

Abstract
Background
The presence of methyl groups on cytosine nucleotides across an organism’s genome (methylation) is a major regulator of genome stability, crossing over, and gene regulation. The capacity for DNA methylation to be altered by environmental conditions, and potentially passed between generations, makes it a prime candidate for transgenerational epigenetic inheritance. Here we conduct the first analysis of the Mimulus guttatus methylome, with a focus on the relationship between DNA methylation and gene expression.
Results
We present a whole genome methylome for the inbred line Iron Mountain 62 (IM62). DNA methylation varies across chromosomes, genomic regions, and genes. We develop a model that predicts gene expression based on DNA methylation (R2 = 0.2). Post hoc analysis of this model confirms prior relationships, and identifies novel relationships between methylation and gene expression. Additionally, we find that DNA methylation is significantly depleted near gene transcriptional start sites, which may explain the recently discovered elevated rate of recombination in these same regions.
Conclusions
The establishment here of a reference methylome will be a useful resource for the continued advancement of M. guttatus as a model system. Using a model-based approach, we demonstrate that methylation patterns are an important predictor of variation in gene expression. This model provides a novel approach for differential methylation analysis that generates distinct and testable hypotheses regarding gene expression.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1668-0) contains supplementary material, which is available to authorized users.
Background
DNA cytosine methylation is an epigenetic modification that acts in conjunction with histone modification and small RNAs to regulate gene expression [1–3] and control transposable elements [4, 5]. In addition, DNA methylation appears to alter mutation rates [6] and to decrease rates of recombination [7]. It is found in organisms spanning the eukaryotic phylogeny [8, 9], and can occur in many sequence contexts. In plants, cytosine methylation can be found in CG, CHG, or CHH contexts, where H is any nucleotide besides G [10]. It appears that much of the methylome is stable within an individual; however, the methylome does exhibit predictable plastic responses to developmental and environmental cues [11, 12].
Recent work has greatly expanded our knowledge of the mechanisms involved in maintaining and modifying DNA methylation in plants [13–18], yet we still do not fully understand how specific patterns of DNA methylation in and near coding sequences control gene expression. In Arabidopsis thaliana, CG DNA methylation in regulatory sequences is negatively correlated with gene expression [3, 19], possibly through limiting promoter accessibility. Contrastingly, gene body CG methylation is elevated in moderate to highly expressed genes [3, 10, 20], potentially though the removal of histone variant H2A.Z [21]. Similar patterns of association between the distribution of plant CG methylation and gene expression have been found in the wild rice [20], tomato [22], and maize [23]. Additionally, Arabidopsis genes within differentially methylated regions tended to be more highly expressed in individuals with increased CG methylation, but lower in individuals with increased non-CG (CHG and CHH) methylation [24]. However, the interaction between gene expression and different forms of DNA methylation in and around genes has not been fully explored. For example, the impact of non-CG methylation on gene expression is especially understudied, despite its established role in regulating transposable elements through pre- and post-transcriptional silencing [25].
The standard method for characterizing genomic patterns of DNA methylation is to classify genes into methylation quantiles and then compare gene expression across these groups [3, 20, 22, 26–29]. Here, we adopt an explicit model-based approach, predicting gene expression from gene methylation and other basic gene-specific features (exon length, intron length, and exon number). We compare the methylome of an inbred line, to gene expression from a distinct recombinant inbred line, and test how well DNA methylation, in combination with other stable genetic factors, predict gene expression across lines and tissue types. The explanatory power of stable epigenetic variation on gene expression is relatively unknown (although see [30] for model-based approaches to predicting gene expression via promoter motifs in Saccharomyces cerevisiae, and [31] for a Sanger sequencing approach to gene expression modeling based on histone and DNA methylation in rice). With the model-based approach presented here, we are able to assess the scale to which constitutive epigenetic variation effects global gene expression, and the patterns of DNA methylation through which this regulation is manifest.
Previous studies of Mimulus guttatus have demonstrated transgenerational epigenetic inheritance [32–35]. Herbivore induced defensive traits can be transmitted between generations, and the observed transcriptional basis of this response [11], has made it a promising model system in the burgeoning field of ecological epigenetics [36–39]. However, along with identifying transmissible epigenetic marks, it is vital to understand the role that stable epigenetic regulation has on gene expression. Here we present the first M. guttatus methylome. We utilize a novel modeling approach to untangle the complex interactions between methylation and gene expression. We show that non-CG gene body methylation may have a significant effect on gene expression despite occurring at relatively low levels. Utilizing a GO term enrichment approach, we demonstrate that certain functional categories are over-represented in genes with high gene body CG methylation. We provide evidence that there are differences in methylation and gene expression between chromosomes, such that mean gene expression is significantly lower across some chromosomes than others. Finally, we look at transcriptional start sites across the genome, where recent evidence suggests increased recombination in M. guttatus [40], and find a corresponding decrease in DNA methylation.
Methods
DNA extraction and bisulfite sequencing
We germinated seeds from the M. guttatus Iron Mountain inbred line, IM62, the line that was sequenced to establish the M. guttatus reference genome [40] ( http://phytozome.jgi.doe.gov). When the second leaf pair of seedlings was just visible we collected leaf tissue from multiple seedlings, flash froze it in liquid nitrogen, and stored it at −80 °C. We preformed DNA extractions using a CTAB protocol [41]. We pooled DNA from multiple seedlings before library construction in order to limit the effects of aberrant intra-individual variation [42]. From this pooled sample we generated sequencing template for whole genome bisulfite sequencing (WGBS) following the PBAT (Post-Bisulfite Adaptor Tagging) protocol [43]. With 1 ng of unmethylated lambda DNA obtained from Promega used as a spike-in control for conversion efficiency, 100 ng of genomic DNA from M. guttatus was treated with bisulfite using EZ DNA Methylation kit from Zymo Research. Two rounds of random primer extension for tagging bisulfite treated DNA with adaptors were performed using primers for single-end library construction as described in [41]. The concentration of templates was determined by qPCR with Library Quantification Kits from KAPA biosystems. A single lane of 100 cycle reactions on HiSeq 2500 was assigned for the library sequencing.
Read mapping
We used the software BMap [43] (http://itolab.med.kyushu-u.ac.jp/BMap/index.html) to map bisulifte treated reads to the M. guttatus v2.0 reference genome (http://phytozome.jgi.doe.gov). In short, BMap first searches candidate genomic loci for each read in two duplicated genome sequences, one with every C in the genome converted to a T (C2T), and one with G to A (G2A), using an approach called adaptive seed [44]. Next BMap creates pairwise alignments between the read and original DNA sequence of every candidate loci, and calculates scores for each alignment allowing mismatches between T in the reads with C in the reference. Finally an alignment with the highest score is reported for each read. We used default parameters for mapping with BMap. Using alignments exported by BMap, methylation status for every cytosine in every read was called, and counts both supporting the methylated and unmethylated state are assigned for every cytosine residue of the reference genome. Methylation levels for CG, CHG and CHH contexts are exported to different files and analyzed independently.
Global methylome analysis
We estimated the number of total and methylated cytosines mapped across the genome on a per-nucleotide basis for the M. guttatus IM62 seedling methylome. Percent methylation was calculated for each 1 kb window across the genome for total methylation, as well as methylation in each of the three sequence contexts. Centromere positions were estimated from characteristic repeat sequences [45].
Gene methylation analysis
Using the M. guttatus v2.0 annotations [46], we calculated the percent methylation in each sequence context for each of the 24,130 annotated genes. Only the 17,043 for which we had gene expression data [32] were used for down-stream analysis. For each annotated gene we defined three regions: up-stream as the 1kb up-stream of the transcriptional start site, gene body as the transcribed portion of the gene, and down-stream as the 1kb downstream of the 3′ UTR. Gene expression values were generated previously by RNAseq from seedling tissue of genetically distinct M. guttatus – a recombinant inbred line derived from cross between divergent populations [32].
In order to determine if gene methylation and expression varied across chromosomes we preformed four ANOVAs with chromosome as an explanatory variable and CG, CHG, CHH, and log-transformed gene expression as response variables.
Gene ontology terms were already assigned to genes [32], and were utilized both to calculate the total number of GO terms per gene, as well as to perform a Fisher’s Exact test to determine what, if any, types of genes were enriched or depleted in our set of highly CG methylated genes, and our set of chromosomes exhibiting significantly reduced gene expression levels.
In order to choose a predictive gene expression model, we included methylation in each of three contexts, percent methylation in gene bodies, up-stream and down-stream regions, intron length (sum of all introns for a gene), exon length (sum of all exons for a gene), number of exons, and interaction terms up to the third degree. Gene length, intron size, and intron number are all known to be positively correlated with gene expression in plants [47], opposite the trend observed in animals [48]. We used a Bayesian information criterion (BIC) [49] to inform our restricted maximum likelihood (REML) model selection (done in order to limit the number of parameters included in our model, and in turn reduce over fitting). Additionally, genes were parsed randomly into thirds, and parameters were tunes for each of these three groups independently. These models were then used to predict gene expression in the remaining to gene groups to provide 3-fold cross-validation [50]. We Z-transformed values to make parameter estimates comparable, making a value of 0 represent the mean value for a variable, with positive or negative deviations reflecting the number of standard deviations a value is from the mean.
We identified transposable elements across the M. guttatus genome from the repeat-masked genome assembly [46]. Genomic repeats larger than 100 base pairs were selected and percent methylation in all three sequence contexts was identified for these repeats.
Results and discussion
Global methylation
Of the 186 million reads generated, 126 million were mapped to the genome (67.7 % mapping, mean read depth = 19, median = 6). This proportion is typical for Mimulus genomic studies eg. [51] given the substantial proportion of the physical genome that is not contained in the v2 reference genome. Mapping to unmethylated lambda DNA confirmed that our PBAT treatment achieved 99.4 % conversion of unmethylated cytosines to thymine. Methylation is most common in a CG context (72 %), intermediate in a CHG context (36.5 %), and lowest in a CHH context (6.1 %) (Fig. 1), with 23 % of total cytosine’s being methylated. The percent of genome methylation found in M. guttatus is higher in all contexts than Oryza sativa [20], Arabiopsis thaliana [8], Brachypodium distachyiom [27], lower in all contexts than Solanum lycopersicum [22], and both higher or lower than Zea mays [26] and Glycine max [52] depending on context (Fig. 1). While CHH methylation levels are higher in M. guttatus than Z. mays and G. max, the opposite is true for CHG methylation. CG methylation is highest in Z. Mays, moderate in M. guttatus, and lowest in G. max (Fig. 1).
Fig. 1.
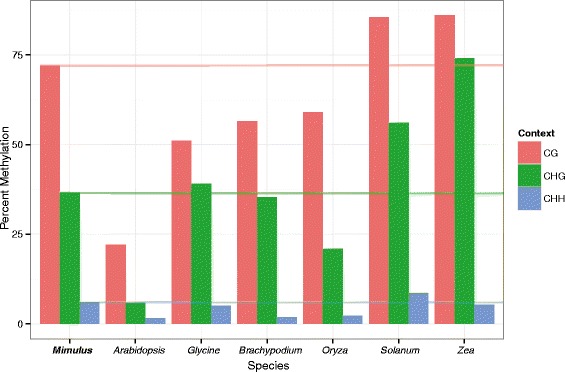
Interspecific comparison of plant DNA methylation levels. A comparison of global DNA methylation levels in CG (red), CHG (green), and CHH (blue) sequence contexts found in Mimulus guttatus compared with those of Arabidopsis thaliana [66], Glycine max [52], Brachypodium distachyiom [27], Oryza sativa [20], Solanum lycopersicum [22], and Zea mays [26]
Approximate positions of centromeres on M. guttatus chromosomes are given by the location and density of centromeric repeats [45]. We confirmed that regions of the genome with high levels of centromeric repeats also tended to have high CG, CHG, and CHH methylation (Fig. 2). We found that gene expression and gene body CG, CHG, CHH methylation varied significantly across chromosomes (log(expression): F13,17042 = 4.43, CG: F13,17042 = 10.85, CHG: F13,17042 = 19.07, CHH: F13,170423 = 6.10, p < 0.001)). Chromosomes that have on average higher levels of methylation tended to also have lower gene expression (Fig. 3). From this result, it is unclear whether certain chromosomes are constitutively more highly methylated and transcriptionally silenced, or whether throughout development epigenetic modification at a whole chromosome scale can change the relative expression of genes across entire chromosomes. It does appear that silenced chromosomes have a higher density of heterochromatic repeats, hinting that certain chromosomes may be condensed throughout development.
Fig. 2.
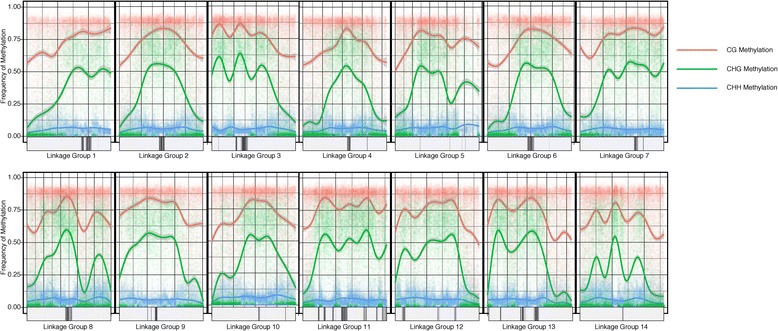
DNA methylation across the Mimulus guttatus genome. DNA methylation across the 14 Mimulus guttatus linkage groups (putative chromosomes) in all three sequence contexts: CG (red), CHG (green), and CHH (blue). Centromeric repeat densities, adapted from [45], are shown along the X-axis (darker bars indicate higher repeat density). Areas with higher repeat density tend to also have higher DNA methylation. A smoother line [67] was fit across 1kb methylation averages
Fig. 3.
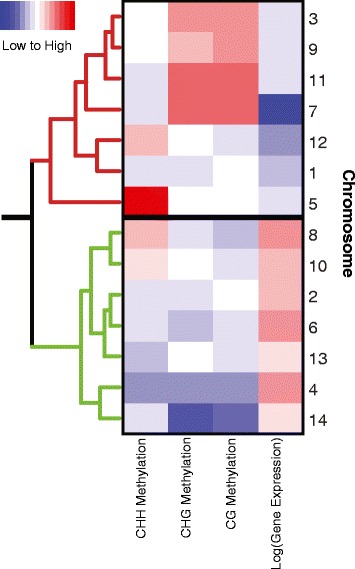
Variation in methylation and expression across chromosomes. A heatmap showing variation in gene expression and methylation across the 14 Mimulus guttatus putative chromosomes. The 14 chromosomes clustered into two large groups, those with generally high methylation and low gene expression (top cluster, red dendogram), and those exhibiting the opposite pattern (bottom cluster, green dendogram). On the heat map, red indicates high values and blue indicates low values of methylation or gene expression
Gene methylation
Methylation was significantly depleted in gene bodies relative to both inter-genic regions and transposable elements in all three-sequence contexts (Table 1). While CG methylation was only modestly reduced in gene bodies relative to intergenic regions (Gene Bodies: 56 %, Intergenic: 75 %), CHG (Gene Bodies: 3.8 %, Intergenic: 45 %) and CHH (Gene Bodies: 1.2 %, Intergenic: 7.2 %) methylation levels were drastically reduced (Table 1). Similar results were found in Oryza sativa [20], Arabidopsis thaliana [53], and Glycine max [52]. Methylation both up-stream and down-stream of gene starts was also reduced relative to genome-wide averages. We found that up-stream regions were elevated in non-CG methylation compared to gene bodies, but that up-stream CG methylation was reduced compared to gene body CG methylation (Table 1).
Table 1.
Mimulus guttatus methylation across sequence contexts and genomic regions
| Proportion of cytosines methylated | |||
|---|---|---|---|
| CG | CHG | CHH | |
| Transposable Elements | 0.73 | 0.36 | 0.063 |
| Gene Body | 0.56 | 0.038 | 0.012 |
| 1st 500bp of Gene Body | 0.28 | 0.032 | 0.019 |
| Up-stream Regulatory | 0.35 | 0.11 | 0.027 |
| Inter-Genic Regions | 0.75 | 0.45 | 0.072 |
| Total | 0.72 | 0.365 | 0.061 |
The methylation levels in all contexts (CG, CHG, CHH) and genic positions (up-stream, down-stream, and gene body) at a given gene were significantly correlated with one another (Fig. 4). These were positive correlations for all cases but two. The two exceptions were negative correlations between up- and down-stream CHH methylation with gene body CG methylation. The most significant positive correlations were found between CHG and CHH or CG methylation levels at both up-stream and down-stream regions, as well as between CHG and CHH gene body methylation. Interestingly, the methylation levels for all three contexts vary greatly across the three gene regions in a fairly unpredictable manner. For instance, correlation between up-stream CG methylation and gene body CG methylation is only r = 0.14. This highlights the disparate functions of regulatory region methylation with that of gene body methylation [54]. The extremely high correlations between CHG and CHH methylation (Fig. 4, r > 0.67) in all three regions is likely due to the involvement of similar enzymatic machinery in the propagation of both types of non-CG methylation [55].
Fig. 4.
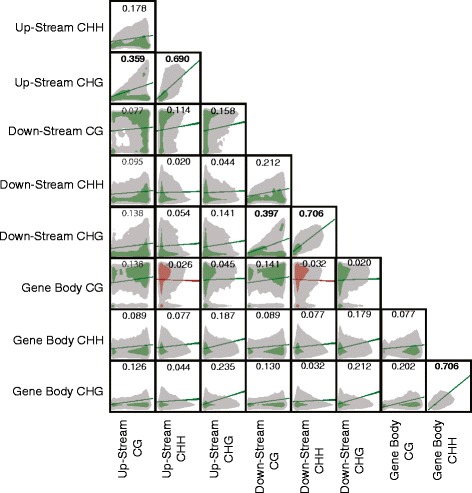
Correlation matrix between forms of methylation at individual genes. Clouds represent density, and lines show the slope of the correlation. Green lines indicate forms of methylation with a positive correlation, while red represents negative correlation. Numbers represent the Pearson correlation (r) value, bolded numbers highlight correlations with an r > 0.35. All correlations were found to be statistically significant (n = 17,038, p < 0.05)
Methylation effect on gene expression
A stepwise cubic polynomial model was selected to predict log(gene expression) based on minimum BIC. Out of a possible 454 parameters, the minimum BIC criterion selected a model with 29 factors that explained (R2) 20.1 % of the variation in log transformed expression values (SS Model: 1764, SS Error: 6981, F28,17042 = 153.6, p < 0.0001, Tables 2, 3 and 4, Fig. 5, Additional file 1: Figure S1). Including all 454 parameters increases R2 only marginally (to 23.3 %), and the minimum calculated R2 calculated in 3-fold cross-validation was 17.9 %. Generally, there is an excess of genes predicted to be expressed at log-transformed values between 1.5 and 2.5, that were actually expressed at levels less than 1.2, as well as genes expressed above 4, which this model never predicts (Additional file 1: Figure S1). It is clear that while gene methylation can modify gene expression, it cannot predict the complete repression, or extremely high expression of some genes. As all parameters were Z-transformed prior to modeling, the effect estimates are comparable across variables (Table 4). In order to maintain both statistical and molecular consistency throughout, both Z-transformed values and raw values are reported. The inclusion of both various forms of DNA methylation and gene architecture (number of exons, exon length, intron length) have not been included in a single model explicitly testing their ability to predict gene expression, but their independent effects have often been looked at in relation to gene expression. While it is hard to compare our integrative analysis on gene expression with prior studies, we generally find the same direction of effect in our data as was found in other plant systems [3]. Trends are thus not Mimulus specific, but likely more general effects of DNA methylation on gene expression in angiosperms. Finally, when discussing the role of various forms of methylation on gene expression we often designate a specific type of methylation as having a positive or negative effect on gene expression. In this context that indicates that there was significant predictive ability for a given type of methylation on gene expression. However, due to the nature of this experimental design we cannot definitively define the arrow of causation.
Table 2.
Summary of REML genetic architecture and methylation fit on log transformed gene expression
| R-Square | 0.201775 |
| R-Square Adj. | 0.200462 |
| Root Mean Square Error | 0.640578 |
| Mean of Response | 2.483507 |
Table 3.
Analysis of variance in gene expression predictive model
| Source | DF | Sum of Squares | Mean Square | F Ratio |
|---|---|---|---|---|
| Model | 28 | 1764.7903 | 63.0282 | 153.6 |
| Error | 17014 | 6981.5210 | 0.4070 | Prob > F |
| C. Total | 17042 | 8746.3113 | p < 0.0001 |
Table 4.
Sorted estimate of parameter effects on log transformed gene expression
| Positive Terms | Estimate | Std Error | t Ratio | Prob > |t| |
|---|---|---|---|---|
| Intron Length | 0.3472 | 0.0117 | 29.75 | <.0001 |
| Gene Body CHG2 | 0.0874 | 0.0082 | 10.64 | <.0001 |
| Number of Exons*Intron Length | 0.0793 | 0.0081 | 9.74 | <.0001 |
| Exon Length | 0.0767 | 0.0102 | 7.55 | <.0001 |
| Exon Length* Intron Length | 0.0553 | 0.0070 | 7.86 | <.0001 |
| Gene Body CG * Exon Length | 0.0392 | 0.0089 | 4.4 | <.0001 |
| Gene Body CG2 * Exon Length | 0.0303 | 0.0069 | 4.37 | <.0001 |
| Up-Stream CHH | 0.0275 | 0.0055 | 4.99 | <.0001 |
| Gene Body CG*Gene Body CHH | 0.0244 | 0.0064 | 3.78 | 0.0002 |
| Down-stream CHH | 0.0185 | 0.0050 | 3.69 | 0.0002 |
| Up-stream CHH* Percent CG | 0.0167 | 0.0051 | 3.28 | 0.0011 |
| Intron Length3 | 0.0105 | 0.0007 | 14.4 | <.0001 |
| Exon Length2 * Number of Exons | 0.0074 | 0.0009 | 8.19 | <.0001 |
| Negative Terms | ||||
| Gene Body CHG | −0.3273 | 0.0197 | −16.58 | <.0001 |
| Intron Length2 | −0.1611 | 0.0076 | −21.18 | <.0001 |
| Gene Body CG2 | −0.0980 | 0.0092 | −10.62 | <.0001 |
| Gene Body CG | −0.0720 | 0.0118 | −6.09 | <.0001 |
| Exon Length * Number of Exons | −0.0662 | 0.0076 | −8.72 | <.0001 |
| Gene Body CHH | −0.0451 | 0.0076 | −5.93 | <.0001 |
| Number of Exons | −0.0308 | 0.0112 | −2.75 | 0.0059 |
| Percent CG3 | −0.0277 | 0.0059 | −4.73 | <.0001 |
| Up-Stream CG | −0.0274 | 0.0054 | −5.06 | <.0001 |
| Exon Length2 | −0.0205 | 0.0033 | −6.28 | <.0001 |
| Gene Body CHG * Exon Length | −0.0198 | 0.0058 | −3.41 | 0.0007 |
| Up-stream CG* Up-stream CHH | −0.0188 | 0.0058 | −3.23 | 0.0012 |
| Up-stream CG* Gene Body CG | −0.0170 | 0.0052 | −3.28 | 0.001 |
| Exon Length * Intron Length * Number of Exons | −0.0118 | 0.0016 | −7.21 | <.0001 |
| Gene Body CHG3 | −0.0063 | 0.0008 | −7.95 | <.0001 |
*Superscripts represent the power to which a term is raised
Fig. 5.
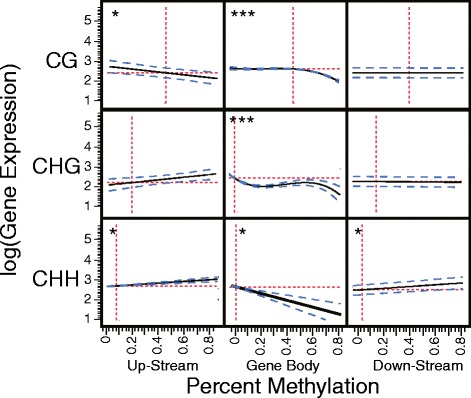
Correlations between DNA methylation and gene expression. A single star represents a significant linear correlation, two stars a significant second-order correlation, and three stars a third order correlation. The red dashed lines represent the means, the black line represents the regression line, and the blue line represents 95 % confidence intervals
Gene body CG methylation
Linear Effects: log(GE) = 2.61 − 0.07mcg = f 1, where mcg is gene body CG methylation and GE is gene expression. Controlling for gene architecture and other forms of methylation, we observe a negative linear effect of gene body CG methylation on gene expression (Figs. 5 and 6a. black line). The effect size of gene body CG methylation (mcg) is −0.07 (Table 3); a gene with mcg = − 1 (32 %) is predicted to have 35 % higher expression than one with mcg = 1 (80 %) (Fig. 6a, black line). Previous studies report that gene body CG methylation is positively correlated with gene expression [3, 10, 19, 20]. While the linear component of the model seems to contradict these previous reports, it cannot be interpreted in isolation. The polynomial and interaction terms indicate that gene body methylation has neither universally positive nor negative effects on gene expression. Traditional methods that looked for associations between gene expression and gene body CG methylation (which find a positive correlation between the two), and modeling methods as applied here followed by only analysis of the simple linear terms (which finds a negative correlation) come up quite short in portraying the role of gene body CG methylation in transcriptional regulation. By considering non-linear effects of methylation on gene expression we can begin to increase our understanding of the role of gene body CG methylation in gene regulation.
Fig. 6.
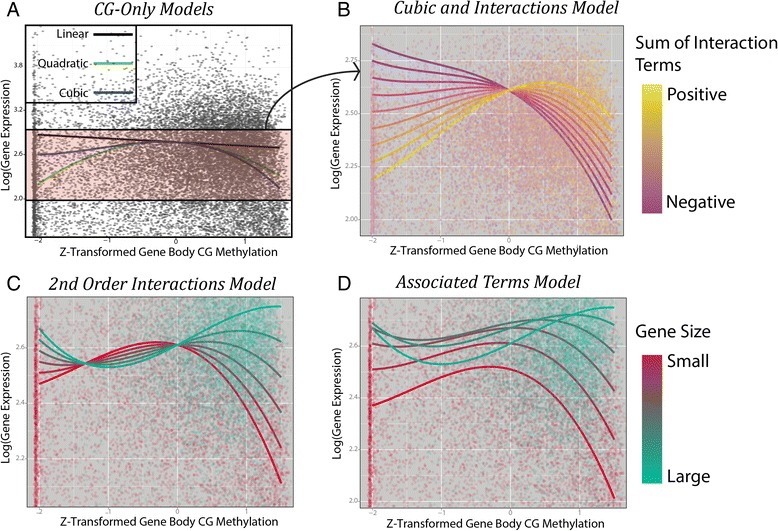
DNA methylation modeling to predict gene expression. A visual depiction of our simplified model showing the effect of gene body CG methylation and an increasing complexity of interaction terms on gene expression. a A scatterplot comparing Z-transformed gene body CG methylation values with log(gene expression) values. The black line shows the linear term, green line includes both the linear and quiadratic term, and the blue line includes linear, quadratic, and cubic terms. b Interaction plot depicting the interaction between gene CG methylation and exonlength, up-stream CHH methylation, gene body CHH methylation, and gene body CG methylation on gene expression. Summed terms across these four terms are considered ranging from −1.6 (dark purple) to 1.6 (yellow). Points represent actual genes CG gene body methylation, gene expression, and their color represents their interaction sum on the same scale as the model colors. c The second order interaction term of squared gene body CG methylation by exon length is added to the model depicted in b. As exon length increases (goes from red to blue) gene body CG methylation is found to have a more positive effect on gene expression. Points represent genes, and colors represent the exon length of these genes on the same scale as the model colors. d The independent effect of exon length on gene expression is added to the model depicted in c. The shape of the lines does not change, however predicted gene expression is altered (the lines move up or down on the y-axis) depending on the predicted effects of exon length on gene expression
Quadratic Effects: f 1 − 0.1m2cg = f 2. The squared gene body CG methylation term has the second largest effect size of any methylation term (after gene body CHG methylation) on gene expression, and leads to a predicted local mcg maximum for gene expression (due to it being a negative parabola, Fig. 6a, green line). This maximum is found at mcg = − 0.35 (47 %). As gene methylation increases or decreases relative to a moderate 45 % methylation, gene expression is expected to decrease (Fig. 6a; green line).
Cubic Effects:f 2 − 0.03m3cg = f 3. The cubed gene body CG methylation term is also negative; compared to our quadratic model, this leads to higher predicted gene expression for genes with lower than average methylation, and lower for genes with higher than average methylation. This slightly lowers the predicted local maximum of gene expression to mcg = − 0.43 (45 %) (Fig. 6a, blue line). These data agree with previous findings that there is a non-linear relationship between gene body CG methylation and gene expression with an intermediate optimum [3].
Interaction terms
Negative Promoter CG Methylation Interaction:f 3−.02mcgucg = f 4. The effect of interaction terms in this model is best understood by comparing expected gene expression across mcg values for a variety of interaction term values. Changes in linear interaction term values (in this case up-stream cg methylation ucg), lead to a change in our linear mcg coefficient. For example, at ucg = 1 (82 %), 0.2mcg is subtracted from our earlier model, we are left with:
At ucg = 1 (82 %) we find that the local maximum for gene expression is at mcg = − 0.62 (40.2 %), while at ucg = − 1 (24.1 %) the local maximum for gene expression is at mcg = − 0.29 (48.8 %). As up-stream CG methylation decreases (Fig. 6b, from purple to yellow lines), gene body CG methylation is expected to have a more positive effect on gene expression.
While it has long been noted that regulatory region methylation is linked with reduced gene expression, here we find evidence that the difference in methylation between these regions also appears to correlate with gene expression. The negative interaction term between up-stream and gene body CG methylation predicts that distinctly different levels of methylation up-stream and within genes tends to correspond with higher levels of gene expression. When gene body CG methylation and regulatory methylation are both high, gene expression tends to be low (Fig. 6b, purple lines at high gene body CG values). However, as either decreases (Fig. 6b, purple lines at low gene body CG values, or yellow lines at high CG methylation values), gene expression is expected to increase.
Three positive interaction terms:f 4 + mcg(.02uchh+.02mchh+.04lexon) = f 5: While only up-stream CG methylation showed a negative interaction with gene body CG methylation, three terms have positive linear interactions: Up-stream CHH methylation, gene body CHH methylation, and exon length. These can be treated in much the same way as our negative interaction term. Depending on the values of these terms, they can offset each other and lead to the removal of any interaction effect. For example if exon length (lexon) = − 1 and up-stream (uchh) and gene body (mchh) CHH methylation = 1 these positive interaction terms cancel out (−.4 + .2 + .2 = 0). However, if we consider them varying in the same direction, they can have a striking effect on the relationship between gene body CG methylation and gene expression. At uchh = mchh = lexon = 1 (and the negative interaction term ucg = 0), we see the local maximum is at a methylation level of mcg = − 0.05 (55.1 %). If our negative interaction term ucg = − 1 , this increases to mcg = + 0.05 (57.3 %) (Fig. 6b, varying the values of our interaction terms, uchh = mchh = lexon = − ucg from −1.6 to 1.6, as the summed interaction term increases (lines become yellow) the local maxima for gene expression does so as well). When uchh = mchh = lexon = − ucg < 0, gene body CG methylation is almost purely repressive. At a summed interaction value less than −0.7 there is no longer a local maximum, and CG methylation has a purely negative effect on gene expression.
Quadratic Interaction Terms: log(GE) = f 5+.03m2cglexon = f 6. Finally the interaction between the quadratic gene body CG methylation term and exon length is included in this model. As our quadratic term increases, not only does the position of the local gene expression maximum increase, so to does the inflection point (the point at which the function changes from concave to convex). Now, at the same linear interaction values tested above (uchh = mchh = lexon = − ucg = 1), our local maximum occurs at mcg = 0.07 (57.8 %) (Fig. 6c). As exon sizes increase, the effect of gene body CG methylation is expected to rapidly become more positive, and peak gene expression is predicted to occur at higher mcg levels. At lexon = 3 (3.5 kb), we find that the local maximum for gene expression occurs at mcg = 0.90 (78.1 %) and at lexon = 4 (6kb) there is no longer a local maximum for mcg, and the highest expected gene expression occurs at mcg approaching 100 % (largest gene size in Fig. 6c). It appears that for genes with smaller exons, moderately methylated genes are most highly expressed, but as genes become larger so to does the level of gene methylation that is associated with more highly expressed genes. Our gene size by gene body CG methylation results confirm a pattern observed by Zilberman et al. [3] in the first genome-wide methylome analysis in Arabidopsis in which found only a marginal relationship between gene size and gene expression, except for the genes in which gene bodies were methylated and then they found a positive relationship between gene size and gene expression.
Individual effects of interaction terms: log(GE) = f 5 + .08lexon − .02l2exon = f 6: Finally, we consider the effect of multiple terms simultaneously. Up until this point we have only included gene body CG methylation effects, and its interaction terms, while not including the independent effects of the term with which it interacts. Independent of gene body CG methylation, we find that gene expression tends to increase as the standardized exon length increases from −1 (500bp) to 2 (2kb), and beyond this point we expect a decline. In the absence of interaction terms, only considering independent effects of gene body CG methylation and exon length, we would estimate that peak gene expression occurs at an exon length of 2kb, and methylation of 45 %. Here we show that the effect of gene body CG methylation on expression is extremely size dependent, and that gene expression is expected to be highest for large highly gene body CG methylated genes, but lowest for small highly gene body CG methylated genes (Fig. 6d). It may be that as exon length increases, gene methylation is necessary to stabilize transcription, while for smaller genes it is not necessary for this purpose, and rather plays a repressive effect due to condensing chromatin near the transcription start site.
In this same way all other independent and interaction terms could be added to this model, parameters considered, and hypotheses tested. As nine distinct parameters are included (with 27 total terms) in this model the results quickly become difficult to conceptualize or visualize, yet through full-model construction, followed by simplification methods as presented above it is possible to decipher complex higher order regulatory interactions. We briefly discuss the effects of the other significant gene size and methylation terms in this model.
Intron length
Intron length shows significant first, second, and third order effects with a gene expression peak at an intron size of approximately 1700 base pairs. Additionally, a positive interaction term with both exon length and number of introns suggests that generally, longer genes with more introns tend to be more highly expressed. Although relatively large genes do tend to be most highly expressed, there are negative quadratic terms for both exon and intron length that suggest after a certain point, increasing exon and intron length should be associated with decreased gene expression.
Non-CG gene body methylation
Gene body CHG methylation had significant linear, quadratic, and cubic independent terms, and an exon length interaction term. Gene body CHG methylation has a negative effect on gene expression across nearly its full range of possible values (Fig. 5), and it appears that it is the increase from no CHG methylation to slight CHG methylation that reduces gene expression. After this point the effect of CHG methylation appears to be minimal. The negative exon length interaction term suggests that long genes with CHG methylation tend to be more significantly repressed than smaller genes.
Gene body CHH methylation was found to have a negative effect on gene expression (Fig. 5), but a positive interaction with gene body CG methylation. Thus, as gene body CHH methylation increases, gene body CG methylation is expected to have a more positive effect on gene expression, but mean gene expression, independent of gene body CG methylation, is expected to decrease. Like CHG methylation, a manual inspection reveals that the jump from no CHH methylation to low levels of CHH methylation leads to a decrease in gene expression, but after this, the effects of increased methylation are minimal.
While it has been suggested that non-CG gene body methylation may be misattributed to genomic regions that are actually pseudogenes or paralogs [52, 56], here we find evidence that in at least some cases these genes are still expressed, albeit at lower levels than non-methylated genes. One possible explanation is that non-CG methylation of genes may be a first step on the path toward pseudogenization [57], whereby genes become targeted by non-CG methylation, gene expression is reduced, mutational constraints become lightened, and eventually the gene becomes entirely non-functional. Additionally, it may be that tightly developmentally controlled small RNAs are responsible for the majority of this methylation, and the use of identical tissue for methylation and gene expression analysis would identify a stronger role of gene body non-CG methylation on gene expression. Finally, even trace amounts of non-CG gene body methylation may be indicative of the presence of small RNAs, and RNA-directed DNA methylation (RdDM) [58]. It could be that the methylation of just a few nucleotides by a single 24nt siRNA is enough to reduce gene expression, without significantly altering the methylation state of the whole gene.
Regulatory region methylation
Along with a negative interaction with gene body CG methylation, up-stream CG methylation also has a direct negative effect on gene expression (Fig. 5) and a negative interaction with up-stream CHH methylation. Not only does up-stream CG methylation limit the positive effect of gene body CG methylation on predicted gene expression, it also directly reduces predicted expression. Up-stream CHH methylation has both a significant positive linear effect on gene expression (Fig. 5), and a positive interaction with gene body CG methylation. The negative interaction term with up-stream CG methylation suggests that while up-stream CHH methylation generally has a positive effect on gene expression, when it is found alongside CG methylation, this effect is negated. While down-stream CHH methylation did not interact with gene body CG methylation, it was also found to have a positive effect on gene expression (Fig. 5).
A previous study in Arabidopsis similarly found that there was a positive correlation between gene expression and regulatory CHH methylation (albeit not in a regression framework) [26]. They posit that as gene expression increases, unstable transcripts are produced as by-products at both the 5′ and 3′ ends of genes. In turn, this lead to the production of small RNAs that can target and cause CHH methylation bracketing highly expressed genes through RNA directed DNA methylation (RdDM). The possibility that increased gene expression causes increased regulatory CHH methylation, and not vice-versa does not introduce bias in this framework, but rather reinforces that our interpretations do not imply causality.
Gene expression modeling overview
While the traditional method of looking for simple associations between methylation state and gene expression has provided some insight into epigenetic regulation, here we demonstrate that modeling approaches can provide additional insight into these systems. We explain a surprisingly high (20.1 %) amount of the variation in log(gene expression) simply through methylation and gene architecture variation. We considered a potential 454 parameters in our model before settling on 29, but it is important to note that many other factors such as presence of enhancers within the gene body and distance to transposable elements, likely also modify the role of methylation on expression. By considering exon and intron length within this model we take the first steps to account for these potential confounding factors of methylation on expression. It is worth stressing that the gene expression and methylome data were not only collected from different individuals, but also different genetic lines, using different vegetative tissue types, and grown under slightly different greenhouse conditions. It is certainly possible that a similar model, tuned across multiple paired methylome and gene expression samples, could predict gene expression with greater precision. This portion of gene expression variation explained represents that which is at least relatively stable across individual genotypes, tissue, and conditions. While here we apply this model to gene expression on a gene-by-gene basis, through altering the response variable to another parameter of a gene, such as it’s mutation rate, gene expression variance, or the tissues in which it is expressed, this model could be extended to look for other roles of DNA methylation on gene function and evolution.
Results from this and other [3, 20, 29, 59] studies suggest that gene body CG methylation needs to be considered to have a quadratic effect on gene expression, and that this effect is highly dependent on exon size. Thus, genes can either be parsed according to exon length prior to estimating the role of gene body CG methylation on expression, or the interaction between exon length and methylation should be considered in the model. Other forms of methylation appear to have a more straightforward role in regulating gene expression, and in some cases it may suffice to predict that, for example, as up-stream CG methylation increases at a gene, its expression will likely decrease.
Gene ontology analysis of genes with high CG gene body methylation
Comparing genes in the top 10 % genome wide for gene body CG methylation with the remainder of the genome, we found numerous gene categories that are either enriched or depleted in our set of highly CG methylated genes. Genes coding for proteins with kinase activity, involved in signal transduction, and nucleotide binding were among those which tended to be highly methylated, while proteins functioning in the thylakoid, plastid, and ribosome, as well as proteins involved in primary metabolism, photosynthesis, and RNA binding tended to be lowly or moderately methylated (Fig. 7). Similar results have been found in Brachypodium, rice [29], and Arabidopsis [3].
Fig. 7.
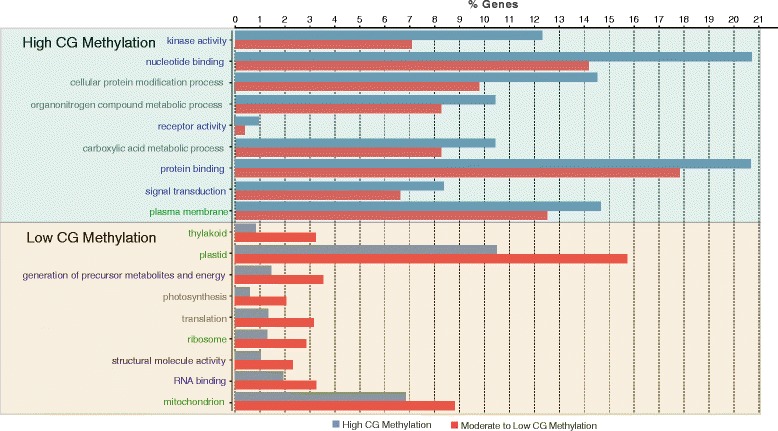
Gene Ontology classes over and underrepresented in highly gene body methylated genes. Genes Ontology terms significantly enriched and depleted in genes in the top 10 % for gene body CG methylation. X-axis shows the percent of genes in both the high CG methylated portion (blue) as well as the remainder of the transcriptome (red) that contained the given GO terms. Text color represents the class of GO term: blue-molecular functions, grey-biological processes, and green-cellular component
Decreased methylation near transcription start sites
We looked for changes in methylation near gene transcription start sites. We found that CG, CHG, and CHH methylation all were significantly depleted at and around gene start sites (Fig. 8). This depletion, along with the negative interaction term between up-stream and gene body CG methylation on gene expression, points towards a role of methylation in epigenetically labeling coding genetic regions. Additionally, recent evidence has shown that in M. guttatus genetic recombination occurs at higher frequency near gene start sites. In other systems it has been shown that DNA methylation is negatively correlated with recombination [7], and it may be that decreased methylation at gene start sites is related to the increase in recombination.
Fig. 8.
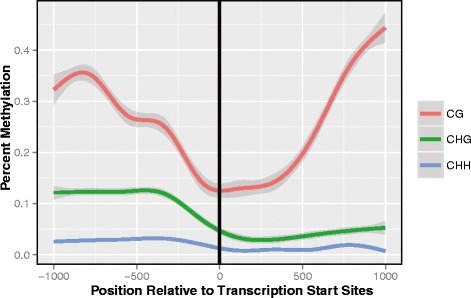
DNA methylation around transcriptional start sites. Around gene start sites, and persisting into the first 500 base pairs of the gene body, we observe a significant drop in DNA methylation. For CG (p-value = 6.45 × 10−55), CHG (p = 3.38 × 10−100), and CHH (p = 4.61 × 10−308) methylation was significantly reduced in the first 500bp of the gene relative to the up-stream regions. Both CG (p = 3.55 × 10−138) and CHG (p = 4.04 × 10−11) methylation then significantly increases over the next 500 bp, while for CHH it continued to decline (p = 8.93 × 10−10)
Decreased methylation near transcription start sites (TSS) was one of the earliest discovered phenomena of gene methylation [3]. However, new evidence in M. guttatus [40] provides us with a novel framework in which to view this pattern. Hellsten et al. [40] identified an approximately two-fold increase in recombination near gene start sites (the beginning of exon 1 being most enriched), and postulated that this may be related to nucleosome depleted open chromatin at these regions as is the case in Arabidopsis [60] and rice [61]. At the time of their publication however, there was no evidence for a similar trend in Mimulus. Here, evidence of depleted methylation near TSS (Table 1; Fig. 8) provides support to the theory that open chromatin (unmethylated) near TSS may increase local recombination rates. It appears that at least in yeast double stranded breaks occur most frequently in open chromatin regions [62], which may explain the observed increase in recombination near transcription start sites. It is likely that the increased recombination near TSS is simply a by-product of the dual forces exerted by DNA methylation, one involved in gene regulation, and another limiting double stranded breaks. The ability for DNA methylation to alter both of these processes provides an interesting link between gene regulation and DNA recombination that may or may not prove to be of evolutionary significance. Further studies linking methylation and recombination at a nucleotide level should further clarify this trend.
Transposable element methylation
We identified 1,411 transposable elements across the genome ranging in copy number from 1 to 2,380 (median copy number = 7). Percent methylation was calculated in each of three sequence contexts. In total, 34 % of the M. guttatus genome was estimated to be of transposable element sequence, and methylation levels within transposable elements were significantly higher than that of genes, and at similar levels to inter-genic regions (Table 1). We did not find there to be a significant copy number effect on TE methylation. Of the top 25 most common transposable elements in the Mimulus genome, six were type 1, and 19 were type 2 transposons (Table 5).
Table 5.
Transposable element frequencies, classes, and methylation
| Percent Methylation | ||||||
|---|---|---|---|---|---|---|
| ID | Copies | CG | CHG | CHH | Family | Class |
| helB8c | 2380 | 0.809 | 0.473 | 0.052 | Helitron | 2 |
| MULE_MITE1c | 674 | 0.627 | 0.263 | 0.102 | MITE | 2 |
| Copia1b | 424 | 0.780 | 0.437 | 0.058 | Copia | 1 |
| helD8b | 402 | 0.712 | 0.357 | 0.055 | Helitron | 2 |
| MULE_MITE2b | 245 | 0.633 | 0.285 | 0.077 | MULE | 2 |
| pogo_MITE2b | 203 | 0.738 | 0.281 | 0.071 | MITE | 2 |
| MULE_MITE16b | 200 | 0.713 | 0.207 | 0.070 | MULE | 2 |
| hAT_MITE1 | 197 | 0.782 | 0.294 | 0.051 | MITE | 2 |
| MULE_na62 | 165 | 0.768 | 0.359 | 0.064 | MULE | 2 |
| MULE_MITE1a | 158 | 0.720 | 0.250 | 0.071 | MULE | 2 |
| LARD4 | 155 | 0.793 | 0.442 | 0.081 | LARD | 1 |
| hAT_na66a | 151 | 0.869 | 0.276 | 0.042 | hAT | 2 |
| Tourist6c | 151 | 0.634 | 0.259 | 0.071 | MITE | 2 |
| MuDR8 | 150 | 0.791 | 0.492 | 0.089 | MuDR | 2 |
| MULE_na13a | 145 | 0.752 | 0.400 | 0.068 | MULE | 2 |
| Copia1a | 143 | 0.717 | 0.374 | 0.045 | Copia | 1 |
| Copia2 | 137 | 0.685 | 0.494 | 0.085 | Copia | 1 |
| SINE1a | 134 | 0.685 | 0.293 | 0.112 | SINE | 1 |
| Gypsy8 | 128 | 0.605 | 0.228 | 0.033 | Gypsy | 1 |
| MULE_na13b | 128 | 0.449 | 0.260 | 0.058 | MULE | 2 |
| helF3c | 119 | 0.737 | 0.362 | 0.067 | Helitron | 2 |
| Jittery7 | 116 | 0.639 | 0.260 | 0.053 | Mu | 2 |
| Toursit4c | 115 | 0.781 | 0.315 | 0.085 | MITE | 2 |
| Gypsy4 | 111 | 0.818 | 0.402 | 0.051 | Gypsy | 1 |
| MULE_MITE25b | 109 | 0.626 | 0.151 | 0.042 | MULE | 2 |
We find that DNA methylation in all contexts is enriched in transposable elements relative to genes, however this is most significant for non-CG methylation (Table 1). This suggests that both RNA dependent DNA methylation (RdDM) is targeting and silencing transposable elements in M. guttatus as is this case in other angiosperms. Found at 2,380 copies, the helB8c family of helitron elements is far and away the most common transposon in the Mimulus genome (more abundant than the next seven TE families combined; Table 5). Helitrons are a relatively newly discovered class of type 2 transposable elements that propagate through a rolling circle mechanism that is still somewhat mysterious [63]. One thing that is clear, is that these elements have been highly successful in propagating across flowering plants, making up 2 % of the Arabidopsis genome [64]; a single family of helitrons makes up 6 % of the maize genome [63], making it the most abundant DNA transposon identified. Here, we provide evidence for the success of these elements across the diversity of flowering plants.
Conclusions
Much remains unknown about the gene regulatory information contained in an organism’s methylome, but here we provide further evidence of complex interactions between gene methylation and expression. DNA methylation may actively alter gene expression, itself be altered by gene expression, or both methylation and expression may be jointly determined by a distinct genetic feature. Still the ability to explain over a fifth of the variation in log transformed gene expression by local DNA methylation, and basic genetic architecture (exon length, intron length, exon number), is promising and has numerous potential applications. Recent efforts have shown that the plant methylome is relatively stable throughout development [65], unlike gene expression. In this way methylation at a gene likely reflects moderately stable epigenetic control of gene expression, while developmentally activated transcription factors and small RNAs may provide highly plastic gene expression control throughout development. Through combining differential methylation analyses across tissue types, environmental treatments, or genetic lines with a modeling approach as described here; our understanding of the role of epigenetic variation in gene regulation can be greatly increased.
Acknowledgments
Nicholas McCool for plant care and DNA extractions. Masahiko Shimizu for laboratory aid, travel assistance, and general support. The University of Kansas Genome Sequencing Facility for preforming sequencing operations. This work was supported by NSF IOS-0951254 to JKK, LCH and AG Scoville. JMC’s travel to the University of Tokyo was supported by an NSF RCN microMorph grant, and a University of Kansas Botany Endowment grant to JMC.
Additional file
Predicted log (gene expression) from cubic polynominal REML model compared to actual log (gene expression). Slope =1.02, R2 = 0.201, df = 28.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JC carried out PBAT library preparation, preformed gene expression modeling, and drafted the manuscript. JK designed code necessary to calculate methylation percentages across genomic regions, coordinated plant grow ups and DNA extraction, and perceived the link between methylation and crossing over at transcription start sites. FM aided in the construction of libraries, and preformed BMap read mapping to the reference genome. TI participated in the coordination of the experiment, aided in the construction of libraries, and provided general support. LH conceived of the study, participated in its design and coordination, and helped draft the manuscript. All authors read and approved the final manuscript.
References
- 1.Flavell R. Inactivation of gene expression in plants as a consequence of specific sequence duplication. Proc Natl Acad Sci. 1994;91(9):3490–3496. doi: 10.1073/pnas.91.9.3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tate PH, Bird AP. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev. 1993;3(2):226–231. doi: 10.1016/0959-437X(93)90027-M. [DOI] [PubMed] [Google Scholar]
- 3.Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2006;39(1):61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
- 4.Lippman Z, Gendrel A-V, Black M, Vaughn MW, Dedhia N, McCombie WR, Lavine K, Mittal V, May B, Kasschau KD. Role of transposable elements in heterochromatin and epigenetic control. Nature. 2004;430(6998):471–476. doi: 10.1038/nature02651. [DOI] [PubMed] [Google Scholar]
- 5.Miura A, Yonebayashi S, Watanabe K, Toyama T, Shimada H, Kakutani T. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature. 2001;411(6834):212–214. doi: 10.1038/35075612. [DOI] [PubMed] [Google Scholar]
- 6.Xia J, Han L, Zhao Z. Investigating the relationship of DNA methylation with mutation rate and allele frequency in the human genome. BMC Genomics. 2012;13(8):1–9. doi: 10.1186/1471-2164-13-S8-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mirouze M, Lieberman-Lazarovich M, Aversano R, Bucher E, Nicolet J, Reinders J, Paszkowski J. Loss of DNA methylation affects the recombination landscape in Arabidopsis. Proc Natl Acad Sci. 2012;109(15):5880–5885. doi: 10.1073/pnas.1120841109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng S, Cokus SJ, Zhang X, Chen P-Y, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci. 2010;107(19):8689–8694. doi: 10.1073/pnas.1002720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huff JT, Zilberman D. Dnmt1-independent CG methylation contributes to nucleosome positioning in diverse eukaryotes. Cell. 2014;156(6):1286–1297. doi: 10.1016/j.cell.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gruenbaum Y, Naveh-Many T, Cedar H, Razin A. Sequence specificity of methylation in higher plant DNA. 1981. [DOI] [PubMed] [Google Scholar]
- 11.Bond DM, Baulcombe DC. Small RNAs and heritable epigenetic variation in plants. Trends Cell Biol. 2014;24(2):100–107. doi: 10.1016/j.tcb.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Kinoshita T, Jacobsen SE. Opening the door to epigenetics in PCP. Plant Cell Physiol. 2012;53(5):763–765. doi: 10.1093/pcp/pcs061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eichten SR, Schmitz RJ, Springer NM. Epigenetics: beyond chromatin modifications and complex genetic regulation. Plant Physiol. 2014;165(3):933–947. doi: 10.1104/pp.113.234211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao X, Jacobsen SE. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc Natl Acad Sci. 2002;99(suppl 4):16491–16498. doi: 10.1073/pnas.162371599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Law JA, Du J, Hale CJ, Feng S, Krajewski K, Palanca AMS, Strahl BD, Patel DJ, Jacobsen SE. Polymerase IV occupancy at RNA-directed DNA methylation sites requires SHH1. Nature. 2013;498(7454):385–9. doi: 10.1038/nature12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11(3):204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leonhardt H, Page AW, Weier H-U, Bestor TH. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell. 1992;71(5):865–873. doi: 10.1016/0092-8674(92)90561-P. [DOI] [PubMed] [Google Scholar]
- 18.Lindroth AM, Cao X, Jackson JP, Zilberman D, McCallum CM, Henikoff S, Jacobsen SE. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science. 2001;292(5524):2077–2080. doi: 10.1126/science.1059745. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW, Chen H, Henderson IR, Shinn P, Pellegrini M, Jacobsen SE, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell. 2006;126(6):1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Zhu J, Hu F, Ge S, Ye M, Xiang H, Zhang G, Zheng X, Zhang H, Zhang S, et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genomics. 2012;13(1):300. doi: 10.1186/1471-2164-13-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coleman-Derr D, Zilberman D. Deposition of histone variant H2A. Z within gene bodies regulates responsive genes. PLoS Genet. 2012;8(10):e1002988. doi: 10.1371/journal.pgen.1002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong S, Fei Z, Chen YR, Zheng Y, Huang M, Vrebalov J, McQuinn R, Gapper N, Liu B, Xiang J, et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat Biotechnol. 2013;31(2):154–159. doi: 10.1038/nbt.2462. [DOI] [PubMed] [Google Scholar]
- 23.Eichten SR, Briskine R, Song J, Li Q, Swanson-Wagner R, Hermanson PJ, Waters AJ, Starr E, West PT, Tiffin P, et al. Epigenetic and genetic influences on DNA methylation variation in maize populations. Plant Cell. 2013;25(8):2783–2797. doi: 10.1105/tpc.113.114793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmitz RJ, Schultz MD, Urich MA, Nery JR, Pelizzola M, Libiger O, Alix A, McCosh RB, Chen H, Schork NJ, et al. Patterns of population epigenomic diversity. Nature. 2013;495(7440):193–198. doi: 10.1038/nature11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saze H, Tsugane K, Kanno T, Nishimura T. DNA methylation in plants: relationship to small RNAs and histone modifications, and functions in transposon inactivation. Plant Cell Physiol. 2012;53(5):766–784. doi: 10.1093/pcp/pcs008. [DOI] [PubMed] [Google Scholar]
- 26.Gent JI, Ellis NA, Guo L, Harkess AE, Yao Y, Zhang X, Dawe RK. CHH islands: de novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013;23(4):628–637. doi: 10.1101/gr.146985.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takuno S, Gaut BS. Gene body methylation is conserved between plant orthologs and is of evolutionary consequence. Proc Natl Acad Sci. 2013;110(5):1797–1802. doi: 10.1073/pnas.1215380110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Q, Eichten SR, Hermanson PJ, Springer NM. Inheritance patterns and stability of DNA methylation variation in maize near-isogenic lines. Genetics. 2014;196(3):667–676. doi: 10.1534/genetics.113.158980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Marowsky NC, Fan C. Divergence of gene body DNA methylation and evolution of plant duplicate genes. PLoS One. 2014;9(10):e110357. doi: 10.1371/journal.pone.0110357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan Y, Guo L, Shen L, Liu JS. Predicting gene expression from sequence: a reexamination. PLoS Comput Biol. 2007;3(11):e243. doi: 10.1371/journal.pcbi.0030243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Wang X, He K, Ma Y, Su N, He H, Stolc V, Tongprasit W, Jin W, Jiang J. High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression. Plant Cell Online. 2008;20(2):259–276. doi: 10.1105/tpc.107.056879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colicchio JM, Monnahan PJ, Kelly JK, Hileman LC. Gene expression plasticity resulting from parental leaf damage in Mimulus guttatus. New Phytol. 2015;205(2):894–906. [DOI] [PubMed]
- 33.Holeski L. Within and between generation phenotypic plasticity in trichome density of Mimulus guttatus. J Evol Biol. 2007;20(6):2092–2100. doi: 10.1111/j.1420-9101.2007.01434.x. [DOI] [PubMed] [Google Scholar]
- 34.Holeski LM, Chase‐Alone R, Kelly JK. The genetics of phenotypic plasticity in plant defense: trichome production in Mimulus guttatus. Am Nat. 2010;175(4):391–400. doi: 10.1086/651300. [DOI] [PubMed] [Google Scholar]
- 35.Scoville AG, Barnett LL, Bodbyl‐Roels S, Kelly JK, Hileman LC. Differential regulation of a MYB transcription factor is correlated with transgenerational epigenetic inheritance of trichome density in Mimulus guttatus. New Phytolo. 2011;191(1):251–263. doi: 10.1111/j.1469-8137.2011.03656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holeski LM, Jander G, Agrawal AA. Transgenerational defense induction and epigenetic inheritance in plants. Trends Ecol Evol. 2012;27(11):618–26. doi: 10.1016/j.tree.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 37.Holeski LM, Zinkgraf MS, Couture JJ, Whitham TG, Lindroth RL. Transgenerational effects of herbivory in a group of long-lived tree species: maternal damage reduces offspring allocation to resistance traits, but not growth. J Ecol. 2013;101(4):1062–1073. doi: 10.1111/1365-2745.12110. [DOI] [Google Scholar]
- 38.Latzel V, Allan E, Bortolini Silveira A, Colot V, Fischer M, Bossdorf O. Epigenetic diversity increases the productivity and stability of plant populations. Nat Commun. 2013;4:2875. doi: 10.1038/ncomms3875. [DOI] [PubMed] [Google Scholar]
- 39.Kilvitis H, Alvarez M, Foust C, Schrey A, Robertson M, Richards C. Ecological Epigenetics. In: Landry CR, Aubin-Horth N, editors. Ecological Genomics. Netherlands: Springer; 2014. pp. 191–210. [Google Scholar]
- 40.Hellsten U, Wright KM, Jenkins J, Shu S, Yuan Y, Wessler SR, Schmutz J, Willis JH, Rokhsar DS. Fine-scale variation in meiotic recombination in Mimulus inferred from population shotgun sequencing. Proc Natl Acad Sci. 2013;110(48):19478–19482. doi: 10.1073/pnas.1319032110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holeski L, Keefover-Ring K, Bowers MD, Harnenz Z, Lindroth R. Patterns of Phytochemical Variation in Mimulus guttatus (Yellow Monkeyflower) J Chem Ecol. 2013;39(4):525–536. doi: 10.1007/s10886-013-0270-7. [DOI] [PubMed] [Google Scholar]
- 42.Hardcastle T. High-throughput sequencing of cytosine methylation in plant DNA. Plant Methods. 2013;9(1):16. doi: 10.1186/1746-4811-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miura F, Enomoto Y, Dairiki R, Ito T. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 2012;40(17):e136. doi: 10.1093/nar/gks454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kiełbasa SM, Wan R, Sato K, Horton P, Frith MC. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011;21(3):487–493. doi: 10.1101/gr.113985.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flagel LE, Willis JH, Vision TJ. The standing pool of genomic structural variation in a natural population of Mimulus guttatus. Genome Biol Evol. 2014;6(1):53–64. doi: 10.1093/gbe/evt199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40(D1):D1178–D1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ren X-Y, Vorst O, Fiers MW, Stiekema WJ, Nap J-P. In plants, highly expressed genes are the least compact. Trends Genet. 2006;22(10):528–532. doi: 10.1016/j.tig.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 48.Castillo-Davis CI, Mekhedov SL, Hartl DL, Koonin EV, Kondrashov FA. Selection for short introns in highly expressed genes. Nat Genet. 2002;31(4):415–418. doi: 10.1038/ng940. [DOI] [PubMed] [Google Scholar]
- 49.Posada D, Buckley TR. Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst Biol. 2004;53(5):793–808. doi: 10.1080/10635150490522304. [DOI] [PubMed] [Google Scholar]
- 50.Kohavi R. A study of cross-validation and bootstrap for accuracy estimation and model selection. IJCAI. 1995;1995:1137–1145. [Google Scholar]
- 51.Kelly JK, Koseva B, Mojica JP. The genomic signal of partial sweeps in Mimulus guttatus. Genome Biol Evol. 2013;5(8):1457–1469. doi: 10.1093/gbe/evt100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmitz RJ, He Y, Valdes-Lopez O, Khan SM, Joshi T, Urich MA, Nery JR, Diers B, Xu D, Stacey G, et al. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 2013;23(10):1663–1674. doi: 10.1101/gr.152538.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shen H, He H, Li J, Chen W, Wang X, Guo L, Peng Z, He G, Zhong S, Qi Y, et al. Genome-wide analysis of DNA methylation and gene expression changes in two Arabidopsis ecotypes and their reciprocal hybrids. Plant Cell. 2012;24(3):875–892. doi: 10.1105/tpc.111.094870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 55.Bartee L, Malagnac F, Bender J. Arabidopsis cmt3 chromomethylase mutations block non-CG methylation and silencing of an endogenous gene. Genes Dev. 2001;15(14):1753–1758. doi: 10.1101/gad.905701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seymour DK, Koenig D, Hagmann J, Becker C, Weigel D. Evolution of DNA methylation patterns in the brassicaceae is driven by differences in genome organization. PLoS Genet. 2014;10(11):e1004785. doi: 10.1371/journal.pgen.1004785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li X, Li W, Wang H, Cao J, Maehashi K, Huang L, Bachmanov AA, Reed DR, Legrand-Defretin V, Beauchamp GK. Pseudogenization of a sweet-receptor gene accounts for cats’ indifference toward sugar. PLoS Genet. 2005;1(1):e3. doi: 10.1371/journal.pgen.0010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wassenegger M. RNA-directed DNA methylation. In: Plant Gene Silencing. Springer; 2000: 83–100.
- 59.Yang H, Chang F, You C, Cui J, Zhu G, Wang L, Zheng Y, Qi J, Ma H. Whole‐genome DNA methylation patterns and complex associations with gene structure and expression during flower development in Arabidopsis. Plant J. 2015;81(2):268–281. doi: 10.1111/tpj.12726. [DOI] [PubMed] [Google Scholar]
- 60.Zhang W, Zhang T, Wu Y, Jiang J. Genome-wide identification of regulatory DNA elements and protein-binding footprints using signatures of open chromatin in Arabidopsis. Plant Cell Online. 2012;24(7):2719–2731. doi: 10.1105/tpc.112.098061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu J, Hu S, Wang J, Wong GK-S, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica) Science. 2002;296(5565):79–92. doi: 10.1126/science.1068037. [DOI] [PubMed] [Google Scholar]
- 62.Pan J, Sasaki M, Kniewel R, Murakami H, Blitzblau Hannah G, Tischfield Sam E, Zhu X, et al. A Hierarchical Combination of Factors Shapes the Genome-wide Topography of Yeast Meiotic Recombination Initiation. Cell, 2011;144(5):719–731. [DOI] [PMC free article] [PubMed]
- 63.Xiong W, He L, Lai J, Dooner HK, Du C. HelitronScanner uncovers a large overlooked cache of Helitron transposons in many plant genomes. Proc Natl Acad Sci. 2014;111(28):10263–10268. doi: 10.1073/pnas.1410068111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hollister JD, Gaut BS. Population and evolutionary dynamics of helitron transposable elements in Arabidopsis thaliana. Mol Biol Evol. 2007;24(11):2515–2524. doi: 10.1093/molbev/msm197. [DOI] [PubMed] [Google Scholar]
- 65.Eichten SR, Vaughn MW, Hermanson PJ, Springer NM. Variation in DNA methylation patterns is more common among maize inbreds than among tissues. Plant Genome 2013, 6(2).
- 66.Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452(7184):215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wickham H. ggplot2: elegant graphics for data analysis: Springer; 2009.