

Abstract
Formylglycine (fGly) is a catalytically essential residue found almost exclusively in the active sites of type I sulfatases. Formed by post-translational oxidation of cysteine or serine side chains, this aldehyde-functionalized residue participates in a unique and highly efficient catalytic mechanism for sulfate ester hydrolysis. The enzymes that produce fGly, formylglycine-generating enzyme (FGE) and anaerobic sulfatase-maturating enzyme (anSME), are as unique and specialized as fGly itself. FGE especially is structurally and mechanistically distinct, and serves the sole function of activating type I sulfatase targets. This review summarizes the current state of knowledge regarding the mechanism by which fGly contributes to sulfate ester hydrolysis, the molecular details of fGly biogenesis by FGE and anSME, and finally, recent biotechnology applications of fGly beyond its natural catalytic function.
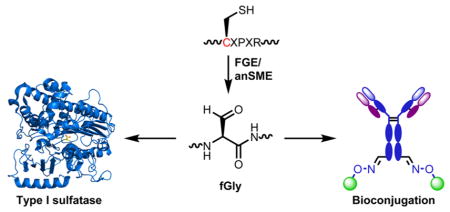
INTRODUCTION
Post-translational modification (PTM) of canonical amino acid side chains is a mechanism for augmenting the chemical diversity of enzymatic catalysis. Many cofactors involved in fundamental metabolic transformations derive from protein backbone or side chain modifications.1 The novel functionalities created—redox moieties, electrophiles, or metal chelators, for example—allow for catalytic mechanisms unattainable with canonical protein chemical groups. A relative newcomer to the family of PTM-derived catalytic cofactors is Cα-formylglycine (fGly, Figure 1A), a modification of Cys or Ser side chains that is essential for the activity of type I sulfatases.2 Unlike the complex pathways for generating many of the peptide-derived quinocofactors,3 the biosynthesis of fGly is accomplished by only one enzymatic step. Astoundingly, type I sulfatases achieve rate enhancements (kcat/ kuncat) of 1026 for alkylsulfate substrates, making them among the most efficient enzymes ever measured, and this can be attributed in part to the unique catalytic properties of fGly.4
Figure 1.

Hydration of fGly to fGly-diol, Pseudomonas aeruginosa arylsulfatase structure and active site architecture (PAS, PDB: 1HDH), and proposed catalytic mechanisms of type I sulfatase. (A) fGly is rapidly hydrated to a geminal diol, fGly-diol, and this form predominates in the sulfatase resting state. (B) fGly diol, yellow; Ca2+, green. (C) Highlighted features: Ca2+, green; metal binding, magenta; substrate binding and catalytic, gray. (D) Following the binding of sulfate ester substrate, fGly-diol is activated for nucleophilic attack at sulfur by aspartate (Asp317). The sulfoenzyme is formed, and desulfation may proceed by one of two pathways, hydrolysis by sulfur (SN2) or, more probable, elimination from the remaining fGly-diol hydroxyl (E2), catalyzed by a histidine base (His115). Catalytic mechanism scheme adapted with alteration from ref 28.
Ubiquitous across all domains of life, sulfatases catalyze the hydrolysis of a vast array of natural and synthetic aryl- and alkylsulfate ester substrates. Three divergent classes of sulfatases have been identified, but the type I family members are the most common and the only class found in eukaryotes. In aerobic organisms, type I sulfatases become active when the formylglycine-generating enzyme (FGE) (also referred to as sulfatase-modifying factor 1, or SUMF1)5,6 catalyzes the oxidation of cysteine to fGly. In humans, 17 sulfatases have been identified, of which 14 have been assigned specific activities in catabolism, signaling, and development.2,7 Human sulfatases are initially translated into the endoplasmic reticulum (ER); some are retained there, while others are targeted to the lysosome, the Golgi, or the cell surface.7 Lysosomal sulfatases act on sulfated glycolipids (sulfatides) and glycosaminoglycans, and their activities are necessary for proper degradation of these glycosides. ER-resident sulfatases, most notably steroid sulfatase (STS; arylsulfatase C, ASC), regulate hormone levels by desulfation of inactive precursors such as dehydroepiandroster-one 3-sulfate and iodothyronine sulfate.2 Secreted sulfatases (Sulf1, Sulf2) modulate the sulfation level of cell-surface heparan sufate, thereby regulating signaling events critical for development and tumor progression.8 The disruption of individual sulfatases causes at least eight pathologies in humans, including six lysosomal storage disorders (e.g., mucopolysaccaridoses, metachromatic leukodystrophy), the bone disease chondrodys-plasia punctate type 1, and skin disorder X-linked ichthyosis.9 Deficiency in FGE causes multiple sulfatase deficiency (MSD), a fatal disorder marked by decreased activity of all sulfatases.10 Microbial sulfatases were historically thought to be utilized for scavenging environmental sulfur, but a growing body of work over the past decade has revealed a much more elaborate role in modulating endosymbiont and host-pathogen interactions by remodeling host sulfation.11 Given the breadth of research on sulfatase biology, we defer to a number of reviews for a thorough appraisal of sulfatase biochemistry and physiology,2,12–14 the genetic basis of FGE and sulfatase disorders in humans,9,15,16 and the pursuit of novel sulfatases for bioengineering applications.17
This review will focus the catalytic function of fGly and mechanisms by which enzymes from various organisms are thought to produce this PTM. Finally, we discuss the use of fGly’s aldehyde functionality as a chemical handle for site-specific protein chemical modification, a biotechnology application of fGly that has undergone recent commercial translation.
FGLY IS AN ESSENTIAL POSTTRANSLATION MODIFICATION OF TYPE I SULFATASES
Type I sulfatases are the predominant mediators of sulfate ester hydrolysis in all domains of life. They are abundant, highly conserved, and require the fGly PTM for catalysis.2 Some sulfatases have been assigned defined biological substrates (e.g., sulfatases that act on the glycosaminoglycans chondroitin and heparan sulfate),16 while most, particularly from microbial sources, have not been characterized at this level of biochemical detail. Thus, many type I sulfatases are annotated as arylsulfatases, simply because they catalyze the hydrolysis of colorimetric arylsulfate substrates in vitro.2
fGly is generated from a cysteine or serine residue embedded in a conserved active site-localized sequence motif, (C/ S)XPXRXXXLTGR; cysteine predominates as the modification target in eukaryotes and aerobic microbes whereas cysteine or serine is found in anaerobes.15,18 The original discovery of fGly was made during biochemical analysis of the fatal congenital illness Multiple Sulfatase Deficiency (MSD).19 The severe physiological defects associated with this condition reflected a combination of those associated with single sulfatase deficiencies.10 Analysis of MSD-derived cell lines revealed diminished activity across the type I sulfatase family, despite the fact that protein levels of specific sulfatases appeared unaltered.10,20 This observation led to speculation that MSD patients lacked a regulatory component or catalytic cofactor required for sulfatase activity. Importantly, cell-fusion complementation of MSD cells with those deficient in a single-sulfatase could restore activity of the deficient sulfatase, confirming the competence of individual MSD sulfatase genes.21 The inability of MSD cells to produce active recombinant sulfatases likewise suggested a defect in a factor required for activity of several sulfatase gene products.22
Direct biochemical characterization of human arylsulfatases A and B (ASA and ASB) revealed a tryptic peptide carrying an unknown modification. At the site where a cysteine residue was predicted, mass spectrometry analysis indicated a novel amino acid that formed an adduct with glycerol and, when treated with NaBH4, yielded a serine residue. fGly was thus identified for the first time as a PTM.19 Subsequently, MSD-derived sulfatases were shown to contain significantly lower amounts of fGly, but the complete loss of sulfatase activity is likely embryonic-lethal in humans.23
FGLY PARTICIPATES DIRECTLY IN SULFATE ESTER HYDROLYSIS
The correlation of fGly deficiency with a loss of sulfatase activity implied a direct role for fGly in enzymatic activity. Absent further information, several modes of reactivity were proposed based on known chemistries of the aldehyde group its electrophilicity as well as the nucleophilicity of the geminal diol formed by aldehyde hydration (Figure 1A). Clarity came from the analysis of several sulfatase crystal structures which support a role for fGly in covalent catalysis.24 The first sulfatase structure, that of human ASB, was solved by Guss and co-workers in 1997, and one year later the structure of ASA was solved by von Figura and coworkers.25,26 A third human sulfatase structure, that of steroid sulfatase (ASC, STS), was later reported by Ghosh and coworkers.27 Notably, despite relatively low sequence homology, these sulfatases adopt a similar fold to alkaline phosphatases, consistent with their related catalytic functions.
The sulfatase structures resolved several uncertainties regarding fGly. First, the fGly geminal diol (fGly-diol) was detected in ASA, suggesting a role as a catalytic nucleophile. Electron density consistent with a sulfated fGly-diol, a putative intermediate in a covalent catalysis mechanism, was observed in both ASB and ASC.25,27 However, it is important to note that the presence of an artifactual phosphate ester instead could not be ruled out. Further evidence came from the 1.3 Å resolution structure of the P. aeruginosa arylsulfatase (PAS), which clearly showed electron density for the fGly-diol at full occupancy (Figure 1B).28 An earlier observation that fGly within native folded ASA and ASB is refractory to NaBH4 reduction is consistent with the presence of the fGly-diol rather than the fGly-aldehyde in the resting enzyme.19 In addition to fGly, the active sites of type I sulfatases contain a divalent cation, usually Ca2+ but also Mg2+, and a side-chain hydrogen bonding network which coordinates fGly and substrate. The metal helps to bind and polarize the substrate, and the fGly-centered hydrogen bonding network provides for general acid–base catalysis. Three aspartates, one of which H-bonds with an fGly-diol hydroxyl, and a glutamine/asparagine constitute the metal-binding residues, leaving two open coordination sites for binding of sulfate and the second fGly-diol hydroxyl. Several acidic residues bind the substrate, and a histidine base is predicted to assist in catalysis. A concise comparison of the corresponding active site residues for seven (of eight total) fGly enzyme crystal structures is provided by Steinfeld and co-workers (Figure 1C).24 The first structure of human sulfamidase (N-sulfoglucosamine sulfohy-drolase, SGSH) was recently reported and overlays closely with previous sulfatases, especially in the active site region, despite low overall sequence identity (between 19 and 25% across fGly enzymes that have been structurally characterized).24
The presence of an active site fGly-diol suggested possible catalytic mechanisms (Figure 1D). Guss (ASB)25 and von Figura (ASA)26 both proposed that one of the hydroxyls of the fGly-diol acts as a nucleophile for initial attack at the substrate sulfur atom, releasing the alcohol coproduct and forming a sulfoenzyme intermediate. The details of their proposed pathways diverge thereafter, though both emphasize acid-base catalysis. In the latter case, deprotonation of the free remaining fGly-diol hydroxyl group by a conserved histidine would allow E2 elimination of sulfate, generating a transient fGly-aldehyde in a scheme abbreviated “SN2-E2.”26 Guss and co-workers proposed instead that SN2 substitution at sulfur by hydroxide or water was responsible for the breakdown of the sulfoenzyme intermediate, which can be designated as the “SN2-SN2” pathway (Figure 1D).25 Consistent with the unequal roles of each hydroxyl group in the geminal diol in either proposed mechanism, only the S stereoisomer of the transesterified enzyme has been observed in crystal structures of ASB and PAS.25,29
Intuitively, one may suppose that the biosynthetic cost of installing the unusual fGly PTM is more than repaid because fGly enables a mechanism unavailable to Ser/Cys/Thr nucleophiles. Replacing cysteine with serine in recombinant sulfatases prevents the conversion to fGly by eukaryotic FGEs and has provided an instructive probe to compare the two proposed mechanisms. In the case of ASA and ASB, the Cys-to-Ser mutant sulfatase will bind substrate and perform the initial S-O cleavage to form the serine sulfoenzyme and release the alcohol coproduct. But, this adduct is a dead-end product that will not release sulfate even after extended time periods.30 If the wild-type α-hydroxy sulfate ester intermediate was resolved by an SN2-type attack of water at sulfur, and not by E2 eliminaton, the serine mutant would be expected to retain at least partial activity. Thus, the E2 pathway is presently favored. Sulfoenzyme formation is detected in only about 20% of the Cys-to-Ser sufatase mutant, however, and this complex is unable to form in crystals. These facts suggest that the fGly-diol contributes to accelerated enzyme sulfation as well, either by the enhanced nucleophilicity of the diol hydroxyl group compared to a primary alcohol or by specific hydrogen bonding features of the active site that are not replicated in the serine mutant.31 Since the serine mutant affects both breakdown and formation of the sulfoenzyme intermediate, its behavior alone is not sufficient to prove the elimination mechanism.
Further chemical and theoretical analysis has yielded a framework for understanding the significance of fGly and its likely reaction pathway. First, the uncatalyzed hydrolysis of the S-O bond of sulfate esters proceeds 106-fold slower than spontaneous hydrolysis of the P-O bond of phosphate esters.4 The high barrier for the uncatalyzed reaction, with a t1/2 = 1018 years,4 may explain the evolutionary significance of fGly’s novel catalytic strategy. Alkaline phosphatase hydrolyzes phosphate esters through a phosphoserine intermediate that releases inorganic phosphate by attack of a Zn+-activated hydroxide.32 This catalytic paradigm may be inadequate for sulfate ester S-O cleavage, or was otherwise impractical or unavailable to sulfatases during evolution, thus requiring the specialized functionality of fGly. Of the two proposed fGly catalyzed pathways, the SN2-E2 mechanism requires only one high energy barrier S-O bond cleavage. The rate of the initial SN2 step may be accelerated by the decreased pKa of the geminal diol hydroxyl group (e.g., acetaldehyde hydrate has a pKa of 13.5733) compared to a conventional hydroxyl group (e.g., 15.9334). A recent theoretical study has suggested that the initial nucleophilic attack of fGly-diol on substrate may be rate-determining, and not the subsequent elimination.35 Perhaps inconsistent with this prediction, a measured βLG of 0 for Vmax but −0.86 for Vmax/ KM for PAS suggests that the alkoxide leaving group is not involved in the rate-determining step but only the first irreversible chemical step.36 For the desulfation step, hydrolysis of the sulfoenzyme intermediate by water in the SN2 pathway may be quite slow compared to E2, again for the reason for S-O bond cleavage. Experimentally, pre-steady-state kinetics of the wild-type enzyme may permit the comparison of the relative sulfation/desulfation rates. Stereochemical characterization of reaction products can also provide valuable clues to the mechanism. In principle, the SN2-E2 mechanism would result in net inversion at sulfate while SN2-SN2 would result in net retention. In an early study, prior to the discovery of fGly, interception of the sulfoenzyme intermediate generated using a chiral sulfate ester with an exogenous nucleophile produced a new sulfate ester with net retention of configuration.37 This observation is consistent with an initial SN2-like step in the sulfatase mechanism but does not inform the course of desulfation.
Other determinants of reactivity may explain the proposed preference of sulfatases for an E2 release pathway, and the differential specificity of sulfatases and phosphatases in general. A recent experimental and theoretical study by Krenske and coworkers affirms earlier theoretical studies,35 which predict a much lower transition state barrier for E2 elimination compared to SN2 substitution to release sulfate (estimated ΔΔH‡ of 26.5 kcal/mol).36 This study estimates a ΔΔH‡ of only 12 kcal/mol for the corresponding phosphoenzyme intermediate, which may account for why fGly is found mostly in sulfatases, and not in alkaline phosphatase. Wild-type ASA is also inhibited by a prohibitively slow rate of elimination/hydrolysis of the corresponding phosphoenzyme intermediate at fGly.29 This is not an inherit characteristic of fGly enzymes, however. PAS can catalyze arylphosphate hydrolysis at a lower catalytic efficiency (kcat/KM) compared to arylsulfates (7.9 × 102 M−1 s−1 vs 4.9 × 107 M−1 s−1), but with dramatic rate acceleration (kcat/kuncat) and catalytic proficiency ((kcat/KM)/kuncat) compared to the uncatalyzed hydrolysis of arylphosphates; PAS is predicted to release phosphate by elimination as with sulfate.35,38,39 In the opposing case of promiscuous sulfatase activity in alkaline phosphatase, the source of large catalytic discrimination for phosphate esters over sulfate esters appears to be complex and only partially related to the difference in formal charge between the two substrates.40 Notably, a family of type I sulfatase/alkaline phosphatase related fGly-dependent enzymes has been recently discovered and structurally characterized. It includes a phosphatase/phosphonatase from Rhizobium leguminosarum termed RlPMH41 and an orthologous but highly promiscuous phosphate/phosphonate/sulfate/sulfonate ester hydrolase from Burkholderia caryophilli (BcPMH).42 RlPMH has a >103-fold preference for phosphonate monoesters/phosphate diesters over sulfate esters and displays burst-phase kinetics, indicating that formation of the phosphoenzyme intermediate is not rate-limiting.41 This enzyme family reflects the apparent catalytic versatility of fGly as tuned by protein context, as well as the shared ancestry of type I sulfatases and alkaline phosphatases.
It is important to note that two other unrelated sulfatase families exist in some prokaryotes, but unlike type I, they proceed with concomitant oxidation of the alcohol coproduct to an aldehyde (type II), or stereochemical inversion of the alcohol coproduct (type III).17 It is clear that such changes would not be suitable for the signaling or catabolic roles of eukaryotic sulfatases. Though further experimental evidence is needed to clarify remaining mechanistic uncertainties, it is clear that type I sulfatases are among the most extraordinary biological catalysts known, with rate enhancements (kcat/kuncat) as high as 1026, and undoubtedly warrant further studies.4
FGE CATALYZES FGLY FORMATION AND IS DYSFUNCTIONAL IN MSD
In 2003, the gene encoding FGE (also termed sulfatase-modifying factor 1 or SUMF1) was identified as responsible for MSD by Ballabio et al.6 and von Figura et al.,5 using genetic and biochemical approaches, respectively. Von Figura and coworkers exploited FGE’s ability to bind tightly but not turn over a peptide containing its recognition motif with serine substituted for cysteine (SXPXR). By affinity enrichment of bovine testis extract, FGE was thus purified for the first time.5 Investigation of human and bacterial FGEs has revealed a new enzyme family defined by both a novel catalytic mechanism and protein fold. Mapping the mutations in SUMF1 alleles from MSD patients onto the hFGE crystal structure, as well as in vivo and in vitro dissection of mutant phenotypes, has provided a rationale for their deleterious effects. MSD mutations result in structural, catalytic, and intracellular stability defects.43–45
Prior to isolation of FGE, fGly formation was studied using intact microsomes and later with reticuloplasm, using an in vitro transcription/translation system.46,47 These studies indicated that fGly generation occurs cotranslationally, and prior to folding. These initial efforts suggested a dependence on thiol reductants, Ca2+ ions, and an alkaline pH optimum.48 Subsequently, it was observed that FGE activity is dependent on O2, presumably as a terminal electron acceptor (cf. Figure 3C).49 Interestingly, O2 availability can limit sulfatase activity in vivo by decreasing FGE activity, and the decrease of sulfatase activity levels can mediate hypoxic response.50
Figure 3.

FGE structure, overall reaction, and speculative mechanistic proposal. (A) Crystal structure of human FGE (PDB: 1Y1E), with active site cysteines (Cys336, Cys341; engaged in partial disulfide bond), the essential serine (Ser333), and Ca2+ ions (green) highlighted. (B) Close-up of human FGE Cys336S (PDB: 2AFY) in complex with substrate as Cys341-LCTPSRA mixed disulfide, with substrate binding residues highlighted. (C) Overall reaction catalyzed by FGE on generic CTPSR substrate. Given the 2e− oxidation of cysteine to formylglycine, O2 is reduced to H2O2. However, as has been proposed, the concomitant input of an additional 2e− and protons would result in the production of 2 H2O instead. (D) Abbreviated summary of proposed mechanism provided by Roeser et al.63 FGEox binds substrate through thiol–disulfide exchange with substrate at Cys341. Cys336 then reacts with O2 to generate a peroxysulfenic acid. Decomposition of this intermediate, and perhaps the input of two external electrons, would regenerate FGEox, release 1 H2O, and form a substrate cysteine sulfenic acid. Base-catalyzed elimination of H2O from substrate sulfenic acid produces a transient thioaldehyde, which gives fGly upon hydrolysis.
MAMMALIAN FGE ACTIVITY IS REGULATED BY PROTEIN PARTNERS
Regulation of mammalian FGE activity occurs spatially and is mediated by protein–protein interactions in the ER and Golgi. Recent work has identified functional interactions with several proteins in the ER and along the secretory pathway including ERp44, PDI, ERGIC-53, and pFGE among others.51–54 FGE lacks a canonical ER retention sequence; instead, the N-terminal region of FGE (residues 34–88 following signal peptide cleavage) forms both noncovalent interactions and a disulfide linkage with ERp44, a trafficking protein that is the primary means for retrieval of FGE from the Golgi to the ER. In fact, this N-terminal region is also involved in activation of sulfatases by an unknown (ER-retention-independent) mechanism. The region contains a cysteine pair (Cys50 and Cys52) that forms intermolecular disulfide bonds both with Erp44 and with other FGE molecules. Only Cys52 is necessary for FGE activity in vivo, but not in vitro,54 while both cysteines are dispensable for retrieval by ERp44 through noncovalent complex formation,51 though an alternate mutational study disputes this finding (Figure 2).52
Figure 2.

Regulation and trafficking of mammalian FGE by protein partners. (A) ERp44 binds the N-terminal extension of FGE and delivers it back to the ER. (B) Protein disulfide isomerase (PDI) retains FGE in the ER through binding and increases catalytic activity, perhaps by acting as a disulfide reductant. (C) ERGIC-53 transports FGE to the Golgi and prevents degradation by the proteasome. (D) A portion of FGE is trafficked to the cell surface and secreted through the secretory pathway; furin and other proteases cleave and thereby inactivate 70–80% of secreted FGE. (E) Secreted FGE binds mannose receptor (MR) through its N-glycan. (F) Extracellular FGE can be trafficked back to the ER in an active state. (G) pFGE interacts with FGE for an unknown purpose, but may chaperone unfolded sulfatases or mediate their interaction with FGE.
Protein disulfide isomerase (PDI) is a well-known ER-resident chaperone that allows nascent polypeptides to reach their preferred disulfide configuration by catalyzing fast exchange with its own redox active disulfide.55 PDI appears to be involved both in retaining FGE in the ER and in promoting activity, and these functions are dependent on catalytically active PDI (Figure 2B).52 ERGIC-53 is an anterograde cargo binding protein which cycles between the ER and the Golgi.52 It functions to traffic FGE from the ER to the cis-Golgi and also seems to protect it from proteasomal degradation (Figure 2C). Once in the cis-Golgi, most FGE is retrieved to the ER via ERp44, with the remaining fraction secreted from the cell.52 It is possible that competition with other ERp44 substrates may regulate FGE secretion by this process. Downstream in the secretory pathway, a fraction of FGE is N-terminally truncated and inactivated by furin and furin-like proteases (Figure 2D). Approximately 20–30% of secreted FGE avoids proteolytic cleavage and remains active.56 Secreted FGE can traffic back to the ER from the cell surface. Indeed, tissue-specific expression of FGE in FGE-null mice has been shown to activate sulfatases in nontransduced tissues, in this way acting as a paracrine agent. Uptake is dependent in part on the mannose receptor and FGE N-glycosylation (Figure 2D–F).57 Taken together, a model for the localization and activity of FGE has emerged (Figure 2). However, it remains of great interest to understand the significance of FGE secretion and reuptake to the ER, as this offers a route for enzyme replacement therapy for treatment of MSD.
Finally, the paralog of FGE (pFGE, encoded by the gene SUMF2) was identified by homology shortly after SUMF1 was identified, and its precise role is the subject of ongoing investigation.6 pFGE displays strong sequence and structural conservation (48% identity) with hFGE, is localized to the ER, and is transcribed at comparable levels to FGE in human tissues, but pFGE lacks FGE activity and is different by several features.58–60 pFGE lacks the active site cysteine pair and functionally critical N-terminal region of FGE. Instead, pFGE has a C-terminal PGEL sequence, which is a noncanonical variant of the ER retention signal KDEL. Based on coimmunoprecipitation data, pFGE forms heterodimers and higher order oligomers with FGE and also interacts with sulfatases, though direct physical evidence for pFGE interactions has remained elusive (Figure 2G). Curiously, pFGE overexpression decreases sulfatase activation in vivo in a dose-dependent manner but does not increase FGE retention in the ER. The meaning of these results is unclear, but it is possible that in appropriate stoichiometry, pFGE may act as a sulfatase-specific chaperone or otherwise mediate the interaction of FGE with sulfatases.58,60
FGE DEFINES A NOVEL PROTEIN FOLD
The first series of crystal structures of human FGE were reported in 2005 by Dierks et al., and FGE was shown to adopt a novel fold, the FGE-fold (Figure 3A).43,61 Subsequently, the crystal structure of the S. coelicolor FGE (scFGE) was solved in our laboratory and overlays closely with hFGE, with an rmsd of 0.65 Å.49 The FGE fold is defined by one domain of low secondary structure content, approximately 30% combined β-sheet and α-helix in both orthologs. hFGE binds two Ca2+ ions and scFGE just one; two structural disulfides and an N-glycosite are particular to the human enzyme. A cysteine pair (C336 and C341), shown to be catalytically essential and separated by four residues in a loop, exists in a distribution of the reduced (dithiol, FGEred) and oxidized (disulfide, FGEox) forms based on both crystal structures and by mass spectrometry (Figure 3A).62,43 A cocrystal structure of hFGE C336S with LCTPSRA substrate was obtained by Roeser et al. in 2006 and defined a shallow substrate binding channel which can bind up to six substrate residues in the consensus peptide sequence in an extended conformation (Figure 3B).63 Additional structures support the existence of a probable O2 binding pocket adjacent to the active site cysteines. The use of halide bound hFGE crystal structures to define the size and environment of this pocket predict that it could accommodate a negatively charged oxidative intermediate in complex with substrate.64 Homologues of FGE have been found in a class of bacteria and bacteriophage diversity-generating retroelement (DGR) variable proteins, which generate adaptive protein variation through an RNA intermediate.65 With an rmsd of 0.98 Å between the catalytic region of FGE and the Treponema denticola TvpA variable region, the FGE-fold has since been classified as a subtype of the C-type lectin fold.66
FGE IS A UNIQUE CYSTEINE-DEPENDENT OXIDASE/OXYGENASE
Many biochemical transformations in both primary and secondary metabolism across aerobic organisms harness the reduction potential of O2. Reduction of molecular oxygen is thermodynamically favorable, but there is a high barrier to its reaction with singlet state organic compounds due to its triplet ground state.67 Most oxidases/oxygenases require reductants of sufficient strength, typically paramagnetic transition metals (mainly Fe2+ and Cu2+), and/or organic cofactors (flavins, pterins, quinones) which generate a stabilized radical following single electron transfer.68 Subsequent one or two electron transfer events are very favorable and rapid, producing potent O2-derived oxidants that may add in to substrate (oxygenase) or become further reduced to H2O or H2O2 (oxidase). Cofactorless oxidases/oxygenases have also been identified. They typically promote the direct oxidation of substrates, without the intermediacy of a metal or cofactor, by targeting O2 to a stabilized carbanion at a specific position of an aryl substrate in a manner comparable to flavin reactivity.69
FGE does not possess a canonical metal or organic redox cofactor and thus has been categorized as a cofactorless oxidase/ oxygenase. But, there is no evidence that FGE utilizes any variation of the strategies above to catalyze reaction with O2. Instead, mechanistic proposals center on the essential redox-active C336/C341 pair located in the active site. The lability of these cysteines to irreversible oxidation by air or H2O2, creating cysteine sulfonic acids, suggests that they may mediate reaction of O2 with the substrate cysteine side chain.49,43 Mutation of either of these cysteines abrogates activity, and they are positioned closely to the substrate cysteine in a cocrystal structure where Cys336 (hFGE) is mutated to serine (Figure 3B).63 The isolation of this complex, and the observation of partially oxidized cysteine side chains in various crystal structures, formed the basis of the proposed mechanism shown in Figure 3D. This scheme invokes a disulfide bonded form of FGE (FGEox) as the enzyme’s resting state. Substrate binding is then followed by disulfide exchange, forming a covalent mixed disulfide bond between enzyme and substrate. Direct reaction of a free cysteine side chain at C336 with O2 is proposed to generate a reactive intermediate, a cysteine oxoform presumably, that then targets the substrate cysteine side chain.
In both the hFGE and scFGE apo crystal structures, Cys336 (hFGE numbering) displays additional electron density that can be modeled as a sulfenic acid, peroxysulfenic acid, free peroxide, or water. Ser333 is likely critical for catalysis due to its role in coordinating such an intermediate. A substrate cysteine sulfenic acid may be generated through the transfer of oxidative species from one of the above reactive derivatives. Elimination of H2O from sulfenic acid would then generate a thioaldehyde intermediate, which would hydrolyze to generate fGly and H2S (Figure 3D). Notably, in vitro biochemical studies have shown that an external reductant is required to achieve high substrate turnover (e.g., GSH or DTT).48 Thus, although FGE activates O2 in a cofactor-independent manner, a reductant may be required to regenerate the enzyme’s active state during each catalytic cycle, or at an intermediate stage.69 It is possible that hFGE relies on PDI as a reducing agent in vivo, but there is no direct evidence for this.52
ANSME CATALYZES O2-INDEPENDENT FGLY FORMATION ON SER/CYS SULFATASES IN ANAEROBIC MICROBES
Given fGly’s impressive catalytic capability, it is not surprising that an O2-independent fGly generating system also exists in anaerobic and facultative anaerobic microbes. A family of radical S-adenosylmethione (RS)-dependent dehydrogenases has been identified to fill this role. AtsB from Klebsiella pneumoniae was identified as an fGly generating enzyme by Dierks and coworkers in 1999 and was previously shown to be required for high activity of recombinant Klebsiella sulfatase in E. coli.70,71 Curiously, the native substrate of this enzyme contains Ser and not Cys in a recognition motif that is otherwise identical to the FGE-dependent sulfatase sequences. Based on biochemical and taxonomic evidence, the discovery of a Cys-sulfatase-specific ortholog from Clostridium perf ringens (cpe) led to the designation of a new enzyme family, the anaerobic sulfatase-maturating enzymes (anSME).18 anSMEcpe can also oxidize Ser-type substrates,72 suggesting a similar reaction pathway for both targeted side chains.73 AtsB, whose native substrates are Ser-type, is 4-fold more active with cysteine-based substrates in vitro. This preference for Cys may originate from increased thiol compared to alcohol reactivity at one or more proposed elementary steps. Spectroscopic characterization revealed that anSME, in addition to the canonical RS [4Fe-4S] cluster, contains two auxiliary [4Fe-4S] clusters as well (Aux I and Aux II).74 Drennan and coworkers recently reported the first crystal structure of anSMEcpe with and without bound substrate (1.6–1.8 Å resolution), showing both Aux clusters in the C-terminal SPASM domain with fully occupied coordination sites, unlike the RS cluster which contains an open Fe-binding site for substrate.75
Mechanistic investigation of anSMEs has revealed many details of this reaction (Figure 4). As with all RS enzymes, a canonical [4Fe-4S]+ cluster reductively cleaves S-adenosylme-thionine (SAM) to generate Met and the 5′-deoxyadenosine radical (5′-dA•). Initial H• abstraction by 5′-dA•, as evidenced by substrate deuterium labeling and stereospecific Cβ-methyl substitution, is performed at the pro-S Cβ-H of Cys/Ser in the consensus sequence.73,76 Deprotonation of the thiol/alcohol Cβradical by an asparate side chain and charge donation into the radical promotes the second substrate oxidation step. The second one-electron oxidation would yield a thioaldehyde intermediate in the case of cysteine, which would hydrolyze to fGly, or would yield fGly directly in the case of serine. Based on the orientation of Aux I and Aux II in relation to the RS cluster, Aux I is the likely electron acceptor for the second oxidation step. Aux II is proximal to bulk solvent and may receive this electron from Aux I and donate it to a soluble carrier. This pathway accounts for the multiple turnover ability of anSME in the presence of flavodoxin without external reductants, meaning that recycling of the second substrate electron to reduce the RS cluster cannot occur directly from AuxI/II.75 Further experimentation will clarify the role of each Aux cluster. Despite the strong conservation between substrates of anSME and FGE, the way each enzyme binds substrate is distinct. FGE-substrate interaction hinges on a hydrophobic pocket for Pro binding and several hydrogen bond partners to Arg (in CXPXR), with only 4 of 12 H-bonds to the substrate backbone.63 However, anSME binds substrate primarily through the substrate backbone, accounting for 14 of 17 H-bonds; this explains the existence of alternative native anSME substrates containing CXAXR without Pro, though Arg side-chain binding is maintained.75
Figure 4.

Mechanism of fGly biosynthesis by the anaerobic sulfatase maturing enzyme (anSME). The reduced radical S-adenosylmethionine (RS) [4Fe-4S]+ cluster first binds and reductively cleaves SAM to produce methionine (Met) and 5′-deoxyadenosyl radical (5′-dA•). The 5′-dA• abstracts the pro-S Cβ hydrogen from the conserved substrate cysteine or serine residue. Thiol deprotonation by an active site aspartyl side chain generates a transient radical anion, which is thought to be oxidized by auxiliary [4Fe-4S]2+ cluster I (Aux I). Auxiliary cluster II may oxidize Aux I and transfer the second substrate electron to a soluble carrier for regeneration of the reduced RS cluster.
SOME ORGANISMS HAVE AS YET UNIDENTIFIED FGLY PRODUCING SYSTEMS
Yet a third means for producing fGly has been observed, with the responsible enzyme(s) remaining elusive. Recombinant expression of type I sulfatases in E. coli has been shown to produce active enzymes with varying proportions of fGly modification.77 Berteau and co-workers have shown that a putative E. coli anSME ortholog was unnecessary for the conversion of recombinant sulfatases, and that an unidentified, O2-dependent, Cys-type fGly-generating enzyme must be responsible for installing the aldehyde. According to sequence data available at the time, as of 2007, approximately 10% of bacterial genomes were predicted to encode arylsulfatase genes without the co-occurrence of obvious FGE or anSME orthologs.78 This is also true for several fungi, and even for the metazoan Caenorhabditis elegans, which contains three sulfatase genes.12,79 C. elegans has been shown to utilize sulfatases for heparan sulfate remodeling, as in humans.80 Thus, it is likely that these organisms all carry a related system for fGly production with important functional consequences. We believe that identification of the putative third type of fGly-generating enzyme may reveal new aspects of fGly biology.
FGLY PROVIDES A GENETICALLY ENCODED, SITE-SPECIFIC PROTEIN BIOCONJUGATION HANDLE
During characterization of the sequence determinants of cotranslational FGE activity, it was reported that the minimal five-residue motif, CXPXR, is efficiently converted when embedded in an otherwise nonnative sequence.81 This observation led us to speculate that the CXPXR sequence could be exploited as a means to genetically encode fGly residues in various proteins of interest. The aldehyde group, in addition to its unique catalytic nature within sulfatases, is also a reactive electrophile with orthogonal reactivity to canonical protein side chain functionalities. We proposed that classical carbonyl-based bioconjugation chemistries, such as oxime and hydrazine formation, could be used to covalently attach various cargo to such “aldehyde tagged” proteins in a site-specific manner. The use of FGE and its five-residue motif to arm recombinant proteins with aldehyde “chemical handles” was first reduced to practice in E. coli expression systems.82 The limited endogenous FGE-like activity in E. coli was found to be saturated by overexpressed proteins, necessitating coexpression of ectopic FGE along with the protein of interest in order to achieve high yields of Cys-to-fGly conversion. The M. tuberculosis FGE was particularly useful in this regard, as the enzyme was found to have relaxed sequence specificity and tolerated single alanine substitutions within the CXPXR motif.83 Subsequently, the method was used to produce aldehyde tagged recombinant proteins in mammalian cell expression systems. As eukaryotic FGE is naturally ER localized, membrane-associated and secreted proteins, both of which traffic through the secretory pathway, were found to be viable targets for aldehyde tagging.84,85
As a site-specific protein modification method, the aldehyde tag has many advantages compared to other options.86,87 It is operationally simple, requiring simple cloning of the CXPXR motif at the desired modification site and expression of the protein of interest in FGE-expressing cells. Once installed, the aldehyde can be reacted with aminooxy or hydrazide reagents to form the corresponding oxime and hydrazone conjugates, respectively (Figure 5A). This straightforward process has been used to site-specifically PEGylate proteins,82 to introduce biophysical probes at discrete locations for single molecule studies of protein dynamics,88,89 to immobilize proteins and phage on solid materials,90–92 to construct artificially glycosy-lated proteins,93,94 and to generate DNA–protein conjugates.95 The aldehyde tag method has also been used in conjunction with other bioconjugation reactions to generate chemically fused protein heterodimers (Figure 5B).96
Figure 5.

Principle and application of aldehyde tag engineering. (A) fGly is installed on a protein target and used as a bioconjugation handle with aminooxy, or modified Pictet-Spengler reagent functionalized molecules. (B) Bifunctional protein fusion of human growth hormone (hGH) and human IgG (hIgG) synthesized by aldehyde tagging each followed by Cu-free azide-alkyne cycloaddition (reproduced with permission from ref 96, copyright Wiley-VCH, 2012). (C) ADC produced site-specifically and with precise drug/antibody ratio, shown with hydrazino Pictet-Spengler ligation product, for recent example of trastuzumab and maytansine.102
The biopharma industry has a growing interest in chemically modified protein therapeutics. Antibody-drug conjugates (ADCs) in particular are highly represented in biopharma pipelines, largely for cancer indications.97 There is increasing awareness of the benefits of generating such constructs via site-specific attachment of drug molecules to the antibody scaffold.98,99 This trend presents an opportunity for clinical applications of fGly-functionalized proteins. However, critical to the therapeutic performance of such constructs is stability of the drug–protein linkage. Oximes and hydrazones can undergo reversible hydrolysis at physiologically relevant pHs, an inherent liability to their use in ADCs. To address this issue, we developed a new C–C bond forming reaction of protein-associated fGly residues with alkoxyamine-functionalized indoles (1, Figure 5A), an adaptation of the classic Pictet-Spengler reaction that we termed the Pictet-Spengler ligation.100 A second-generation version, the hydrazino Pictet-Spengler ligation (2, Figure 5A) proceeds readily at neutral pH to give stable fGly conjugates.101 This chemistry has recently been employed to construct homogeneous ADCs comprising the antimitotic natural product maytansine linked to the anti-Her2 antibody trastuzumab (Figure 5C).102 The use of fGly as a chemical handle for these ADCs enabled precise control over the site and stoichiometry of drug conjugation, thereby facilitating drug optimization.
CONCLUSION
Many questions remain regarding the molecular details of fGly biogenesis and its role in catalysis. But in the two decades since fGly was first discovered, sulfatases and the enzymes that activate them have become widely appreciated as lessons in enzyme evolution and catalytic strategy. As well, the orthogonal reactivity of the aldehyde group with respect to canonical protein functionality has enabled biotechnology applications of fGly that are approaching clinical translation. The scope of FGE as a tool for protein engineering has yet to be fully explored. Interesting questions abound, such as the amenability of the enzyme to targeted engineering for enhanced performance or altered substrate specificity. As well, the intriguing discovery of fGly outside the type I sulfatase family as required for the phosphatase/phosphonatase RlPMH and multisubstrate hydro-lase BcPMH hints that fGly could be more widespread across microbial proteomes. And the search for additional FGE-like enzymes is far from over. Organisms as familiar as E. coli and C. elegans generate fGly on recombinant proteins and produce fGly-dependent sulfatases respectively, but their FGE-like machineries remain undefined. We anticipate a rich future for fundamental and applied studies of this small but powerful PTM.
Acknowledgments
We thank J. Klinman, D. Fox, and P. Agarwal for critical reading of the manuscript.
Footnotes
The authors declare no competing financial interest.
References
- 1.Okeley NM, van der Donk WA. Novel cofactors via post-translational modifications of enzyme active sites. Chem Biol. 2000;7:R159–R171. doi: 10.1016/s1074-5521(00)00140-x. [DOI] [PubMed] [Google Scholar]
- 2.Hanson SR, Best MD, Wong CH. Sulfatases: Structure, Mechanism, Biological Activity, Inhibition, and Synthetic Utility. Angew Chem, Int Ed. 2004;43:5736–5763. doi: 10.1002/anie.200300632. [DOI] [PubMed] [Google Scholar]
- 3.Klinman JP, Bonnot F. Intrigues and Intricacies of the Biosynthetic Pathways for the Enzymatic Quinocofactors: PQQ, TTQ, CTQ, TPQ, and LTQ. Chem Rev. 2013;114:4343–4365. doi: 10.1021/cr400475g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edwards DR, Lohman DC, Wolfenden R. Catalytic Proficiency: The Extreme Case of S–O Cleaving Sulfatases. J Am Chem Soc. 2011;134:525–531. doi: 10.1021/ja208827q. [DOI] [PubMed] [Google Scholar]
- 5.Dierks T, Schmidt B, Borissenko LV, Peng J, Preusser A, Mariappan M, von Figura K. Multiple Sulfatase Deficiency Is Caused by Mutations in the Gene Encoding the Human Cα-Formylglycine Generating Enzyme. Cell. 2003;113:435–444. doi: 10.1016/s0092-8674(03)00347-7. [DOI] [PubMed] [Google Scholar]
- 6.Cosma MP, Pepe S, Annunziata I, Newbold RF, Grompe M, Parenti G, Ballabio A. The Multiple Sulfatase Deficiency Gene Encodes an Essential and Limiting Factor for the Activity of Sulfatases. Cell. 2003;113:445–456. doi: 10.1016/s0092-8674(03)00348-9. [DOI] [PubMed] [Google Scholar]
- 7.Wiegmann EM, Westendorf E, Kalus I, Pringle TH, Lübke T, Dierks T. Arylsulfatase K, a Novel Lysosomal Sulfatase. J Biol Chem. 2013;288:30019–30028. doi: 10.1074/jbc.M113.499541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buono M, Visigalli I, Bergamasco R, Biffi A, Cosma MP. Sulfatase modifying factor 1–mediated fibroblast growth factor signaling primes hematopoietic multilineage development. J Exp Med. 2010;207:1647–1660. doi: 10.1084/jem.20091022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diez-Roux G, Ballabio A. Sulfatases and Human Disease. Annu Rev Genomics Hum Genet. 2005;6:355–379. doi: 10.1146/annurev.genom.6.080604.162334. [DOI] [PubMed] [Google Scholar]
- 10.Austin JH. Studies in metachromatic leukodystrophy: Xii. multiple sulfatase deficiency. Arch Neurol. 1973;28:258–264. doi: 10.1001/archneur.1973.00490220066010. [DOI] [PubMed] [Google Scholar]
- 11.Ulmer JE, Vilén EM, Namburi RB, Benjdia A, Beneteau J, Malleron A, Bonnaffé D, Driguez PA, Descroix K, Lassalle G, Le Narvor C, Sandström C, Spillmann D, Berteau O. Characterization of Glycosaminoglycan (GAG) Sulfatases from the Human Gut Symbiont Bacteroides thetaiotaomicron Reveals the First GAG-specific Bacterial Endosulfatase. J Biol Chem. 2014;289:24289–24303. doi: 10.1074/jbc.M114.573303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sardiello M, Annunziata I, Roma G, Ballabio A. Sulfatases and sulfatase modifying factors: an exclusive and promiscuous relationship. Hum Mol Genet. 2005;14:3203–3217. doi: 10.1093/hmg/ddi351. [DOI] [PubMed] [Google Scholar]
- 13.Bojarová P, Williams SJ. Sulfotransferases, sulfatases and formylglycine-generating enzymes: a sulfation fascination. Curr Opin Chem Biol. 2008;12:573–581. doi: 10.1016/j.cbpa.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh D. Human sulfatases: A structural perspective to catalysis. Cell Mol Life Sci. 2007;64:2013–2022. doi: 10.1007/s00018-007-7175-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Figura K, Schmidt B, Selmer T, Dierks T. A novel protein modification generating an aldehyde group in sulfatases: its role in catalysis and disease. BioEssays. 1998;20:505–510. doi: 10.1002/(SICI)1521-1878(199806)20:6<505::AID-BIES9>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 16.Dierks T, Schlotawa L, Frese MA, Radhakrishnan K, von Figura K, Schmidt B. Molecular basis of multiple sulfatase deficiency, mucolipidosis II/III and Niemann-Pick C1 disease -Lysosomal storage disorders caused by defects of non-lysosomal proteins. Biochim Biophys Acta. 2009;1793:710–725. doi: 10.1016/j.bbamcr.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 17.Toesch M, Schober M, Faber K. Microbial alkyl-and aryl-sulfatases: mechanism, occurrence, screening and stereo-selectivities. Appl Microbiol Biotechnol. 2014;98:1485–1496. doi: 10.1007/s00253-013-5438-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berteau O, Guillot A, Benjdia A, Rabot S. A New Type of Bacterial Sulfatase Reveals a Novel Maturation Pathway in Prokaryotes. J Biol Chem. 2006;281:22464–22470. doi: 10.1074/jbc.M602504200. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt B, Selmer T, Ingendoh A, Vonfigura K. A novel amino-acid modification in sulfatases that is defective in Multiple Sulfatase Deficiency. Cell. 1995;82:271–278. doi: 10.1016/0092-8674(95)90314-3. [DOI] [PubMed] [Google Scholar]
- 20.Fluharty AL, Stevens RL, Davis LL, Shapiro LJ, Kihara H. Presence of arylsulfatase A (ARS A) in multiple sulfatase deficiency disorder fibroblasts. Am J Hum Genet. 1978;30:249–255. [PMC free article] [PubMed] [Google Scholar]
- 21.Horwitz AL. Genetic complementation studies of multiple sulfatase deficiency. Proc Natl Acad Sci U S A. 1979;76:6496–6499. doi: 10.1073/pnas.76.12.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rommerskirch W, von Figura K. Multiple sulfatase deficiency: catalytically inactive sulfatases are expressed from retrovirally introduced sulfatase cDNAs. Proc Natl Acad Sci U S A. 1992;89:2561–2565. doi: 10.1073/pnas.89.7.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlotawa L, Ennemann EC, Radhakrishnan K, Schmidt B, Chakrapani A, Christen HJ, Moser H, Steinmann B, Dierks T, Gartner J. SUMF1 mutations affecting stability and activity of formylglycine generating enzyme predict clinical outcome in multiple sulfatase deficiency. Eur J Hum Genet. 2011;19:253–261. doi: 10.1038/ejhg.2010.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sidhu NS, Schreiber K, Propper K, Becker S, Uson I, Sheldrick GM, Gartner J, Kratzner R, Steinfeld R. Structure of sulfamidase provides insight into the molecular pathology of mucopolysaccharidosis IIIA. Acta Crystallogr, Sect D: Biol Crystallogr. 2014;70:1321–1335. doi: 10.1107/S1399004714002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bond CS, Clements PR, Ashby SJ, Collyer CA, Harrop SJ, Hopwood JJ, Guss JM. Structure of a human lysosomal sulfatase. Structure. 1997;5:277–289. doi: 10.1016/s0969-2126(97)00185-8. [DOI] [PubMed] [Google Scholar]
- 26.Lukatela G, Krauss N, Theis K, Selmer T, Gieselmann V, von Figura K, Saenger W. Crystal structure of human arylsulfatase A: the aldehyde function and the metal ion at the active site suggest a novel mechanism for sulfate ester hydrolysis. Biochemistry. 1998;37:3654–3664. doi: 10.1021/bi9714924. [DOI] [PubMed] [Google Scholar]
- 27.Hernandez-Guzman FG, Higashiyama T, Pangborn W, Osawa Y, Ghosh D. Structure of Human Estrone Sulfatase Suggests Functional Roles of Membrane Association. J Biol Chem. 2003;278:22989–22997. doi: 10.1074/jbc.M211497200. [DOI] [PubMed] [Google Scholar]
- 28.Boltes I, Czapinska H, Kahnert A, von Bülow R, Dierks T, Schmidt B, von Figura K, Kertesz MA, Usón I. 1.3 Å Structure of Arylsulfatase from Pseudomonas aeruginosa Establishes the Catalytic Mechanism of Sulfate Ester Cleavage in the Sulfatase Family. Structure. 2001;9:483–491. doi: 10.1016/s0969-2126(01)00609-8. [DOI] [PubMed] [Google Scholar]
- 29.Chruszcz M, Laidler P, Monkiewicz M, Ortlund E, Lebioda L, Lewinski K. Crystal structure of a covalent intermediate of endogenous human arylsulfatase A. J Inorg Biochem. 2003;96:386–392. doi: 10.1016/s0162-0134(03)00176-4. [DOI] [PubMed] [Google Scholar]
- 30.Recksiek M, Selmer T, Dierks T, Schmidt B, von Figura K. Sulfatases, Trapping of the Sulfated Enzyme Intermediate by Substituting the Active Site Formylglycine. J Biol Chem. 1998;273:6096–6103. doi: 10.1074/jbc.273.11.6096. [DOI] [PubMed] [Google Scholar]
- 31.von Bulow R, Schmidt B, Dierks T, von Figura K, Uson I. Crystal structure of an enzyme-substrate complex provides insight into the interaction between human arylsulfatase A and its substrates during catalysis. J Mol Biol. 2001;305:269–277. doi: 10.1006/jmbi.2000.4297. [DOI] [PubMed] [Google Scholar]
- 32.Cleland WW, Hengge AC. Enzymatic Mechanisms of Phosphate and Sulfate Transfer. Chem Rev. 2006;106:3252–3278. doi: 10.1021/cr050287o. [DOI] [PubMed] [Google Scholar]
- 33.Bell RP, Onwood DP. Acid strengths of the hydrates of formaldehyde, acetaldehyde and chloral. Trans Faraday Soc. 1962;58:1557–1561. [Google Scholar]
- 34.Murto J. Nucleophilic Reactivity of Alkoxide Ions toward 2,4-Dinitro-fluorobenzene and the Acidity of Alcohols. Acta Chem Scand. 1964;18:1043–1053. [Google Scholar]
- 35.Marino T, Russo N, Toscano M. Catalytic Mechanism of the Arylsulfatase Promiscuous Enzyme from Pseudomonas Aeruginosa. Chem— Eur J. 2013;19:2185–2192. doi: 10.1002/chem.201201943. [DOI] [PubMed] [Google Scholar]
- 36.Williams SJ, Denehy E, Krenske EH. Experimental and Theoretical Insights into the Mechanisms of Sulfate and Sulfamate Ester Hydrolysis and the End Products of Type I Sulfatase Inactivation by Aryl Sulfamates. J Org Chem. 2014;79:1995–2005. doi: 10.1021/jo4026513. [DOI] [PubMed] [Google Scholar]
- 37.Chai CL, Loughlin WA, Lowe G. The stereochemical course of sulphuryl transfer catalysed by arylsulphatase II from Aspergillus oryzae. Biochem J. 1992;287:805–812. doi: 10.1042/bj2870805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olguin LF, Askew SE, O’Donoghue AC, Hollfelder F. Efficient Catalytic Promiscuity in an Enzyme Superfamily: An Arylsulfatase Shows a Rate Acceleration of 1013 for Phosphate Monoester Hydrolysis. J Am Chem Soc. 2008;130:16547–16555. doi: 10.1021/ja8047943. [DOI] [PubMed] [Google Scholar]
- 39.Luo J, van Loo B, Kamerlin SC. Examining the promiscuous phosphatase activity of Pseudomonas aeruginosa arylsulfatase: a comparison to analogous phosphatases. Proteins. 2012;80:1211–1226. doi: 10.1002/prot.24020. [DOI] [PubMed] [Google Scholar]
- 40.Andrews LD, Zalatan JG, Herschlag D. Probing the Origins of Catalytic Discrimination between Phosphate and Sulfate Monoester Hydrolysis: Comparative Analysis of Alkaline Phosphatase and Protein Tyrosine Phosphatases. Biochemistry. 2014;53:6811–6819. doi: 10.1021/bi500765p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jonas S, van Loo B, Hyvönen M, Hollfelder F. A New Member of the Alkaline Phosphatase Superfamily with a Formylglycine Nucleophile: Structural and Kinetic Characterisation of a Phosphonate Monoester Hydrolase/Phosphodiesterase from Rhizobium leguminosarum. J Mol Biol. 2008;384:120–136. doi: 10.1016/j.jmb.2008.08.072. [DOI] [PubMed] [Google Scholar]
- 42.van Loo B, Jonas S, Babtie AC, Benjdia A, Berteau O, Hyvönen M, Hollfelder F. An efficient, multiply promiscuous hydrolase in the alkaline phosphatase superfamily. Proc Natl Acad Sci U S A. 2010;107:2740–2745. doi: 10.1073/pnas.0903951107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dierks T, Dickmanns A, Preusser-Kunze A, Schmidt B, Mariappan M, von Figura K, Ficner R, Rudolph MG. Molecular Basis for Multiple Sulfatase Deficiency and Mechanism for Formylglycine Generation of the Human Formylglycine-Generating Enzyme. Cell. 2005;121:541–552. doi: 10.1016/j.cell.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 44.Annunziata I, Bouche V, Lombardi A, Settembre C, Ballabio A. Multiple sulfatase deficiency is due to hypomorphic mutations of the SUMF1 gene. Hum Mutat. 2007;28:928. doi: 10.1002/humu.9504. [DOI] [PubMed] [Google Scholar]
- 45.Schlotawa L, Radhakrishnan K, Baumgartner M, Schmid R, Schmidt B, Dierks T, Gartner J. Rapid degradation of an active formylglycine generating enzyme variant leads to a late infantile severe form of multiple sulfatase deficiency. Eur J Hum Genet. 2013;21:1020–1023. doi: 10.1038/ejhg.2012.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dierks T, Schmidt B, von Figura K. Conversion of cysteine to formylglycine: a protein modification in the endoplasmic reticulum. Proc Natl Acad Sci U S A. 1997;94:11963–11968. doi: 10.1073/pnas.94.22.11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dierks T, Lecca MR, Schmidt B, von Figura K. Conversion of cysteine to formylglycine in eukaryotic sulfatases occurs by a common mechanism in the endoplasmic reticulum. FEBS Lett. 1998;423:61–65. doi: 10.1016/s0014-5793(98)00065-9. [DOI] [PubMed] [Google Scholar]
- 48.Fey J, Balleininger M, Borissenko LV, Schmidt B, von Figura K, Dierks T. Characterization of post-translational formylglycine formation by luminal components of the endoplasmic reticulum. J Biol Chem. 2001;276:47021–47028. doi: 10.1074/jbc.M108943200. [DOI] [PubMed] [Google Scholar]
- 49.Carlson BL, Ballister ER, Skordalakes E, King DS, Breidenbach MA, Gilmore SA, Berger JM, Bertozzi CR. Function and Structure of a Prokaryotic Formylglycine-generating Enzyme. J Biol Chem. 2008;283:20117–20125. doi: 10.1074/jbc.M800217200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhattacharyya S, Tobacman JK. Hypoxia reduces arylsulfatase B activity and silencing arylsulfatase B replicates and mediates the effects of hypoxia. PLoS One. 2012;7:e33250. doi: 10.1371/journal.pone.0033250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mariappan M, Radhakrishnan K, Dierks T, Schmidt B, von Figura K. ERp44 mediates a thiol-independent retention of formylglycine-generating enzyme in the endoplasmic reticulum. J Biol Chem. 2008;283:6375–6383. doi: 10.1074/jbc.M709171200. [DOI] [PubMed] [Google Scholar]
- 52.Fraldi A, Zito E, Annunziata F, Lombardi A, Cozzolino M, Monti M, Spampanato C, Ballabio A, Pucci P, Sitia R, Cosma MP. Multistep, sequential control of the trafficking and function of the multiple sulfatase deficiency gene product, SUMF1 by PDI, ERGIC-53 and ERp44. Hum Mol Genet. 2008;17:2610–2621. doi: 10.1093/hmg/ddn161. [DOI] [PubMed] [Google Scholar]
- 53.Gande SL, Mariappan M, Schmidt B, Pringle TH, von Figura K, Dierks T. Paralog of the formylglycine-generating enzyme – retention in the endoplasmic reticulum by canonical and noncanonical signals. FEBS J. 2008;275:1118–1130. doi: 10.1111/j.1742-4658.2008.06271.x. [DOI] [PubMed] [Google Scholar]
- 54.Mariappan M, Gande SL, Radhakrishnan K, Schmidt B, Dierks T, von Figura K. The Non-catalytic N-terminal Extension of Formylglycine-generating Enzyme Is Required for Its Biological Activity and Retention in the Endoplasmic Reticulum. J Biol Chem. 2008;283:11556–11564. doi: 10.1074/jbc.M707858200. [DOI] [PubMed] [Google Scholar]
- 55.Wilkinson B, Gilbert HF. Protein disulfide isomerase. Biochim Biophys Acta. 2004;1699:35–44. doi: 10.1016/j.bbapap.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 56.Ennemann EC, Radhakrishnan K, Mariappan M, Wachs M, Pringle TH, Schmidt B, Dierks T. Proprotein Convertases Process and Thereby Inactivate Formylglycine-generating Enzyme. J Biol Chem. 2013;288:5828–5839. doi: 10.1074/jbc.M112.405159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zito E, Buono M, Pepe S, Settembre C, Annunziata I, Surace EM, Dierks T, Monti M, Cozzolino M, Pucci P, Ballabio A, Cosma MP. Sulfatase modifying factor 1 trafficking through the cells: from endoplasmic reticulum to the endoplasmic reticulum. EMBO J. 2007;26:2443–2453. doi: 10.1038/sj.emboj.7601695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mariappan M, Preusser-Kunze A, Balleininger M, Eiselt N, Schmidt B, Gande SL, Wenzel D, Dierks T, von Figura K. Expression, localization, structural, and functional characterization of pFGE, the paralog of the Cα-formylglycine-generating enzyme. J Biol Chem. 2005;280:15173–15179. doi: 10.1074/jbc.M413698200. [DOI] [PubMed] [Google Scholar]
- 59.Dickmanns A, Schmidt B, Rudolph MG, Mariappan M, Dierks T, von Figura K, Ficner R. Crystal structure of human pFGE, the paralog of the Calpha-formylglycine-generating enzyme. J Biol Chem. 2005;280:15180–15187. doi: 10.1074/jbc.M414317200. [DOI] [PubMed] [Google Scholar]
- 60.Zito E, Fraldi A, Pepe S, Annunziata I, Kobinger G, Di Natale P, Ballabio A, Cosma MP. Sulphatase activities are regulated by the interaction of sulphatase-modifying factor 1 with SUMF2. EMBO Rep. 2005;6:655–660. doi: 10.1038/sj.embor.7400454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roeser D, Dickmanns A, Gasow K, Rudolph MG. De novo calcium/sulfur SAD phasing of the human formylglycine-generating enzyme using in-house data. Acta Crystallogr, Sect D: Biol Crystallogr. 2005;61:1057–1066. doi: 10.1107/S0907444905013831. [DOI] [PubMed] [Google Scholar]
- 62.Preusser-Kunze A, Mariappan M, Schmidt B, Gande SL, Mutenda K, Wenzel D, von Figura K, Dierks T. Molecular Characterization of the Human Cα-formylglycine-generating Enzyme. J Biol Chem. 2005;280:14900–14910. doi: 10.1074/jbc.M413383200. [DOI] [PubMed] [Google Scholar]
- 63.Roeser D, Preusser-Kunze A, Schmidt B, Gasow K, Wittmann JG, Dierks T, von Figura K, Rudolph MG. A general binding mechanism for all human sulfatases by the formylglycine-generating enzyme. Proc Natl Acad Sci U S A. 2006;103:81–86. doi: 10.1073/pnas.0507592102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roeser D, Schmidt B, Preusser-Kunze A, Rudolph MG. Probing the oxygen-binding site of the human formylglycine-generating enzyme using halide ions. Acta Crystallogr, Sect D: Biol Crystallogr. 2007;63:621–627. doi: 10.1107/S0907444907009961. [DOI] [PubMed] [Google Scholar]
- 65.Alayyoubi M, Guo H, Dey S, Golnazarian T, Brooks GA, Rong A, Miller JF, Ghosh P. Structure of the Essential Diversity-Generating Retroelement Protein bAvd and Its Functionally Important Interaction with Reverse Transcriptase. Structure. 2013;21:266–276. doi: 10.1016/j.str.2012.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Le Coq J, Ghosh P. Conservation of the C-type lectin fold for massive sequence variation in a Treponema diversity-generating retroelement. Proc Natl Acad Sci U S A. 2011;108:14649–14653. doi: 10.1073/pnas.1105613108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ho RN, Liebman J, Valentine J. Overview of the Energetics and Reactivity of Oxygen. In: Foote C, Valentine J, Greenberg A, Liebman J, editors. Active Oxygen in Chemistry. Springer; Netherlands: 1995. pp. 1–23. [Google Scholar]
- 68.Klinman JP. How Do Enzymes Activate Oxygen without Inactivating Themselves? Acc Chem Res. 2007;40:325–333. doi: 10.1021/ar6000507. [DOI] [PubMed] [Google Scholar]
- 69.Fetzner S, Steiner R. Cofactor-independent oxidases and oxygenases. Appl Microbiol Biotechnol. 2010;86:791–804. doi: 10.1007/s00253-010-2455-0. [DOI] [PubMed] [Google Scholar]
- 70.Murooka Y, Ishibashi K, Yasumoto M, Sasaki M, Sugino H, Azakami H, Yamashita M. A sulfur- and tyramine-regulated Klebsiella aerogenes operon containing the arylsulfatase (atsA) gene and the atsB gene. J Bacteriol. 1990;172:2131–2140. doi: 10.1128/jb.172.4.2131-2140.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Szameit C, Miech C, Balleininger M, Schmidt B, von Figura K, Dierks T. The iron sulfur protein AtsB is required for post-translational formation of formylglycine in the Klebsiella sulfatase. J Biol Chem. 1999;274:15375–15381. doi: 10.1074/jbc.274.22.15375. [DOI] [PubMed] [Google Scholar]
- 72.Benjdia A, Subramanian S, Leprince J, Vaudry H, Johnson MK, Berteau O. Anaerobic sulfatase-maturating enzymes, first dual substrate radical S-adenosylmethionine enzymes. J Biol Chem. 2008;283:17815–17826. doi: 10.1074/jbc.M710074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Grove TL, Ahlum JH, Qin RM, Lanz ND, Radle MI, Krebs C, Booker SJ. Further Characterization of Cys-Type and Ser-Type Anaerobic Sulfatase Maturating Enzymes Suggests a Commonality in the Mechanism of Catalysis. Biochemistry. 2013;52:2874–2887. doi: 10.1021/bi400136u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grove TL, Lee KH, St Clair J, Krebs C, Booker SJ. In vitro characterization of AtsB, a radical SAM formylglycine-generating enzyme that contains three [4Fe-4S] clusters. Biochemistry. 2008;47:7523–7538. doi: 10.1021/bi8004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goldman PJ, Grove TL, Sites LA, McLaughlin MI, Booker SJ, Drennan CL. X-ray structure of an AdoMet radical activase reveals an anaerobic solution for formylglycine post-translational modification. Proc Natl Acad Sci U S A. 2013;110:8519–8524. doi: 10.1073/pnas.1302417110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Benjdia A, Leprince J, Sandström C, Vaudry H, Berteau O. Mechanistic Investigations of Anaerobic Sulfatase-Maturating Enzyme: Direct Cβ H-Atom Abstraction Catalyzed by a Radical AdoMet Enzyme. J Am Chem Soc. 2009;131:8348–8349. doi: 10.1021/ja901571p. [DOI] [PubMed] [Google Scholar]
- 77.Dierks T, Miech C, Hummerjohann J, Schmidt B, Kertesz MA, von Figura K. Post-translational formation of formylglycine in prokaryotic sulfatases by modification of either cysteine or serine. J Biol Chem. 1998;273:25560–25564. doi: 10.1074/jbc.273.40.25560. [DOI] [PubMed] [Google Scholar]
- 78.Benjdia A, Dehò G, Rabot S, Berteau O. First evidences for a third sulfatase maturation system in prokaryotes from E. coli aslB and ydeM deletion mutants. FEBS Lett. 2007;581:1009–1014. doi: 10.1016/j.febslet.2007.01.076. [DOI] [PubMed] [Google Scholar]
- 79.Landgrebe J, Dierks T, Schmidt B, von Figura K. The human SUMF1 gene, required for post-translational sulfatase modification, defines a new gene family which is conserved from pro- to eukaryotes. Gene. 2003;316:47–56. doi: 10.1016/s0378-1119(03)00746-7. [DOI] [PubMed] [Google Scholar]
- 80.Townley RA, Bülow HE. Genetic Analysis of the Heparan Modification Network in Caenorhabditis elegans. J Biol Chem. 2011;286:16824–16831. doi: 10.1074/jbc.M111.227926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frese MA, Dierks T. Formylglycine aldehyde Tag–protein engineering through a novel post-translational modification. ChemBioChem. 2009;10:425–427. doi: 10.1002/cbic.200800801. [DOI] [PubMed] [Google Scholar]
- 82.Carrico IS, Carlson BL, Bertozzi CR. Introducing genetically encoded aldehydes into proteins. Nat Chem Biol. 2007;3:321–322. doi: 10.1038/nchembio878. [DOI] [PubMed] [Google Scholar]
- 83.Rush JS, Bertozzi CR. New Aldehyde Tag Sequences Identified by Screening Formylglycine Generating Enzymes in Vitro and in Vivo. J Am Chem Soc. 2008;130:12240–12241. doi: 10.1021/ja804530w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu P, Shui W, Carlson BL, Hu N, Rabuka D, Lee J, Bertozzi CR. Site-specific chemical modification of recombinant proteins produced in mammalian cells by using the genetically encoded aldehyde tag. Proc Natl Acad Sci U S A. 2009;106:3000–3005. doi: 10.1073/pnas.0807820106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rabuka D, Rush JS, deHart GW, Wu P, Bertozzi CR. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat Protoc. 2012;7:1052–1067. doi: 10.1038/nprot.2012.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rabuka D. Chemoenzymatic methods for site-specific protein modification. Curr Opin Chem Biol. 2010;14:790–796. doi: 10.1016/j.cbpa.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rashidian M, Dozier JK, Distefano MD. Enzymatic Labeling of Proteins: Techniques and Approaches. Bioconjugate Chem. 2013;24:1277–1294. doi: 10.1021/bc400102w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shi X, Jung Y, Lin LJ, Liu C, Wu C, Cann IKO, Ha T. Quantitative fluorescence labeling of aldehyde-tagged proteins for single-molecule imaging. Nat Methods. 2012;9:499–503. doi: 10.1038/nmeth.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ghoneim MK, Spies M. Direct Correlation of DNA Binding and Single Protein Domain Motion via Dual Illumination Fluorescence Microscopy. Nano Lett. 2014;14:5920–5931. doi: 10.1021/nl502890g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cho H, Jaworski J. A portable and chromogenic enzyme-based sensor for detection of abrin poisoning. Biosens Bioelectron. 2014;54:667–673. doi: 10.1016/j.bios.2013.11.058. [DOI] [PubMed] [Google Scholar]
- 91.Kwak EA, Jaworski J. Controlled surface immobilization of viruses via site-specific enzymatic modification. J Mater Chem B. 2013;1:3486–3493. doi: 10.1039/c3tb20526f. [DOI] [PubMed] [Google Scholar]
- 92.Cho H, Jaworski J. Enzyme directed formation of un-natural side-chains for covalent surface attachment of proteins. Colloids Surf, B. 2014;122:846–850. doi: 10.1016/j.colsurfb.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 93.Hudak JE, Yu HH, Bertozzi CR. Protein glycoengineering enabled by the versatile synthesis of aminooxy glycans and the genetically encoded aldehyde tag. J Am Chem Soc. 2011;133:16127–16135. doi: 10.1021/ja206023e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Smith EL, Giddens JP, Iavarone AT, Godula K, Wang LX, Bertozzi CR. Chemoenzymatic Fc Glycosylation via Engineered Aldehyde Tags. Bioconjugate Chem. 2014;25:788–795. doi: 10.1021/bc500061s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liang SI, McFarland JM, Rabuka D, Gartner ZJ. A Modular Approach for Assembling Aldehyde-Tagged Proteins on DNA Scaffolds. J Am Chem Soc. 2014;136:10850–10853. doi: 10.1021/ja504711n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hudak JE, Barfield RM, de Hart GW, Grob P, Nogales E, Bertozzi CR, Rabuka D. Synthesis of Heterobifunctional Protein Fusions Using Copper-Free Click Chemistry and the Aldehyde Tag. Angew Chem, Int Ed. 2012;51:4161–4165. doi: 10.1002/anie.201108130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chari RVJ, Miller ML, Widdison WC. Antibody–Drug Conjugates: An Emerging Concept in Cancer Therapy. Angew Chem, Int Ed. 2014;53:3796–3827. doi: 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- 98.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, Lu Y, Meng YG, Ng C, Yang J, Lee CC, Duenas E, Gorrell J, Katta V, Kim A, McDorman K, Flagella K, Venook R, Ross S, Spencer SD, Lee Wong W, Lowman HB, Vandlen R, Sliwkowski MX, Scheller RH, Polakis P, Mallet W. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925–932. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 99.Agarwal P, Bertozzi CR. Site-specific antibody-drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjugate Chem. 2014 doi: 10.1021/bc5004982. Online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Agarwal P, van der Weijden J, Sletten EM, Rabuka D, Bertozzi CR. A Pictet-Spengler ligation for protein chemical modification. Proc Natl Acad Sci U S A. 2013;110:46–51. doi: 10.1073/pnas.1213186110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Agarwal P, Kudirka R, Albers AE, Barfield RM, de Hart GW, Drake PM, Jones LC, Rabuka D. Hydrazino-Pictet-Spengler Ligation as a Biocompatible Method for the Generation of Stable Protein Conjugates. Bioconjugate Chem. 2013;24:846–851. doi: 10.1021/bc400042a. [DOI] [PubMed] [Google Scholar]
- 102.Drake PM, Albers AE, Baker J, Banas S, Barfield RM, Bhat AS, de Hart GW, Garofalo AW, Holder P, Jones LC, Kudirka R, McFarland J, Zmolek W, Rabuka D. Aldehyde Tag Coupled with HIPS Chemistry Enables the Production of ADCs Conjugated Site-Specifically to Different Antibody Regions with Distinct in Vivo Efficacy and PK Outcomes. Bioconjugate Chem. 2014;25:1331–1341. doi: 10.1021/bc500189z. [DOI] [PMC free article] [PubMed] [Google Scholar]