

Abstract
We present a plug-and-play radiosynthesis platform and accompanying computer software based on modular subunits that can easily and flexibly be configured to implement a diverse range of radiosynthesis protocols. Modules were developed that perform: (i) reagent storage and delivery, (ii) evaporations and sealed reactions, and (iii) cartridge-based purifications. The reaction module incorporates a simple robotic mechanism that removes tubing from the vessel and replaces it with a stopper prior to sealed reactions, enabling the system to withstand high pressures and thus provide tremendous flexibility in choice of solvents and temperatures. Any number of modules can rapidly be connected together using only a few fluidic connections to implement a particular synthesis, and the resulting system is controlled in a semi-automated fashion by a single software interface. Radiosyntheses of 2-[18F]fluoro-2-deoxy-D-glucose ([18F]FDG), 1-[18F]fluoro-4-nitrobenzene ([18F]FNB), and 2′-deoxy-2′-[18F]fluoro-1-β-D-arabinofuranosyl cytosine (D-[18F]FAC) were performed to validate the system and demonstrate its versatility.
Keywords: Automated radiosynthesis, high-pressure reactions, PET, D-[18F]FAC, nucleoside analogs, plug-and-play reconfiguration
1 Introduction
Increasing interest in several 18F-radiolabeled nucleoside analog probes for positron emission tomography (PET) has created the need for a radiosynthesizer for routine production that is capable of the challenging synthesis conditions. Production of analogs such as 2′-deoxy-2′-[18F]fluoro-5-methyl-1-β-D-arabinofuranosyluracil ([18F]FMAU), 2′-deoxy-2′-[18F]fluoro-5-ethyl-1-β-D-arabinofuranosyluracil ([18F]FEAU) (Shields 2006; Vallabhajosula 2007), and 1-(2′-deoxy-2′-[18F]fluoroarabinofuranosyl) cytosine ([18F]FAC) (Laing et al. 2009; Radu et al. 2008; Shu et al. 2010) involves reactions at high temperatures in volatile solvents (and therefore high vapor pressures) to achieve sufficient yields for downstream imaging. Estimating from physical properties of the solvent (Chemical Engineering Research Information Center (CHERIC) 2013), the [18F]FMAU synthesis reported by Mangner et al. (2003) involves a pressure of 139 psig [957 kPag] during the coupling step (in chloroform at 150°C), and the D[18F]FAC synthesis described by Radu et al. (2008) involves estimated pressures of 121 psig [834 kPag] during fluorination (in acetonitrile at 165°C) and 101 psig [695 kPag] during the coupling step (in 1,2-dichloroethane at 160°C). In addition to the production of radiofluorinated nucleoside analogs, there is evidence that the synthesis of other tracers can be improved by performing reactions at substantially higher temperatures and pressures than typically used (Lu et al. 2009).
Since currently available radiosynthesizers are limited to heating solvents not far above their boiling points (Ungersboeck et al. 2011), significant compromises in reaction conditions (such as reduced temperature or alternative solvents) are needed to adapt the above syntheses to automated platforms (Chin et al. 2007; Paolillo et al. 2009). Unfortunately, these changes and other strategies, such as milder synthesis pathways (Anderson et al. 2010; Li et al. 2011), seem to have an adverse effect on yield. We believe that the significant efforts that are generally spent to modify radiochemistry to make it more amenable to automation in current systems should be balanced with efforts in making it easier and more practical to work under more extreme conditions. To this end, we previously developed a first-generation reaction module (Amarasekera et al. Under review; Satyamurthy et al. 2010) capable of reliably performing sealed reactions at pressures up to at least 150 psig [1.03 MPag] without solvent loss. Addressing the fundamental source of limitations in other synthesizers, this module ensured all tubing was removed from the reaction vessel and replaced with a stopper during the high-pressure steps.
Though suitable for performing challenging syntheses with good radiochemical yield, this reaction module was not automated. For example, the robotic motions to manipulate the oil bath and stoppers were manually activated via switches on the controller, and liquid transfers and reagent additions were driven manually via syringes connected to tubing outside the hot cell. In this paper, we present a second-generation radiosynthesis platform consisting of several modules: an improved, more compact, version of the first-generation high-pressure reaction module, a module for reagent storage and delivery, and a module for cartridge purification. With only a few fluidic connections required between them, any number of modules can be assembled in a plug-and-play fashion to implement a system with the necessary capabilities for a wide range of different radiosyntheses. The resulting system can immediately be operated via a user-friendly software interface in a semi-automated fashion without any configuration of the software. To demonstrate the performance and versatility, 2-[18F]fluoro-2-deoxy-D-gluocise ([18F]FDG), 1-[18F]fluoro-4-nitrobenzene ([18F]FNB), and D-[18F]FAC were synthesized as model compounds.
2 Materials and Methods
2.1 Synthesis Apparatus
The radiosynthesis platform comprises three different types of modules that can be assembled to match the requirements (i.e., number of vessels, number of reagents, and type of purification steps) of the intended radiosynthesis (Figure 1). In addition to a Pressurized Reaction Module (PRM) based on our previous work (Amarasekera et al. Under review), we have developed two additional modules: (i) a Reagent Delivery Module (RDM) for storing reagents and delivering a selected reagent on demand to a reaction module, and (ii) a Cartridge Purification Module (CPM) for storing diluent, wash, and eluent solutions, and for delivering the crude sample and these solutions on demand through a cartridge for purification. These additional modules reduce the need for manual intervention and thereby facilitate automation of syntheses requiring the high-pressure modules. By connecting modules in a plug-and-play fashion, diverse syntheses of varying complexity from single-vessel syntheses such as [18F]FDG to multi-vessel syntheses such as D-[18F]FAC can be readily implemented in a short time.
Figure 1.

Photographs of the PRM, RDM, and CPM modules and controllers. Note that some tubing has been omitted for clarity.
2.1.1 Pressurized Reaction Module (PRM)
The PRM is designed to perform heated reactions under sealed conditions as well as evaporations to remove solvent. The module was engineered to maintain seal integrity of reactions even at high pressures (at least 150 psig [1.0 MPag]), such as those generated when reactions are performed in volatile organic solvents under superheated conditions. Compatibility with high pressures is achieved by linearly translating the vial to one of three positions or “stations”. Each position is customized for a different step in the synthesis protocol: a chemically-resistant “stopper” when sealed conditions are needed, and various tubing interfaces when other operations (reagent addition, transfer, evaporation) are needed (Figure 2). With this mechanism, there are no valves or tubes exposed to the high-pressure vapor during sealed reactions. While our first-generation system (Amarasekera et al. Under review) exhibited repeatable and reliable performance in the hands of skilled radiochemists, we made a number of engineering modifications to enable the system to be moved into a variety of laboratory settings by providing increased automation, modular construction, and greater flexibility for synthesis development. Furthermore, the system was miniaturized (decreased in height from 58 cm to 27 cm) to facilitate installation in a mini-cell rather than a hot-cell. (Dimensions are 27 cm × 9.5 cm × 27 cm height × width × depth.)
Figure 2.

(a) Schematic of pressurized reaction module (PRM). The reaction vessel is contained within a heating/cooling jacket that controls its temperature. Pneumatic actuators acting in the vertical direction and a stepper motor in the horizontal direction allow the vessel to be positioned at different fluidic interfaces (labeled “1”, “2” and “3”). A flat gasket material provides a gas-tight seal at each interface. Station “1” provides one or more lines for incoming reagents as well as a nitrogen stream and vacuum to facilitate evaporations. Though only one reagent line is shown, typically two are used: one for [18F]fluoride or product of previous step, and one for reagents that must be added to complete the current step. Station “2” provides a seal only and is for pressurized reactions. Station “3” provides nitrogen supply and a dip tube for transferring reagents out of the vessel into the next reaction or purification module. (b) Corresponding photograph of the PRM. The insets at top show details of the fluidic interfaces and gaskets.
2.1.1.1 Reactor Temperature Control
The reaction vessel (Wheaton 5 mL glass V-vial) is held within a chamber comprising an aluminum jacket lined with a compressible thermally-conductive silicone heat transfer material to ensure good thermal contact to the vessel despite vial-to-vial size variation. Resistive heating elements with feedback control and compressed air cooling (through channels embedded in the metal jacket) are used to rapidly control temperature. In contrast with an oil bath heater, the heating jacket is more compact, less messy, and enables consecutive evaporation and reaction steps at arbitrary temperatures to be performed in a single PRM. Heating was tested up to a maximum setpoint of 240°C. Two narrow slots were machined into the chamber to provide the ability to view the vial contents by eye or with a small camera during heating and evaporation processes.
2.1.1.2 Reactor Mixing
A rotating magnet, powered by a DC motor, is mounted adjacent to the heating jacket to enable the use of a magnetic stir bar in the reaction vessel. Pulse-width-modulation control of the motor voltage provides speed adjustment. Unlike mixing based on bubbling of inert gas, the stir bar allows solutions to be continually mixed during sealed reactions.
2.1.1.3 Robotic Motion and Reaction Vessel Stations
To seal the vessel at any of the three fluidic interface “stations”, the rim of the vessel pressed up against a flat, rigidly-mounted rubber gasket by pneumatic actuators. Compared to a conventional stopper, the use of a flat seal requires less positioning accuracy, and permits a wider variety of sealing materials to be used (i.e. any elastomer available in sheet form). We found PTFE-coated silicone (Cannon Gasket) to provide the best performance in terms of compatibility with high temperatures and aggressive reagents, and minimal loss of solvent volume or radioactivity during heating. These inexpensive gaskets were replaced after each synthesis. The use of pneumatic actuators provides high sealing force in a compact system and the ability to adjust the sealing force by varying the pressure supplied to the actuators. This feature was useful as a means to accommodate different gasket materials with different thicknesses and elasticity.
Movement in the horizontal direction between stations is controlled by a stepper motor (Anaheim Automation) with an integrated encoder for closed-loop feedback control of position, speed, and acceleration (AllMotion). Splashing of the reaction vessel contents during motion has never been observed. The present design ensures that all tubing connected to the various stations remains fixed in place, thus reducing the risk of cutting or damage to the tubing during motion.
Each PRM has three different stations (Figure 2a). The vessel is moved to station 1 for either adding reagents or evaporating solvents. Fluid connections to station 1 include: (i) transfer line from previous module or [18F]fluoride source, (ii) transfer line(s) for additional reagents from a reagent delivery module or manual addition from a point outside the hot cell (e.g. for sensitive reagents), (iii) a vacuum line to draw away vapor during evaporations, and (iv) a nitrogen line to deliver a flow of gas to accelerate vapor removal during evaporations. Nitrogen flow was found necessary; otherwise, substantial condensed vapor would remain at the top of the vessel and gasket seal. The vessel is moved to station 2 for performing sealed reactions. There are no connections to this station, allowing for high temperature and high pressure reactions to be safely performed without loss of vapor or liquid. Finally, the vessel is moved to station 3 for performing transfer out of the vessel to another module or product collection vial. Station 3 has two connections: (i) dip tube leading to transfer line to deliver product to next module through an optional purification cartridge and (ii) a nitrogen gas inlet to drive the liquid out of the reaction vessel into the dip tube. We elected to have the transfer dip-tube in a separate station than the evaporation station to prevent an occasional issue where a substantial portion of radioactive products(s) would stick to the transfer dip-tube during evaporation and subsequently be left behind when the vessel was moved to another station. The user needs only connect the [18F]fluoride (or previous module) input line, the reagent input line, and the product output line; all other lines and valves are internal to the PRM and are not configured by the user.
With these capabilities, the PRM can perform several “unit operations”, including (1) reagent addition, (2) evaporation of solvent, (3) sealed reaction, or (4) transfer of product out of the reaction vessel. Figure 3 illustrates a generalized procedure for a one-pot synthesis in the PRM, showing the sequences of events needed to accomplish each of these unit operations. (More detail is given in Table S1 of the Supporting Information.)
Figure 3.

Generalized sequence of steps to perform a one-pot synthesis or one step of a multi-step reaction. (Row 1) Ensure reaction vessel is in position 1. Load starting solution (e.g. [18F]fluoride solution eluted from QMA cartridge, or the product of a previous reaction step) and eliminate solvent by evaporation. Load next reactant. (Row 2) Move reaction vessel to position 2 and perform reaction. (Row 3) Move reaction vessel to position 3 and transfer product through sep-pak to trap on resin. (Row 4) Move reaction vessel to position 1 and add eluent into vessel. (Row 5) Move reaction vessel to position 3 and transfer through sep-pak to elute product into next module.
2.1.2 Reagent Delivery Module (RDM)
The reagent delivery module comprises eight reagent reservoirs that can be pre-loaded prior to the synthesis, stored, and then transferred by inert gas under software control to a reaction vessel during the synthesis as needed. The reservoirs are affixed to a manifold with a common output fluid path (Figure 4) that is typically connected to the reagent inlet line of a PRM. To deliver the contents of a particular reservoir during the synthesis, the corresponding liquid valve and inert gas valve are opened, as illustrated in Figure 4 and described in detail in Table S2 of the Supporting Information.
Figure 4.

Design and operation of reagent delivery module (RDM). (a) Schematic of fluid path. The actual module has 8 reagent reservoirs but only 4 are shown for clarity. Reagent reservoirs are connected to a common path leading to a single output connection. Due to a small amount of carryover in the fluid path after reagent delivery, care must be taken in selection of reagent positions to mitigate effects of cross-contamination (or multiple RDMs can be used). Remaining panels show operating states: (b) User loads reagents through check-valves into reagent reservoirs during setup prior to synthesis. These reagents are stored in the reservoirs until needed. (c) Reagent #2 is delivered to the common delivery line with nitrogen pressure.
We explored an alternative design of this module with reservoir bodies themselves integrated into a more complex manifold. The manifold was machined from PEEK to provide compatibility for storage of diverse reagents. Though the design achieved a substantial reduction in tubing and number of fittings, which should improve reliability, and demonstrated significantly reduced carryover (<0.5%), the inability to observe reagent levels within the reservoirs proved to be a significant drawback in practice.
2.1.3 Cartridge Purification Module (CPM)
The CPM was designed as a flexible module to perform various cartridge purification protocols. Prior to the synthesis, the operator loads the module with pre-metered volumes of diluent solution (e.g. water), wash solution, and eluent solution, and installs a preconditioned cartridge. A typical solid-phase extraction can be performed as follows: (1) dilute the sample by loading it into a reservoir containing diluent solution and bubbling nitrogen to mix, (2) flow diluted sample through a cartridge to trap desired compound, (3) wash the cartridge, and (4) elute the desired compound from the cartridge to the output. The output could be connected to a collection vial, an HPLC system, or to the reagent inlet line in Station 1 of a PRM. A schematic of the module and details of operation are illustrated in Figure 5 and described in detail in Table S3 of the Supporting Information.
Figure 5.

Design and operation of cartridge purification module (CPM). (a) Schematic of fluid path. Reagent reservoirs are connected to a common path leading through the cartridge to waste and output valves. Remaining panels show operating states: (b) User loads reagents through check-valves into reservoirs during setup prior to synthesis: (i) diluent solution to dilute sample prior to passing through cartridge, (ii) wash solution to wash cartridge after trapping, and (iii) eluent solution to elute from cartridge to output. (c) Sample is loaded via a tube connected to the diluents loading port. It is flowed into the diluent solution and followed by nitrogen bubbling to induce mixing. (d) Diluted sample is flowed through the cartridge to trap the desired species. (e) Cartridge is washed by flowing wash solution through to waste. (f) Purified compound is eluted from cartridge with eluent solution to output.
Though not used for experiments described here, the CPM was designed with an additional feature to allow the eluate from the cartridge to be loaded into a dedicated mixing reservoir where it can be thoroughly mixed by nitrogen bubbling and then subsequently pushed to the output with inert gas. The purpose of this feature was to allow the strong concentration gradient in the eluate to be eliminated as may be needed if the downstream module was a capillary microreactor, for example.
2.1.4 Control Architecture and Software
We first investigated a control architecture based on the concept of operating each module in a standalone fashion with a compact hand-held touchscreen interface outside the hotcell. While we found this paradigm useful in certain circumstances (e.g. when a CPM connected to a manual synthesis apparatus), we found that manipulating multiple touchscreen interfaces was cumbersome in syntheses requiring multiple modules. Additionally, because each module required its own control interface external to the cell, and each control interface required a wiring harness to transport user commands and power, cable management and organization became difficult.
We thus developed an improved architecture to facilitate control and programming of any number of modules via a single personal computer (PC). Each module contains a microcontroller board (Arduino Mega) and a small custom circuit board for serial communication and electrical connection to the actuators and sensors. Modules (acting as slave devices) communicate with the PC (acting as master controller) over a multidrop RS-485 bus using a simple, human-readable communication protocol based on the MojoBus protocol (Wright and Jellman 2007). We wrote a graphical user interface in the python programming language to show the system state and permit manual control of low-level functionality (e.g. opening or closing valves) as well as manual activation of unit operations or “macros” that represent a more intuitive, functional interaction with the module. The microcontroller in each module provides a flexible means for future expansion of the set of unit operations and commands known to each module or even expansion of the communication protocol itself.
Only minimal electronics are located within each module, a design strategy that keeps the overall size of the system small for placement in a mini-cell or hot cell, and reduces potential problems of electronics in high radiation fields. Modules are daisy-chained together (Figure 6) with connections for power, serial communication, and sources of inert gas (for driving liquids through the system and for pneumatic actuators), and compressed air (for reactor cooling). Outside the cell, there is a power supply and a master computer that communicates with the modules over the serial bus. Daisy-chaining minimizes the number of cables and tubes that need to pass from controllers outside the hot cell to the inside. When the system is first turned on, the PC automatically detects which modules are connected through a device enumeration protocol. All detected modules are then displayed in a tabbed interface such that user can select a particular module and have access to status information as well as buttons to activate low-level commands or macros for that module (see Supporting Information, Figure S2 and Tables S1–S3). After assembly of modules, the newly configured system can instantly be used from the PC in a semi-automated fashion with no updates needed by the user. To enable syntheses to be automated, a simple recording and playback feature was developed in the software. It allows the system to record the sequence and timings of commands as the user operates the system via the tabbed interface and then to play back the sequence at a future time.
Figure 6.

Control system architecture. Modules are connected to pressure supply, power supply and master controller in a bus-like / daisy-chain fashion to minimize the number of connections that must cross the hot-cell boundary. Each module contains a small amount of communication circuitry and drivers (“Controller”) to facilitate connection in this fashion. The fluid path is formed by connecting “Modules” together in series via tubing. In cases where the RDM cannot be used without extensive cross-contamination concerns, reagents can be added manually vial optional lines installed to outside of hot cell.
2.2 Radiosyntheses
2.2.1 Reagents
Anhydrous acetonitrile (99.8%), anhydrous dimethyl sulfoxide (99.85%), anhydrous hexane (98%), anhydrous ethyl acetate (99.8%), hydrochloric acid (1N), sodium methoxide (0.5M in methanol), anhydrous 1,2-dichloroethane (99.8%), dichloromethane (99.5%), anhydrous toluene (99.8%), hydrobromic acid (33 wt. % in acetic acid), ammonium acetate, ammonium phosphate monobasic, potassium carbonate, 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane (Kryptofix K222), 1-fluoro-4-nitrobenzene (non-radioactive reference standard for FNB), and 1,4-dinitrobenzene were purchased from Sigma Aldrich. 1,3,4,6-tetra-O-acetyl-2-O-trifluoro-methanesulfonyl-beta-D-mannopyranose (mannose triflate), 2-trifluoromethasulfonyl-1,3,5, tri-O-benzoyl-α-D-ribofuranose and bis(trimethylsilyl)cytosine were purchased from ABX, Germany. The non-radioactive D-FAC reference standard was purchased from Moravek Biochemicals and Radiochemicals Company (Brea, CA). All chemicals were used without further purification.
No-carrier-added [18F]fluoride was produced in an RDS-111 cyclotron by proton bombardment of 18O-enriched water ([18O]H2O, Rotem, Inc.). After bombardment, before each synthesis, ~20–1500 mCi of the target water is flowed through a QMA ion exchange cartridge (WAT023525, Waters Corporation) to trap the [18F]fluoride (with ~99% efficiency). The QMA cartridge is preconditioned with 6 mL 1M KHCO3 followed by 12 mL H2O and dried with nitrogen at 5 psig. The [18F]fluoride was then released from this cartridge by eluting with K2CO3 (1 mg in 0.4 mL H2O).
2.2.2 Chromatography and Analytical Methods
Analytical high-performance liquid chromatography (HPLC) was performed with a Smartline system (Knauer, Germany) comprised of a quaternary gradient HPLC pump, solvent degasser, and UV detector. UV absorption was monitored at 254 nm, and radioactivity was monitored with a gamma detector (PET Metabolite Detector, Bioscan, Inc., Washington, DC).
Semi-preparative HPLC was performed with a Knauer 501 HPLC pump, K2501 UV detector, and a B-FC-1000 flow-count gamma detector (Bioscan). Control and data acquisition were provided by a GINA system (Raytest USA, Inc., Wilmington, NC) and analysis was performed with the GINA Star software (Raytest).
Thin layer chromatography (TLC) was performed on precoated silica gel plates (Baker-flex® IB2-F, J.T. Baker). TLC plates were scanned using a miniGITA Star radio-TLC scanner (Raytest). Data acquisition and analysis were performed using a GINA system (Raytest) and corresponding GINA Star TLC software (Raytest).
Radioactivity of vials and other components (for characterization purposes) was measured in a dose calibrator (CRC-25 PET, Capintec, Ramsey, NJ).
2.2.3 Synthesis of 2-[18F]Fluoro-2-deoxy-D-glucose ([18F]-FDG)
[18F]FDG was synthesized according to the method of Hamacher et al. (1986) using a configuration of one RDM and one PRM module (Figure 7, and Supporting Information Table S4). The reservoirs of the RDM were preloaded with the following solutions: (1) 1 mL MeCN, (2) 1 mL MeCN, (3) mannose triflate (10 mg in 1.5 mL MeCN), (4) 1.5 mL 1N HCl, (5) 4 mL H2O, (6) 4 mL H2O. Purification was performed with an assembly of cartridges (Padgett et al. 1989) comprising a custom-packed Econo-column (15 cm long, 0.7 cm diameter; BioRad Laboratories, Hercules, CA) followed by a C18 cartridge (Sep-Pak® Plus C-18, WAT020515, Waters Corporation, Milford, MA) and an alumina cartridge (Sep-Pak® Plus Alumina N, 020510, Waters Corporation) cartridge. The Econo-column was packed with 12 cm of an ion retardation resin (AG11-A8, 50–100 mesh, BioRad Laboratories) and 1 cm of a strong cation exchange resin (AG50W-X8, 50–100 mesh size, BioRad Laboratories), pre-equilibrated with sterile H2O. After packing, 20 mL H2O was passed through the column to eliminate air. The C-18 cartridge was preconditioned by flowing 5 mL anhydrous EtOH followed by 10 mL sterile H2O. The alumina cartridge was conditioned with 10 mL sterile H2O. Finally, 20 mL sterile H2O was passed through the whole assembly.
Figure 7.
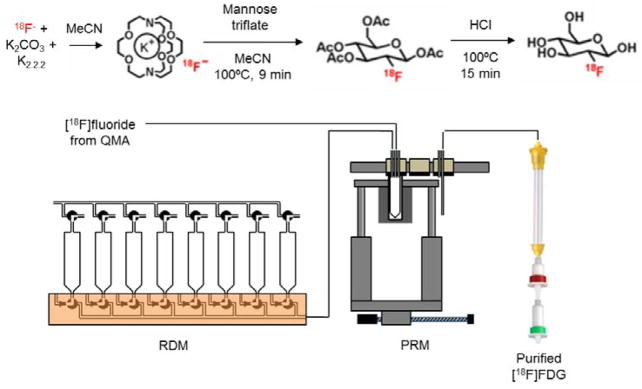
(Top) 2-step, 1-pot synthesis scheme for [18F]FDG. (Bottom) Module configuration to perform this synthesis.
20–1500 mCi of [18F]fluoride solution eluted from the QMA was added at Station 1 of the PRM into the reaction vessel preloaded with K222 (10 mg in 1 mL MeCN). The solvent was evaporated at 105°C for 7 min and then the vessel was cooled to 40°C. Azeotropic distillation was performed next, first adding 1 mL anhydrous MeCN to the vessel and evaporating at 105°C for 7 min, then adding 1 mL additional MeCN and evaporating at 105°C for 3 min. Next, mannose triflate (10 mg in 1.5 mL MeCN) was added, then the reaction vessel moved to Station 2 and heated at 100°C for 9 min under sealed conditions to perform the radiofluorination. After cooling the vessel to 40°C, it was moved to Station 1 and the solvent was reduced during 5 min at 100°C (with nitrogen flow and vacuum). 1.5 mL of 1N HCl was added to the concentrated solution, then the vessel moved to Station 2 and heated at 100°C for 15 min under sealed conditions to perform the hydrolysis reaction. After cooling the vessel to 40°C, it was moved to Station 3 and the 1–2 mL of crude [18F]FDG was transferred directly into the assembly of purification cartridges. The radiochemically pure [18F]FDG is flushed out of the assembly by the addition and subsequent transfer of two portions of water (~4.0 mL each) to the vessel. A small sample was taken for quality control. The radiochemical purity was confirmed by radio-TLC (mobile phase: MeCN:H2O, 95:5 v/v).
2.2.4 Synthesis of 1-[18F]fluoro-4-nitrobenzene ([18F]FNB)
We adapted the [18F]FNB syntheses described by Collins et al. (1992) and VanBrocklin et al. (2001) to a system consisting of one RDM, one PRM, and one CPM module (Figure 8, and Supporting Information Table S5). The reservoirs of the RDM were preloaded with the following solutions: (1) [18F]fluoride solution (20 μL [18O]H2O containing ~20 mCi [18F]fluoride, 1 mg K2CO3, 10 mg K2.2.2, plus 975 μL MeCN); (2) 1 mL MeCN, (3) 1 mL MeCN, (4) 1,4-dinitrobenzene (10mg in 0.5mL DMSO), (5) 1 mL H2O, (6) 1 mL H2O. The Strata-X 33 cartridge (500 mg, Phenomenex, Torrance, CA) was preconditioned with 10 mL EtOH followed by 10 mL of H2O and installed on the CPM. The CPM reservoirs were filled as follows: 10 mL H2O as diluent, 10 mL H2O as wash solution, 1 mL MeOH as eluent.
Figure 8.
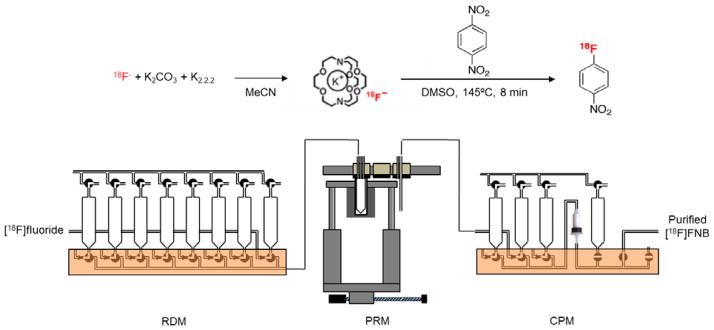
(Top) 1-step, 1-pot synthesis scheme for [18F]FNB. (Bottom) Module configuration to perform this synthesis.
First, the [18F]fluoride solution was delivered via Station 1 into the vessel of the PRM. Solvent was evaporated by heating to 110°C for 5min. Next, azeotropic distillation was performed by adding MeCN and evaporating at 110°C for 5 min. This was repeated an additional time with 3 min evaporation time. Next, the precursor solution was added. The vessel was moved to Station 2, and then heated to 145°C for 8 min under sealed conditions, followed by cooling to 60°C. The vessel was moved back to Station 1 where 1 mL H2O was added. The crude product was transferred to the CPM for purification after moving the vessel to Station 3. The vessel was then moved back to Station 1 where an additional 1 mL H2O was added to rinse the reaction vessel and transfer to the cartridge after moving the vessel to Station 3. The sample, diluted with 10 mL H2O in the CPM was flushed through the cartridge to waste, followed by washing of the cartridge with another 10 mL H2O. Finally, the purified [18F]FNB was eluted off of the cartridge and out of the CPM with 1 mL MeOH into an empty collection vial.
A small sample was taken from the collection vial for analysis by analytical HPLC (Luna C18 column 25cm × 0.46 cm, 5μm particle size; flow rate 1 mL/min; mobile phase: MeCN/H2O (50:50 v/v) with 0.2% trifluoroacetic acid (TFA)). The retention time of the reference standard was 7.0 min. Radiochemical identity was also confirmed by radio-TLC (mobile phase: EtOAc:hexane (20:80 v/v)).
2.2.5 Synthesis of 2′-deoxy-2′-[18F]fluoro-B-D-arabinofuranosyl cytosine (D[18F]FAC)
For the synthesis of D-[18F]FAC as described in Radu et al. (2008), we used three PRMs configured as shown in Figure 9 (see also Supporting Information Table S6). Cartridge purifications needed between reaction steps were not performed with CPMs but rather with the simplified inline system (containing only a single liquid valve) depicted in Figure 3. The crude product is delivered through the transfer line of Station 3 through the cartridge to waste, followed by the addition of eluent solution at Station 1 to the reaction vessel and subsequent transfer from Station 3 through the cartridge to the next PRM. Since several of the reagents are very sensitive to atmospheric exposure and require preparation immediately prior to use, all reagents were delivered manually via tubing connected to the outside of the hot cell to Station 1 of each of the PRMs. The focus of this validation was on the ability of the PRMs to withstand the harsh reaction conditions.
Figure 9.

(a) 4-step, 3-pot synthesis scheme for D-[18F]FAC. (b) Module configuration to perform this synthesis. Reagents in this case are added manually from outside the hot cell.
The fluorination reaction was performed in PRM#1. 20–1500 mCi of [18F]fluoride solution eluted from the QMA was added at Station 1 of the PRM into the reaction vessel preloaded with K222 (10 mg in 1 mL MeCN). The solvent was evaporated at 105°C for 7 min. Azeotropic distillation was performed next, first adding 1 mL MeCN to the vessel and evaporating at 105°C for 7 min, then adding 1 mL additional MeCN and evaporating at 105°C for 3 min. The precursor [2-O-[Trifluoromethyl)sulfonyl]-1,3,5-tri-O-benzoyl-α-D-arabinofuranose (10 mg in 1mL MeCN) was added, then the reaction vessel moved to Station 2, and heated at 160°C for 15 min under sealed conditions. After cooling, the vessel was moved to Station 3 and the fluorinated product loaded into a silica cartridge (Sep-Pak® Classic Silica cartridge, WAT051900, Waters Corporation) preconditioned with 8 mL hexane. The vessel was moved back to Station 1, 2mL EtOAc was added, then the vessel was moved to Station 3 and the contents transferred to elute the silica cartridge into PRM #2. The solvent was reduced by evaporation at 80°C for 2 min in PRM#2. The protected fluorinated intermediate was confirmed by radio-TLC (mobile phase MeCN:H2O, 95:5 v/v).
After complete evaporation of the EtOAc solvent, 100μL of HBr in acetic acid was added, followed by 400μL of 1,2-dichloroethane. The reaction vessel was then moved to Station 2, and the contents heated at 80°C for 10 min under sealed conditions. The volume was slightly reduced by moving the vessel to Station 1 and evaporating at 52°C for 1 min. Next, 700μL of toluene was added to the reaction mixture and the solvent evaporated at 60°C for 1 min.
Bis(trimethylsilylcytosine) (30 mg in 1mL of 1,2-dichloroethane) was added to the reaction vessel in PRM#2. The vessel was moved to Station 2, then heated at 158°C for 30 min under sealed conditions. After cooling to 40°C, the vessel was moved to Station 3, and the reaction mixture loaded into a silica cartridge (Sep-Pak Vac Silica cartridge 6cc/500mg, WAT043400, Waters Corporation), preconditioned with 8mL of hexane. Large tubing (1/8″ OD) and a large size cartridge were used because of the possibility of precipitate after this reaction step, which can clog small tubing and cartridges. Next, the vessel was returned to Station 1, and 1 mL 90:10 (v/v) CH2Cl2/MeOH was added. The vessel was moved to Station 3, and the contents transferred through the cartridge to elute the trapped product into PRM #3. The solvent was evaporated at 80°C for 2 min. This procedure was repeated a total of 3 times. Next, 500 μL of NaOMe (0.5M in methanol) was added to the vessel, the vessel then moved to Station 2 and heated for 5 min at 100°C under sealed conditions. After cooling to 40°C, the vessel was moved to Station 1, then the product mixture was neutralized by adding 250 μL 1N HCl. The neutral mixture was then diluted in 2.5 mL HPLC semi-preparative mobile phase (1:99 (v/v) EtOH/ammonium phosphate monobasic 10mM).
The 3mL reaction mixture was injected into the Phenomenex Gemini C18 semi-preparative column (25cm × 1cm) at a flow rate of 5 mL/min with mobile phase 1:99 v/v EtOH / 10 mM NH4H2PO4. Using a remote-controlled stream selector, peaks were distributed into different fractions. The fraction containing the radiochemically pure D[18F]FAC was eluted off the column with a retention time of approximately 17–18 min. Identity and radiochemical purity of collected fractions was confirmed by co-injection with D-FAC reference standard into the analytical HPLC with a Phenomenex Luna C18 column (25 cm × 0.46 cm, 5 μm particle size) at a flow rate of 1 mL/min and mobile phase 1:9 (v/v) EtOH / 50 mM NH4OAc. Both eluted with a retention time of about 4.5 min.
2.2.5.1 MicroPET Imaging with D-[18F]FAC
The fraction from the semi-preparative HPLC containing radiochemically pure D-[18F]FAC was concentrated by rotary evaporation to a volume of ~600 μL. A C57Bl6 mouse was lethally irradiated then transplanted with hematopoietic stem cell (HSC) enriched bone marrow type IV. Eight weeks post-transplantation, the mouse was injected with D-[18F]FAC (~200 μCi diluted in 100 μL saline), followed by 60 min unconscious uptake and 10 min static PET scan (Focus 220 microPET scanner) to visualize dCK activity and 10 min CT scan (Micro CAT II CT).
3 Results and Discussion
The plug-and-play platform affords advantages in flexibility and expandability similar to existing modular systems such as ModularLab (Eckert & Ziegler Eurotope GmbH 2013). However, instead of using low-level building blocks such as valve banks, sets of reagent vials, or individual heated reaction vessels, our system uses higher-level building blocks based on conceptual tasks the radiochemist performs. These larger building blocks require very few fluidic connections between them. In particular, the RDM has a single output, the PRM has two inputs (i.e., one for product of previous reaction, one for reagent addition), and the CPM has one input (crude product) and one output (purified product). Furthermore, these larger building blocks can be operated in an intuitive manner based on high-level commands or “unit operations” (e.g. “Add reagent #3”, “React for 15 min at 165°C”, or “Trap crude product on cartridge”, etc.) rather than low-level operations such as switching individual valves. We expect this approach to reduce the setup and operating complexity of modular systems.
The performance of each individual module is characterized in Section 2 of the Supporting Information. As a validation of the overall plug-and-play platform and a demonstration of its versatility, we assembled three different configurations of the PRM, RDM, and CPM modules and synthesized [18F]FDG, [18F]FNB, and D-[18F]FAC. [18F]FDG is a two-step, single-vessel synthesis with a simple cartridge purification, [18F]FNB is a one-step, single-vessel synthesis with cartridge purification, and D-[18F]FAC is four-step, three-vessel synthesis with two intermediate cartridge purification steps and a final semi-preparative HPLC purification step. The synthesis of D[18F]FAC involves high pressures during the sealed fluorination and coupling reactions. With limitless ability to expand the system with any number of modules, combined with the ability to perform sealed reactions involving high pressures, we expect the platform to be suitable for the production of large classes of 18F, 11C, and radiometal-labeled PET tracers beyond what is presented here.
Though the well-known synthesis of [18F]FDG does not involve any challenging reaction conditions such as high pressures, it serves well as a validation of the simple module configuration in Figure 7. The decay-corrected radiochemical yield of the non-optimized synthesis was 51±11% (n=11) and radiochemical purity was >95%, which compares favorably with typical literature reports (Yu 2006). Successful and repeatable synthesis of [18F]FDG suggests proper functioning of the unit operations of the RDM and PRM.
With the module configuration of Figure 8 involving a CPM in addition to an RDM and PRM, [18F]FNB was readily synthesized in a semi-automated fashion with a decay-corrected radiochemical yield of 71.0±4.5% (n=5). The yield is comparable to typical reports in the literature (Collins et al. 1992; Vanbrocklin et al. 2001). The CPM enabled remote control of the cartridge purification, including crude product dilution, trapping, washing, and eluting steps, and resulted in a radiochemical purity of >95%.
As a further demonstration of the versatility of the system, we assembled the configuration of PRM modules in Figure 9 to produce D-[18F]FAC. The overall decay-corrected radiochemical yield after semi-preparative HPLC purification was found to be 28.4±6.0% (n=4), with radiochemical purity >99% confirmed by analytical radio-HPLC, and a synthesis time of ~4 hr. Though the synthesis has not been extensively optimized, the radiochemical yield is comparable to that reported in Amarasekera et al. (Under review). An example HPLC chromatogram of typical crude and purified product are shown in Figure 10. Specific activity was found to be ~1 Ci/μmol. A PET/CT scan with this tracer (Figure 11) showed the expected signal in the bone marrow, spleen, and thymus due to immune cell repopulation. (It is known that HSCs use nucleoside salvage including dCK during expansion and hematopoiesis.) Some background signal is seen in the gut and liver, with probe excretion in the kidneys and bladder. The biodistribution is very similar to studies using D-[18F]FAC produced with our first-generation modules.
Figure 10.
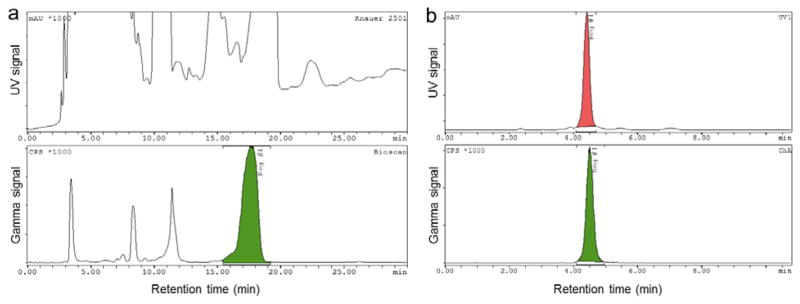
(a) Example semi-preparative radio-HPLC chromatogram (top: UV; bottom: gamma) of the D-[18F]FAC crude product. The desired fraction (β-isomer, highlighted) is collected and analyzed via analytical HPLC. (b) Example analytical radio-HPLC chromatogram (top: UV; bottom: gamma) of the selected fraction co-injected with the D-FAC reference standard, indicating >99% radiochemical purity of the purified D-[18F]FAC at a retention time of ~4.5 min.
Figure 11.

Coronal (left) and sagittal (right) slices of PET-CT scan of mouse injected with D-[18F]FAC produced in the plug-and-play system to visualize dCK activity. The C57Bl6 mouse was lethally irradiated then transplanted with HSC-enriched bone marrow type IV. It is known that HSCs use nucleoside salvage during expansion and hematopoiesis, and expected signal is observed in the bone marrow, spleen, and thymus due to repopulation. Additionally, some background is seen in the gut and liver.
1-(2′-deoxy-2′-[18F]fluoro-β-D-arabinofuranosyl)thymine (D-[18F]FMAU) was also synthesized on the system using the same protocol as for D-[18F]FAC (but replacing the cytosine precursor with the uracil precursor) with a decay-corrected radiochemical yield of 28±7% (n=3). With optimization, we expect to be able to reach the yields reported in Mangner et al. (2003).
In addition to enabling sealed reactions involving high pressures to be performed, the robotic mechanism of the reaction modules also allows the vessel to be easily and conveniently opened at any time during the synthesis to perform radioactivity measurements with a dose calibrator or to take samples for radio-TLC or radio-HPLC analysis. The feature is especially valuable for synthesis development and optimization but is rather unavailable in most existing commercial systems. Examples of how this capability can help to pinpoint the most significant opportunities to improve the radiochemical yield are given in Section 5 of the Supporting Information.
4 Conclusions
Using robotics to remove tubing from the reaction vessel and replace it with a stopper prior to reaction steps enables reproducible, high-yield synthesis of 18F-labeled nucleoside analogs despite the challenging high-pressure conditions in some reaction steps (Amarasekera et al. Under review). We have developed a second-generation, plug-and-play radiosynthesis platform based on this concept that is more compact and reduces manual intervention. This platform includes a Reagent Delivery Module that can store and dispense reagents to the reaction vessel on demand and a Cartridge Purification Module that stores diluent, wash, and eluent solutions to perform diverse cartridge purifications. Operation of any number of modules is controlled via a user-friendly PC-based software interface. As a validation of the platform and demonstration of its versatility we demonstrated the radiosyntheses of [18F]FDG using an RDM and PRM, [18F]FNB using an RDM, PRM, and CPM, and D-[18F]FAC using three PRMs followed by HPLC purification. The system could be quickly reconfigured for different syntheses with a minimum of fluidic connections, and each configuration could be immediately operated without any programming required. [18F]FDG, [18F]FNB, and D-[18F]FAC represent complexities ranging from 1-vessel to 3-vessel syntheses, represent different purification requirements from simple “filtering” through a cartridge, to solid-phase extraction, to HPLC, and represent different pressure requirements from minimal to pressures well over 100 psig [690 kPag]. We expect this platform to be capable of carrying out large classes of syntheses involving many PET radioisotopes, while providing features that facilitate probe development and optimization.
Supplementary Material
Highlights.
A flexible, plug-and-play radiosynthesis platform based on modules was developed
Reaction, reagent delivery, and purification modules can be readily configured for diverse syntheses
The reaction module is capable of high-pressure sealed reactions without solvent loss
Any number of modules can be combined and controlled in a semi-automated fashion using a personal computer
Syntheses of [18F]FDG, [18F]FNB, and D-[18F]FAC demonstrate versatility
Acknowledgments
We thank Nagichettiar Satyamurthy for invaluable contributions to design of the system, design and analysis of experiments, and insightful comments on the manuscript. We also thank Robert W. Silverman for extensive assistance with design and implementation of electronics, Yuliang Deng, Hong-Dun Lin, Yi-Chun Chen, and Chiyun Xia for assistance in design, assembly, and testing of early-generation prototypes, Arion Chatziioannou and David Stout for valuable suggestions on the design and setup of the synthesizer platform, and Waldemar Ladno and Melissa McCracken for assistance with animal imaging. This work was supported in part by the Department of Energy (DE-FG02-06ER64249 and DE-SC0001249), the National Cancer Institute (Nanosystems Biology Cancer Center, grant number U54CA119347), and the Industry-University Cooperative Research Program (UC Discovery Grant number bio07-10665).
References
- Amarasekera B, Marchis PD, Bobinski KP, Radu CG, Czernin J, Barrio JR, Van Dam RM. High-pressure, Compact, Modular Radiosynthesizer for Production of Positron Emitting Biomarkers. Under review. [DOI] [PubMed] [Google Scholar]
- Anderson H, Pillarsetty N, Cantorias M, Lewis JS. Improved Synthesis of 2′-deoxy-2′-[18F]-fluoro-1-[beta]-d-arabinofuranosyl-5-iodouracil ([18F]-FIAU) Nuclear Medicine and Biology. 2010;37 (4):439–442. doi: 10.1016/j.nucmedbio.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemical Engineering Research Information Center (CHERIC) Korea Thermophysical Properties Data Bank (KDB) Pure Component Properties. 2013 http://www.cheric.org/research/kdb/hcprop/cmpsrch.php.
- Chin FT, Namavari M, Levi J, Subbarayan M, Ray P, Chen X, Gambhir SS. Semiautomated Radiosynthesis and Biological Evaluation of [18F]FEAU: A Novel PET Imaging Agent for HSV1-tk/sr39tk Reporter Gene Expression. Molecular Imaging and Biology. 2007;10 (2):82–91. doi: 10.1007/s11307-007-0122-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins M, Lasne M-C, Barré L. Rapid Synthesis of N,N_-disubstituted Piperazines. Application to the Preparation of No Carrier Added 1-(4-[18F]fluorophenyl)piperazine and of an [18F]-selective Ligand of Serotoninergic Receptors (5HT2 Antagonist) Journal of the Chemical Society, Perkin Transactions. 1992;1(23):3185–3188. [Google Scholar]
- Eckert & Ziegler Eurotope GmbH. Modular-Lab Standard. Modular-Lab Standard. 2013 http://www.ezag.com/home/products/radiopharma/radiosynthesis-technology/modular-lab-standard.html.
- Hamacher K, Coenen HH, Stocklin G. Efficient Stereospecific Synthesis of No-Carrier-Added 2-[18F]-Fluoro-2-Deoxy-D-Glucose Using Aminopolyether Supported Nucleophilic Substitution. J Nucl Med. 1986;27 (2):235–238. [PubMed] [Google Scholar]
- Laing RE, Walter MA, Campbell DO, Herschman HR, Satyamurthy N, Phelps ME, Czernin J, Witte ON, Radu CG. Noninvasive Prediction of Tumor Responses to Gemcitabine Using Positron Emission Tomography. Proceedings of the National Academy of Sciences. 2009;106 (8):2847–2852. doi: 10.1073/pnas.0812890106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Cai H, Conti PS. Automated Synthesis of 2′-deoxy-2′-[18F]fluoro-5-methyl-1-β-d-arabinofuranosyluracil ([18F]-FMAU) Using a One Reactor Radiosynthesis Module. Nuclear Medicine and Biology. 2011;38 (2):201–206. doi: 10.1016/j.nucmedbio.2010.08.010. [DOI] [PubMed] [Google Scholar]
- Lu S, Giamis AM, Pike VW. Synthesis of [18F]fallypride in a Micro-reactor: Rapid Optimization and Multiple-production in Small Doses for micro-PET Studies. Current Radiopharmaceuticals. 2009;2 (1):1–13. doi: 10.2174/1874471010902010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangner TJ, Klecker RW, Anderson L, Shields AF. Synthesis of 2′-deoxy-2′-[18F]fluoro-[beta]-D-arabinofuranosyl Nucleosides, [18F]FAU, [18F]FMAU, [18F]FBAU and [18F]FIAU, as Potential PET Agents for Imaging Cellular Proliferation: Synthesis of [18F]labelled FAU, FMAU, FBAU, FIAU. Nuclear Medicine and Biology. 2003;30 (3):215–224. doi: 10.1016/s0969-8051(02)00445-6. [DOI] [PubMed] [Google Scholar]
- Padgett HC, Schmidt DG, Luxen A, Bida GT, Satyamurthy N, Barrio JR. Computer-controlled Radiochemical Synthesis: A Chemistry Process Control Unit for the Automated Production of Radiochemicals. International Journal of Radiation Applications and Instrumentation. Part A. Applied Radiation and Isotopes. 1989;40 (5):433–445. doi: 10.1016/0883-2889(89)90213-x. [DOI] [PubMed] [Google Scholar]
- Paolillo V, Riese S, Gelovani JG, Alauddin MM. A Fully Automated Synthesis of [18F]-FEAU and [18F]-FMAU Using a Novel Dual Reactor Radiosynthesis Module. Journal of Labelled Compounds and Radiopharmaceuticals. 2009;52 (13):553–558. [Google Scholar]
- Radu CG, Shu CJ, Nair-Gill E, Shelly SM, Barrio JR, Satyamurthy N, Phelps ME, Witte ON. Molecular Imaging of Lymphoid Organs and Immune Activation Using Positron Emission Tomography with a New 18F-labeled 2′-deoxycytidine Analog. Nature Medicine. 2008;14 (7):783–788. doi: 10.1038/nm1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyamurthy N, Barrio J, Amarasekera B, Van Dam RM, Olma S, Williams D, Eddings M, Shen C. Modular Radiochemistry Synthesis System. WO/2010/021719 International Patent Application. 2010
- Shields AF. Labeled Pyrimidines in PET Imaging. In: Valk PE, Delbeke D, Bailey DL, Townsend DW, Maisey MN, editors. Positron Emission Tomography. Springer; London: 2006. pp. 375–385. [Google Scholar]
- Shu CJ, Campbell DO, Lee JT, Tran AQ, Wengrod JC, Witte ON, Phelps ME, Satyamurthy N, Czernin J, Radu CG. Novel PET Probes Specific for Deoxycytidine Kinase. J Nucl Med. 2010;51 (7):1092–1098. doi: 10.2967/jnumed.109.073361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungersboeck J, Philippe C, Mien LK, Haeusler D, Shanab K, Lanzenberger R, Spreitzer H, et al. Microfluidic Preparation of [18F]FE@SUPPY and [18F]FE@SUPPY:2 -- Comparison with Conventional Radiosyntheses. Nuclear Medicine and Biology. 2011;38 (3):427–434. doi: 10.1016/j.nucmedbio.2010.09.009. [DOI] [PubMed] [Google Scholar]
- Vallabhajosula S. 18F-Labeled Positron Emission Tomographic Radiopharmaceuticals in Oncology: An Overview of Radiochemistry and Mechanisms of Tumor Localization. Seminars in Nuclear Medicine. 2007;37 (6):400–419. doi: 10.1053/j.semnuclmed.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Vanbrocklin HF, O’Neil JP, Hom DL, Gibbs AR. Synthesis of [18F]fluoroanilines: Precursors to [18F]fluoroanilinoquinazolines. Journal of Labelled Compounds and Radiopharmaceuticals. 2001;44 (S1):S880–S882. [Google Scholar]
- Wright E, Jellman JK. Beginner’s Robotics on $50 a Month. SERVO Magazine. 2007 Mar [Google Scholar]
- Yu S. Review of 18F-FDG Synthesis and Quality Control. Biomedical Imaging and Intervention Journal. 2006;2 (4):e57. doi: 10.2349/biij.2.4.e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.