP has been shown to increase progenitor white adipose levels, body weight and overall body size in rodents exposed prenatally. Like viclozolin, 4-NP effects on adipogenesis in the perinatal period confer transgenerational inheritance of the obesogenic effects observable in F2 offspring, consistent with genome reprogramming through an epigenetic process (107). The proadipogenic effect of 4-NP in these studies was associated with a decrease in ERα in adipose tissue, consistent with its weak endocrine disrupting activity and to the induction of genes related to fatty acid metabolism and lipogenesis (e.g. Ppar-γ, Srebp-1, Lpl and Fas). With the recognized overlap in signaling molecules between the endocrine and the immune system, Han et al. recently reported that 4-NP may be acting as an immune disruptor. In their studies, 4-NP induced COX-2 protein and gene expression in the murine macrophage cell line RAW264.7 and significantly increased PGE2 production. 4-NP was further shown to activate the Akt/MAP kinases/CRE signaling response elements involved in the activation of COX-2 expression (108,109). This observation is the first insight on a potential mechanism for the observed lung inflammation and asthma in mice exposed to 4-NP. And while limited, the recent findings from the Cadet laboratory suggesting an effect of 4-NP on pro-inflammatory cytokines in a model of inflammatory bowel disease raise important concerns about 4-NP as a common environmental chemical that mimic an inflammatory state. As such, given the iniquitousness of 4-NP and evidence favoring transgenerational transmission of exposure effects, there is sufficient evidence to recommend the investigation of cancer risks associated with 4-NP exposures.
Atrazine
The triazine herbicide atrazine is widely used in agricultural to control the unwanted growth of grasses and broadleaf weeds. Being one of the most commonly used pesticides in the world (110), atrazine is widespread in the environment and a frequently detected contaminant in waterways. Like BPA and other chemicals, there are scientific indications that atrazine has endocrine-disrupting potential (110,111), causing mammary gland tumors in rodents (112) and altering male reproduction (113). The mechanism(s) of action associated with reproductive/endocrine disruption do not seem to be receptor-mediated, as there is no detectable interaction with AhR or ER (112,114,115), although there may be with AR (115). In a recent study by Jin et al., both atrazine and its major metabolite diaminochlorotriazine (116,117) induced changes in the anti-oxidant capacity of the liver and decreased the transcription of genes involved in testosterone production (111), supporting that oxidative stress may contribute to alterations in reproductive capacity. Indeed, in vitro experiments using interstitial Leydig cells support that suppression of oxidative stress by the flavonoid quertcetin prevents atrazine-induced toxicity by attenuating oxidative stress partially by modulating the NFκB pathway (118). One of the reputed actions of atrazine is the regulation of NO production (119), an important bioactive molecule which can have profound impact on cancer development by contributing to angiogenesis, suppressing apoptosis, and limiting the host immune response to the tumor itself (120). Although atrazine is considered to be a weak mutagen with low oncogenic potential [see recent re-evaluation by the EPA (121)], the immunotoxic potential of atrazine raises concerns regarding cancer susceptibility (119). In swine granulosa cells, there was induction of both NO and VEGF by atrazine, supporting that, in this context, atrazine may have the potential to contribute to angiogenesis during cancer development (122). In a mouse model, administration of atrazine also caused features of immunotoxicity, including an inhibitory effect on both cell-mediated and humoral immunity (123), findings that may have important implications for the development of lymphoma due to a reduction in immune defense mechanisms. Of the effects noted, there was a significant decrease in NO production by peritoneal macrophages (123), phagocytic cells whose production and release of NO are important cytotoxic elements in immune surveillance and inhibition of tumor growth (124). Whether changes in NO levels are reflective of induction/inhibition of iNOS expression in mammalian systems is not known. Atrazine also significantly decreased cytokine production (e.g. TNFα, IFN-γ) (123,125) as well as impaired lymphocyte proliferation and natural killer cell function (74).
Pthalates
As a group, the widely used chemical plasticizers known collectively as ‘phthalates’ and the esters of phthalic acid used to soften vinyl products are of significant concern simply as a result of the level and ubiquitous nature of exposure to these chemicals. Humans are exposed through multiple routes that include food and drink, inhalation, skin absorption and even medical procedures such as blood transfusions. Body burden studies suggest that diethylhexyl phthalate (DEHP), a high molecular species used in plastic wrapping of foods, is a major source of exposure for humans as a result of contamination from the packaging, an effect made greater with microwave heating. Health concerns related to phthalates have focused largely on reproductive health and, specifically, spermatogenesis. As with other environmental exposures, there is particular concern for early life exposures where pthalates are largely accepted as weak anti-androgens that exhibit metabolite-specific effects on testosterone synthesis by Leydig cells. High levels among children from toy products as well as exposure to breakdown products of the smaller molecular weight diethyl phthalate in personal skin care items such as lotion and soap have attracted the most concern.
More recently, an interest in the effects of phthalates and related metabolites on inflammation has emerged where the focus has been on risk of asthma (126). Research on asthma evolved from the observation of a ‘meat-wrappers’ asthma linked to heating of polyvinyl chloride film or the heating of price labels on foods (127). This and other population studies have suggested phthalates act as immune disruptors (126). While findings across in vitro and in vivo studies confirm effects of phthalates on macrophages, lymphocytes, eosinophils and neutrophils, no consistent effect has emerged, and the actual consequence of exposure appears to be contextually dependent. For example, chronic exposure to airborne DEHP increased the numbers of eosinophils, lymphocytes and neutrophils in the lung and lavage fluid, but only at very high (not human exposure-related) concentrations (128). In a separate study of the major metabolite of DEHP (MEHP), exposure at much lower doses showed similar pro-inflammatory effects, indicating the importance of metabolism in effect dose (128). This result, in part confirms studies showing acute airway irritation and increased macrophages in lavage fluid at high occupational but not low exposure levels (129). However, and paradoxically, in a human challenge study with immune biomarkers, exposure of allergic subjects to house dust containing low DEHP induced granulocyte colony stimulating factor, IL-5 and IL-6, whereas exposure to high DEHP suppressed granulocyte colony stimulating factor and IL-6 (130). These findings have led to the conclusion that phthalates exhibit immune disrupting activity that includes adjuvant effects on the proinflammatory Th2 responses as well as immunomodulatory and immunosuppressive effects depending on the conditions of exposure (dose, duration, tissue type, development). These complex and often paradoxical observations have made translation to humans a challenge but do not dismiss the potential relevance of these exposures in human diseases.
The immune disrupting nature of phthalates is evident in The Comparative Toxicogenomics Database. Recently, Singh et al. (131) found that five of the top ten toxicity networks disrupted by phthalates involved inflammation, with evidence for pathogenic effects for prostate, uterus, ovary and breast, all sites of common human cancers. Consistent with the evidence observed for endocrine disruptors, phthalates disrupt gene expression in a pattern very similar to that of BPA, where the compounds exhibit a high degree of sharing of effects on interacting genes and proteins in an immune-disrupting signature. The latter has been suggested as a potential tool for future research efforts to characterize the inflammatory potential of a compound.
Cross-talk between tumor-promoting inflammation and the other hallmarks of cancer
The carcinogenicity of low-dose exposures to chemical mixtures in our environment probably depends, in large part, on the capacity of such exposures to act on several tumor-promoting mechanisms and/or to disrupt innate tumor defence mechanisms. Thus, characterizing the potential of chemical combinations as ‘carcinogenic’ will ultimately involve investigating mixture effects across the range of mechanisms known to be relevant in tumor development. Accordingly, we undertook a thorough cross-validation activity to illustrate the importance of the prioritized target sites for disruption that were identified by this team (i.e. across multiple aspects of cancer biology) to illustrate the extent to which the prototypical chemical disruptors that we identified may act to disrupt other mechanisms relevant to carcinogenesis.
TAI has been identified as an epithelial-stroma interaction that enables tumor development by acting across the cancer hallmarks (6). Herein, we have identified six common environmental chemicals for which current evidence supports their role as putative ‘immune disruptors’. In other words, exposures to these chemicals are hypothesized to act in tumorigenesis by deregulating and promoting inflammation. For each chemical, we identified a single ‘high priority’ target molecule as a putative mediator of cellular and molecular events linking chemical exposure to carcinogenesis. The chemicals we have identified are (i) bisphenol A (BPA), (ii) PBDE, (iii) vinclozolin, (iv) NP, (v) phthalates and (vi) atrazine, with their main priority targets being the estrogen receptor, iNOS, NFκB, IL-6, COX-2 and TNFα, respectively.
As will be recognized, there is a strong relationship between the prioritized targets, inflammation, and a number of the other cancer hallmarks. The prioritized chemicals proposed here to promote inflammation have also been shown to act on a number of the other hallmarks of cancer that in some reports are complementary to the effects observed for TAI and for others are contrary or are inconsistent across reports. Exceptions are a lack, or limited study, of effect of these chemicals on tumor evasion of the immune system and on the tumor microenvironment. Given that inflammation contributes directly to changes in the microenvironment, which includes immune system evasion, these chemicals may act directly on the microenvironment and/or the function of anti-tumor immune cells. Details of the selected chemicals, the prioritized target, and affected pathways are presented below in support of the summary results shown in Tables 2 and 3.
Table 2.
Cross-validation of target pathways
| TAI targets | Deregulated metabolism | Evasion of anti- growth signalling | Angiogenesis | Genetic instability | Immune system evasion | Resistance to cell death | Replicative immortality | Sustained proliferative signalling | Tissue invasion and metastasis | Tumor microenvironment |
|---|---|---|---|---|---|---|---|---|---|---|
| Estrogen receptor (132) | − (133,134) | − (135,136) | − (137) | + (138) | ND | + (139–141) | + (142, 143) | + (144) | +/− (145–149) | + (150) |
| iNOS (151) | ND | + (152–154) | + (155) | + (156) | ND | − (157) | ND | + (158) | + (159–162) | + (163) |
| NFκB | + (164) | − (165) | ND | ND | ND | − (166) | + (163) | + (167) | ND | + (168) |
| IL-6 | + (169) | + (170–172) | ND | ND | ND | + (173–176) | + (177, 178) | + (178) | + (179–181) | + (7) |
| COX-2 | + (182) | − (183–185) | + (186) | + (187) | ND | + (188,189) | + (163, 190) | + (191) | + (187) | + (192) |
| TNF-α | + (193) | + (194,195) | + (196) | ND | + (unpublished) | +/− (197–199) | + (163) | + (200) | + (201–203) | + (7) |
Target pathways for TAI were cross-validated for effects in other cancer hallmark pathways. Targets that were found to have opposing actions in a particular hallmark (i.e. anti-carcinogenic) were noted as ‘−’ while targets that were found to have promoting actions in a particular hallmark (i.e. carcinogenic) were noted ‘+’ effects. In instances where reports on relevant actions in other hallmarks showed both pro-carcinogenic and anti-carcinogenic potential, +/− was used. Finally, in instances where no literature support was found to document the relevance of a target in a particular aspect of cancer biology, we documented this as not determined (ND).
Table 3.
Cross-validation of disruptors in the cancer hallmarks
| Disruptor | Deregulated metabolism (180,204–210) | Evade anti- growth signalling (104,211,212) | Angiogenesis (213–215) | Genetic instability (216,217),(218, 219) | Immune evasion (220) | Resist cell death (208,221–228) | Replicative immortality (229) | Sustained proliferative signaling (224,230–234) | Tissue invasion/metastasis (231,235–238) | Tumor micro- environment |
|---|---|---|---|---|---|---|---|---|---|---|
| BPA (239) | + | + | + | + | ND | +/− | + | + | + | ND |
| PBDEs | + | ND | ND | + | ND | +/− | ND | +/− | ND | ND |
| Vinclozolin | ND | + | ND | + | ND | ND | ND | ND | ND | ND |
| Nonylphenol | ND | + | ND | + | ND | ND | ND | ND | ND | ND |
| Phthalates | + | + | + | + | ND | +/− | ND | + | + | ND |
| Atrazine | +/− | ND | ND | + | + | ND | ND | ND | ND | ND |
Disruptors of TAI were cross-validated for effects in other cancer hallmark pathways. Disruptors that were found to have opposing actions in a particular hallmark were noted as ‘−’ while disruptors that were found to have promoting actions in a particular hallmark were noted ‘+’ effects. In instances where reports on relevant actions in other hallmarks were mixed, +/− was used. Finally, in instances where no literature support was found to document the relevance of a chemical in a particular aspect of cancer’s biology, we documented this as not determined (ND).
Bisphenol A
Treatment of cell lines with BPA results in a number of cellular and molecular changes, including those associated with the cancer hallmarks and inflammation as already discussed. However, there is no clear singular molecular target of BPA, and the role of BPA in human disease remains controversial. Consistently, BPA deregulates metabolism by disrupting the activity of respiratory chain complex II (204–207). At low exposure levels, BPA has been shown to activate the mammalian target of rapamycin (mTOR) pathway, an intracellular signalling pathway that integrates the growth signals, such as insulin and insulin-like growth factors, to promote survival (211,240). Treatment of cells with BPA blocks the induction of p53, thereby mediating evasion of anti-growth signals (211) and promoting angiogenesis (213). BPA has also been shown to promote genetic instability through anti-estrogenic activity (216) and upregulation of hTERT, an indicator of replicative immortality (241). In breast cancer cell lines, BPA exposure promotes a sustained proliferative signal (230). In other studies, tumor cell invasion and metastasis were shown to be promoted by BPA exposure (235–237). In contrast, BPA has also been reported to induce apoptosis and cytotoxicity in HL-60 and ovarian granulosa cells (221,222), effects that are more consistent with an anti-cancer activity.
BPA has been extensively studied as an endocrine disruptor given its binding affinity for ERβ is greater than that of ERα. This topic has been reviewed extensively (239). Of note is that, similar to the ambiguous relationships described earlier, ER and BPA display paradoxical effects in tumor development that are context-dependent. For example, loss of ERα promotes hepatocarcinogenesis (133), and activation of ERβ impairs mitochondrial oxidative metabolism, thereby suppressing tumor growth (134). The loss of ERα also showed the same effect when its mechanism was antagonized by binding to p53 in evasion of anti-growth signalling (135,136). The introduction of ERβ into malignant cells inhibits their growth and prevents tumor expansion by inhibiting angiogenesis (137). In contrast, ER signalling can promote genetic instability by promoting DNA double strand breaks and chromosomal aberrations (138). Estrogen also promotes resistance to cell death by preventing p53-dependent apoptosis, as well as stimulating cell growth and inhibiting apoptosis (139–141). Estrogen downregulates YPEL3, a growth suppressive gene, and activates hTERT transcription and replicative immortality via binding of ligand-activated ERα (142,143). Ligand-bound ERs can either bind directly to estrogen response elements in the promoters of target genes or they can interact with other transcription factor complexes like Fos/Jun (AP-1-responsive elements) in sustained proliferative signalling (144). There is also a potential crosstalk between ERβ and AR in the tumor microenvironment (150). Plausibly, the ERβ has effects in tissue invasion and metastasis, in which ERβ ligation could protect tumor cells from acquiring aggressive epithelial to mesenchymal transition features by blocking loss of e-cadherin and translocation of β-catenin to the nucleus (145). These paradoxical effects of ER are well known in the breast cancer field, where synthetic estrogens, for which BPA was originally studied, prevent tumorigenesis in one tissue while promoting it in another (e.g. tamoxifen in breast and endometrium, respectively) (146–148). Short exposure to BPA induces ERα and/or ERβ loading to DNA changing target gene transcription (132,242).
Polybrominated diphenyl ethers
PBDEs represent another class of chemicals which have been reported to disrupt glucose and lipid metabolism (208) promote genetic instability (218). PBDEs have been reported to be both pro-apoptotic in one context (223) yet anti-apoptotic in the presence of 17β-estradiol in the MCF7 breast tumor cell line (224). The putative target of PBDE, iNOS, has been associated with the accumulation of p53 in a feedback mechanism that both protects the genome from DNA damage but also results in p53-mediated transrepression of iNOS (149,152–154). In the absence of p53 activated iNOS fails to return to basal levels due to the lack of transrepression. This may partially explain high rates of tumor development in p53 knockout mice (151). Elevated intracellular levels of NO are genotoxic to cells and promote genetic instability (156), with strong evidence for iNOS as a contributing factor in angiogenesis and tumor invasion and metastasis—hallmarks that often occur later in tumorigenesis when p53 is more probably to be lost (155,157,159–161), as well as in sustained proliferative signalling (158). As a product of inflammation, NO plays a major role in wound healing type inflammation (i.e. a macrophage prominent inflammatory response) and acts as a permissive factor in tumor invasion and metastasis (243).
Vinclozolin
Like BPA, vinclozolin is considered an endocrine disruptor with activity for AR as well as ER and progesterone receptor. As with most endocrine disruptors, a number of cellular and molecular activities have been attributed to vinclozolin. With regard to the cancer hallmarks, vinclozolin has been shown to promote the evasion of anti-growth signals (104), induce oxidative damage leading to inflammation, and cause DNA damage and genetic instability (217). In rats, in utero exposure to vinclozolin for 5 days did not impair prostate gland development but decreased AR expression in the pubertal prostate. Exposed animals develop a prostatitis during puberty that has been mechanistically linked to phosphorylation and nuclear translocation of NFκB, with subsequent induction of pro-inflammatory NFκB-dependent genes (IL-8 and transforming growth factor-β1). Of note, early life exposure to vinclozolin persists into adulthood with evidence of epigenetic deregulation of NFκB, which resulted in inflammation in the prostate. Findings of heritable alterations and transgenerational effects on reproductive, immune, and neurologic systems raise concern about the transmission of new traits associated with carcinogenesis, for which little research has been conducted. In the rat prostatitis model, exposure to vinclozolin alone was insufficient for tumor development, suggesting that the exposure is not genotoxic in nature. However, like other endocrine disruptors, vinclozolin induces a spectrum of molecular and cellular effects, including increased apoptotic germ cell numbers in the testis of pubertal and adult animals (244). Effects on NFκB, particularly if transmissible across generations, are noteworthy, given the well documented role that unrepressed NFκB plays in tumorigenesis (163,164). NFκB has also been reported to promote sustained proliferative signalling (167) and, via its critical role in propagating a wound healing type inflammatory mechanism, has potent effects on the tumor microenvironment (168). On activation, the NFκB signalling pathway decreases p53 stabilization (165) and inhibits TNFα-induced apoptosis, promoting resistance to cell death (166)—all critical hallmarks in the evolution of cancers.
Nonylphenol
As with the other endocrine disruptors, NP exerts estrogenic action and stimulates proliferation in estrogen responsive ovarian cancer PEO4 cells (219). Derivatives of NP, such as 4-NP, exhibit genotoxic affects in Saccharomyces cerevisiae supporting a role in genetic instability (245). In contrast, NP in other models has been shown to exhibit anti-cancer properties including triggering, inducing, or enhancing apoptosis in various tumor cells (225). NP has been shown to induce expression of a pro-inflammatory cytokine, TNF-α, and to suppress regulatory cytokines, including IL-10, IFN-α and IFN-β (246).
The regulatory cytokine IL-6 has been linked to a number of the cancer hallmarks (247). NP exposure that results in chronic activation of IL-6 thus, has potential to act on a number of the tumor hallmarks through local and systemic effects on metabolism (169), growth signalling (170–172), cell death mechanisms (173–176), enhancement of replicative immortality by altering telomerase activity (177,248), and chronic exposure that leads to sustained proliferative signals (178). Effects of IL-6 on tissue invasion and metastasis have been shown for a number of cancers including ovarian (179), melanoma (179) and head and neck tumor metastasis (180). As with NFκB, IL-6 promotes a wound healing type inflammatory response contributing suppression of immune effectors with potent effects on the tumor microenvironment, leading to greater permissiveness and tumor invasion (7).
Phthalates
Phthalates have been shown to act on a number of the cancer hallmarks, though we found no studies investigating effects on replicative immortality or tumor microenvironment. Evidence that exposure of hepatocytes to diisononyl phthalate increases proliferation, palmitoyl-CoA oxidase activity, and levels of enzymes involved in β- and ω-oxidation of fatty acids dependent on another nuclear receptor, PPARα, support effects on metabolism (214). In addition, benzyl butyl phthalate, a common plasticizer use in manufacturing polyvinyl chloride and recognized developmental toxicant, has been shown to increase angiogenesis in vivo (238,249). Phthalates have been reported to promote tumor growth and invasion of cancer cells in vitro via regulation of cyclin D, PPARα and AhR (231,232,250). Direct effects of phthalates on p53 have been proposed that would support effects of phthalates on evasion of anti-growth signals, though this is controversial (251,252). A recent study by Lee et al. (212), reported growth promoting effects of di-n-buthyl phthalate in the LNCaP mouse xenograft model of prostate cancer that was in part mediated by reduction of Smad. This observation was similar to that observed with estradiol and was found to be reversible with an ER antagonist. These findings suggest that phthalates may act on tumor growth by disrupting important crosstalk between TGF-β and ER signals leading to evasion of anti-growth signals. In addition, exposure of human oropharangeal and nasal mucosa cells to the phthalates di-n-buthyl phthalate and diisobutyl phthalate increases DNA strand breaks and the possibility of increasing genetic instability (253). As with a number of environmental chemicals, phthalates exhibit a wide array of both anti-carcinogenic and carcinogenic activities. For example, phthalate esters induce apoptosis in bovine testicular pluripotent stem cells through an AhR-mediated mechanism (226) and inhibit tamoxifen-induced apoptosis in MCF7 human breast cancer cells (254). Reported induction of COX-2 expression by these chemicals would support a protumorigenic action. Overexpression of COX-2 has been consistently shown to contribute to a number of the hallmarks of cancer including effects on metabolism (182), enhancement of cell motility and invasiveness (187), promotion of resistance to pro-apoptotic activity associated with NFκB activation (188–190). Genetic instability via chromosomal aberrations is a common phenotype associated with COX-2 overexpression (255), as is the induction of proangiogenic signaling pathways (186), sustained proliferative signalling (191), and effects on the tumor microenvironment (192). Nevertheless, the effects of COX-2 are inhibited in the presence of wildtype p53 and, as with a number of the other chemicals, the effect of exposure in terms of carcinogenic potential is probably dependent on the cellular type and molecular context in which the exposure occurs (e.g. p53 wildtype or mutant background) (183–185).
Triazine herbicides
The triazine herbicide atrazine has been extensively studied for its effects in promoting tumor immune system evasion (220) and sustained proliferative signalling via the ERα signalling pathway (233). Atrazine significantly increases the formation of micronuclei and DNA strand breaks in erythrocytes of Carassius auratus, a model fish species (256). Interestingly, atrazine exposure has been reported to decrease cancer by suppressing prostate carcinogenesis through metabolic deregulation that manifests as bodyweight reduction (209). In contrast, atrazine exhibits carcinogenic effects by promoting obesity and insulin resistance by blocking the activities of oxidative phosphorylation complexes I and III (210). The effects of atrazine on female reproductive health have been extensively studied with exposure being associated with deregulated sterodoigenesis and angiogenesis (215). There are no studies specifically addressing anti-cancer hallmark properties, but a number of studies have demonstrated that triazine derivatives suppress replicative immortality (229,257,258). For effects on inflammation, the proposed target molecule is TNFα (197–199). An extensive body of work on TNFα supports its role in promoting evasion of anti-growth signals (158,193–196,200–202,259,260), promotion of angiogenesis (196), direct effects on promoting myeloid suppressors cell and tumor immune system evasion (unpublished data) replicative immortality (163), and sustained proliferative signalling (200,260). In addition, TNFα promotes epithelial to mesenchymal transition and tumor invasiveness in breast and colon cancer cell lines (201–203) as well as alters the local metabolic and microenvironment including promoting tumor-associated fibroblasts (7,193).
Future directions
While there is clearly intriguing evidence for a role of environmental chemicals to act as immune disruptors, there is a large paucity of information for how such effects on the immune system would ultimately influence individual cancer risk in humans. Given the potential transgenerational inheritance of some exposures, there are new concerns that traditional exposure association studies would entirely fail to capture any relationship between the exposure and long-term, multigenerational impacts of chemically exposed individuals. Because of the tremendous paucity of information on the role of immune disruption and risk of cancer, the co-authors identified specific areas for future research that included identifying promising novel technologies to expand our understanding of the effects of exposures, as well as, using observations from the past to guide future experimental design considerations. These have been broken down into a few key areas that, while no means comprehensive, should serve to stimulate discussion and innovation in the field.
Systematic approaches to study the role of immune disruptors (stimuli) in carcinogenesis
Given the crucial status of host tissue immune cell responses in cancer, systematic understanding of the host (resident) immune and non-immune interactions with those of recruited cells with environmental toxicants are important when deciding on accurate risk assessment formulations.
The following is a proposed list of priorities for future directions in understanding the role of environmental immune disruptors in alteration of immune dynamics, inflammation and individual cancer susceptibility.
Need for systematic approaches to characterize the nature of specific chemicals that would alter immune response profiles toward cellular growth and site-specific cancers.
Experimental models to gain a better understanding of the nature of heterogeneities in immune response profiles, since the extent of chemical exposure and access to inter-epithelial and sub-epithelial cells may produce significantly different outcomes.
More comprehensive modeling of the cumulative processes of immune disruptors (e.g. mixtures, low-doses) that promote chronic inflammation in host tissues that lead to disruption of cellular compartments in the multistep carcinogenesis toward angiogenesis and metastasis.
In line with item (iii), attention to time-course kinetics of developmental phases of immune response alterations during early stages of tumorigenesis and angiogenesis that might be preventable, reversible or correctable.
Experimental constructs to identify key interactions/players between stimuli and host immune and non-immune responses in tissues considering the influence of the resident (local) and recruited cell compositions in future studies of inflammation and cancer (e.g. Kupfer myeloid cell in the liver, inflammation and HCC).
More complete monitoring of the early changes of immune dynamics (e.g. induction of mediators such as histamine, heparin, PGs, enzymes and neurotransmitters) that reflect important changes in early responses that include neurologic stress response effectors that are also prone to disruption by environmental chemicals as early and preventable pro-inflammation/pro-tumor targets.
Incorporation of information on genetic background (gene polymorphisms), sex-specific effects and life-span exposure periods as modifiers of susceptibility to environmental immune toxicants in carcinogenesis.
The hypothalmic-pituitary-adrenal axis, stress, chemicals and inflammation
Given that the hypothalmic-pituitary-adrenal (HPA) axis is an endocrine-driven system, it is highly plausible that individual chemical exposures such as atrazine and other chlorotriazine herbicides (and metabolites) may have the potential to disrupt and deregulate the immune system including the cellular makeup and chemokine/cytokine composition of the inflammatory response mechanism (261). The HPA-axis operates in a cascade-fashion whereby relatively small secretions of corticotrophin releasing hormone in the hypothalamus result in more substantial secretions of adrenocorticotropic hormone from the pituitary leading to substantial changes in adrenal output of cortisol. The cumulative effects of combinations of low doses of endocrine-disrupting chemicals that mimic and/or impact corticotrophin releasing hormone or adrenocorticotropic hormone through indirect mechanisms are thus a concern given the impact of cortisol on a number of cytokine and chemokine molecules including macrophage migration inhibiting factor (MIF). As an example, MIF is a potent pro-inflammatory cytokine that binds to the CD74 molecule on immune cells and activates acute immune responses; coupling the HPA axis to inflammation (262,263). Similarly, the cumulative effects of other chemicals such as PBDE that act directly on the adrenal gland (264,265) could further; disrupt this system While transgenerational effects such as those that have been demonstrated for BPA, may act by disrupting the HPA-axis, cause adrenal abnormalities, and alter the basal levels of circulating hormones in rodent offspring exposed in utero (266,267). These observations strongly suggest that such chemical exposures can have long-lasting and profound effects on the HPA stress axis, which in turn lead to altered sensitivity to immune perturbagens; an evolving field that has received little attention in cancer biology and risk assessment.
Since MIF and a number of other HPA-induced immune molecules have been separately implicated in tumor growth and progression (268–272), basic research is needed to screen and identify environmentally relevant chemicals that have disruptive potential for different aspects of the entire system. Moreover, empirical research is needed to determine whether or not low dose combinations of common chemical exposures deregulate the HPA-axis, and impact adrenal output such that MIF and other pro-inflammatory cytokines are not well controlled (i.e., instead become tumor-promoting). This is a critically important area in need of research given the consistent and unexplained higher risk of more life-threatening cancers in poor populations that experience a disproportionate burden of exposure to environmental chemicals and to stressful living conditions.
Environmental chemicals, the human microbiome, inflammation and cancer
It is increasingly clear that the human microbiome shapes the immune system and plays a major role throughout life in the health of immune responses. A question in need of further study is whether or not chemical exposures (low-dose, mixed) influence the composition of our microbiome and, if so, to what effect on our immune system and long-term cancer risk (273).
Ongoing and planned epidemiology cohorts, environmental chemicals and cancer
There are currently several large prospective population-based cohort studies underway that will facilitate the evaluation of cancer risks from exposures to environmental contaminants that occur individually and in combination with other exposures and modifying factors. These studies include the UK Biobank, the Canadian Partnership for Tomorrow Project (CPT) and the European Prospective Investigation into Cancer and Nutrition (EPIC) (274,275). These studies, which are prospective in design and biomarker based, may allow for the evaluation of intermediate inflammatory effects of exposures to environmental chemicals and mixtures across genetically diverse populations and the eventual evaluation of associated cancer risks. Although sufficiently powered for the more common cancers, such as breast, lung, colorectal and prostate, it is probably that sample sizes will not be sufficient for the evaluation of risks for some of the less common cancers that may be induced or promoted via inflammatory response mechanisms. Also, given the evidence of transgenerational mechanisms of inheritance, consideration of off-spring cohorts, may ultimately prove useful and possibly necessary to establish the long-term risks of cancer with certain exposures and thought should be given to how population cohorts will translate the findings from the early life exposure evidence in experimental models to humans.
Advanced technologies in the study of environmental chemicals and cancer
There is a need to integrate more experimental platforms that facilitate the study of the dynamic and complex response of system perturbation. This is true for all the Hallmarks, as noted in the capstone that accompanies this article. One such example includes evolving techniques to study cellular protein dynamics upon stressor binding in three dimension to investigate latent stages and early stages of carcinogenesis involving inflammation, immune system evasion and/or tumor microenvironment related pathways. Combinations of high-quality imaging and high-dimension omics in complex culture systems aided by computational techniques with knowledge from pathway-based databases and structure-based virtual ligand screenings of environmental toxins (276,277) offer systems biology approaches to assess the permutable nature of tissue/cell responses to environmental chemicals. Emerging techniques hold significant promise as results of pathway and systems perturbation will guide development of novel animal models that, perhaps coupled with live animal monitoring strategies, would promote work to obtain much needed information on immediate, early and late exposure-related effects across the human lifespan on organs and tissues including detection of subtle perturbation leading to later life cancer risk or cancer risk in subsequent generations. Success in such efforts has the potential to enhance discovery and development of novel tissue/body fluid biomarkers as surrogates for risk stratification, risk prediction and cancer surveillance in humans. Ultimately, it is imaginable that integration of such information would advance prevention by eliminating potentially harmful exposures, establishing safe exposure levels and for those adversely exposed individuals who might be particularly vulnerable, insights on disease prevention.
Summary
Here, we have provided a molecular and cellular mechanistic framework on which to consider the role of common environmental chemicals as cancer enabling through activity as immune disruptors. While limited and mixed, the experimental evidence to date strongly suggests a role for common environmental chemicals as perturbagens of key immune and non-immune cell target molecules that have been mechanistically linked with tumor-associated immune responses, tumor invasion and metastasis. These observations warrant attention given the widespread use and exposures to a number of these chemicals. This is made particularly compelling by the emerging evidence that in utero and early-life exposures may lead to disordered immune responses in adulthood and lead to heritable, epigenetic modifications in the immune responses of subsequent generations. It is important to point out, that few studies have been conducted that relate chemically induced disturbances on inflammatory to the ability of a chemical to contribute to carcinogenesis. In the absence of active research, the role of chemical exposures acting in carcinogenesis by disrupting TAI is unknown. As such, we focused on the few examples of chemicals and their putative target molecules that have been mechanistically linked with TAI. It is important to recognize that the target molecules identified here exhibit a plurality of function and derive from immune and non-immune cells, including for example COX2, TNFα, NFκB. Many of these molecules are also overexpressed by epithelial tumor cells as well as associated immune cells in the microenvironment where they exhibit an array of cellular and molecular effects—not all of which are to elicit an inflammatory response. This plurality probably explains the high degree of complementarity between the target molecules of environmental chemicals that we identified as important in TAI with those identified for other cancer hallmarks. The potential for action across the cancer hallmarks speaks strongly to the potential role that environmental chemicals may play in disrupting biological systems as opposed to acting on a single target molecule or single cell type. With the rate at which new chemicals have and continue to enter the human environment, the paucity of research and of innovation in methods to study the effects of chemical mixtures in systems perturbation in cancer etiology represents a significant research gap. Importantly, with the tremendous benefits that these chemicals provide to society—facilitation of mass food production and distribution, use in medicine and medical devices, clothing, jobs generation and numerous other impactful contributions to society—there is a real need to fully understand how these chemicals act on human biological. This includes conducting experiments that consider effects across the lifespan of the exposed to better clarify the role of in utero and early life exposure on cancer and other diseases for both current and future generations. More comprehensive integration of knowledge on chemical effects on and across biological systems, considering the multifactorial pathogenesis of carcinogenesis will both inform the industry and the public on the safe use of these chemicals as single agents and as they occur in complex mixtures in the environment.
Funding
National Institute of Environmental Health Sciences (P30 ES000210 to W.H.B.).
Acknowledgments
T.G. was supported by a grant from Fundamental Oriented Research (RFO) to the Alma Mater Studiorum University of Bologna, Bologna, Italy and thanks the Fondazione Cassa di Risparmio di Bologna and the Fondazione Banca del Monte di Bologna e Ravenna for supporting the Center for Applied Biomedical Research, S.Orsola-Malpighi University Hospital, Bologna, Italy.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- 5-LOX
5-lipoxygenase
- AhR
aryl hydrocarbon receptor
- AR
androgen receptor
- BPA
bisphenol A
- COX
cyclooxygenase
- DEHP
diethylhexyl phthalate
- HCC
hepatocellular cancer
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- MIF
migration inhibiting factor
- MMP
matrix metalloproteinase
- NO
nitric oxide
- NP
nonylphenol
- PBDE
polybrominated diphenyl ether
- PPAR
peroxisome proliferator-activated receptor
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- STAT
signal transducer and activator of transcription family
- TAI
tumor-associated inflammation
- TNF
tumor necrosis factor
References
- 1. Hanahan D., et al. (2000) The hallmarks of cancer. Cell, 100, 57–70. [DOI] [PubMed] [Google Scholar]
- 2. Khatami M. (2014) Chronic inflammation: synergistic interactions of recruiting macrophages (TAMs) and eosinophils (Eos) with host mast cells (MCs) and tumorigenesis in CALTs. M-CSF, suitable biomarker for cancer diagnosis! Cancers (Basel)., 6, 297–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Khatami M. (2008) ‘Yin and Yang’ in inflammation: duality in innate immune cell function and tumorigenesis. Expert Opin. Biol. Ther., 8, 1461–1472. [DOI] [PubMed] [Google Scholar]
- 4. Khatami M. (2011) Unresolved inflammation: ‘immune tsunami’ or erosion of integrity in immune-privileged and immune-responsive tissues and acute and chronic inflammatory diseases or cancer. Expert Opin. Biol. Ther., 11, 1419–1432. [DOI] [PubMed] [Google Scholar]
- 5. Colotta F., et al. (2009) Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis, 30, 1073–1081. [DOI] [PubMed] [Google Scholar]
- 6. Hanahan D., et al. (2011) Hallmarks of cancer: the next generation. Cell, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 7. Candido J., et al. (2013) Cancer-related inflammation. J. Clin. Immunol., 33 (suppl. 1), S79–S84. [DOI] [PubMed] [Google Scholar]
- 8. Costa A., et al. (2014) The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol., 25, 23–32. [DOI] [PubMed] [Google Scholar]
- 9. Roberts R.A., et al. (2010) Toxicological and pathophysiological roles of reactive oxygen and nitrogen species. Toxicology, 276, 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Naik E., et al. (2011) Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med., 208, 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kongara S., et al. (2012) The interplay between autophagy and ROS in tumorigenesis. Front. Oncol., 2, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Danelli L., et al. (2015) Mast cells boost myeloid-derived suppressor cell activity and contribute to the development of tumor-favoring microenvironment. Cancer Immunol. Res., 3, 85–95. [DOI] [PubMed] [Google Scholar]
- 13. Grimm E.A., et al. (2013) Molecular pathways: inflammation-associated nitric-oxide production as a cancer-supporting redox mechanism and a potential therapeutic target. Clin. Cancer Res., 19, 5557–5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parke D.V., et al. (1996) Chemical toxicity and reactive oxygen species. Int. J. Occup. Med. Environ. Health, 9, 331–340. [PubMed] [Google Scholar]
- 15. Vallyathan V., et al. (1998) Reactive oxygen species: their relation to pneumoconiosis and carcinogenesis. Environ. Health Perspect., 106 (suppl. 5), 1151–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Azad N., et al. (2008) Inflammation and lung cancer: roles of reactive oxygen/nitrogen species. J. Toxicol. Environ. Health. B. Crit. Rev., 11, 1–15. [DOI] [PubMed] [Google Scholar]
- 17. Pourazar J., et al. (2005) Diesel exhaust activates redox-sensitive transcription factors and kinases in human airways. Am. J. Physiol. Lung Cell. Mol. Physiol., 289, L724–L730. [DOI] [PubMed] [Google Scholar]
- 18. Mazzarella G., et al. (2007) Effects of diesel exhaust particles on human lung epithelial cells: an in vitro study. Respir. Med., 101, 1155–1162. [DOI] [PubMed] [Google Scholar]
- 19. Hartz A.M., et al. (2008) Diesel exhaust particles induce oxidative stress, proinflammatory signaling, and P-glycoprotein up-regulation at the blood-brain barrier. FASEB J., 22, 2723–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ma J.Y., et al. (2002) The dual effect of the particulate and organic components of diesel exhaust particles on the alteration of pulmonary immune/inflammatory responses and metabolic enzymes. J. Environ. Sci. Health. C. Environ. Carcinog. Ecotoxicol. Rev., 20, 117–147. [DOI] [PubMed] [Google Scholar]
- 21. Zhao H., et al. (2009) Reactive oxygen species- and nitric oxide-mediated lung inflammation and mitochondrial dysfunction in wild-type and iNOS-deficient mice exposed to diesel exhaust particles. J. Toxicol. Environ. Health. A, 72, 560–570. [DOI] [PubMed] [Google Scholar]
- 22. Kisley L.R., et al. (2002) Genetic ablation of inducible nitric oxide synthase decreases mouse lung tumorigenesis. Cancer Res., 62, 6850–6856. [PubMed] [Google Scholar]
- 23. Moran E.M. (2002) Epidemiological and clinical aspects of nonsteroidal anti-inflammatory drugs and cancer risks. J. Environ. Pathol. Toxicol. Oncol., 21, 193–201. [PubMed] [Google Scholar]
- 24. Wakabayashi K. (2000) NSAIDs as cancer preventive agents. Asian Pac. J. Cancer Prev., 1, 97–113. [PubMed] [Google Scholar]
- 25. Baglole C.J., et al. (2006) More than structural cells, fibroblasts create and orchestrate the tumor microenvironment. Immunol. Invest., 35, 297–325. [DOI] [PubMed] [Google Scholar]
- 26. Nataraj C., et al. (2001) Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J. Clin. Invest., 108, 1229–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tilley S.L., et al. (2001) Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J. Clin. Invest., 108, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu Z., et al. (2011) Targeting the inflammatory pathways to enhance chemotherapy of cancer. Cancer Biol. Ther., 12, 95–105. [DOI] [PubMed] [Google Scholar]
- 29. Bollrath J., et al. (2009) IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep., 10, 1314–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nguyen D.P., et al. (2014) Inflammation and prostate cancer: the role of interleukin 6 (IL-6). BJU Int., 113, 986–992. [DOI] [PubMed] [Google Scholar]
- 31. Grivennikov S., et al. (2009) IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell, 15, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carpenter R.L., et al. (2014) STAT3 target genes relevant to human cancers. Cancers (Basel)., 6, 897–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang H.F., et al. (2014) STAT3 in cancer-friend or foe? Cancers (Basel)., 6, 1408–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. He G., et al. (2011) NF-κB and STAT3—key players in liver inflammation and cancer. Cell Res., 21, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Feldmann M., et al. (2008) Role of cytokines in rheumatoid arthritis: an education in pathophysiology and therapeutics. Immunol. Rev., 223, 7–19. [DOI] [PubMed] [Google Scholar]
- 36. Croft M. (2014) The TNF family in T cell differentiation and function—unanswered questions and future directions. Semin. Immunol., 26, 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ataie-Kachoie P., et al. (2014) Gene of the month: interleukin 6 (IL-6). J. Clin. Pathol., 67, 932–937. [DOI] [PubMed] [Google Scholar]
- 38. Rose-John S., et al. (2006) Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J. Leukoc. Biol., 80, 227–236. [DOI] [PubMed] [Google Scholar]
- 39. Taniguchi K., et al. (2014) IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol., 26, 54–74. [DOI] [PubMed] [Google Scholar]
- 40. Tsukamoto H., et al. (2013) Myeloid-derived suppressor cells attenuate TH1 development through IL-6 production to promote tumor progression. Cancer Immunol. Res., 1, 64–76. [DOI] [PubMed] [Google Scholar]
- 41. Zarogoulidis P., et al. (2014) Interleukin-8 and interleukin-17 for cancer. Cancer Invest., 32, 197–205. [DOI] [PubMed] [Google Scholar]
- 42. Campbell L.M., et al. (2013) Rationale and means to target pro-inflammatory interleukin-8 (CXCL8) signaling in cancer. Pharmaceuticals (Basel)., 6, 929–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grivennikov S.I., et al. (2012) Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature, 491, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schneider C., et al. (2011) Cyclooxygenases and lipoxygenases in cancer. Cancer Metastasis Rev., 30, 277–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Greene E.R., et al. (2011) Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat., 96, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bishayee K., et al. (2013) 5-lipoxygenase antagonist therapy: a new approach towards targeted cancer chemotherapy. Acta Biochim. Biophys. Sin. (Shanghai)., 45, 709–719. [DOI] [PubMed] [Google Scholar]
- 47. Gilmartin A.G., et al. (2014) Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat. Chem. Biol., 10, 181–187. [DOI] [PubMed] [Google Scholar]
- 48. Datta K., et al. (1999) The 5-lipoxygenase-activating protein (FLAP) inhibitor, MK886, induces apoptosis independently of FLAP. Biochem. J., 340 (Pt 2), 371–375. [PMC free article] [PubMed] [Google Scholar]
- 49. Schuller H.M., et al. (2002) The cyclooxygenase inhibitor ibuprofen and the FLAP inhibitor MK886 inhibit pancreatic carcinogenesis induced in hamsters by transplacental exposure to ethanol and the tobacco carcinogen NNK. J. Cancer Res. Clin. Oncol., 128, 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kim K.H., et al. (2014) Diverse influences of androgen-disrupting chemicals on immune responses mounted by macrophages. Inflammation, 37, 649–656. [DOI] [PubMed] [Google Scholar]
- 51. Kuan Y.H., et al. (2012) Proinflammatory activation of macrophages by bisphenol A-glycidyl-methacrylate involved NFκB activation via PI3K/Akt pathway. Food Chem. Toxicol., 50, 4003–4009. [DOI] [PubMed] [Google Scholar]
- 52. Tian X., et al. (2003) Bisphenol A promotes IL-4 production by Th2 cells. Int. Arch. Allergy Immunol., 132, 240–247. [DOI] [PubMed] [Google Scholar]
- 53. Wang K.H., et al. (2013) Bisphenol A at environmentally relevant doses induces cyclooxygenase-2 expression and promotes invasion of human mesenchymal stem cells derived from uterine myoma tissue. Taiwan. J. Obstet. Gynecol., 52, 246–252. [DOI] [PubMed] [Google Scholar]
- 54. Valentino R., et al. (2013) Bisphenol-A impairs insulin action and up-regulates inflammatory pathways in human subcutaneous adipocytes and 3T3-L1 cells. PLoS One, 8, e82099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wiseman S.B., et al. (2011) Polybrominated diphenyl ethers and their hydroxylated/methoxylated analogs: environmental sources, metabolic relationships, and relative toxicities. Mar. Pollut. Bull., 63, 179–188. [DOI] [PubMed] [Google Scholar]
- 56. Koike E., et al. (2014) Penta- and octa-bromodiphenyl ethers promote proinflammatory protein expression in human bronchial epithelial cells in vitro . Toxicol. In Vitro, 28, 327–333. [DOI] [PubMed] [Google Scholar]
- 57. Peltier M.R., et al. (2012) Polybrominated diphenyl ethers enhance the production of proinflammatory cytokines by the placenta. Placenta, 33, 745–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Walker D.M., et al. (2011) Transgenerational neuroendocrine disruption of reproduction. Nat. Rev. Endocrinol., 7, 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cowin P.A., et al. (2008) Early-onset endocrine disruptor-induced prostatitis in the rat. Environ. Health Perspect., 116, 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Y.Q., et al. (2008) Elevation of inducible nitric oxide synthase and cyclooxygenase-2 expression in the mouse brain after chronic nonylphenol exposure. Int. J. Mol. Sci., 9, 1977–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yoshitake J., et al. (2008) Suppression of NO production and 8-nitroguanosine formation by phenol-containing endocrine-disrupting chemicals in LPS-stimulated macrophages: involvement of estrogen receptor-dependent or -independent pathways. Nitric Oxide, 18, 223–228. [DOI] [PubMed] [Google Scholar]
- 62. Liu X, et al. (2014) Nonylphenol regulates cyclooxygenase-2 expression via Ros-activated NF-κB pathway in sertoli TM4 cells. Environ. Toxicol. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 63. Kuo C.H., et al. (2014) Epigenetic regulation in allergic diseases and related studies. Asia Pac. Allergy, 4, 14–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Han E.H., et al. (2010) Upregulation of cyclooxygenase-2 by 4-nonylphenol is mediated through the cyclic amp response element activation pathway. J. Toxicol. Environ. Health. A, 73, 1451–1464. [DOI] [PubMed] [Google Scholar]
- 65. Desvergne B., et al. (2009) PPAR-mediated activity of phthalates: A link to the obesity epidemic? Mol. Cell. Endocrinol., 304, 43–48. [DOI] [PubMed] [Google Scholar]
- 66. Jacobson-Dickman E, et al. (2009) The influence of endocrine disruptors on pubertal timing. Curr. Opin. Endocrinol. Diabetes Obes., 16, 25–30. [DOI] [PubMed] [Google Scholar]
- 67. Vetrano A.M., et al. (2010) Inflammatory effects of phthalates in neonatal neutrophils. Pediatr. Res., 68, 134–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yavaşoğlu N.Ü., et al. (2014) Induction of oxidative stress and histological changes in liver by subacute doses of butyl cyclohexyl phthalate. Environ. Toxicol., 29, 345–353. [DOI] [PubMed] [Google Scholar]
- 69. Win-Shwe T.T., et al. (2013) Expression levels of neuroimmune biomarkers in hypothalamus of allergic mice after phthalate exposure. J. Appl. Toxicol., 33, 1070–1078. [DOI] [PubMed] [Google Scholar]
- 70. Bornehag C.G., et al. (2010) Phthalate exposure and asthma in children. Int. J. Androl., 33, 333–345. [DOI] [PubMed] [Google Scholar]
- 71. Hayes T.B., et al. (2011) Demasculinization and feminization of male gonads by atrazine: consistent effects across vertebrate classes. J. Steroid Biochem. Mol. Biol., 127, 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Abarikwu S.O., et al. (2012) The protective effects of quercetin on the cytotoxicity of atrazine on rat Sertoli-germ cell co-culture. Int. J. Androl., 35, 590–600. [DOI] [PubMed] [Google Scholar]
- 73. Devos S., et al. (2003) Inhibition of cytokine production by the herbicide atrazine. Search for nuclear receptor targets. Biochem. Pharmacol., 65, 303–308. [DOI] [PubMed] [Google Scholar]
- 74. Zhao S., et al. (2013) Sub-acute exposure to the herbicide atrazine suppresses cell immune functions in adolescent mice. Biosci. Trends, 7, 193–201. [PubMed] [Google Scholar]
- 75. Michałowicz J. (2014) Bisphenol A—sources, toxicity and biotransformation. Environ. Toxicol. Pharmacol., 37, 738–758. [DOI] [PubMed] [Google Scholar]
- 76. Hoekstra E.J., et al. (2013) Release of bisphenol A from polycarbonate: a review. Crit. Rev. Food Sci. Nutr., 53, 386–402. [DOI] [PubMed] [Google Scholar]
- 77. Huang Y.Q., et al. (2012) Bisphenol A (BPA) in China: a review of sources, environmental levels, and potential human health impacts. Environ. Int., 42, 91–99. [DOI] [PubMed] [Google Scholar]
- 78. Flint S., et al. (2012) Bisphenol A exposure, effects, and policy: a wildlife perspective. J. Environ. Manage., 104, 19–34. [DOI] [PubMed] [Google Scholar]
- 79. Ho S.M., et al. (2006) Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res., 66, 5624–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Stoker T.E., et al. (1999) Prepubertal exposure to compounds that increase prolactin secretion in the male rat: effects on the adult prostate. Biol. Reprod., 61, 1636–1643. [DOI] [PubMed] [Google Scholar]
- 81. Rogers J.A., et al. (2013) Review: endocrine disrupting chemicals and immune responses: a focus on bisphenol-A and its potential mechanisms. Mol. Immunol., 53, 421–430. [DOI] [PubMed] [Google Scholar]
- 82. Yan H., et al. (2008) Exposure to bisphenol A prenatally or in adulthood promotes T(H)2 cytokine production associated with reduction of CD4CD25 regulatory T cells. Environ. Health Perspect., 116, 514–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kharrazian D. (2014) The potential roles of bisphenol A (BPA) pathogenesis in autoimmunity. Autoimmune Dis., 2014, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Glass C.K., et al. (2010) Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat. Rev. Immunol., 10, 365–376. [DOI] [PubMed] [Google Scholar]
- 85. Bonefeld-Jørgensen E.C., et al. (2007) Endocrine-disrupting potential of bisphenol A, bisphenol A dimethacrylate, 4-n-nonylphenol, and 4-n-octylphenol in vitro: new data and a brief review. Environ. Health Perspect., 115 (suppl. 1), 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Qin X.Y., et al. (2011) Xenoestrogens down-regulate aryl-hydrocarbon receptor nuclear translocator 2 mRNA expression in human breast cancer cells via an estrogen receptor alpha-dependent mechanism. Toxicol. Lett., 206, 152–157. [DOI] [PubMed] [Google Scholar]
- 87. Veldhoen M., et al. (2010) The aryl hydrocarbon receptor: fine-tuning the immune-response. Curr. Opin. Immunol., 22, 747–752. [DOI] [PubMed] [Google Scholar]
- 88. Quintana F.J., et al. (2008) Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature, 453, 65–71. [DOI] [PubMed] [Google Scholar]
- 89. Kerkvliet N.I. (2012) TCDD: an environmental immunotoxicant reveals a novel pathway of immunoregulation—a 30-year odyssey. Toxicol. Pathol., 40, 138–142. [DOI] [PubMed] [Google Scholar]
- 90. Setoguchi K., et al. (2001) Peroxisome proliferator-activated receptor-gamma haploinsufficiency enhances B cell proliferative responses and exacerbates experimentally induced arthritis. J. Clin. Invest., 108, 1667–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chinetti G., et al. (1998) Activation of proliferator-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J. Biol. Chem., 273, 25573–25580. [DOI] [PubMed] [Google Scholar]
- 92. Gosset P., et al. (2001) Peroxisome proliferator-activated receptor gamma activators affect the maturation of human monocyte-derived dendritic cells. Eur. J. Immunol., 31, 2857–2865. [DOI] [PubMed] [Google Scholar]
- 93. Jones D.C., et al. (2002) Nuclear receptor peroxisome proliferator-activated receptor alpha (PPARalpha) is expressed in resting murine lymphocytes. The PPARalpha in T and B lymphocytes is both transactivation and transrepression competent. J. Biol. Chem., 277, 6838–6845. [DOI] [PubMed] [Google Scholar]
- 94. Raikwar H.P., et al. (2005) PPARgamma antagonists exacerbate neural antigen-specific Th1 response and experimental allergic encephalomyelitis. J. Neuroimmunol., 167, 99–107. [DOI] [PubMed] [Google Scholar]
- 95. Sun C., et al. (2006) Single laboratory validation of a method for the determination of bisphenol A, bisphenol A diglycidyl ether and its derivatives in canned foods by reversed-phase liquid chromatography. J. Chromatogr. A, 1129, 145–148. [DOI] [PubMed] [Google Scholar]
- 96. Riu A., et al. (2011) Characterization of novel ligands of ERα, Erβ, and PPARγ: the case of halogenated bisphenol A and their conjugated metabolites. Toxicol. Sci., 122, 372–382. [DOI] [PubMed] [Google Scholar]
- 97. Trudel D., et al. (2011) Total consumer exposure to polybrominated diphenyl ethers in North America and Europe. Environ. Sci. Technol., 45, 2391–2397. [DOI] [PubMed] [Google Scholar]
- 98. Schuhmacher M., et al. (2013) Levels of PCDD/Fs, PCBs and PBDEs in breast milk of women living in the vicinity of a hazardous waste incinerator: assessment of the temporal trend. Chemosphere, 93, 1533–1540. [DOI] [PubMed] [Google Scholar]
- 99. Frederiksen M., et al. (2009) Patterns and concentration levels of polybrominated diphenyl ethers (PBDEs) in placental tissue of women in Denmark. Chemosphere, 76, 1464–1469. [DOI] [PubMed] [Google Scholar]
- 100. Frederiksen M., et al. (2010) Placental transfer of the polybrominated diphenyl ethers BDE-47, BDE-99 and BDE-209 in a human placenta perfusion system: an experimental study. Environ. Health, 9, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Leung A.O., et al. (2010) Body burdens of polybrominated diphenyl ethers in childbearing-aged women at an intensive electronic-waste recycling site in China. Environ. Sci. Pollut. Res. Int., 17, 1300–1313. [DOI] [PubMed] [Google Scholar]
- 102. Rider C.V., et al. (2010) Cumulative effects of in utero administration of mixtures of reproductive toxicants that disrupt common target tissues via diverse mechanisms of toxicity. Int. J. Androl., 33, 443–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Skinner M.K., et al. (2007) Epigenetic transgenerational actions of vinclozolin on the development of disease and cancer. Crit. Rev. Oncog., 13, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Anway M.D., et al. (2008) Transgenerational effects of the endocrine disruptor vinclozolin on the prostate transcriptome and adult onset disease. Prostate, 68, 517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Careghini A., et al. (2014) Bisphenol A, nonylphenols, benzophenones, and benzotriazoles in soils, groundwater, surface water, sediments, and food: a review. Environ. Sci. Pollut. Res., 22, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pasquini L., et al. (2014) Occurrence of eight household micropollutants in urban wastewater and their fate in a wastewater treatment plant. Statistical evaluation. Sci. Total Environ., 481, 459–468. [DOI] [PubMed] [Google Scholar]
- 107. Zhang H.Y., et al. (2014) Perinatal exposure to 4-nonylphenol affects adipogenesis in first and second generation rats offspring. Toxicol. Lett., 225, 325–332. [DOI] [PubMed] [Google Scholar]
- 108. Zhou H.R., et al. (2003) Rapid, sequential activation of mitogen-activated protein kinases and transcription factors precedes proinflammatory cytokine mRNA expression in spleens of mice exposed to the trichothecene vomitoxin. Toxicol. Sci., 72, 130–142. [DOI] [PubMed] [Google Scholar]
- 109. Shin S.G., et al. (2005) Suppression of inducible nitric oxide synthase and cyclooxygenase-2 expression in RAW 264.7 macrophages by sesquiterpene lactones. J. Toxicol. Environ. Health. A, 68, 2119–2131. [DOI] [PubMed] [Google Scholar]
- 110. Hayes T.B., et al. (2010) Atrazine induces complete feminization and chemical castration in male African clawed frogs (Xenopus laevis). Proc. Natl. Acad. Sci. U. S. A., 107, 4612–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Jin Y., et al. (2014) Exposure of mice to atrazine and its metabolite diaminochlorotriazine elicits oxidative stress and endocrine disruption. Environ. Toxicol. Pharmacol., 37, 782–790. [DOI] [PubMed] [Google Scholar]
- 112. Cooper R.L., et al. (2007) Atrazine and reproductive function: mode and mechanism of action studies. Birth Defects Res. B. Dev. Reprod. Toxicol., 80, 98–112. [DOI] [PubMed] [Google Scholar]
- 113. Stanko J.P., et al. (2010) Effects of prenatal exposure to a low dose atrazine metabolite mixture on pubertal timing and prostate development of male Long-Evans rats. Reprod. Toxicol., 30, 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Casa-Resino I, et al. (2014) Chlorotriazines do not activate the aryl hydrocarbon receptor, the oestrogen receptor or the thyroid receptor in in vitro assays. Altern. Lab. Anim., 42, 25–30. [DOI] [PubMed] [Google Scholar]
- 115. Danzo B.J. (1997) Environmental xenobiotics may disrupt normal endocrine function by interfering with the binding of physiological ligands to steroid receptors and binding proteins. Environ. Health Perspect., 105, 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ross M.K., et al. (2006) Determination of atrazine and its metabolites in mouse urine and plasma by LC-MS analysis. Anal. Biochem., 351, 161–173. [DOI] [PubMed] [Google Scholar]
- 117. Barr D.B., et al. (2007) Assessing exposure to atrazine and its metabolites using biomonitoring. Environ. Health Perspect., 115, 1474–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Abarikwu S.O., et al. (2013) Quercetin decreases steroidogenic enzyme activity, NF-κB expression, and oxidative stress in cultured Leydig cells exposed to atrazine. Mol. Cell. Biochem., 373, 19–28. [DOI] [PubMed] [Google Scholar]
- 119. Chen J.Y., et al. (2013) Immunotoxicity of atrazine in Balb/c mice. J. Environ. Sci. Health. B., 48, 637–645. [DOI] [PubMed] [Google Scholar]
- 120. Choudhari S.K., et al. (2013) Nitric oxide and cancer: a review. World J. Surg. Oncol., 11, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Boffetta P., et al. (2013) Atrazine and cancer: a review of the epidemiologic evidence. Eur. J. Cancer Prev., 22, 169–180. [DOI] [PubMed] [Google Scholar]
- 122. Basini G., et al. (2012) Atrazine disrupts steroidogenesis, VEGF and NO production in swine granulosa cells. Ecotoxicol. Environ. Saf., 85, 59–63. [DOI] [PubMed] [Google Scholar]
- 123. Angst E., et al. (2013) The flavonoid quercetin inhibits pancreatic cancer growth in vitro and in vivo . Pancreas, 42, 223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Prabhu V., et al. (2010) Nitric oxide: pros and cons in tumor progression. Immunopharmacol. Immunotoxicol., 32, 387–392. [DOI] [PubMed] [Google Scholar]
- 125. Hooghe R.J., et al. (2000) Effects of selected herbicides on cytokine production in vitro . Life Sci., 66, 2519–2525. [DOI] [PubMed] [Google Scholar]
- 126. Tsai M.J., et al. (2012) The association between phthalate exposure and asthma. Kaohsiung J. Med. Sci., 28(7 suppl), S28–S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sokol W.N., et al. (1973) Meat-wrapper’s asthma. A new syndrome? JAMA, 226, 639–641. [PubMed] [Google Scholar]
- 128. Larsen S.T., et al. (2007) Airway inflammation and adjuvant effect after repeated airborne exposures to di-(2-ethylhexyl)phthalate and ovalbumin in BALB/c mice. Toxicology, 235, 119–129. [DOI] [PubMed] [Google Scholar]
- 129. Larsen S.T., et al. (2004) Effects of mono-2-ethylhexyl phthalate on the respiratory tract in BALB/c mice. Hum. Exp. Toxicol., 23, 537–545. [DOI] [PubMed] [Google Scholar]
- 130. Deutschle T., et al. (2008) A controlled challenge study on di(2-ethylhexyl) phthalate (DEHP) in house dust and the immune response in human nasal mucosa of allergic subjects. Environ. Health Perspect., 116, 1487–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Singh S., et al. (2012) Bisphenol A and phthalates exhibit similar toxicogenomics and health effects. Gene, 494, 85–91. [DOI] [PubMed] [Google Scholar]
- 132. Routledge E.J., et al. (2000) Differential effects of xenoestrogens on coactivator recruitment by estrogen receptor (ER) alpha and ERbeta. J. Biol. Chem., 275, 35986–35993. [DOI] [PubMed] [Google Scholar]
- 133. Hong E.J., et al. (2013) Loss of estrogen-related receptor α promotes hepatocarcinogenesis development via metabolic and inflammatory disturbances. Proc. Natl. Acad. Sci. U. S. A., 110, 17975–17980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Manente A.G., et al. (2013) Estrogen receptor β activation impairs mitochondrial oxidative metabolism and affects malignant mesothelioma cell growth in vitro and in vivo . Oncogenesis, 2, e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Konduri S.D., et al. (2010) Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc. Natl. Acad. Sci. U. S. A., 107, 15081–15086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Rasti M., et al. (2012) p53 Binds to estrogen receptor 1 promoter in human breast cancer cells. Pathol. Oncol. Res., 18, 169–175. [DOI] [PubMed] [Google Scholar]
- 137. Hartman J., et al. (2006) Estrogen receptor beta inhibits angiogenesis and growth of T47D breast cancer xenografts. Cancer Res., 66, 11207–11213. [DOI] [PubMed] [Google Scholar]
- 138. Williamson L.M., et al. (2011) Estrogen receptor α-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis, 32, 279–285. [DOI] [PubMed] [Google Scholar]
- 139. Bailey S.T., et al. (2012) Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc. Natl. Acad. Sci. U. S. A., 109, 18060–18065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lewis-Wambi J.S., et al. (2009) Estrogen regulation of apoptosis: how can one hormone stimulate and inhibit? Breast Cancer Res., 11, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Scull C.M., et al. (2011) Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arterioscler. Thromb. Vasc. Biol., 31, 2792–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Tuttle R., et al. (2012) Novel senescence associated gene, YPEL3, is repressed by estrogen in ER+ mammary tumor cells and required for tamoxifen-induced cellular senescence. Int. J. Cancer, 130, 2291–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Kyo S., et al. (2008) Understanding and exploiting hTERT promoter regulation for diagnosis and treatment of human cancers. Cancer Sci., 99, 1528–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Heldring N., et al. (2007) Estrogen receptors: how do they signal and what are their targets. Physiol. Rev., 87, 905–931. [DOI] [PubMed] [Google Scholar]
- 145. Goulioumis A.K., et al. (2009) Estrogen receptor-beta expression in human laryngeal carcinoma: correlation with the expression of epithelial-mesenchymal transition specific biomarkers. Oncol. Rep., 22, 1063–1068. [DOI] [PubMed] [Google Scholar]
- 146. Wik E., et al. (2013) Lack of estrogen receptor-α is associated with epithelial-mesenchymal transition and PI3K alterations in endometrial carcinoma. Clin. Cancer Res., 19, 1094–1105. [DOI] [PubMed] [Google Scholar]
- 147. Dhasarathy A., et al. (2007) The transcription factor snail mediates epithelial to mesenchymal transitions by repression of estrogen receptor-alpha. Mol. Endocrinol., 21, 2907–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Giretti M.S., et al. (2008) Extra-nuclear signalling of estrogen receptor to breast cancer cytoskeletal remodelling, migration and invasion. PLoS One, 3, e2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Park S.H., et al. (2008) Estrogen regulates Snail and Slug in the down-regulation of E-cadherin and induces metastatic potential of ovarian cancer cells through estrogen receptor alpha. Mol. Endocrinol., 22, 2085–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Grubisha M.J., et al. (2013) Local endocrine, paracrine and redox signaling networks impact estrogen and androgen crosstalk in the prostate cancer microenvironment. Steroids, 78, 538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ambs S., et al. (1998) Up-regulation of inducible nitric oxide synthase expression in cancer-prone p53 knockout mice. Proc. Natl. Acad. Sci. U. S. A., 95, 8823–8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Goodman J.E., et al. (2004) Nitric oxide and p53 in cancer-prone chronic inflammation and oxyradical overload disease. Environ. Mol. Mutagen., 44, 3–9. [DOI] [PubMed] [Google Scholar]
- 153. Forrester K., et al. (1996) Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc. Natl. Acad. Sci. U. S. A., 93, 2442–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Ying L., et al. (2007) Nitric oxide inactivates the retinoblastoma pathway in chronic inflammation. Cancer Res., 67, 9286–9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Vakkala M., et al. (2000) Inducible nitric oxide synthase expression, apoptosis, and angiogenesis in in situ and invasive breast carcinomas. Clin. Cancer Res., 6, 2408–2416. [PubMed] [Google Scholar]
- 156. Liu R.H., et al. (1995) Potential genotoxicity of chronically elevated nitric oxide: a review. Mutat. Res., 339, 73–89. [DOI] [PubMed] [Google Scholar]
- 157. Lechner M., et al. (2005) Inducible nitric oxide synthase (iNOS) in tumor biology: the two sides of the same coin. Semin. Cancer Biol., 15, 277–289. [DOI] [PubMed] [Google Scholar]
- 158. Dong J., et al. (2014) Function of inducible nitric oxide synthase in the regulation of cervical cancer cell proliferation and the expression of vascular endothelial growth factor. Mol. Med. Rep., 9, 583–589. [DOI] [PubMed] [Google Scholar]
- 159. Sun M.H., et al. (2005) Expressions of inducible nitric oxide synthase and matrix metalloproteinase-9 and their effects on angiogenesis and progression of hepatocellular carcinoma. World J. Gastroenterol., 11, 5931–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Song Z.J., et al. (2002) Relationship between the expression of iNOS,VEGF,tumor angiogenesis and gastric cancer. World J. Gastroenterol., 8, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Singh S., et al. (2011) Nitric oxide: role in tumour biology and iNOS/NO-based anticancer therapies. Cancer Chemother. Pharmacol., 67, 1211–1224. [DOI] [PubMed] [Google Scholar]
- 162. Brennan P.A., et al. (2008) Inducible nitric oxide synthase: correlation with extracapsular spread and enhancement of tumor cell invasion in head and neck squamous cell carcinoma. Head Neck, 30, 208–214. [DOI] [PubMed] [Google Scholar]
- 163. Wu X.Q., et al. (2013) Feedback regulation of telomerase reverse transcriptase: new insight into the evolving field of telomerase in cancer. Cell. Signal., 25, 2462–2468. [DOI] [PubMed] [Google Scholar]
- 164. Mimeault M., et al. (2014) Altered gene products involved in the malignant reprogramming of cancer stem/progenitor cells and multitargeted therapies. Mol. Aspects Med., 39, 3–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Tergaonkar V., et al. (2002) p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell, 1, 493–503. [DOI] [PubMed] [Google Scholar]
- 166. Van Antwerp D.J., et al. (1996) Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science, 274, 787–789. [DOI] [PubMed] [Google Scholar]
- 167. Kahn P.J., et al. (2013) Higher-dose Anakinra is effective in a case of medically refractory macrophage activation syndrome. J. Rheumatol., 40, 743–744. [DOI] [PubMed] [Google Scholar]
- 168. Mantovani A. (2010) Molecular pathways linking inflammation and cancer. Curr. Mol. Med., 10, 369–373. [DOI] [PubMed] [Google Scholar]
- 169. Kim J., et al. (2014) STAT3 regulation by S-nitrosylation: implication for inflammatory disease. Antioxid. Redox Signal., 20, 2514–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Urashima M., et al. (1996) Interleukin-6 promotes multiple myeloma cell growth via phosphorylation of retinoblastoma protein. Blood, 88, 2219–2227. [PubMed] [Google Scholar]
- 171. Urashima M., et al. (1997) Role of CDK4 and p16INK4A in interleukin-6-mediated growth of multiple myeloma. Leukemia, 11, 1957–1963. [DOI] [PubMed] [Google Scholar]
- 172. Steiner H., et al. (2003) Accelerated in vivo growth of prostate tumors that up-regulate interleukin-6 is associated with reduced retinoblastoma protein expression and activation of the mitogen-activated protein kinase pathway. Am. J. Pathol., 162, 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Lichtenstein A., et al. (1995) Interleukin-6 inhibits apoptosis of malignant plasma cells. Cell. Immunol., 162, 248–255. [DOI] [PubMed] [Google Scholar]
- 174. Biffl W.L., et al. (1995) Interleukin-6 suppression of neutrophil apoptosis is neutrophil concentration dependent. J. Leukoc. Biol., 58, 582–584. [DOI] [PubMed] [Google Scholar]
- 175. Biffl W.L., et al. (1996) Interleukin-6 delays neutrophil apoptosis. Arch. Surg., 131, 24–9; discussion 29. [DOI] [PubMed] [Google Scholar]
- 176. Xu F.H., et al. (1998) Interleukin-6-induced inhibition of multiple myeloma cell apoptosis: support for the hypothesis that protection is mediated via inhibition of the JNK/SAPK pathway. Blood, 92, 241–251. [PubMed] [Google Scholar]
- 177. Yamagiwa Y., et al. (2006) Interleukin-6 decreases senescence and increases telomerase activity in malignant human cholangiocytes. Life Sci., 78, 2494–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Chua A.C., et al. (2013) Dietary iron enhances colonic inflammation and IL-6/IL-11-Stat3 signaling promoting colonic tumor development in mice. PLoS One, 8, e78850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Touboul C., et al. (2013) Mesenchymal stem cells enhance ovarian cancer cell infiltration through IL6 secretion in an amniochorionic membrane based 3D model. J. Transl. Med., 11, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Yadav A., et al. (2011) IL-6 promotes head and neck tumor metastasis by inducing epithelia–mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res., 9, 1658–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Na Y.R., et al. (2013) Interleukin-6-induced Twist and N-cadherin enhance melanoma cell metastasis. Melanoma Res., 23, 434–443. [DOI] [PubMed] [Google Scholar]
- 182. Fosslien E. (2008) Cancer morphogenesis: role of mitochondrial failure. Ann. Clin. Lab. Sci., 38, 307–329. [PubMed] [Google Scholar]
- 183. Tang H.Y., et al. (2006) Resveratrol-induced cyclooxygenase-2 facilitates p53-dependent apoptosis in human breast cancer cells. Mol. Cancer Ther., 5, 2034–2042. [DOI] [PubMed] [Google Scholar]
- 184. Subbaramaiah K., et al. (1999) Inhibition of cyclooxygenase-2 gene expression by p53. J. Biol. Chem., 274, 10911–10915. [DOI] [PubMed] [Google Scholar]
- 185. Corcoran C.A., et al. (2005) Cyclooxygenase-2 interacts with p53 and interferes with p53-dependent transcription and apoptosis. Oncogene, 24, 1634–1640. [DOI] [PubMed] [Google Scholar]
- 186. Yao L., et al. (2011) The function and mechanism of COX-2 in angiogenesis of gastric cancer cells. J. Exp. Clin. Cancer Res., 30, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Singh B., et al. (2005) COX-2 overexpression increases motility and invasion of breast cancer cells. Int. J. Oncol., 26, 1393–1399. [PubMed] [Google Scholar]
- 188. Kern M.A., et al. (2006) Cyclooxygenase-2 inhibition induces apoptosis signaling via death receptors and mitochondria in hepatocellular carcinoma. Cancer Res., 66, 7059–7066. [DOI] [PubMed] [Google Scholar]
- 189. Nzeako U.C., et al. (2002) COX-2 inhibits Fas-mediated apoptosis in cholangiocarcinoma cells. Hepatology, 35, 552–559. [DOI] [PubMed] [Google Scholar]
- 190. Liu L., et al. (2011) Activation of telomerase by seminal plasma in malignant and normal cervical epithelial cells. J. Pathol., 225, 203–211. [DOI] [PubMed] [Google Scholar]
- 191. Li W., et al. (2014) Cyclooxygenase 2 promoted the tumorigenecity of pancreatic cancer cells. Tumour Biol., 35, 2271–2278. [DOI] [PubMed] [Google Scholar]
- 192. Xiang X., et al. (2009) Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer, 124, 2621–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Straus D.S. (2013) TNFα and IL-17 cooperatively stimulate glucose metabolism and growth factor production in human colorectal cancer cells. Mol. Cancer, 12, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Chau B.N., et al. (2004) Tumor necrosis factor alpha-induced apoptosis requires p73 and c-ABL activation downstream of RB degradation. Mol. Cell. Biol., 24, 4438–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Han J., et al. (2013) Nuclear expression of β-catenin promotes RB stability and resistance to TNF-induced apoptosis in colon cancer cells. Mol. Cancer Res., 11, 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Montrucchio G., et al. (1994) Tumor necrosis factor alpha-induced angiogenesis depends on in situ platelet-activating factor biosynthesis. J. Exp. Med., 180, 377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. van den Berg J.M., et al. (2001) Divergent effects of tumor necrosis factor alpha on apoptosis of human neutrophils. J. Leukoc. Biol., 69, 467–473. [PubMed] [Google Scholar]
- 198. Polunovsky V.A., et al. (1994) Induction of endothelial cell apoptosis by TNF alpha: modulation by inhibitors of protein synthesis. Exp. Cell Res., 214, 584–594. [DOI] [PubMed] [Google Scholar]
- 199. Rolfe M., et al. (1997) Tumour necrosis factor alpha (TNF alpha) suppresses apoptosis and induces DNA synthesis in rodent hepatocytes: a mediator of the hepatocarcinogenicity of peroxisome proliferators? Carcinogenesis, 18, 2277–2280. [DOI] [PubMed] [Google Scholar]
- 200. Nair S., et al. (2013) Obesity and the endometrium: adipocyte-secreted proinflammatory TNF α cytokine enhances the proliferation of human endometrial glandular cells. Obstet. Gynecol. Int., 2013, 368543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Bates R.C., et al. (2003) Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol. Biol. Cell, 14, 1790–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Wang H., et al. (2013) Epithelial-mesenchymal transition (EMT) induced by TNF-α requires AKT/GSK-3β-mediated stabilization of snail in colorectal cancer. PLoS One, 8, e56664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Dong R., et al. (2007) Role of nuclear factor kappa B and reactive oxygen species in the tumor necrosis factor-alpha-induced epithelial–mesenchymal transition of MCF-7 cells. Braz. J. Med. Biol. Res., 40, 1071–1078. [DOI] [PubMed] [Google Scholar]
- 204. Jiang Y, et al. (2013) Prenatal exposure to bisphenol A at the reference dose impairs mitochondria in the heart of neonatal rats. J. Appl. Toxicol., 34, 1012–1022. [DOI] [PubMed] [Google Scholar]
- 205. Lee H.S., et al. (2012) Set, a putative oncogene, as a biomarker for prenatal exposure to bisphenol A. Asian Pac. J. Cancer Prev., 13, 2711–2715. [DOI] [PubMed] [Google Scholar]
- 206. Betancourt A., et al. (2014) Alterations in the rat serum proteome induced by prepubertal exposure to bisphenol a and genistein. J. Proteome Res., 13, 1502–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Fillon M. (2012) Getting it right: BPA and the difficulty proving environmental cancer risks. J. Natl. Cancer Inst., 104, 652–655. [DOI] [PubMed] [Google Scholar]
- 208. Nash J.T., et al. (2013) Polybrominated diphenyl ethers alter hepatic phosphoenolpyruvate carboxykinase enzyme kinetics in male Wistar rats: implications for lipid and glucose metabolism. J. Toxicol. Environ. Health. A, 76, 142–156. [DOI] [PubMed] [Google Scholar]
- 209. Kandori H., et al. (2005) Influence of atrazine administration and reduction of calorie intake on prostate carcinogenesis in probasin/SV40 T antigen transgenic rats. Cancer Sci., 96, 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Lim S., et al. (2009) Chronic exposure to the herbicide, atrazine, causes mitochondrial dysfunction and insulin resistance. PLoS One, 4, e5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Dairkee S.H., et al. (2013) Bisphenol-A-induced inactivation of the p53 axis underlying deregulation of proliferation kinetics, and cell death in non-malignant human breast epithelial cells. Carcinogenesis, 34, 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Lee H.R., et al. (2014) The estrogen receptor signaling pathway activated by phthalates is linked with transforming growth factor-β in the progression of LNCaP prostate cancer models. Int. J. Oncol., 45, 595–602. [DOI] [PubMed] [Google Scholar]
- 213. Durando M., et al. (2011) Prenatal exposure to bisphenol A promotes angiogenesis and alters steroid-mediated responses in the mammary glands of cycling rats. J. Steroid Biochem. Mol. Biol., 127, 35–43. [DOI] [PubMed] [Google Scholar]
- 214. Valles E.G., et al. (2003) Role of the peroxisome proliferator-activated receptor alpha in responses to diisononyl phthalate. Toxicology, 191, 211–225. [DOI] [PubMed] [Google Scholar]
- 215. Basini G., et al. (2012) Atrazine disrupts steroidogenesis, VEGF and NO production in swine granulosa cells. Ecotoxicol. Environ. Saf., 85, 59–63. [DOI] [PubMed] [Google Scholar]
- 216. Allard P., et al. (2010) Bisphenol A impairs the double-strand break repair machinery in the germline and causes chromosome abnormalities. Proc. Natl. Acad. Sci. U. S. A., 107, 20405–20410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Radice S., et al. (1998) Adaptation to oxidative stress: effects of vinclozolin and iprodione on the HepG2 cell line. Toxicology, 129, 183–191. [DOI] [PubMed] [Google Scholar]
- 218. Helleday T., et al. (1999) Brominated flame retardants induce intragenic recombination in mammalian cells. Mutat. Res., 439, 137–147. [DOI] [PubMed] [Google Scholar]
- 219. Lü B, et al. (2010) Effects of nonylphenol on ovarian cancer cells PEO4 c-myc and p53 mRNA and protein expression. Toxicol. Environ. Chem., 92, 1303–1308. [Google Scholar]
- 220. Pinchuk L.M., et al. (2007) In vitro atrazine exposure affects the phenotypic and functional maturation of dendritic cells. Toxicol. Appl. Pharmacol., 223, 206–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Terasaka H., et al. (2005) Cytotoxicity and apoptosis-inducing activity of bisphenol A and hydroquinone in HL-60 cells. Anticancer Res., 25(3B), 2241–2247. [PubMed] [Google Scholar]
- 222. Xu J., et al. (2002) Bisphenol A induces apoptosis and G2-to-M arrest of ovarian granulosa cells. Biochem. Biophys. Res. Commun., 292, 456–462. [DOI] [PubMed] [Google Scholar]
- 223. Gregoraszczuk E.L., et al. (2012) Polybrominated diphenylethers (PBDEs) act as apoptotic factors in the corpus luteum in addition to having a short-term stimulatory effect on progesterone secretion by luteal cells. Toxicol. Mech. Methods, 22, 131–138. [DOI] [PubMed] [Google Scholar]
- 224. Kwiecińska P., et al. (2011) Combinatory effects of PBDEs and 17β-estradiol on MCF-7 cell proliferation and apoptosis. Pharmacol. Rep., 63, 189–194. [DOI] [PubMed] [Google Scholar]
- 225. Sabbieti M.G., et al. (2011) 4-nonylphenol triggers apoptosis and affects 17-β-estradiol receptors in calvarial osteoblasts. Toxicology, 290, 334–341. [DOI] [PubMed] [Google Scholar]
- 226. Wang S.W., et al. (2013) Androgen receptor-mediated apoptosis in bovine testicular induced pluripotent stem cells in response to phthalate esters. Cell Death Dis., 4, e907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Aoki M., et al. (2004) Nonylphenol enhances apoptosis induced by serum deprivation in PC12 cells. Life Sci., 74, 2301–2312. [DOI] [PubMed] [Google Scholar]
- 228. Choi M.S., et al. (2014) Nonylphenol-induced apoptotic cell death in mouse TM4 Sertoli cells via the generation of reactive oxygen species and activation of the ERK signaling pathway. J. Appl. Toxicol., 34, 628–636. [DOI] [PubMed] [Google Scholar]
- 229. Riou J.F., et al. (2002) Cell senescence and telomere shortening induced by a new series of specific G-quadruplex DNA ligands. Proc. Natl. Acad. Sci. U. S. A., 99, 2672–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Pupo M., et al. (2012) Bisphenol A induces gene expression changes and proliferative effects through GPER in breast cancer cells and cancer-associated fibroblasts. Environ. Health Perspect., 120, 1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Hsieh T.H., et al. (2012) Phthalates induce proliferation and invasiveness of estrogen receptor-negative breast cancer through the AhR/HDAC6/c-Myc signaling pathway. FASEB J., 26, 778–787. [DOI] [PubMed] [Google Scholar]
- 232. Park M.A., et al. (2012) Cell growth of BG-1 ovarian cancer cells is promoted by di-n-butyl phthalate and hexabromocyclododecane via upregulation of the cyclin D and cyclin-dependent kinase-4 genes. Mol. Med. Rep., 5, 761–766. [DOI] [PubMed] [Google Scholar]
- 233. Albanito L., et al. (2008) G-protein-coupled receptor 30 and estrogen receptor-alpha are involved in the proliferative effects induced by atrazine in ovarian cancer cells. Environ. Health Perspect., 116, 1648–1655. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 234. Li Z.H., et al. (2012) Effects of decabrominated diphenyl ether (PBDE-209) in regulation of growth and apoptosis of breast, ovarian, and cervical cancer cells. Environ. Health Perspect., 120, 541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Zhu H., et al. (2010) Environmental endocrine disruptors promote invasion and metastasis of SK-N-SH human neuroblastoma cells. Oncol. Rep., 23, 129–139. [PubMed] [Google Scholar]
- 236. Jenkins S., et al. (2011) Chronic oral exposure to bisphenol A results in a nonmonotonic dose response in mammary carcinogenesis and metastasis in MMTV-erbB2 mice. Environ. Health Perspect., 119, 1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Derouiche S., et al. (2013) Bisphenol A stimulates human prostate cancer cell migration via remodelling of calcium signalling. Springerplus, 2, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Hsieh T.H., et al. (2012) n-Butyl benzyl phthalate promotes breast cancer progression by inducing expression of lymphoid enhancer factor 1. PLoS One, 7, e42750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Vandenberg L.N., et al. (2009) Bisphenol-A and the great divide: a review of controversies in the field of endocrine disruption. Endocr. Rev., 30, 75–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Goodson W.H., III, et al. (2011) Activation of the mTOR pathway by low levels of xenoestrogens in breast epithelial cells from high-risk women. Carcinogenesis, 32, 1724–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Takahashi A., et al. (2004) Bisphenol A from dental polycarbonate crown upregulates the expression of hTERT. J. Biomed. Mater. Res. B. Appl. Biomater., 71, 214–221. [DOI] [PubMed] [Google Scholar]
- 242. Stossi F., et al. (2014) Defining estrogenic mechanisms of bisphenol A analogs through high throughput microscopy-based contextual assays. Chem. Biol., 21, 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Wu Y., et al. (2014) Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett., 345, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Uzumcu M., et al. (2004) Effect of the anti-androgenic endocrine disruptor vinclozolin on embryonic testis cord formation and postnatal testis development and function. Reprod. Toxicol., 18, 765–774. [DOI] [PubMed] [Google Scholar]
- 245. Frassinetti S., et al. (2011) Genotoxicity of 4-nonylphenol and nonylphenol ethoxylate mixtures by the use of Saccharomyces cerevisiae D7 mutation assay and use of this text to evaluate the efficiency of biodegradation treatments. Ecotoxicol. Environ. Saf., 74, 253–258. [DOI] [PubMed] [Google Scholar]
- 246. Hung C.H., et al. (2013) Environmental alkylphenols modulate cytokine expression in plasmacytoid dendritic cells. PLoS One, 8, e73534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Wang S.W., et al. (2014) The IL-6/JAK/STAT3 pathway: potential therapeutic strategies in treating colorectal cancer (Review). Int. J. Oncol., 44, 1032–1040. [DOI] [PubMed] [Google Scholar]
- 248. Akiyama M., et al. (2002) Cytokines modulate telomerase activity in a human multiple myeloma cell line. Cancer Res., 62, 3876–3882. [PubMed] [Google Scholar]
- 249. Catalano M.G., et al. (2012) Histone deacetylase inhibition modulates E-cadherin expression and suppresses migration and invasion of anaplastic thyroid cancer cells. J. Clin. Endocrinol. Metab., 97, E1150–E1159. [DOI] [PubMed] [Google Scholar]
- 250. Rusyn I., et al. (2006) Modes of action and species-specific effects of di-(2-ethylhexyl)phthalate in the liver. Crit. Rev. Toxicol., 36, 459–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Sabbieti M.G., et al. (2009) Involvement of p53 in phthalate effects on mouse and rat osteoblasts. J. Cell. Biochem., 107, 316–327. [DOI] [PubMed] [Google Scholar]
- 252. Erkekoğlu P., et al. (2011) Induction of ROS, p53, p21 in DEHP- and MEHP-exposed LNCaP cells-protection by selenium compounds. Food Chem. Toxicol., 49, 1565–1571. [DOI] [PubMed] [Google Scholar]
- 253. Kleinsasser N.H., et al. (2000) Phthalates demonstrate genotoxicity on human mucosa of the upper aerodigestive tract. Environ. Mol. Mutagen., 35, 9–12. [DOI] [PubMed] [Google Scholar]
- 254. Kim I.Y., et al. (2004) Phthalates inhibit tamoxifen-induced apoptosis in MCF-7 human breast cancer cells. J. Toxicol. Environ. Health. A, 67, 2025–2035. [DOI] [PubMed] [Google Scholar]
- 255. Singh B., et al. (2007) Cyclooxygenase-2 expression induces genomic instability in MCF10A breast epithelial cells. J. Surg. Res., 140, 220–226. [DOI] [PubMed] [Google Scholar]
- 256. Cavas T. (2011) In vivo genotoxicity evaluation of atrazine and atrazine-based herbicide on fish Carassius auratus using the micronucleus test and the comet assay. Food Chem. Toxicol., 49, 1431–1435. [DOI] [PubMed] [Google Scholar]
- 257. Gomez D., et al. (2003) Resistance to senescence induction and telomere shortening by a G-quadruplex ligand inhibitor of telomerase. Cancer Res., 63, 6149–6153. [PubMed] [Google Scholar]
- 258. Gomez D., et al. (2004) Telomerase downregulation induced by the G-quadruplex ligand 12459 in A549 cells is mediated by hTERT RNA alternative splicing. Nucleic Acids Res., 32, 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Pastor D.M., et al. (2010) Tumor necrosis factor alpha induces p53 up-regulated modulator of apoptosis expression in colorectal cancer cell lines. Dis. Colon Rectum, 53, 257–263. [DOI] [PubMed] [Google Scholar]
- 260. Zhang Z., et al. (2013) Interleukin-17A- or tumor necrosis factor α-mediated increase in proliferation of T cells cocultured with synovium-derived mesenchymal stem cells in rheumatoid arthritis. Arthritis Res. Ther., 15, R169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Laws S.C., et al. (2009) Chlorotriazine herbicides and metabolites activate an ACTH-dependent release of corticosterone in male Wistar rats. Toxicol. Sci., 112, 78–87. [DOI] [PubMed] [Google Scholar]
- 262. Calandra T., et al. (1995) MIF as a glucocorticoid-induced modulator of cytokine production. Nature, 377, 68–71. [DOI] [PubMed] [Google Scholar]
- 263. Bucala R. (1996) MIF rediscovered: cytokine, pituitary hormone, and glucocorticoid-induced regulator of the immune response. FASEB J., 10, 1607–1613. [DOI] [PubMed] [Google Scholar]
- 264. Lee E., et al. (2010) Evaluation of liver and thyroid toxicity in Sprague-Dawley rats after exposure to polybrominated diphenyl ether BDE-209. J. Toxicol. Sci., 35, 535–545. [DOI] [PubMed] [Google Scholar]
- 265. Martin P.A., et al. (2007) Immunotoxicity of the commercial polybrominated diphenyl ether mixture DE-71 in ranch mink (Mustela vison). Environ. Toxicol. Chem., 26, 988–997. [DOI] [PubMed] [Google Scholar]
- 266. Chen F., et al. (2014) Sex differences in the adult HPA axis and affective behaviors are altered by perinatal exposure to a low dose of bisphenol A. Brain Res., 1571, 12–24. [DOI] [PubMed] [Google Scholar]
- 267. Panagiotidou E., et al. (2014) Perinatal exposure to low-dose bisphenol A affects the neuroendocrine stress response in rats. J. Endocrinol., 220, 207–218. [DOI] [PubMed] [Google Scholar]
- 268. Bifulco C., et al. (2008) Tumor growth-promoting properties of macrophage migration inhibitory factor. Curr. Pharm. Des., 14, 3790–3801. [DOI] [PubMed] [Google Scholar]
- 269. Bach J.P., et al. (2009) The role of macrophage inhibitory factor in tumorigenesis and central nervous system tumors. Cancer, 115, 2031–2040. [DOI] [PubMed] [Google Scholar]
- 270. Babu S.N., et al. (2012) Macrophage migration inhibitory factor: a potential marker for cancer diagnosis and therapy. Asian Pac. J. Cancer Prev., 13, 1737–1744. [DOI] [PubMed] [Google Scholar]
- 271. Conroy H., et al. (2010) Inflammation and cancer: macrophage migration inhibitory factor (MIF)—the potential missing link. QJM, 103, 831–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Simpson K.D., et al. (2012) Macrophage migration inhibitory factor promotes tumor growth and metastasis by inducing myeloid-derived suppressor cells in the tumor microenvironment. J. Immunol., 189, 5533–5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Schwabe R.F., et al. (2013) The microbiome and cancer. Nat. Rev. Cancer, 13, 800–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Elliott P., et al. (2008) The UK Biobank sample handling and storage protocol for the collection, processing and archiving of human blood and urine. Int. J. Epidemiol., 37, 234–244. [DOI] [PubMed] [Google Scholar]
- 275. Riboli E., et al. (2002) European Prospective Investigation into Cancer and Nutrition (EPIC): study populations and data collection. Public Health Nutr., 5, 1113–1124. [DOI] [PubMed] [Google Scholar]
- 276. Bisson W.H. (2012) Editorial: computational chemogenomics in drug design and discovery. Curr. Top. Med. Chem., 12, 1867–1868. [DOI] [PubMed] [Google Scholar]
- 277. Benninghoff A.D., et al. (2011) Estrogen-like activity of perfluoroalkyl acids in vivo and interaction with human and rainbow trout estrogen receptors in vitro . Toxicol. Sci., 120, 42–58. [DOI] [PMC free article] [PubMed] [Google Scholar]