

Abstract
Background:
Multiple myeloma SET domain (MMSET)/nuclear receptor binding SET domain 2 (NSD2) is a lysine histone methyltransferase (HMTase) and bona fide oncoprotein found aberrantly expressed in several cancers, suggesting potential role for novel therapeutic strategies. In particular, MMSET/NSD2 is emerging as a target for therapeutic interventions against multiple myeloma, especially t(4;14) myeloma that is associated with a significantly worse prognosis than other biological subgroups. Multiple myeloma is the second most common hematological malignancy in the United States, after non-Hodgkin lymphoma and remains an incurable malignancy. Thus, effective therapeutic strategies are greatly needed. HMTases inhibitors are scarce and no NSDs inhibitors have been isolated.
Methods:
We used homology modeling, molecular modeling simulations, virtual ligand screening, computational chemistry software for structure-activity relationship and performed in vitro H3K36 histone lysine methylation inhibitory assay using recombinant human NSD2-SET and human H3.1 histone.
Results:
Here, we report the discovery of LEM-06, a hit small molecule inhibitor of NSD2, with an IC50 of 0.8 mM against H3K36 methylation in vitro.
Conclusions:
We propose LEM-06 as a hit inhibitor that is useful to further optimize for exploring the biology of NSD2. LEM-06 derivatives may pave the way to specific NSD2 inhibitors suitable for therapeutic efforts against malignancies.
Keywords: Epigenomics, Drug design, Histone-Lysine N-Methyltransferase, NSD2 protein, Antagonists & inhibitors
INTRODUCTION
A rapidly increasing amount of evidence highlights the importance of epigenetic deregulation in numerous carcinogenetic events.1 Nuclear receptor binding SET domain 1 (NSD1), NSD2/multiple myeloma SET domain (MMSET)/Wolf-Hirschhorn syndrome candidate 1 (WHSC1), and NSD3/WHSC1L1 compose the NSD family and are classified into the histone methyltransferase (HMTase) KMT3 family. The NSDs are histone modifiers involved in maintaining the chromatin. The NSDs predominantly methylate histone H3 lysine 36 (H3K36) and histone H4 lysine 20 (H4K20) at the chromatin, in vivo.2–4 NSD1, NSD2, and NSD3 are likely to operate in distinct pathways since NSD2 and NSD3 do not make substantial contribution to overgrowth phenotypes observed in NSD1 gene alterations.5 However, it remains unclear whether any of the NSD family members can substitute each other in pathways.
The NSDs are instrumental in the development and progression of numerous cancers and are considered valuable drug-targets, especially NSD2.3,6–16 The amplification of NSD1 is found in multiple myeloma, lung cancer, neuroblastomas, and glioblastomas. The amplification of either NSD1 or NSD2 triggers the cellular transformation to cancer formation. NSD3 plays a role in lung cancer and is found amplified in breast cancer cell lines and primary breast carcinomas.17,18 NSD2/MMSET is associated with tumor aggressiveness or prognosis in most types of cancers including prostate cancer and multiple myeloma.10–13,19,20 NSD2 is overexpressed in solid tumors especially in breast cancer, myeloma and glioblastoma, resulting in aberrantly high global levels of H3K36me2.3,6–16 Overexpression of NSD2 in prostate cancer causes aberrations in gene transcription leading to a metastatic phenotype.9 NSD2/MMSET is a focal point for therapeutic interventions against multiple myeloma and especially for t(4;14) myeloma, which has a worse prognosis than other biological subgroups.20 Multiple myeloma remains the second most common hematological cancer after non-Hodgkin lymphoma. Unfortunately, no effective cure exists against multiple myeloma and effective therapeutic strategies are sorely needed.
Chromatin remodelling inhibitors targeting DNA methyltransferases (DNMTs), histone methyltransferases, and deacetylases (HDACs) are being pursued for both cancer chemotherapies and cancer chemoprevention. Several DNA methylation and histone deacetylase inhibitors are currently in clinical trial stages.21–25 Amongst the large family of HMTases, several such as EZH2, DOT1L are therapeutic targets, but HMTases inhibitors are still scarce and very few compounds have been reported to be selective and specific.26 Notably, GlaxoSmithKline Inc. (Brentford, United Kingdom) and Epizyme Inc. (Epizyme Inc., Cambridge, MA, USA) have made significant progress on the discovery of HMTase DOT1L and EZH2 potent inhibitors.27–30 DOT1L is distinct from other HMTases as it does not contain a canonical SET domain.31,32 The HMTase inhibitors, BIX-01294 and BIX-01338, have been shown to be effective on G9a with an IC50 of 3 μM and 5 μM, respectively.33 In addition, Chaetocin inhibits Su(var)3–9 with an IC50 of 0.8 μM.33 Importantly, Liu et al.34 completed a seminal work on the G9a inhibitors such as UNC0224, with an IC50 of 15 nM. Both Epizyme Inc., GlaxoSmithKline Inc. along with research groups on G9a and EZH2 have reported HMTase inhibitors with IC50 in the nano molar range.
The catalytic mechanism of lysine-HMTase has been established and it proceeds through a linear SN2 nucleophilic attack between the cofactor S-Adenosyl methionine (SAM) and the lysine-NH3 substrate.1 The SAM binds into a small cavity immediately adjacent to the histone-tail large binding groove where the lysine substrate extends deep inside a channel at the interface between both binding areas.15 Previously, we demonstrated that the SET domain of NSD1 accommodates a 7-amino acid peptide, similarly as it was further identified in SET8.26,35 In addition, we demonstrated the opening mechanism of the SET domain of NSD1 through rotation of a small loop at the interface between the SET and postSET subdomain.26 This regulatory-loop is likely to participate in both the substrate recognition and the catalytic mechanism by acting as a seat belt for the lysine-substrate. The regulatory-loop sits on top of the lysine-substrate strongly anchoring the histone-tail in the SET domain.35 The histone-tail binding area involves areas from both the SET and postSET subdomains (Fig. 1). The SET domain sequence is highly conserved across the NSDs, thus it is likely that NSD2 and NSD3 proceed through the same mechanism as described for NSD1 (Fig. 2). Noteworthy, the NSDs are phylogenically distinct from other known HMTases (Fig. 2).
Figure 1.

Architecture of nuclear receptor binding SET domain 2 (NSD2)/multiple myeloma SET domain (MMSET) and model of the opened SET domain with LEM-06. (A) Schematic of the primary structure of NSD2: PWWP domain; PHD zinc fingers domain; SET histone methyl transferase (HMTase) with the preSET and postSET domains. The regulatory loop closing onto the histone-binding site is indicated in red. (B) Commassie staining on sodium dodecylsulfate-polyacrylamide gel electrophoresis gel of the recombinant expressed NSD2-SET after purification. (C) Molecular surface of the model of the opened NSD2-SET bound with the computed most favourable conformation of LEM-06 during virtual ligand screening. Electrostatic surface is coloured as follow: blue: positive charges; red: negative charges with unit +5/−5 kb.T.ec−1.
Figure 2.

Sequences alignment of the preSET, SET and postSET subdomains of nuclear receptor binding SET domain 1 (NSD1), NSD2 and NSD3. Boxed in blue are the regions involved in histone-tails binding. Boxed in red are the regions responsible for the S-Adenosyl methionine. The multiple sequence alignment was done with CLUSTAL 2.1
The inhibition of HMTases can be achieved either by targeting the SAM binding pocket or the histone-tail binding cleft. In EZH2, the inhibitor GSK343 is a SAM competitor that has good inhibition for EZH1 and non-negligible inhibition for both SET7 and PRMT3.28 Interestingly, EZH1 and EZH2 have SET domain sequences closely related to the NSDs and it may be valuable to assay the selectivity of EZH2 inhibitors against the NSDs, especially NSD2 (Fig. 2).28 MCTP39 is a SAM competitor for NSD2 recently patented.36
The SAM binding pocket is partially buried in the crystal structure of the SET domain in NSD1 and the key residues stabilizing SAM are highly conserved across the NSDs (Fig. 2).26 In contrast, the larger and more accessible histone-tail binding area possesses greater sequence heterogeneity, compared to the SAM binding pocket (Fig. 2). The residues involved in stabilizing the binding of histone substrates are less conserved amongst NSDs unlike the SAM binding site (Fig. 2).1,26 This was previously observed in the crystal structure of the HMTase G9a-like protein with the inhibitor BIX-01294, an histone-tail mimetic inhibitor targeting the histone tail binding domain.37 In addition, the involvement of the specific flexible regulatory loop in the binding of histone-tail into the NSDs SET domain, further suggest that the histone-tail binding cleft may be exploited for achieving specific and selective inhibition in the case of NSD2.
Taken together, we hypothesized that the selective inhibition of the NSDs may be reached with small molecules targeting the histone-tail site over the more conserved SAM binding site. In a bid to complement existing inhibitory strategies targeting the SAM binding pocket, we screened molecules for the binding to the histone-tail binding region of NSD2-SET. In this study, we identified an NSD2 inhibitor for the histone-tail binding groove using virtual ligand screening on the molecular model of the opened conformation of the histone-tail binding domain of NSD2-SET.
MATERIALS AND METHODS
1. Cloning
The SET domain of human NSD2 gene (NSD2-SET, 873 bp, 2844–3717 nt; 291 a.a., 948–1239 a.a) was amplified by polymerase chain reaction (PCR) using human liver cDNA library (Takara Bio Inc., Otsu, Japan) as template. The forward and reverse primers are PK162, 5′-GGCAGCCATATG(NdeI)CAGGGGGTCAGAGGGATC GGAAGAG-3′ and PK163, 5′-GAAGCACTCGAG(XhoI)CTCTGACTG CCTCTTCCCTTCCCC-3′, respectively. The PCR-amplified NSD2-SET DNA fragment was digested with NdeI and XhoI and inserted into the multi cloning site of the protein expression Intein-tagging vector, pTYB2 (New England Biolabs, Ipswich, MA, USA). The sequence was verified by sequencing.
2. Protein expression and purification
Escherichia coli expression strain, BL21, transformed with pTYB2 plasmid harbouring NSD2-SET was grown in LB medium containing 100 μg/mL ampicillin and the expression of recombinant NSD2-SET was induced with 250 μM isopropyl 1-thio-D-galactopyranoside (IPTG) for 4 hours at 15°C. E. coli cells were harvested and lysed by freeze-and-thaw method and incubation in buffer A (20 mM Tris [pH 8.0], 500 mM NaCl, and 0.1 mM ethylenediaminetetraacetic acid) containing 0.1% Triton X-100 and 10 mM phenylmethanesulfonylfluoride along with 20 cycles of sonication on ice. The resulting cell extract containing NSD2-SET-Intein-chitin-binding domain fusion protein was loaded onto an affinity column of chitin beads and washed with 100-column volumes of buffer A with 0.1% Triton X-100, followed by 20-column volumes of buffer A without Triton X-100. To remove bacterial chaperones bound to the recombinant proteins, the recombinant NSD2-SET-bound chitin beads were washed with 10-bed volumes of buffer A containing 10 mM adenosine triphosphate and 2.5 mM MgCl2. NSD2-SET proteins was cleaved off from the chitin beads by incubation in buffer A with 50 mM 2-mercaptoethanol at 4°C for 48 hours, eluted in buffer A, concentrated using Amicon Ultra centrifugal filters and then used for methyltransferase assays. A small portion of purified NSD2-SET was resolved on a sodium dodecylsulfate-polyacrylamide gel electrophoresis. Coomassie staining gel showed soluble and pure NSD2-SET at around expected molecular weight of 33.2 kDa (Fig. 1B). The resulting recombinant NSD2-SET was proven to be stable and retained catalytic properties for an extended period of time when stored at −80°C.
3. Compounds
Compounds were prepared in dimethyl sulfoxide (DMSO) > 99.9% pure (D8418; Sigma-Aldrich, St. Louis, MO, USA).
4. Histone methyltransferase inhibitor assay
HMTase activity of NSD2-SET on H3K36 was measured by colorimetric quantification kits (Epigentek, Farmingdale, NY, USA) following the manufacturer’s protocol. Purified recombinant NSD2-SET (4.0 μg, final 2.42 μM) was preincubated for 20 minutes with a recombinant purified human histone H3.1 (5 μM) (New England Biolabs) and indicated concentrations of inhibitors/DMSO or DMSO alone (0 mM inhibitor) in C2 buffer provided by the assay kit. The recombinant histone used for this assay is not modified at any potential modification site during the production in E. coli and have been checked by electrospray ionization time-of-flight mass spectrometry, according to the manufacturer (New England Biolabs). The pre-incubated mix was added with a methyl group donor (Adomet) (50 μM), immediately transferred to and incubated in the strip wells for 120 minutes at room temperature in the dark. The mono-methylated H3K36 (H3K36me1) was captured by anti-mono-H3K36 antibody attached to the bottom of the strip wells. Excess of purified NSD2-SET, histone H3, inhibitors, and Adomet was thoroughly washed away and the labelled detection antibody followed by colour development reagent detected the captured antibody-H3K36me1. The strip wells were analyzed with an ELISA plate reader at 450 nm. The level of H3K36me1 is proportional to the intensity of the absorbance. Assays were done in triplicate individual experiments. The results were normalized against the control that does not contain any enzymes.
5. Molecular modeling of the closed conformation of NSD2-SET
The crystal structure of the SET domain of NSD1 (Protein Data Bank [PDB] ID: 3OOI - a.a. 1850–2080) served as the template to build the corresponding SET domain of NSD2 (a.a. 971–1202) by homology modelling. The sequence of NSD2-SET (a.a. 971–1202) shares 75.9% identity and 90.1% similarity with the template NSD1-SET (PDB: 3OO1). After a careful multiple-sequence alignment with ClustalW V2.0.9, one hundred models were generated with Modeler V9.5. The best model, according to the intrinsic Modeler function, was subjected to side-chains positioning using the SCWRL4 program. Stereochemistry was checked with PROCHECK. The model was manually inspected with COOT. Models of NSD2-SET has an H-factor of 18.1% (e.g. accurate). The quality and accuracy of the set of models were assessed by the H-factor, a novel quality metric for homology modelling we recently introduced.38,39 Due to the high sequence conservation of the SET domain between NSD1 and NSD2, the modelling of NSD2-SET represents an ideal case.
6. Molecular modeling of the opened conformation of NSD2-SET
The histone tail H3K36 (a.a. 32–38) peptide was manually built in COOT. A structural overlay between NSD1-SET (PDB: 3OOI) and the crystal structure of SET8 bound to histone H4 peptide (PDB: 3F9Y) was used to manually place and orientate the H3K36 (a.a. 32–38) peptide into the histone binding site of the model of the closed conformation of NSD2. The structural overlay was done using the SSM algorithm in COOT. The complex peptide-NSD2-SET complex was fully solvated with a water box (140 Å × 100 Å × 110 Å) using VMD 1.9. Energy minimizations and molecular dynamics (MD) were performed with NAMD v2.8 using the CHARMM force field. A constant temperature was maintained by using a Langevin damping at 300 K, and the pressure was held constant at 1 atmosphere with a Langevin piston. Electrostatic forces were taken into account using the particle mesh Ewald method with a 10 Å cut-off distance. The solvated complex peptide-NSD2-SET was subjected to a total of 100,000 steps (100 ns) of MD and a total of 30,000 steps (30 ns) of energy minimizations. After the MD simulations, the solvated complex was manually checked for ideal stereochemistry with COOT. The MD trajectory was analyzed with VMD 1.9. The peptide H3K36 (a.a. 32–38) and water molecules were deleted from the peptide-NSD2-SET complex leaving NSD2-SET in an open conformation suitable for virtual ligand screening.
7. Virtual ligand screening and selection of the compounds
AutoDock Vina was used to perform the dockings with the ‘Clean Drug-Like compounds’ downloaded from the ZINC docking database version 11 (free database).40 The missing hydrogen atoms on the opened NSD2-SET were added using PDB2PQR program in conjunction with the CHARMM force field.41 Ligands were prepared with the AutoDock tools (ADT) v1.5.4 r29. The docking grid sizes and positions were manually set using ADT. The grid dimension (22 ×20 × 20 Å) was large enough to cover the opened NSD2-SET and centered onto the catalytic channel identified in NSD1-SET.26 A subset of 40,325 molecules (190 Da < MW < 500 Da) was docked and the top 3,000 results were analysed and filtered with FieldView from Cresset (Litlington, UK). The top docking solutions were manually inspected using COOT and PyMOL.
RESULTS AND DISCUSSION
NSD2/MMSET is a key therapeutic target against multiple myeloma, a malignancy with a very low survival rate and no cure.6,7,10–13,19,20 Unfortunately, HMTase inhibitors remain rare and only one NSD2 inhibitor MCTP39, a SAM competitor has been identified.42–45 In this work, using a structure-based discovery approach, we identified a novel small molecule named LEM-06 that inhibits, in vitro, the H3K36 HMTase activity of NSD2.
The structure of NSD2 is unknown and the closest crystal structure solved is the apo SET domain of NSD1. Following our previous study on NSD1-SET, we build by homology modelling, a model of NSD2-SET exploiting the crystal structure of NSD1-SET as both share high sequence identity (75.9%) and similarity (90.1%).26,46 Previously, we studied the movement and role of the regulatory loop located at the interface between the SET and postSET domain of NSD1-SET (Fig. 1A and 1C).26 In a closed conformation, the binding of H3 or H4 tails is sterically prevented. However, this is not observed in both H3K9 and H3K4-specific HMTases.46–48 Therefore, we elected to model NSD2-SET in an opened conformation using long range MD simulations that we previously described for NSD1 (Fig. 1C).26 We inserted a 7-mer H3 peptide (a.a. 32–38) underneath the regulatory loop at the interface of the SET and postSET sub-domains, in the closed conformation of NSD2-SET. The complex NSD2-SET-H3K36 (a.a. 32–38) was forced deliberately in a sterically unstable conformation. Energy minimizations and long MD simulations relieved the steric constraints and forced the SET domain to accommodate the substrate (Fig. 1C). The MD simulations allowed the complex to stabilize into a stable conformation similarly observed for NSD1-SET.26 The regulatory loop of NSD2-SET underwent significant displacements, with a rotation −45° and a translation ∼6 Å at the tip, that open a binding groove largely negatively charged suitable for the docking of H3 or H4 tails.26 Both NSD1-SET and NSD2-SET have recognition sequence covering at least 7 amino-acids in par with the H4K20 HMTase SET8.35
BIX-01294 is a histone-tail mimetic targeting the SET domain of G9a like protein HMTase.37 Exploiting the findings of the crystal structure of BIX-01294 bounds into the histone-tail cleft of GLP, we designed a virtual ligand screening strategy centered on the histone-tail binding pocket of the opened conformation of NSD2-SET. The HMTase inhibitors BIX-01294, BIX-01338, and chaetocin have molecular weights of 492 Da, 535 Da, and 696 Da, respectively. BIX-01294, BIX-01338 and chaetocin have positive electrostatic fields with steric hindrance matching the binding groove for histone tails. However, molecules of over 500 Da may have unfavorable pharmacokinetic parameters. Therefore, we filtered the compounds by molecular weight of less than −500 Da, yielding a subset of 40,325 molecules used for docking. The 40,325 docking results were ranked according to the best docking energy of the best-docked conformer. We applied screening filters on the top 3,000 molecules to ensure that the selected compounds have electrostatic positive field matching the histone-tail binding groove of NSD2-SET (priority #1), a proper structural fit and binding interactions in the SET domain upon manual inspection with COOT (priority #2), none or low number of violations of the Lipinski’s rule of 5 (priority #3), a total polar surface area < 100 Å2 (priority #4) and molar refractivity (priority #5). Molecules with heavy atoms were not considered. Out of 20 handpicked molecules (internally named LEM-XX), nine were eventually hand picked and subjected to in vitro assay against H3K36 mono-methylation (H3K36me1).
The selected molecules were tested for inhibition of H3K36me1 by recombinant NSD2-SET with recombinant un-modified human histone H3.1 and compound LEM-06 inhibited NSD2-SET in vitro with an IC50 of 890 μM. LEM-06 in vitro IC50 value is slightly above other HMTase inhibitors such as MCTP39, TMDC 1c, AMI-1, AMI-5, AMI-6, AMI-9 and GSK343 IC50 range (Table 126,38,49–53, Fig. 3 and 4).
Table 1.
Lysine- and arginine-HMTase inhibitors
| Compound | HMTase | Target | IC50 | Reference |
|---|---|---|---|---|
| UNC0224 | Lysine HMTase | G9a | 15 nM | 51 |
| UNC0321 | Lysine HMTase | G9a | 6 nM | 51 |
| BIX-01294 | Lysine HMTase | G9a | 2.7 μM | 52 |
| EPZ-5676 | Lysine HMTase | DOT1L | 1.3 μM–3.5 nM | 26 |
| GSK343 | Lysine HMTase | EZH2 | 0.2 μM | 49 |
| EPZ-6438 | Lysine HMTase | EZH2 | 2.5 nM | 50 |
| MCTP39 | Lysine HMTase | MMSET/NSD2 | 3 μM | 38 |
| BIX-01294 | Lysine HMTase | EHMT2 | 2.7 μM | 53 |
| TMDC 1c | Lysine HMTase | SET7/9 | 10 μM | 52 |
| SAH | HMTase | Non specific | 0.1–20 μM | 52 |
| Cmp5 | Arginine HMTase | Human PRMT1 | 55 μM | 52 |
| AMI-1 | Arginine HMTase | Human PRMT1 | 8.8 μM | 51 |
| AMI-5 | Arginine HMTase | Human PRMT1 | 1.4 μM | 51 |
| AMI-6 | Arginine HMTase | Human PRMT1 | 5.1 μM | 51 |
| AMI-9 | Arginine HMTase | Human PRMT1 | 280 nM | 51 |
HMTase, histone methyltransferase.
Figure 3.

Dose-response curves of LEM-06 against nuclear receptor binding SET domain 2 (NSD2)-SET H3K36 mono-methylation activity. The dose-response curve and the IC50 calculation of LEM-06 were done using GraphPad Prism 6.0. Assays were done in triplicate individual experiments. The results were normalized against the control that does not contain any enzymes and only average data points are plotted.
Figure 4.
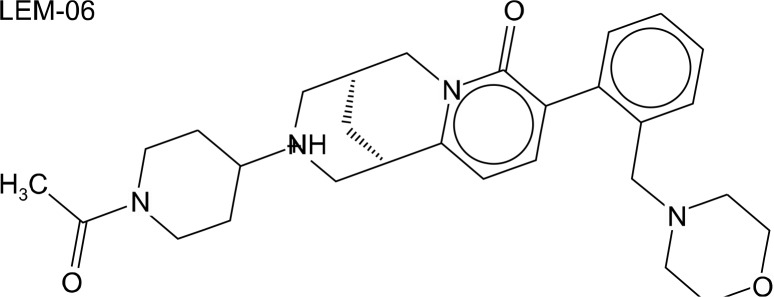
Chemical structure of LEM-06. LEM-06 molecular weight is 492.7 Da; IC50 = 0.89 mM (NSD2-SET in vitro); Docking energy = −10.3 kcal/mol; wcLogP = 2.4, total polar surface area = 58.7 (Å2); number of rotated bonds = 4; number of violations of the Lipinski’s rule of five = 0; ZINC database ID: 20503683.
The weaker inhibition by LEM-06 could perhaps be explained by a weak stabilization in the SET domain with three hydrogen bonds for LEM-06 (Fig. 5). One possible explanation for a reduced inhibition by LEM-06 may lie in the molar refractivity of the tested molecules to effectively compete for the binding with histone H3.1.
Figure 5.

Molecular details of the binding of LEM-06 into the histone-tail binding groove of nuclear receptor binding SET domain 2 Dashed yellow lines indicate hydrogen bonds.
In summary, we report the discovery of LEM-06, a novel NSD2 inhibitor against H3K36 methylation and effective in vitro with an IC50 = 0.8 mM. LEM-06 is likely to bind to the histone binding pocket, but further studies such as protein X-ray crystallography are needed to confirm and fully define its binding characteristics. We propose LEM-06 as a hit molecule to further derivate for investigating the biology of NSD2/MMSET. LEM-06 derivatives may eventually lead to specific NSD2/MMSET inhibitors suitable against malignancies that include multiple myeloma.
Acknowledgments
This research was supported by Kyungpook National University Research Fund, 2011.
Footnotes
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
- 1.Morishita M, di Luccio E. Cancers and the NSD family of histone lysine methyltransferases. Biochim Biophys Acta. 2011;1816:158–63. doi: 10.1016/j.bbcan.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 2.Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol. 2012;13:115–26. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, et al. MMSET regu-lates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470:124–8. doi: 10.1038/nature09658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lucio-Eterovic AK, Singh MM, Gardner JE, Veerappan CS, Rice JC, Carpenter PB. Role for the nuclear receptor-binding SET domain protein 1 (NSD1) methyltransferase in coordinating lysine 36 methylation at histone 3 with RNA polymerase II function. Proc Natl Acad Sci U S A. 2010;107:16952–7. doi: 10.1073/pnas.1002653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Douglas J, Coleman K, Tatton-Brown K, Hughes HE, Temple IK, Cole TR, et al. Childhood Overgrowth Collaboration Evaluation of NSD2 and NSD3 in overgrowth syndromes. Eur J Hum Genet. 2005;13:150–3. doi: 10.1038/sj.ejhg.5201298. [DOI] [PubMed] [Google Scholar]
- 6.Asangani IA, Ateeq B, Cao Q, Dodson L, Pandhi M, Kunju LP, et al. Characterization of the EZH2-MMSET histone methyl-transfer-ase regulatory axis in cancer. Mol Cell. 2013;49:80–93. doi: 10.1016/j.molcel.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brito JL, Walker B, Jenner M, Dickens NJ, Brown NJ, Ross FM, et al. MMSET deregulation affects cell cycle progression and adhe-sion regulons in t(4;14) myeloma plasma cells. Haematologica. 2009;94:78–86. doi: 10.3324/haematol.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998;92:3025–34. [PubMed] [Google Scholar]
- 9.Ezponda T, Popovic R, Shah MY, Martinez-Garcia E, Zheng Y, Min DJ, et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene. 2013;32:2882–90. doi: 10.1038/onc.2012.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hudlebusch HR, Santoni-Rugiu E, Simon R, Ralfkiær E, Rossing HH, Johansen JV, et al. The histone methyltransferase and puta-tive oncoprotein MMSET is overexpressed in a large variety of human tumors. Clin Cancer Res. 2011;17:2919–33. doi: 10.1158/1078-0432.CCR-10-1302. [DOI] [PubMed] [Google Scholar]
- 11.Kassambara A, Klein B, Moreaux J. MMSET is overexpressed in cancers: link with tumor aggressiveness. Biochem Biophys Res Commun. 2009;379:840–5. doi: 10.1016/j.bbrc.2008.12.093. [DOI] [PubMed] [Google Scholar]
- 12.Kim JY, Kee HJ, Choe NW, Kim SM, Eom GH, Baek HJ, et al. Multiple-myeloma-related WHSC1/MMSET isoform RE-IIBP is a histone methyltransferase with transcriptional repression activity. Mol Cell Biol. 2008;28:2023–34. doi: 10.1128/MCB.02130-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kassambara A, Klein B, Moreaux J. MMSET is overexpressed in cancers: link with tumor aggressiveness. Biochem Biophys Res Commun. 2009;379:840–5. doi: 10.1016/j.bbrc.2008.12.093. [DOI] [PubMed] [Google Scholar]
- 14.Marango J, Shimoyama M, Nishio H, Meyer JA, Min DJ, Sirulnik A, et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood. 2008;111:3145–54. doi: 10.1182/blood-2007-06-092122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinez-Garcia E, Popovic R, Min DJ, Sweet SM, Thomas PM, Zamdborg L, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117:211–20. doi: 10.1182/blood-2010-07-298349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang P, Guo L, Duan ZJ, Tepper CG, Xue L, Chen X, et al. Histone methyltransferase NSD2/MMSET mediates constitutive NF-κB signaling for cancer cell proliferation, survival, and tumor growth via a feed-forward loop. Mol Cell Biol. 2012;32:3121–31. doi: 10.1128/MCB.00204-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Angrand PO, Apiou F, Stewart AF, Dutrillaux B, Losson R, Chambon P. NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines. Genomics. 2001;74:79–88. doi: 10.1006/geno.2001.6524. [DOI] [PubMed] [Google Scholar]
- 18.Rosati R, La Starza R, Veronese A, Aventin A, Schwienbacher C, Vallespi T, et al. NUP98 is fused to the NSD3 gene in acute mye-loid leukemia associated with t(8;11)(p11.2;p15) Blood. 2002;99:3857–60. doi: 10.1182/blood.V99.10.3857. [DOI] [PubMed] [Google Scholar]
- 19.Xie Z, Chng WJ. MMSET: role and therapeutic opportunities in multiple myeloma. Biomed Res Int. 2014;2014:636514. doi: 10.1155/2014/636514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mirabella F, Wu P, Wardell CP, Kaiser MF, Walker BA, Johnson DC, et al. MMSET is the key molecular target in t(4;14) myeloma. Blood Cancer J. 2013;3:e114. doi: 10.1038/bcj.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–68. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 22.Spannhoff A, Sippl W, Jung M. Cancer treatment of the future: in-hibitors of histone methyltransferases. Int J Biochem Cell Biol. 2009;41:4–11. doi: 10.1016/j.biocel.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 23.Mack GS. To selectivity and beyond. Nat Biotechnol. 2010;28:1259–66. doi: 10.1038/nbt.1724. [DOI] [PubMed] [Google Scholar]
- 24.Brueckner B, Kuck D, Lyko F. DNA methyltransferase inhibitors for cancer therapy. Cancer J. 2007;13:17–22. doi: 10.1097/PPO.0b013e31803c7245. [DOI] [PubMed] [Google Scholar]
- 25.Kristensen LS, Nielsen HM, Hansen LL. Epigenetics and cancer treatment. Eur J Pharmacol. 2009;625:131–42. doi: 10.1016/j.ejphar.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 26.Morishita M, di Luccio E. Structural insights into the regulation and the recognition of histone marks by the SET domain of NSD1. Biochem Biophys Res Commun. 2011;412:214–9. doi: 10.1016/j.bbrc.2011.07.061. [DOI] [PubMed] [Google Scholar]
- 27.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110:7922–7. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, et al. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med Chem Lett. 2012;3:1091–6. doi: 10.1021/ml3003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122:1017–25. doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Basavapathruni A, Jin L, Daigle SR, Majer CR, Therkelsen CA, Wigle TJ, et al. Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem Biol Drug Des. 2012;80:971–80. doi: 10.1111/cbdd.12050. [DOI] [PubMed] [Google Scholar]
- 31.Min J, Feng Q, Li Z, Zhang Y, Xu RM. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003;112:711–23. doi: 10.1016/S0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 32.Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spannhoff A, Hauser AT, Heinke R, Sippl W, Jung M. The emerging therapeutic potential of histone methyltransferase and demethylase inhibitors. ChemMedChem. 2009;4:1568–82. doi: 10.1002/cmdc.200900301. [DOI] [PubMed] [Google Scholar]
- 34.Liu F, Chen X, Allali-Hassani A, Quinn AM, Wasney GA, Dong A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J Med Chem. 2009;52:7950–3. doi: 10.1021/jm901543m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kudithipudi S, Dhayalan A, Kebede AF, Jeltsch A. The SET8 H4K20 protein lysine methyltransferase has a long recognition sequence covering seven amino acid residues. Biochimie. 2012;94:2212–8. doi: 10.1016/j.biochi.2012.04.024. [DOI] [PubMed] [Google Scholar]
- 36.Chinnaiyan AM, Lnu S, Cao Q, Asangani I. inventors; the Regents of the University of Michigan, assignee. Compositions and methods for inhibiting mmset. European patent EP2536431 A2. 2012 Dec 26.
- 37.Chang Y, Zhang X, Horton JR, Upadhyay AK, Spannhoff A, Liu J, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat Struct Mol Biol. 2009;16:312–7. doi: 10.1038/nsmb.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.di Luccio E, Koehl P. A quality metric for homology modeling: the H-factor. BMC Bioinforma. 2011;12:48. doi: 10.1186/1471-2105-12-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.di Luccio E, Koehl P. The H-factor as a novel quality metric for homology modeling. J Clin Bioinforma. 2012;2:18. doi: 10.1186/2043-9113-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–82. doi: 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oberoi H, Allewell NM. Multigrid solution of the nonlinear Poisson-Boltzmann equation and calculation of titration curves. Biophys J. 1993;65:48–55. doi: 10.1016/S0006-3495(93)81032-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao Y, Chen P, Diao J, Cheng G, Deng L, Anglin JL, et al. Selective inhibitors of histone methyltransferase DOT1L: design, synthesis, and crystallographic studies. J Am Chem Soc. 2011;133:16746–9. doi: 10.1021/ja206312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Upadhyay AK, Rotili D, Han JW, Hu R, Chang Y, Labella D, et al. An analog of BIX-01294 selectively inhibits a family of histone H3 lysine 9 Jumonji demethylases. J Mol Biol. 2012;416:319–27. doi: 10.1016/j.jmb.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan Y, Wang Q, Paulk J, Kubicek S, Kemp MM, Adams DJ, et al. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem Biol. 2012;7:1152–7. doi: 10.1021/cb300139y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chinnaiyan AM, Lnu S, Cao Q, Asangani I. inventors; the Regents of the University of Michigan, assignee. Compositions and methods for inhibiting MMSET. World patent WO2011103028 A3. 2011 Dec 8.
- 46.Qiao Q, Li Y, Chen Z, Wang M, Reinberg D, Xu RM. The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. J Biol Chem. 2011;286:8361–8. doi: 10.1074/jbc.M110.204115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu H, Min J, Lunin VV, Antoshenko T, Dombrovski L, Zeng H, et al. Structural biology of human H3K9 methyltransferases. PLoS One. 2010;5:e8570. doi: 10.1371/journal.pone.0008570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Southall SM, Wong PS, Odho Z, Roe SM, Wilson JR. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks. Mol Cell. 2009;33:181–91. doi: 10.1016/j.molcel.2008.12.029. [DOI] [PubMed] [Google Scholar]
- 49.Malmquist NA, Moss TA, Mecheri S, Scherf A, Fuchter MJ. Small-molecule histone methyltransferase inhibitors display rapid antimalarial activity against all blood stage forms in Plasmodium falciparum. Proc Natl Acad Sci U S A. 2012;109:16708–13. doi: 10.1073/pnas.1205414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ho AS, Turcan S, Chan TA. Epigenetic therapy: use of agents targeting deacetylation and methylation in cancer management. Onco Targets Ther. 2013;6:223–32. doi: 10.2147/OTT.S34680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yost JM, Korboukh I, Liu F, Gao C, Jin J. Targets in epigenetics: inhibiting the methyl writers of the histone code. Curr Chem Genomics. 2011;5(Suppl 1):72–84. doi: 10.2174/1875397301005010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Copeland RA, Moyer MP, Richon VM. Targeting genetic alterations in protein methyltransferases for personalized cancer therapeutics. Oncogene. 2013;32:939–46. doi: 10.1038/onc.2012.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov. 2009;8:724–32. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]