

Abstract
The Leucine rich repeat kinase 2 (LRRK2) gene is genetically and biochemically linked to several diseases that involve innate immunity. LRRK2 protein is highly expressed in phagocytic cells of the innate immune system, most notably in myeloid cells capable of mounting potent pro-inflammatory responses. Knockdown of LRRK2 protein in these cells reduces pro-inflammatory responses. However, the effect of LRRK2 pathogenic mutations that cause Parkinson's disease on myeloid cell function is not clear but could provide insight into LRRK2-linked disease. Here, we find that rats expressing G2019S LRRK2 have exaggerated pro-inflammatory responses and subsequent neurodegeneration after lipopolysaccharide injections in the substantia nigra, with a marked increase in the recruitment of CD68 myeloid cells to the site of injection. While G2019S LRRK2 expression did not affect immunological homeostasis, myeloid cells expressing G2019S LRRK2 show enhanced chemotaxis both in vitro in two-chamber assays and in vivo in response to thioglycollate injections in the peritoneum. The G2019S mutation enhanced the association between LRRK2 and actin-regulatory proteins that control chemotaxis. The interaction between G2019S LRRK2 and actin-regulatory proteins can be blocked by LRRK2 kinase inhibitors, although we did not find evidence that LRRK2 phosphorylated these interacting proteins. These results suggest that the primary mechanism of G2019S LRRK2 with respect to myeloid cell function in disease may be related to exaggerated chemotactic responses.
Introduction
The leucine rich repeat kinase 2 (LRRK2) gene was initially characterized as part of a newly defined Ras-of-Complex family member and encodes a large ∼286-kDa protein with a number of protein–protein interaction domains (1). The LRRK2 protein harbors two enzymatically active domains, a GTPase domain and a tyrosine-like serine/threonine kinase domain, separated by a linking domain known as COR (C-terminal of Ras domain). The LRRK2 gene has been linked to disease susceptibility in several inflammatory disorders. Familial and genome-wide association studies have implicated LRRK2 prominently in the susceptibility to Parkinson's disease (PD), Crohn's disease (CD) and leprosy (2–7). While many of the LRRK2 genetic variants that are associated with disease are of unknown function, PD-linked familial mutations in LRRK2 have yielded some insight into how mutations lead to disease. PD-linked mutations in LRRK2 cluster within the ‘catalytic core’ of the protein in the kinase, GTPase and COR domains (8). The most common is the G2019S mutation in the kinase domain and is believed to confer increased kinase activity of the protein (8,9). This increased kinase activity has been linked to cellular toxicity and dysfunction in diverse model systems, although the substrates and pathways through which LRRK2 acts remain unclear (10–17).
Following the genetic implication of LRRK2 in inflammatory disease, the expression level of LRRK2 protein has been found to be highest in myeloid cells of the innate immune system (18–22). Myeloid cells are a diverse class of cells that arise from hematopoietic stem cells of the bone marrow that spawn common myeloid progenitors. The progenitors then can differentiate into a wide variety of blood cells including erythrocytes, megakaryocytes and innate immune cells including monocytes, macrophages, neutrophils, dendritic cells and eosinophils (23–25). LRRK2 expression is particularly high or exclusively expressed in a subclass of myeloid cells that are CD14+ [part of the lipopolysaccharide (LPS) receptor complex] and CD16+ (binds Fc regions of antibodies and is associated with mature cell phenotypes) (22). Myeloid cells known to express LRRK2 have diverse functions in the innate immune system such as secreting cytokines and chemokines, presenting antigen to adaptive immune cells, phagocytizing debris, pathogens and dying cells, and recognizing and moving to sites of danger through chemotaxis.
Knockdown or knockout of LRRK2 expression and inhibition of LRRK2 kinase activity has implicated a role for LRRK2 in some myeloid cell effector functions. RNAi knockdown of expression or pharmacological inhibition of LRRK2 has been shown to decrease the release of secreted cytokines such as TNF after pro-inflammatory stimuli in response to a number of pro-inflammatory agonists (18,21,26). LRRK2 kinase inhibition has also been shown to decrease phagocytosis of pathogenic particles (27). Pharmacological inhibition of LRRK2 also decreases cell chemotaxis in cultured microglia cells and fibroblasts (21,28). The impairment of chemotaxis owing to loss of LRRK2 can also be supported through studies of knockout of LRRK2 orthologues GbpC and ROCO4 (29–31). However, mice and rats lacking LRRK2 also have systemic changes in immune cell homeostasis (32,33), including deficits in white-blood cell counts. In addition, widely used LRRK2 kinase inhibitors have significant off-target effects (34), making it difficult to be able to understand the role of LRRK2 in myeloid cells using these models and tools.
While previous studies have focused on how loss of LRRK2 expression or activity influences cells of innate immunity, only a few studies have evaluated the effects of pathogenic missense LRRK2 mutations. Using mice that express the R1441G pathogenic mutation, increased production of pro-inflammatory cytokines were detected in stimulated primary microglial cells (35). Several receptors, including toll like receptors (TLRs), scavenger receptors and various chemokine receptors, underlie these pro-inflammatory processes and can be utilized to determine cell type and activation state (36,37). LPS is a canonical pro-inflammatory stimulus that elicits several of the effector functions of myeloid cells by binding to TLR4/CD14 complexes present in CD14+ cells known to express high LRRK2 levels (22). A direct LPS injection to the brain induces inflammatory responses that involve myeloid cell recruitment and activation and subsequent dopaminergic neurodegeneration (38,39). LRRK2 knockout rats are protected from the effects of LPS-induced neurodegeneration (18), but the effects of pathogenic LRRK2 mutations on LPS-induced neurodegeneration and myeloid cell activation are not known.
In this study, we use transgenic rats and mice that over-express G2019S LRRK2 or wild-type (WT) LRRK2 to explore myeloid cell responses altered by G2019S LRRK2 expression. Through a combination of in vitro approaches using isolated primary cultured cells as well as several in vivo models, we find that G2019S LRRK2 expression enhances chemotactic responses to a number of stimuli but fails to alter other components of myeloid cell function affected by the loss of LRRK2 expression. Our findings revealed that in activated myeloid cells, the G2019S mutation robustly increases the association of LRRK2 with the actin-regulatory network in a kinase dependent manner. The actin-regulatory network that interacts with LRRK2 mediates chemotactic responses in myeloid cells. The role of LRRK2 in disease pathogenesis may be explained in part by its role in regulating myeloid cell chemotactic responses.
Results
G2019s LRRK2 expression enhances LPS-induced brain myeloid cell responses
Genetic knockout or knockdown of LRRK2 both in vivo and in vitro produces an attenuated pro-inflammatory response to LPS (18,21). The effect of pathogenic LRRK2 mutations on these myeloid cell-driven responses has not previously been studied. Recently a line of Sprague–Dawley rats that carry a human-derived bacterial artificial chromosome (hBAC) encoding the LRRK2 gene recombineered with a G2019S-pathogenic mutation were developed by CJ Li and distributed in partnership with Cornell University and the Michael J. Fox Foundation. We verified transgenic over-expression of G2019S LRRK2 in thioglycollate-elicited primary macrophages (TEPMs) cultured from these animals (Supplementary Material, Fig. S1A and B). The G2019S LRRK2 protein is also expressed in TH substantia nigra pars compacta (SNpc) neurons and other brain nuclei susceptible to neurodegeneration from LPS exposure. In contrast, LRRK2 expression is not normally detected in TH cells of the SNpc in WT (nTg) rats (40).
To examine myeloid cell responses in the LRRK2-G2019S rats compared with non-transgenic littermate controls (nTg), we injected LPS or saline control (PBS) directly in the right SNpc to induce a robust and selective loss of dopaminergic neurons (38,39,41). After intracranial LPS injection, a panel of antibodies including IBA-1 (microglial morphology), CD68 (mature myeloid lineage phagocytic cells) and tyrosine hydroxylase (dopaminergic neurons) were used to evaluate myeloid cell responses and their effects on dopaminergic neuron survival, respectively. Confocal analysis showed an increase in the intensity and proportion of CD68+/IBA1+ cells in G2019S LRRK2 rats compared with non-transgenic littermate controls (Fig. 1A). Commensurate with the apparent increase in CD68+ cells in the ipsilateral SNpc were reduced numbers of TH cells (Fig. 1B and C). These observations are consistent with an increased pro-inflammatory (M1) response in the G2019S LRRK2 animals in response to LPS stimulation.
Figure 1.

Enhanced neuroinflammation in G2019S LRRK2 transgenic rats. 10- to 12-week-old non-transgenic littermate controls (WT nTg, n = 10) and G2019S LRRK2 (GS-LRRK2, n = 13) rats were unilaterally injected with 5 µg ultra-pure lipopolysaccharide (LPS) into the right substantia nigra pars compacta (SNpc). Animals were sacrificed 2 weeks post-injection. (A) Representative confocal images of the SNpc stained for ionized calcium-binding adapter molecule 1 (IBA1) and cluster of differentiation protein 68 (CD68), both markers of myeloid cells, along with tyrosine hydroxylase (TH) expressed in dopaminergic neurons. White boxes in the merge panels represent the area for ‘Zoom’ panels. Scale bars for images are 100 and 25 µm for ‘Zoom’ panels. (B) Representative bright-field images of DAB-stained coronal brain sections from WT and GS-LRRK2 rats after LPS injection. TH staining and (C) CD68 staining is shown with Nissl staining for contrast. (D) Unbiased stereological quantification of TH+ neurons and (E) CD68+ cells in the SNpc in WT and G2019S LRRK2 LPS-injected rats. (F) Unbiased stereological quantification of Nissl+ neurons. (G) Volume calculation of the tissue area encompassing CD68 cell immunoreactivity. Significance was calculated with one-way ANOVA with Tukey's post hoc comparisons or two-tailed unpaired t-test (F), bars represent group means and error bars are S.E.M. * < 0.05, **P < 0.01, ***P < 0.001.
Previously, we found that LRRK2 KO rats were resistant to LPS-induced dopaminergic neurodegeneration compared with WT rats (18). Stereological counts to TH cells in the SNpc revealed that the G2019S LRRK2 rats have exacerbated dopaminergic neurodegeneration in response to LPS exposure (Fig. 1D). The G2019S LRRK2 rats had a ∼60% increase in TH neuron degeneration as compared with nTg controls. Stereological counts of CD68+ cells in adjacent serial sections demonstrated that there were more CD68+ cells recruited to the SNpc of G2019S LRRK2 rats compared with that of nTg animals (Fig. 1E). Additionally, the increased number of CD68+ cells occupied a larger volume in the GS-LRRK2 rat (Fig. 1G). To ensure TH neurons were actually lost rather than decreased in TH expression, we performed unbiased stereology for Nissl neurons on the same sections used to quantify TH cell loss (Fig. 1F).
To determine whether there were differences between nTg and G2019S LRRK2 rats unrelated to LPS exposure (e.g. basal states of inflammation, differential effect of the surgery, etc.), we analyzed by confocal analysis and stereology TH and CD68 cells in a separate cohort of animals injected with only saline (PBS, Supplementary Material, Figs S1C and S2A and B). We did not observe differences in the number of TH cells between G2019S LRRK2 animals and nTg controls in either the injected or un-injected sides (Supplementary Material, Fig. S2). There was no significant CD68 reactivity in either the injected or un-injected sides in these animals. Overall, these data suggest an increase in pro-inflammatory responses in the G2019S LRRK2 animals when challenged with LPS but that the G2019S LRRK2 rats have normal baselines in the SNpc. Other reports published during the course of our study demonstrate that other G2019S LRRK2 rat transgenic strains also do not show neurodegenerative phenotypes or abnormal numbers of TH SNpc neurons at baselines (42,43).
G2019s LRRK2 expression does not affect secretion of pro-inflammatory factors or phagocytosis
To determine the effects of G2019S LRRK2 expression on critical aspects of myeloid cell function, we isolated TEPM cells from transgenic and non-transgenic littermate controls. WT LRRK2 BAC mice that express LRRK2 protein at levels comparable with that in G2019S LRRK2 BAC mice were used to control for the effects of over-expression of LRRK2 protein. BAC mice were developed by Zhenyu Yue and licensed for distribution through Mount Sinai Hospital and the Michael J. Fox foundation. We find that WT LRRK2 and G2019S LRRK2 over-expression is closely matched in TEPM cells (Fig. 2A). Additionally, we examined the phosphorylation state of the LRRK2 residues Ser910, Ser935 and Ser1292 from whole-cell TEPM lysate. We did not observe any changes in Ser910 or Ser935 phosphorylation between G2019S LRRK2 and WT LRRK2 TEPMs. As expected, Ser-(P)-910 and Ser-(P)-935 did not increase with the G2019S LRRK2 mutation consistent with previous results (13). We did observe a robust increase in Ser-(P)-1292 levels in the G2019S LRRK2 TEPMs, consistent with increased kinase activity associated with the G2019S LRRK2 mutation (11).
Figure 2.

Pathogenic G2019S LRRK2 enhances chemotaxis but not phagocytosis or secretion of inflammatory cytokines and chemokines. Thioglycollate-elicited peritoneal macrophages (TEPM) cells from adult male non-transgenic littermate controls (WT, nTg), WT LRRK2 BAC and G2019S LRRK2 (GS-LRRK2) BAC mice were cultured. At least three animals per strain per experiment were used. (A) Representative western blots showing LRRK2 over-expression in TEPMs as well as phosphorylation levels of S910, S935 and S1292 from G2019S LRRK2 and WT LRRK2 animals compared with nTgs. (B) TEPMs were allowed to rest overnight after plating and then stimulated with LPS (100 ng ml−1) for 6 h. No differences in TNF secretion could be detected as measured by ELISA (B), or with 32 other cytokines and chemokines measured by multiplex bead arrays (also see Supplementary Material, Fig. S3A–E for other representative secreted factors measured). (C) Fluorescent Zymosan beads were added to TEPM cultures, and the number of beads internalized per TEPM was determined by fluorescent microscopy (see Supplementary Material, Fig. S4A and B for related histograms and images). No differences in the number of beads phagocytized per group could be observed. (D) TEPM cells were plated into the top well of Boyden chambers in the presence of 100 µm ADP in the bottom chamber, and the number of cells migrating to the bottom chamber over 36 h was measured. Significantly more G2019S LRRK2 TEPM cells migrated to the bottom well. (E) Representative images of TEPM nuclei visualized with DAPI stain in the bottom chamber of the chemotaxis assay. (F) Immediately after addition of TEPM cells to Boyden chambers, 5 µm SRI 29451 (451) and 1 µm HG-10-102-01 (Gnt) LRRK2 kinase inhibitors were added to the top and bottom well. (G) Representative micrographs. Significance was calculated with one-way ANOVA with Tukey's post hoc comparisons. Bars represent group means, and error bars are S.E.M. *P < 0.05, NS represents not significant.
To test the role of G2019S and WT LRRK2 over-expression on cytokines and chemokines secreted by TEPMs, cells were derived from non-transgenic littermate controls (nTg), G2019S LRRK2 mice and WT LRRK2 BAC mice. Cells were counted and plated at equivalent densities into tissue culture wells. Supernatants were collected from LPS-exposed or saline-treated wells and analyzed for a variety of cytokines and chemokines (Fig. 2B, Supplementary Material, Fig. S3). With all secreted factors measured, no significant differences could be observed between groups, showing that the transgenic over-expression of WT LRRK2 or expression of G2019S LRRK2 does not affect this aspect of myeloid cell inflammation.
LRRK2 and G2019S LRRK2 pathogenic function have been implicated in phagocytosis, endocytosis, lysosomal function and vesicle recycling (14,27,44–51). To determine whether LPS-stimulated G2019S LRRK2-expressing TEPMs had increased phagocytosis compared with WT LRRK2 or nTg TEPMs, zymosan beads were applied to cultured cells plated at equivalent density, and uptake of the beads determined by microscopy. A similar number of beads were phagocytized between the three groups of TEPM cells (Fig. 2C), indicating this aspect of myeloid cell function was unaffected by G2019S LRRK2 expression. Histograms revealed comparable distributions and numbers of TEPMs that have high phagocytic activity compared with cells in culture with low phagocytic activity (Supplementary Material, Fig. S4).
G2019s LRRK2 increases myeloid cell chemotaxis in a kinase-dependent manner
Myeloid cell chemotactic response is critical in a diversity of diseases and model systems that involve both innate and adaptive immune responses (52). The chemotactic ability of TEPMs cultured from nTg, WT LRRK2 BAC and G2019S LRRK2 BAC mice were tested in Boyden chambers for differential migration through an 8.0-µm-pore membrane. With the addition of 100 µm ADP to the bottom well, a robust stimulation of chemotaxis occurs (21,53). G2019S LRRK2 TEPMs had an approximate 2-fold increase in bottom-well migration (Fig. 2D and E). The over-expression of WT LRRK2 had no effect compared with cells from non-transgenic littermate controls.
We previously defined the potency of second-generation LRRK2 kinase inhibitors SRI 29451 (451) and HG-10-102-01 (Gnt) in TEPMs (34,54). To test whether increased chemotaxis in G2019S LRRK2 TEPMs was kinase dependent, we again utilized the two-chamber assay but in the presence of SRI 29451 and HG-10-102-01. Both LRRK2 kinase inhibitors blocked the enhanced chemotaxis associated with G2019S LRRK2 expression (Fig. 2F and G). These results show that the increased chemotaxis caused by G2019S LRRK2 expression is dependent on LRRK2 kinase activity.
In the light of the finding that increased numbers of CD68 cells were recruited to the brain in response to LPS injection in G2019S LRRK2 BAC rats (Fig. 1) and enhanced ADP-induced chemotaxis in Boyden chamber assays in cells that express G2019S LRRK2, we next sought to measure chemotactic responses in vivo in a second model of myeloid cell chemotaxis with a stimulant other than LPS or ADP. Thioglycollate-induced peritonitis (TIP) is a robust model of inflammation used to measure acute chemotactic responses of immune cells into the peritoneal cavity (55, 56). At 16 h post-thioglycollate exposure, the majority of cells (>80%) recruited to the peritoneal cavity are monocytes, macrophages and neutrophils (57–59). Flow cytometry revealed that G2019S LRRK2 BAC mice have ∼2-fold more cells recruited to the peritoneum than WT LRRK2 BAC and nTg mice (Fig. 3A). No difference was observed between nTg mice and animals over-expressing WT LRRK2. Classification of the TIP cells showed that there was a ∼2-fold increase in both recruited monocytes/macrophages (CD11b+ Ly6G(A18)+) cells and neutrophils (CD11b+ Ly6G(A18)−). Gating strategies for flow cytometry are given in Supplementary Material, Figure S5. These results indicate that the G2019S mutation in LRRK2 is associated with a substantial increase in chemotaxis of innate immune cells that may include monocytes, macrophages and neutrophils.
Figure 3.
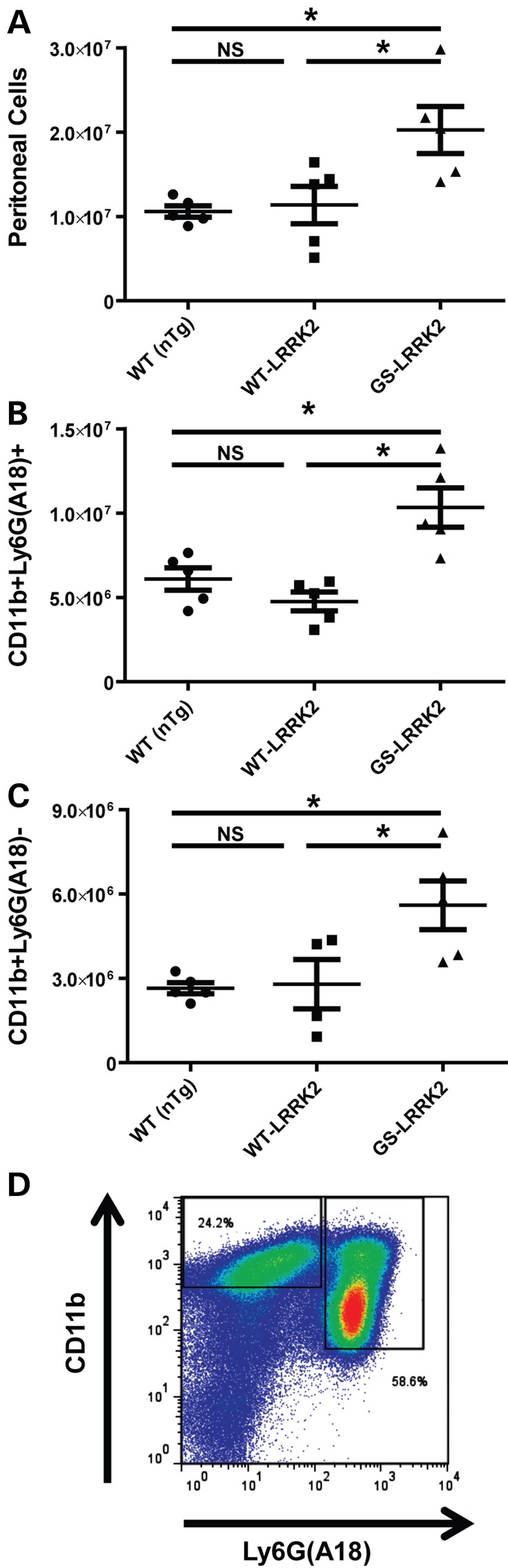
G2019S LRRK2 expression increases chemotaxis of innate immune cells in thioglycollate-elicited peritonitis (TIP). Cells from at least five male adult mice each of WT (non-transgenic, nTg), WT LRRK2 BAC and G2019S(GS)-LRRK2 were lavaged 16 h after thioglycollate exposure. Red blood cells were lysed, and the remaining cells analyzed as follows: (A) Raw TIP cell counts isolated per animal, and (B and C) sub classification of TIP cells by flow cytometry. Cell were stained for Ly6G (A18 clone) and CD11b. Quantification of CD11b+ Ly6G(A18)+ cells, likely neutrophils, and quantification of CD11b+ Ly6G(A18)− cells, likely monocytes/macrophages, are given. (D) Representative flow cytometry plot used in part to calculate (B and C). As expected, >80% of TIP cells are represented by these populations (see Supplementary Material, Fig. S6). *P < 0.05, one-way ANOVA with Tukey's post hoc test, and NS represents not significant (P > 0.5). Lines are group mean and error is shown as S.E.M.
LRRK2 KO animals have previously been shown to have several alterations in blood chemistry, including a decrease in white-blood cells under basal conditions (32,33,60). In order to test whether baseline differences in immunological homeostasis exist in G2019S LRRK2 mice, we performed a complete blood chemistry panel (Table 1). As opposed to LRRK2 KO rodents, no differences were observed in any tested element of blood chemistry between nTg, WT LRRK2 and G2019S LRRK2 BAC mice.
Table 1.
Complete blood count of non-transgenic littermates (nTg, n = 5), WT LRRK2 BAC (n = 5) and G2019S(GS)-LRRK2 BAC mice (n = 5)
| WT* | WT LRRK2a | GS-LRRK2a | |
|---|---|---|---|
| RBC (M/µl) | 6.02 ± 1.48 | 6.69 ± 0.39 | 6.15 ± 0.70 |
| HCT (%) | 26.96 ± 8.03 | 30.66 ± 5.22 | 30.07 ± 4.49 |
| HGB (g/dl) | 9.16 ± 2.09 | 10.1 ± 0.66 | 9.45 ± 0.82 |
| MCV (fl) | 44.41 ± 4.15 | 45.58 ± 4.27 | 48.85 ± 5.69 |
| MCH (pg) | 15.27 ± 0.63 | 15.15 ± 0.93 | 15.35 ± 0.36 |
| MCHC (g/dl) | 34.77 ± 4.74 | 33.63 ± 5.44 | 31.75 ± 3.76 |
| RDW (%) | 30.73 ± 3.65 | 31.20 ± 1.60 | 33.45 ± 3.86 |
| WBC (K/µl) | 3.38 ± 1.95 | 5.28 ± 1.61 | 4.18 ± 1.42 |
| NEU (µl) | 460 ± 330 | 795 ± 442 | 795 ± 282 |
| NEU (%) | 15.5 ± 6.82 | 19.33 ± 11.07 | 20.25 ± 9.32 |
| MONO (µl) | 253 ± 146 | 395 ± 59 | 469 ± 278 |
| MONO (%) | 7.5 ± 2.83 | 8.17 ± 2.78 | 10.75 ± 2.98 |
| LYM (%) | 73.00 ± 8.33 | 70.00 ± 10.31 | 67.75 ± 11.67 |
| LYM (µl) | 2396 ± 1246 | 3682 ± 1642 | 2872 ± 1174 |
Data are presented as mean ± the standard deviation. Differential WBC counts are also presented.
aDifferences between groups were calculated with one-way ANOVA and were not significant (P > 0.05).
RBC, red blood cells; HCT, hematocrit; HGB, hemoglobin; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; RDW, red blood cell distribution; WBC, white-blood cell count; NEU, neutrophil; MONO, monocyte; LYM, lymphocyte.
Although the number of circulating white-blood cells is comparable between the mouse strains used in this study, the basal activation (i.e. polarization) state of G2019S LRRK2-expressing myeloid cells could be different, priming them for enhanced chemotactic responses (e.g. M1 polarization). Further analysis of peritoneal white-blood cells using flow cytometry demonstrated that the percentages of CD11b+ Ly6G(A18)+ or CD11b+ Ly6G(A18)− cells in white-blood-cell populations are equivalent between all strains of mice (Supplementary Material, Fig. S6). MHCII expression is often used to assess the activation state of macrophages (37,61–63). We examined the basal activation state of the G2019S LRRK2 CD11b+ Ly6G(A18)− cells using median-fluorescence intensity (MFI) of MHCII reactivity (Supplementary Material, Fig. S6D and E). No significant differences in MHCII MFI were observed, suggesting the G2019S LRRK2 cells are not more activated under basal conditions.
Despite the lack of activation under basal conditions, an increase in myeloid cell chemotaxis caused by G2019S LRRK2 expression could be explained in part by an increase in surface expression of critical chemotaxis receptors such as CCR2 or CCR5. CCR2 is an essential component of myeloid cell chemotaxis in both in vivo LPS models and in TIP (64–66). CCR5 is important temporally for the recruitment of macrophages to the site of inflammation, as CCR5 knockout animals show delayed macrophage chemotaxis in TIP for 36 h (67). Using flow cytometry, the predominant populations of cells in the peritoneum, CD11b+ Ly6G(A18)− and CD11b+ Ly6G(A18)+ cells, were examined for the expression of CCR2 or CCR5 (gating strategy shown in Supplementary Material, Fig. S7). According to MFI calculations from flow cytometry, the surface expression of CCR2 was comparable between all groups, although a trend (P = ∼0.06) towards increased surface expression of CCR5 was detected (Fig. 4). Together with unaltered chemokine levels secreted from these cells whether or not G2019S LRRK2 is expressed (Supplementary Material, Fig. S3), these results suggest that the differences in chemokine receptors are unlikely to account for the enhanced chemotaxis associated with G2019S LRRK2 expression.
Figure 4.

G2019S LRRK2 expression does not affect CCR2 or CCR5 surface expression in TIP. Cells from at least six male adult mice from WT (non-transgenic, nTg) or G2019S (GS)-LRRK2 were lavaged 16h after thioglycollate exposure. Red blood cells were lysed and the remaining cells analyzed. Representative flow histograms indicating (A) CCR2 expression in comparison with isotype (ISO) control signal. Median fluorescent intensity (MFIs) calculations of from (B) CD11b+ Ly6G(A18) and (C) CD11b+ Ly6G(A18)+. (D) CCR5 expression in comparison with isotype (ISO) control signal. (E and F) CCR5 in CD11b+ Ly6G(A18) and CD11b+ Ly6G(A18)+ cells. NS represents not significant, two-way unpaired t-test with Bonferroni correction for multiple comparison (P > 0.05), bars show group mean and error bars are S.E.M.
The G2019S LRRK2 mutation enhances LRRK2 association with actin-regulatory proteins responsible for myeloid cell mobility
In fibroblasts and other cell lines, LRRK2 forms protein complexes with components of the actin cytoskeleton (31,68–70). LRRK2-positive protein complexes derived from polarized myeloid cells have not been studied previously. Using M1-polarized TEPMs, we utilized the N-terminal FLAG epitope tag engineered into the BAC transgenic mice to immunoprecipitate LRRK2 protein complexes to a high level of purity. Immunoprecipitates from non-transgenic littermate controls that do not express proteins with the FLAG epitope tag were processed in parallel to identify proteins unrelated to LRRK2-positive protein complexes. A Coomassie-stained reducing gel of G2019S LRRK2 protein complexes is shown in Figure 5C. a total of 394 proteins were detected in complex with LRRK2 through identification of high-confidence peptides via tandem mass spectrometry. However, many of these proteins were identified as interactions of low specificity or selectivity through input of the 394 proteins into a database ranking contaminants common to affinity purifications identified by mass spectrometry [www.CRAPome.org, see reference (71)]. Proteins rarely identified in protein complexes across the 411 affinity purification experiments included in the database (<10% chance, see Supplementary Material, Table S1 and Fig. 6) filtered 213 proteins. Twenty-seven other proteins that include immunoglobulin from the experiment, poorly annotated proteins and ambiguous peptides and/or database entries were also filtered from the LRRK2-interacting set and were not included in further analysis (see Supplementary Material, Table S1).
Figure 5.

Definition of the LRRK2 interactome in macrophages. (A) Gene-ontology (GO) process and network analysis to detect enrichment of terms from proteins identified in LRRK2-positive protein complexes (see Supplementary Material, Table S1 post-filter and Methods). P-values were calculated from Wilcoxon signed rank tests and corrected for multiple testing by Benjamini–Hochberg analysis. (B) String-DB analysis of LRRK2 proteins in complex that segregate into the actin-regulatory network and actin-cytoskeleton effector proteins, known to control myeloid cell motility. (C) Representative Coomassie-stained gel of immunoprecipitated proteins from M1-activated (LPS treated, 100 ng ml−1) TEPM from WT (non-transgenic, nTg) and G2019S(GS)-LRRK2 mice. Immunoprecipitates on beads were washed in parallel between nTg and G2019S LRRK2 FLAG-pull-downs so that the only protein bands visible in the nTg pull-down were derivative from the beads (i.e. FLAG antibody). Labeled bands include LRRK2 protein, and the heavy-chain and light-chain from the FLAG resin (HC and LC, respectively).
Figure 6.

G2019S LRRK2 enhances interactions with actin regulatory and effector proteins in a kinase-dependent manner. (A) Representative western blots of selected components from LRRK2-positive protein complexes immunoprecipitated from M1 polarized TEPMs. Blots are representative of three independent experiments. (B) Quantification of western blots from (A). Immunoprecipitates from the G2019S LRRK2 pull-down are compared with WT LRRK2 pull-downs that are represented by a dashed line. Fold changes in intensity are indicated (G2019S LRRK2 relative to WT LRRK2). (C) TEPMs were treated with 500 nm PF-06447475 (PF-475) or equivalent vehicle DMSO (0.00005%) for 24 h. Western blots are representative of three independent experiments. (D) Quantification of western blots from (C). Immunoprecipitates from the G2019S LRRK2 + PF-06447475 pull-downs are compared with the G2019S LRRK2 + DMSO pull-downs, represented by a dashed line. Fold changes in intensity are indicated (G2019S LRRK2 + PF-06447475 relative to G2019S LRRK2 + DMSO). Significance was determined by two-tailed one-sample t-tests comparing each fold change against an expected value for no change in the interaction. Bars represent group means, and error bars are S.E.M. *P < 0.05, ***P < 0.001.
Input of the list of the 154 LRRK2-interacting proteins into gene ontology and String-db analysis revealed a coordinated network of actin-regulatory proteins known to be in complex with one another. These actin-regulatory proteins comprise the actin-effector network, as indicated graphically in Figure 5B. Input of the 154 genes into MetaCORE GeneGO analysis corroborated a strong enrichment for the interactome related to actin-cytoskeleton control, and gene-ontology networks related to the actin cytoskeleton (Fig. 5A).
To confirm the mass spectrometry identifications, we repeated independent immunoprecipitation experiments from at least three TEPM cell lysates derived from at least three mice in each group of nTg, WT and G2019S LRRK2 BAC mice. Evaluation of the LRRK2 protein complexes confirmed the interaction between LRRK2 and myosin 1f (Myo1f), myosin IIa (Myh9), Rac1, actin-related complex 3 (Arpc3) and actin-related complex 2 (Arp2) (Fig. 6A and B). For these interactions, G2019S LRRK2 showed a striking enhancement of interaction (Fig. 6B), even though both WT LRRK2 and G2019S LRRK2 can be precipitated at equivalent concentrations. Further, these actin-regulatory proteins were expressed at equivalent concentrations in the TEPMs irrespective of G2019S LRRK2 expression. We did not identify any proteins exclusively interacting with G2019S LRRK2 and not WT LRRK2, or vice-versa.
To test whether the enhanced G2019S LRRK2-actin-regulatory component interactions were kinase dependent, we utilized a recently described highly potent and selective third-generation LRRK2 kinase inhibitor PF-06447475 (72). TEPMs expressing G2019S LRRK2 were pretreated with 500 nm PF-06447475 or equivalent concentration of DMSO (0.00005%), and LRRK2 protein complexes were isolated with FLAG-immunoprecipitation. LRRK2 kinase inhibition did not affect the amount of LRRK2 that could be immunoprecipitated from the TEPMs. Evaluation of the LRRK2 protein complexes showed that PF-06447475 application significantly decreased LRRK2 interactions with actin-regulatory proteins (Fig. 6C and D). These results suggest that the enhanced association of G2019S LRRK2 with actin-regulatory proteins is dependent on LRRK2 kinase activity.
Because the kinase activity associated with G2019S LRRK2 appears to control the increased chemotaxis and interaction with actin-regulatory proteins, we next sought to determine whether G2019S LRRK2 was phosphorylating components of the actin-regulatory complex. We selected Rac1 as the representative interacting protein in the ‘actin-effector network’ and Arpc3 as the representative protein in the ‘actin-regulatory network’ (Fig. 5B). Addition of Rac1 and Arpc3 recombinant proteins together with WT LRRK2 and G2019S LRRK2 in in vitro kinase assays demonstrated that LRRK2 cannot efficiently phosphorylate these proteins (Fig. 7). In these assays, we observed a robust ∼3-fold increase in autophosphorylation in G2019S LRRK2 compared with WT LRRK2, and this activity was completely blocked with the addition of 500 nm PF-06447475 to kinase reactions (Fig. 7C). As a positive control for LRRK2 trans-phosphorylation of protein substrates, we performed additional in vitro kinase assays with ArfGAP. Consistent with previous observations, LRRK2 robustly phosphorylated ArfGAP and this was completely blocked by LRRK2 kinase inhibitors (12). These results suggest that the G2019S LRRK2 mutation enhances the association of LRRK2 with Rac1 and Arpc3, but the mutation is unlikely to affect phosphorylation of these proteins.
Figure 7.

LRRK2 does not phosphorylate actin-regulatory proteins. (A) Representative autoradiographs and Coomassie-stained gels post-in vitro kinase assays. Recombinant WT LRRK2 and G2019S(GS)-LRRK2 were used with and without 500 nm PF-06447475 (475). Recombinant Rac1 or Arpc3 were added to the in vitro kinase assays of ∼10-fold abundance relative to LRRK2 protein. (B) Known in vitro kinase substrate ArfGAP was analyzed in tandem. (C) Liquid scintillation quantification (∼80% counter efficiency at the time of recording) was performed for recombinant proteins excised from the indicated Coomassie band as in (A and B). Background levels recorded are indicated by the gray line (∼35 C.P.M.). Significance was determined by one-way ANOVA with Tukey's post hoc test. Bars represent group means, and error bars are S.E.M. ***P < 0.001.
Discussion
In this study, we have utilized several model systems and experimental approaches to help understand the effects of pathogenic G2019S LRRK2 expression on myeloid cell function. Because studies in LRRK2 KO rodents suggest that LRRK2 is critical to myeloid cell function in innate immunity, we hypothesized G2019S LRRK2 expression may alter fundamental aspects in myeloid cell responses to pro-inflammatory stimuli. We discovered that most canonical myeloid cell functions were identical whether or not the cells expressed G2019S LRRK2, and blood chemistry of G2019S LRRK2 mice was unperturbed. However, we detected striking increases in chemotaxis in several different model systems owing to G2019S LRRK2 expression. The action of G2019S LRRK2 expression and associated kinase activity on chemotaxis was revealed through diverse chemoattractants including LPS, ADP and thioglycollate. G2019S LRRK2-expressing myeloid cells appeared more mobile. Potentially related to this result, we found that the G2019S LRRK2 mutation stabilized the association of LRRK2 in a kinase-dependent manner with the actin-regulatory network that is known to control the actin cytoskeleton responsible for myeloid cell mobility (73–87).
In other models and cell systems, LRRK2 has been indirectly implicated in regulating elements of the actin cytoskeleton and cell motility (68,70,88–91). Knockout of the LRRK2 homolog GbpC in Dictyostelium not only reduces chemotaxis but disrupts interaction and phosphorylation of Myh9 (29,92). Other proteomic screens of LRRK2 complexes using similar unbiased approaches have identified Myo1f and Arp2 as major LRRK2 interactors that we also observe in TEPMs (68). Arp2 expression increases the speed of cell migration or motility through increasing actin filament branching at the leading edge of the cell (73–78,93). Cells from Myo1f KO mice have been shown to have decreased motility (82,87), and Myh9 expression is likewise critical for cell migration and motility (79,81,83–86,94,95). Fibroblasts derived from mice or patients with LRRK2 mutations have enhanced actin dynamics and differential LRRK2 interactions with cytoskeltal elements, as well as increased fibroblast motility (28,69).
Through the usage of newly developed efficacious LRRK2 kinase inhibitors, we find that the enhanced chemotaxis in G2019S LRRK2-expressing myeloid cells and the formation of G2019S LRRK2 protein complexes with actin-regulatory components are dependent on kinase activity. Rather than G2019S LRRK2 trans-phosphorylating these actin-regulatory components, G2019S LRRK2 may serve as a scaffolding component of the actin-regulatory protein network. Although LRRK2 cellular localization does not appear to be dramatically altered after LPS stimulation (96), kinase active G2019S LRRK2 may preferentially interact with select cellular compartments (46). It is possible that LRRK2 may help recruit the necessary actin-regulatory proteins into complexes that promote cell motility and chemotaxis.
LRRK2 is expressed in many cell types including neurons in the brain. The same actin-regulatory protein complexes that regulate chemotaxis in myeloid cells may serve different functions in neurons. For example, over-expression of G2019S LRRK2 in neurons causes the retraction of neurite outgrowths (10). This phenotypic could potentially be caused by G2019S LRRK2 regulation of the actin cytoskeleton. Indeed, proteomic screens for LRRK2 interactors from both neurons and immortalized cell lines have revealed similar LRRK2-interacting proteins as with the LRRK2 interactome defined here in TEPMs (68). Future in-depth examinations of the exact functional changes in the actin cytoskeleton owing to G2019S LRRK2 expression in diverse cell types may help clarify this apparent aspect of LRRK2 function in health and disease.
Regardless of the exact mechanism underlying G2019S LRRK2 regulation of chemotaxis, it is widely appreciated that myeloid cell chemotaxis is a critical aspect of inflammatory responses occurring in many disease processes. In CD, pro-inflammatory myeloid cells such as macrophages can drive inflammation central to pathology underlying disease (97–100). In Mycobacterium leprae infection, macrophage recruitment to granulomas is essential to contain infected cells and prevent disease progression (101–103). In PD, neuroimaging studies reveal an abnormal abundance of activated myeloid cells throughout much of the brain (104), and this is confirmed in pathological studies that show pro-inflammatory myeloid cells in affected brain regions (105).
LRRK2 is linked to Parkinson's, Crohn's and Hansen's (leprosy) disease through genome-wide association studies. In typical Caucasian PD populations, the prevalence of the G2019S mutation is relatively low (1–5%), although in certain populations such as Ashkenazi Jews and North African Arabs, G2019S mutation may occur in as many as 30% of cases (7,106–108). These high rates of mutations in LRRK2 might be attributable to founder effects (109). However, with respect to G2019S LRRK2 effects on myeloid cells, a positive-selection scenario may be envisaged where an increase in chemotaxis and subsequent innate immune responsiveness may be protective against a pathogen challenge. For example, numerous studies support the conclusion that enhanced myeloid cell chemotaxis contributes to reduced mortality rates associated with sepsis (110–113). While exaggerated chemotactic responses may offer protection from sepsis and possibly other infectious diseases, the same response may enhance neurodegeneration. The available tools and LRRK2 transgenic models are well suited to test this and related hypotheses with respect to LRRK2 function in innate immunity.
Materials and Methods
Animals
All protocols were approved by the local Animal Care and Use Committee. Mouse FLAG-G2019S LRRK2 BAC (B6.Cg-Tg(Lrrk2*G2019S)2Yue/J) and mouse FLAG-WT LRRK2 BAC (B6.Cg-Tg(Lrrk2)6Yue/J) were developed in the laboratory of Zhenyu Yue and obtained from the Jackson Laboratory (The Jackson Laboratory, Bar Harbor, ME, USA). These mice have been previously described (114,115). Mice strains used in this study were maintained on the C57BL/6J strain, and upon study completion sentinels positive for G2019S LRRK2, WT LRRK2, or non-transgenic littermate controls were subjected to genome scanning for C57 substrain identification (Jackson Laboratories SNP panel). Mice showed approximately >99% identity to the 6J substrain from the analysis of ∼1500 SNPs; <1% of SNPs were assigned to substrains other than 6J or 6N. Genotyping was accomplished with the forward primer GAC TAC AAA GAC GAT GAC GAC AAG and the reverse primer CTA CCA CCA CCC AGA TAA TGT C using Phusion DNA Polymerase (NEB). As the primer pair does not distinguish the WT LRRK2 BAC strain from the G2019S LRRK2 BAC strain, for select breeding pairs, genetic identity was confirmed by using the forward primer TAT CTC CAC TCA GCC ATG ATT ATT TAC CG and the reverse primer TTG AGG GCA CTG ATG GTC CAC TG to produce a 209-bp DNA product that will be digested in half with the restriction enzyme MfeI (NEB) if the DNA is G2019S positive. Hemizygous BAC positive mice were utilized for the experiments described in this study. Mice (and rats, described later) were maintained on an ad libitum diet with a 12-h light and 12-h dark cycle and were housed according to AAALAC density guidelines.
The human BAC-G2019S LRRK2 (NTac:SD-Tg(LRRK2*G2019S)571Cjli, Taconic Farms Sprague–Dawley outbred) rats were developed originally at Cornell University by CJ Li and supported for distribution and licensing by the Michael J. Fox Foundation for PD research. Hemizygous BAC positive rats were utilized for the experiments described in this study. We have previously evaluated LRRK2 expression and localization in these strains of rats (40,43). Genotyping of NTac:SD-Tg(LRRK2*G2019S)571Cjli was accomplished with the forward primer GAT AGG CGG CTT TCA TTT TTC C and the reverse primer ACT CAG GCC CCA AAA ACG AG using Phusion Taq DNA polymerase according to manufacturer's instructions (NEB).
For intracranial LPS administration, transgenic rats were selected as the model system as opposed to mice because rats show less variability in neurodegenerative phenotypes after LPS exposure as compared with mice (18,38,39,116–126). One caveat to the use of the rat is that we have failed to detect endogenous LRRK2 expression in the SNpc of the nTg rat as compared with robust expression of G2019S LRRK2 in the SNpc of transgenic rats, so it is not possible to calculate the fold increase of LRRK2 in the SNpc cells owing to transgenic expression (40). All other experiments utilized mice where a WT-LRRK2 strain controlled for the effects of over-expression of LRRK2 protein.
Animal surgeries
Intracranial or control (saline only) injections were conducted under isoflurane anesthesia using a digital stereotaxic frame (David Kopf) with a thermal adjustable height stage (Physitemp). All rats received a single unilateral 2μl injection over the course of 20min using a 32-gauge custom needle (Hamilton) with a 110° bevel fitted to a gas-tight syringe and controlled by a Nanomite digital pump (Harvard Apparatus). A concentration of 2 µl of solution containing ultrapurified LPS (20 000 Endotoxin Units, Invivogen, Santa Barbra, CA, USA) diluted into saline was injected into the right rat SNpc at empirically derived coordinates (4.65 mm anterior/posterior, 2.25 mm medial/lateral and 7.45 mm dorsal/ventral). Scalp incisions were closed by suture, and animals were monitored for successful recovery, with food and water consumption expected in the first few hours post-surgery.
To isolate TEPMs, mice were injected in the peritoneal cavity with 1.5 ml of 4% thioglycollate broth. After 72h (or at the indicated time point), animals were anesthetized under isoflurane and the peritoneal cavity exposed. Ten microliters of ice-cold PBS was injected into the peritoneal cavity through a 28-gauge needle. The fluid containing cells was then withdrawn through an 18-gauge needle. Cells in solution were pelleted through centrifugation at 1000g for 10 min at room temperature. Red blood cells were lysed through resuspension of the cell pellet into 1 ml of red blood cell lysis buffer (Ammonium-Chloride-Potassium lysing buffer, ACK, Invitrogen) for 2 min at room temp. Cells were then centrifuged at 500g for 5 min and resuspended into 5 mls of ice-cold phosphate-buffered saline, PBS, pH 7.4. Cell number was counted using a Z2 Particle Counter (Beckman Coulter, Brea, CA, USA) set between 4 and 10 µm of recording diameter. Appropriate numbers of cells were plated into 10% fetal bovine serum (FBS) in DMEM with glutamine and 100 µg/ml penicillin/streptomycin. Cells were maintained in a humidified chamber set to 5% CO2.
Confocal and immunohistochemistry analysis
For immunofluorescence and confocal analysis, brain sections were prepared from animals terminally anesthetized with isoflurane and transcardially perfused in 0.9% saline with 10 U/ml heparin followed by ice-cold 4% paraformaldehyde (PFA). Brains were dissected and post-fixed at 4°C for 2 h in 4% PFA and then transferred to 30% sucrose in PBS at 4°C for cryopreservation. Once saturated in sucrose, brains were flash-frozen in isopentane, cooled on dry ice to −55°C and stored at −80°C until further processing. Frozen brains were embedded with tissue-freezing medium on a sledge microtome chuck and sections cut to 40 µm (Leica). Sections were rinsed three times with Tris-buffered saline (TBS). All rinses and primary and secondary antibody diluents were in TBS. To allow antigen retrieval, all sections were incubated with 10 mm sodium citrate, pH 6.0, containing 0.05% Tween-20 for 30 min at 37°C with agitation. Following three consecutive 5-min washes, nonspecific antigens were blocked by incubating sections for 1 h in 5% normal serum (Equitech-Bio) from the host of the indicated secondary antibody containing 0.1% Triton X-100 at 4°C with agitation. Primary antibodies were diluted in 5% normal serum and incubated on sections with agitation for 24 h at 4°C. Sections were washed three times for 5 min and then incubated in secondary antibody in 5% normal serum 18 h at 4°C with agitation. The next day, sections were washed three times and for immunofluorescence were mounted to SuperFrost slides (Fisher) and cover-slipped with Prolong Gold (Life Technologies). Confocal images were captured by a single observer blinded to the experimental conditions (drug treatment group and genotype status) using a Leica TCS-SP5 laser-scanning confocal microscope. The Leica LASAF software, Adobe Photoshop (contrast, brightness and color adjustments) and Adobe Illustrator were used to create figures and process images.
For DAB immunohistochemistry, sections were prepared according to the above-mentioned procedure except that after antigen retrieval, sections were quenched in 0.3% hydrogen peroxide (Sigma) in methanol for 30 min. After final washing steps from secondary antibody incubations, the sections were incubated in Avidin–Biotin Complex reagent (Vector Labs) for 30 min, washed 3 times again, and then developed in ImmPACT substrate (Vector Labs, Burlingame, CA, USA) for 2–5 min. The sections were placed in distilled water to terminate the DAB development reaction, rinsed 3 times in TBS and mounted with 25% ethanol in PBS onto SuperFrost glass slides (Fisher). Following air drying, the slides were dehydrated in ascending alcohols and three changes of xylene and cover-slipped with Permount (Fisher). Images from sections stained with DAB were captured using an Olympus BX61 microscope.
ELISA
Enzyme-linked immunoassays were accomplished by first plating 80 000 TEPM into treated wells of 24-well plates and cells maintained at 37°C in a water-jacketed incubator with 5% CO2. Cells were allowed to rest overnight and then washed twice with complete TPEM media to remove non-adherent cells and allowed to rest overnight again. TEPMs were treated for 6 h with 100 ng of LPS (500 endotoxin units) added per well. After 6 h, the media was removed from the cells and snap-frozen on dry ice. Dilutions of media to achieve signal in linear range of ELISA and multiplex ELISA kits were determined empirically prior to analysis. Ready-Set-GO anti mouse TNF ELISA (eBioscience, San Diego, CA, USA) was run according to the manufacturer's instructions. Samples were also analyzed using MILLIPLEX mouse cytokine/chemokine 32-plex magnetic bead ELISA (EMD Millipore, Billerica, MA, USA) according to the manufacturer's instructions.
Phagocytosis assay
A total of 250 000 TEPM cells per genotype were plated in 10-cm tissue culture-treated dishes into complete media (10% FBS in DMEM). Cells were allowed to rest overnight and then washed twice with complete media to remove non-adherent cells. FITC-labeled Zymosan beads (Life Technologies, Carlsbad, CA, USA) were prepared by resuspending the lyophilized beads in 1 ml of sterile PBS, pH 7.4. Beads were then sonicated at 70% power three times for 10 s (Fisher Scientific 500 Sonic Dismembrator with micro sonic probe) to break up clumps of beads. Beads were applied to TEPM cells at a 10:1 bead to cell ratio in serum-free DMEM for 1 h. Plates were then washed with 37°C PBS five times, and then cells fixed with 1% PFA. FITC beads not internalized were quenched with the application of Sudan Black-B reagent (Invitrogen). Nuclei were labeled with the application of 10 μm Hoescht-33342 solution in PBS for 10 min. Combined fluorescent and phase-contrast images of TEPM with beads were acquired on a Zeiss Axioobserver live cell imager (Carl Zeiss, Thornwood, NY, USA) fitted with a Colibri 2 LED cool imager and a high-speed MR2 camera (Carl Zeiss). Images were acquired using computer-assisted Mark and Find software (Axiovision v4.8), and images were analyzed by an investigator blinded to genotype and treatment condition. The number of beads internalized per TEPM was scored.
Boyden chambers chemotaxis assays
Chemotaxis assays were performed as previously reported (21). A total of 10 000 TEPM cells were plated in the upper well of an 8.0-µm-pore tissue culture insert. Cells were allowed to rest for 6 h and then the insert moved into a new well of a treated 24-well plate. In the bottom of the chamber, complete TEPM media was supplemented with 100 µm ADP. TEPM cells were allowed to migrate for 36 h during incubation at 37°C in a humidified chamber with 5% CO2. The number of cells migrating to the bottom well was determined by supplementing to 10 μm Hoescht-33342 and recording nuclei through automated cell-counting software Mark and Find and Automatic Measurement Program, Axiovision v4.8 (Carl Zeiss, Thornwood, NY, USA). For chemotaxis assays that utilized LRRK2 inhibitors, 5 µm SRI 29451 and 1 µm HG-10-102-01 was added to both the top and bottom well of the Boyden Chambers. All other processes were the same as mentioned earlier.
Flow cytometry and cell counts
TEPM were acquired and prepared as described earlier for flow cytometry experiments except the cells were collected 18 h (instead of 72 h) post-thioglycollate injection. Total cell numbers were counted by hemocytometer prior to flow cytometry analysis. Cells were stained for 30 min on ice with APC-labeled Ly6G (A18 clone), CD11b labeled with Alexa Fluor® 488, F4/80 antigen labeled with PE and MHC Class II (I-A/I-E) Biotin–Streptavidin PerCP (all antibodies eBioscience). For chemokine receptor experiments, cells were stained for 30 min at 37°C with CD11b labeled with Alexa Fluor® 488, APC-labeled Ly6G (A18 clone), 7-aminoactinomycin D (7-AAD) and CCR2 labeled with PE (R&D Systems, Minneapolis, MN, USA). Cells were then washed and analyzed on a FACS Caliber Flow Cytometer (BD Bioscience, San Jose, CA, USA). Isotype-matched, fluorescently conjugated antibodies of irrelevant specificity were used as controls.
Results were analyzed using FlowJo Software (Tree Star, Inc., Ashland, OR, USA).
Complete blood chemistry
Non-transgenic, WT LRRK2 and G2019S LRRK2 mice were placed under isoflurane anesthesia. 0.5 ml of blood was drawn through intra-cardiac puncture through 1-ml syringe fitted with a 21-gauge needle. Samples were immediately placed into heparinized vacuum tubes. Samples were analyzed within 24 h on an automated hematology analyzer (Idexx Laboratories, Fremont, CA, USA) by Animal Labs of Birmingham, LLC. Differential counts were performed manually by a licensed veterinary technician.
Immunoprecipitations
Two million TEPM cells per genotype were plated into 15-cm treated culture dishes and were allowed to rest overnight and then washed (as described earlier) to remove non-adherent cells. Cells were then treated with 100 ng/ml of LPS (500 endotoxin units per ml), or saline control, for 6 h. After 6 h, cells were removed from the plate by scraping into ice-cold PBS, pH 7.4, pelleted and then lysed into a solution of 1% Triton X-100, Tris–HCl pH 7.4, and 1× complete protease and phosphatase inhibitors (Roche, Basel, Switzerland). Cell solutions were sonicated at 10% power for 10 s, incubated for 1 h at 4°C while rotating, and lysates clarified by centrifugation at 20 000g for 20 min at 4°C. Pellets were discarded and 200 µl of M2 anti-FLAG magnetic beads (Sigma, St Louis, MO, USA) were washed three times in lysis buffer and then added to lysates overnight on a rotating wheel. Beads were washed two times in lysis buffer and then two times in lysis buffer supplemented with 350 mm NaCl. Protein complexes were eluted from the beads by adding 2× Laemmli Buffer with 5% beta-mercaptoethanol and heated to 70°C with shaking at 1400 rpm. The kinase inhibitor PF-06447475 was obtained by courtesy of Warren Hirst, Pfizer, Inc. For immunoprecipitations with PF-06447475, all procedures were the same, except TEPMs were treated with 500 nm PF-06447475 for 24 h prior to LPS addition. Additionally, 500 nm PF-06447475 was added to the cell lysate during the FLAG-immunoprecipitation to ensure that LRRK2 remained inhibited.
Western blots
Equal volumes of lysates per genotype condition were loaded onto and separated on 4–20% Tris–Glycine gradient gels (BioRad, Hercules, CA, USA). Proteins were transferred to PVDF membranes at 30 volts overnight. Membranes were blocked in 5% BSA in TBS-T and then incubated in primary antibodies overnight at 4°C. Primary antibodies were as follows: 1:2000 LRRK2 N241A/34 (Antibodies, Inc., Davis, CA, USA), 1:2500 Myosin IIa (Myh9) (Cell Signaling Technology, Danvers, MA, USA), 1:5000 Arp2 (Cell Signaling Technology), 1:2500 myosin 1f (Sigma). Membranes were washed, then incubated with appropriate secondary antibodies and imaged using Li-COR Odyssey imaging system (Li-COR Biosciences, Lincoln, NE, USA) or ECL.
Mass spectrometry
Lysates were denatured and separated on a 7.5% Tris–glycine gel (BioRad) and stained with Bio-Safe colloidal Coomassie (BioRad). Bands were excised and enzymatically digested with Trypsin Gold (Promega, Madison, WI, USA) overnight according to the manufacturer's instructions, followed by acidification to pH 3–4 with 10% formic acid for analysis by liquid chromatography-mass spectrometry (LC-MS). Each peptide digest fraction was analyzed using a linear trap quadropole XL (LTQ XL) ion trap mass spectrometer equipped with a nano-electrospray source, and a Surveyor Plus binary high-pressure liquid chromatography (HPLC) pump (Thermo Scientific, Rockford, IL, USA) using a split flow configuration. Separations were carried out using a 100 μm × 13 cm pulled-tip C-18 column (Jupiter C-18 300 A, 5 µm). The HPLC was set up with two mobile phases that included solvent A (0.1% FA in ddH2O) and solvent B (0.1% FA in 85% ddH2O/15% ACN) and was programed as follows: 15 min at 0% B (2 µl/min, load and desalt), 100 min at 0–50% B (∼0.5 nl/min, analyze) and 20 min at 0% B (2 µl/min, equilibrate). During the first 15 min of loading and desalting, the source was set at 0.0 volts. The LTQ XL was operated in data-dependent triple-play mode, with a survey scan range of 300–1200 m/z, followed by an ultra-zoom scan used for charge-state determination (∼20 k resolution at 400 m/z) and an MS2 scan, both carried out with 2.0 Da isolation widths on the three top most intense ions. MS data were collected in profile mode for all scan types. Charge state screening and dynamic exclusion were enabled with a minimum signal intensity of 2000, a repeat count of 2 and exclusion duration of 90 s for ions of ±1.5 m/z of the parent ion. The automatic gain control settings were 3 × 104, 5 × 103 and 1 × 104 ions for survey, zoom and collision-induced dissociation (CID) modes, respectively. For CID, the activation time, activation Q and normalized collision energy were set at 30 ms, 0.25 and 35%, respectively. The spray voltage was set at 1.9 kV following the first 15 min of loading, with a capillary temperature of 170°C. XCalibur RAW files were centroided and converted to MzXML, and the mgf files were then created using both ReAdW and MzXML2Search, respectively (http://sourceforge.net/projects/sashimi/). The data were searched using SEQUEST (v27 rev12, .dta files), set for two missed cleavages, a precursor mass window of 0.45 Da, tryptic enzyme, variable modification M at 15.9949 and static modifications C at 57.0293. Searches were performed with a mouse subset of the UniRef100 database, which included common contaminants such as digestion enzymes and human keratins. Identified peptides were filtered, grouped and quantified using ProteoIQ v2.3.04 (Premierbiosoft, Palo Alto, CA, USA). Only peptides with charge state of ≥2+ and a minimum peptide length of six amino acids were accepted for analysis. ProteoIQ incorporates the two most common methods for statistical validation of large proteome datasets, false discovery rate (FDR) and protein probability (127–129). Relative quantification was performed via spectral count, and spectral count abundances were normalized between samples (130–132). The FDR was set at <1% cutoff, with a total group probability of ≥0.7 and peptides of ≥2 assigned per protein. In order to identify proteins likely to randomly or nonspecifically interact with LRRK2 protein complexes, all proteins found through LC/MS/MS were filtered through the Contaminant Repository for Affinity Purification (CRAPome.org) database. Proteins that had a >10% chance of random interaction or were not found owing to improper or incomplete annotation were excluded from analysis.
Kinase assays
Kinase assays were performed as previously described (34,133). Briefly, 200 nm recombinant Δ970 WT LRRK2 or G2019S LRRK2 (Invitrogen, Carlsbad, CA, USA) was added to kinase reactions with 1 µm Rac1 or Arpc3 (Novus Biologicals, Littleton, CO, USA) in a buffer with 50 mm Tris (pH 7.5), 150 mm NaCl and 10 mm MgCl2 with 100 μm ATP. Kinase reactions were run for 30 min at 30°C and terminated with the addition of 2× Lamelli buffer. Kinase reactions were electrophoresed on 4–20% SDS–PAGE gels (BioRad), stained with Coomassie and dehydrated. Gels were then exposed to BioMax (Kodak, Rochester, NY, USA) film and developed. Phosphate incorporation was quantified by cutting out protein bands from coomassie-stained gels and beta particles counted on a liquid scintillation counter (Beckman Coulter).
Statistical analysis, unbiased quantifications and system analysis
Statistical analysis was performed using GraphPad Prism 5.0 software (GraphPad Software, Inc., LaJolla, CA, USA). A P-value of <0.05 was considered significant. To two-way unpaired t-tests were used with Bonferroni corrections as appropriate. For comparisons of the more than two groups, a one-way ANOVA test with Tukey's post hoc analysis was used. For groups showing non-normal distribution as determined by a Kolmogorov–Smirnov test for normality, a Kruskal–Wallis ANOVA was selected with Dunn's multiple comparisons corrections to compare groups. Correlations were determined by Pearson coefficients.
Stereological estimation of the total TH and CD68 cells in the SNpc was performed using an optical fractionator probe (Microbrightfield, Willston, VT, USA) by an investigator blinded to experimental condition and animal genotype. Optical fractionator and dissector probes were used to count grid sizes of 50 µm2 with density adjusted for analysis of at least 100 objects per sample.
Gene-ontology analysis and network analysis was performed using MetaCore GeneGO software (Thomson Reuters, New York, NY, USA) and STRING v9.1 (www.string-db.org).
Supplementary Material
Funding
We thank the benevolence of John A. and Ruth R. Jurenko, as well as NIH/NINDS grants F31NS081963 to M.S.M and R01 NS064934 to A.B.W.
Supplementary Material
Acknowledgements
We thank Southern Research, Robert Galemmo and Mark Suto for providing LRRK2 small molecule inhibitors SRI-451 and HG-10-102-01. We thank Pfizer, Inc., Warren Hirst and Paul Galatsis for providing the LRRK2 small molecule inhibitor PF-06447475.
Conflict of Interest statement. None declared.
References
- 1.Bosgraaf L., Van Haastert P.J. (2003) Roc, a Ras/GTPase domain in complex proteins. Biochim. Biophys. Acta., 1643, 5–10. [DOI] [PubMed] [Google Scholar]
- 2.Van Limbergen J., Wilson D.C., Satsangi J. (2009) The genetics of Crohn's disease. Annu. Rev. Genomics Hum. Genet., 10, 89–116. [DOI] [PubMed] [Google Scholar]
- 3.Zhang F.R., Huang W., Chen S.M., Sun L.D., Liu H., Li Y., Cui Y., Yan X.X., Yang H.T., Yang R.D., et al. (2009) Genomewide association study of leprosy. N. Engl. J. Med., 361, 2609–2618. [DOI] [PubMed] [Google Scholar]
- 4.Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R.J., Calne D.B., et al. (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron, 44, 601–607. [DOI] [PubMed] [Google Scholar]
- 5.Paisan-Ruiz C., Jain S., Evans E.W., Gilks W.P., Simon J., van der Brug M., Lopez de Munain A., Aparicio S., Gil A.M., Khan N., et al. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron, 44, 595–600. [DOI] [PubMed] [Google Scholar]
- 6.Satake W., Nakabayashi Y., Mizuta I., Hirota Y., Ito C., Kubo M., Kawaguchi T., Tsunoda T., Watanabe M., Takeda A., et al. (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet., 41, 1303–1307. [DOI] [PubMed] [Google Scholar]
- 7.Nalls M.A., Pankratz N., Lill C.M., Do C.B., Hernandez D.G., Saad M., DeStefano A.L., Kara E., Bras J., Sharma M., et al. (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat. Genet., 46, 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.West A.B. (2014) Ten years and counting: moving leucine-rich repeat kinase 2 inhibitors to the clinic. Mov. Disord., 30, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West A.B., Moore D.J., Biskup S., Bugayenko A., Smith W.W., Ross C.A., Dawson V.L., Dawson T.M. (2005) Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl Acad. Sci. USA, 102, 16842–16847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacLeod D., Dowman J., Hammond R., Leete T., Inoue K., Abeliovich A. (2006) The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron, 52, 587–593. [DOI] [PubMed] [Google Scholar]
- 11.Sheng Z., Zhang S., Bustos D., Kleinheinz T., Le Pichon C.E., Dominguez S.L., Solanoy H.O., Drummond J., Zhang X., Ding X., et al. (2012) Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of pd mutations. Sci. Trans. Med., 4, 1–12. [DOI] [PubMed] [Google Scholar]
- 12.Stafa K., Trancikova A., Webber P.J., Glauser L., West A.B., Moore D.J. (2012) GTPase activity and neuronal toxicity of Parkinson's disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet., 8, e1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nichols R.J., Dzamko N., Morrice N.A., Campbell D.G., Deak M., Ordureau A., Macartney T., Tong Y., Shen J., Prescott A.R., et al. (2010) 14–3–3 binding to LRRK2 is disrupted by multiple Parkinson's disease-associated mutations and regulates cytoplasmic localization. Biochem. J., 430, 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacLeod D.A., Rhinn H., Kuwahara T., Zolin A., Di Paolo G., McCabe B.D., Marder K.S., Honig L.S., Clark L.N., Small S.A., et al. (2013) RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron, 77, 425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith W.W., Pei Z., Jiang H., Dawson V.L., Dawson T.M., Ross C.A. (2006) Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci., 9, 1231–1233. [DOI] [PubMed] [Google Scholar]
- 16.Smith W.W., Pei Z., Jiang H., Moore D.J., Liang Y., West A.B., Dawson V.L., Dawson T.M., Ross C.A. (2005) Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl Acad. Sci. USA, 102, 18676–18681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., van der Brug M.P., Beilina A., Blackinton J., Thomas K.J., et al. (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis., 23, 329–341. [DOI] [PubMed] [Google Scholar]
- 18.Daher J.P., Volpicelli-Daley L.A., Blackburn J.P., Moehle M.S., West A.B. (2014) Abrogation of alpha-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc. Natl Acad. Sci. USA, 111, 9289–9294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gardet A., Benita Y., Li C., Sands B.E., Ballester I., Stevens C., Korzenik J.R., Rioux J.D., Daly M.J., Xavier R.J., et al. (2010) LRRK2 is involved in the IFN-gamma response and host response to pathogens. J. Immunol., 185, 5577–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hakimi M., Selvanantham T., Swinton E., Padmore R.F., Tong Y., Kabbach G., Venderova K., Girardin S.E., Bulman D.E., Scherzer C.R., et al. (2011) Parkinson's disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J. Neural. Transm., 118, 795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moehle M.S., Webber P.J., Tse T., Sukar N., Standaert D.G., DeSilva T.M., Cowell R.M., West A.B. (2012) LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci., 32, 1602–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thevenet J., Pescini Gobert R., Hooft van Huijsduijnen R., Wiessner C., Sagot Y.J. (2011) Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS One, 6, e21519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alvarez-Errico D., Vento-Tormo R., Sieweke M., Ballestar E. (2014) Epigenetic control of myeloid cell differentiation, identity and function. Nat. Rev. Immunol., 15, 7–17. [DOI] [PubMed] [Google Scholar]
- 24.Chistiakov D.A., Sobenin I.A., Orekhov A.N., Bobryshev Y.V. (2015) Myeloid dendritic cells: development, functions, and role in atherosclerotic inflammation. Immunobiology, 14, 279–284. [DOI] [PubMed] [Google Scholar]
- 25.Crook K.R., Liu P. (2014) Role of myeloid-derived suppressor cells in autoimmune disease. World J. Immunol., 4, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim B., Yang M.S., Choi D., Kim J.H., Kim H.S., Seol W., Choi S., Jou I., Kim E.Y., Joe E.H. (2012) Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS One, 7, e34693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marker D.F., Puccini J.M., Mockus T.E., Barbieri J., Lu S.M., Gelbard H.A. (2012) LRRK2 kinase inhibition prevents pathological microglial phagocytosis in response to HIV-1 Tat protein. J. Neuroinflammation, 9, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caesar M., Zach S., Carlson C.B., Brockmann K., Gasser T., Gillardon F. (2013) Leucine-rich repeat kinase 2 functionally interacts with microtubules and kinase-dependently modulates cell migration. Neurobiol. Dis., 54, 280–288. [DOI] [PubMed] [Google Scholar]
- 29.Bosgraaf L., Waijer A., Engel R., Visser A.J., Wessels D., Soll D., van Haastert P.J. (2005) RasGEF-containing proteins GbpC and GbpD have differential effects on cell polarity and chemotaxis in Dictyostelium. J. Cell Sci., 118, 1899–1910. [DOI] [PubMed] [Google Scholar]
- 30.Cha I., Lee S.H., Jeon T.J. (2010) Chemoattractant-mediated Rap1 activation requires GPCR/G proteins. Mol. Cells, 30, 563–567. [DOI] [PubMed] [Google Scholar]
- 31.Kicka S., Shen Z., Annesley S.J., Fisher P.R., Lee S., Briggs S., Firtel R.A. (2011) The LRRK2-related Roco kinase Roco2 is regulated by Rab1A and controls the actin cytoskeleton. Mol. Biol. Cell, 22, 2198–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baptista M.A., Dave K.D., Frasier M.A., Sherer T.B., Greeley M., Beck M.J., Varsho J.S., Parker G.A., Moore C., Churchill M.J., et al. (2013) Loss of leucine-rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS One, 8, e80705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herzig M.C., Kolly C., Persohn E., Theil D., Schweizer T., Hafner T., Stemmelen C., Troxler T.J., Schmid P., Danner S., et al. (2011) LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum. Mol. Genet., 20, 4209–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Z., Galemmo R.A., Jr, Fraser K.B., Moehle M.S., Sen S., Volpicelli-Daley L.A., DeLucas L.J., Ross L.J., Valiyaveettil J., Moukha-Chafiq O., et al. (2014) Unique functional and structural properties of the LRRK2 protein ATP-binding pocket. J. Biol. Chem., 289, 32937–32951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gillardon F., Schmid R., Draheim H. (2012) Parkinson's disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience, 208, 41–48. [DOI] [PubMed] [Google Scholar]
- 36.Moehle M.S., West A.B. (2014) M1 and M2 immune activation in Parkinson's Disease: Foe and ally? Neuroscience, 14, 975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinez F.O., Gordon S. (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep, 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castano A., Herrera A.J., Cano J., Machado A. (2002) The degenerative effect of a single intranigral injection of LPS on the dopaminergic system is prevented by dexamethasone, and not mimicked by rh-TNF-alpha, IL-1beta and IFN-gamma. J. Neurochem., 81, 150–157. [DOI] [PubMed] [Google Scholar]
- 39.Castano A., Herrera A.J., Cano J., Machado A. (1998) Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J. Neurochem., 70, 1584–1592. [DOI] [PubMed] [Google Scholar]
- 40.West A.B., Cowell R.M., Daher J.P., Moehle M.S., Hinkle K.M., Melrose H.L., Standaert D.G., Volpicelli-Daley L.A. (2014) Differential LRRK2 expression in the cortex, striatum, and substantia nigra in transgenic and nontransgenic rodents. J. Comp. Neurol., 522, 2465–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim W.G., Mohney R.P., Wilson B., Jeohn G.H., Liu B., Hong J.S. (2000) Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J. Neurosci., 20, 6309–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee J.W., Tapias V., Di Maio R., Greenamyre J.T., Cannon J.R. (2015) Behavioral, neurochemical, and pathologic alterations in bacterial artificial chromosome transgenic G2019S leucine-rich repeated kinase 2 rats. Neurobiol. Aging, 36, 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walker M.D., Volta M., Cataldi S., Dinelle K., Beccano-Kelly D., Munsie L., Kornelsen R., Mah C., Chou P., Co K., et al. (2014) Behavioral deficits and striatal DA signaling in LRRK2 p.G2019S transgenic rats: a multimodal investigation including PET neuroimaging. J. Parkinsons Dis., 4, 483–498. [DOI] [PubMed] [Google Scholar]
- 44.Matta S., Van Kolen K., da Cunha R., van den Bogaart G., Mandemakers W., Miskiewicz K., De Bock P.J., Morais V.A., Vilain S., Haddad D., et al. (2012) LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron, 75, 1008–1021. [DOI] [PubMed] [Google Scholar]
- 45.Stafa K., Tsika E., Moser R., Musso A., Glauser L., Jones A., Biskup S., Xiong Y., Bandopadhyay R., Dawson V.L., et al. (2014) Functional interaction of Parkinson's disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum. Mol. Genet., 23, 2055–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biskup S., Moore D.J., Celsi F., Higashi S., West A.B., Andrabi S.A., Kurkinen K., Yu S.W., Savitt J.M., Waldvogel H.J., et al. (2006) Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol., 60, 557–569. [DOI] [PubMed] [Google Scholar]
- 47.Higashi S., Moore D.J., Yamamoto R., Minegishi M., Sato K., Togo T., Katsuse O., Uchikado H., Furukawa Y., Hino H., et al. (2009) Abnormal localization of leucine-rich repeat kinase 2 to the endosomal-lysosomal compartment in lewy body disease. J. Neuropathol. Exp. Neurol., 68, 994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orenstein S.J., Kuo S.H., Tasset I., Arias E., Koga H., Fernandez-Carasa I., Cortes E., Honig L.S., Dauer W., Consiglio A., et al. (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci., 16, 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manzoni C., Mamais A., Dihanich S., Abeti R., Soutar M.P., Plun-Favreau H., Giunti P., Tooze S.A., Bandopadhyay R., Lewis P.A. (2013) Inhibition of LRRK2 kinase activity stimulates macroautophagy. Biochim. Biophys. Acta., 1833, 2900–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alegre-Abarrategui J., Christian H., Lufino M.M., Mutihac R., Venda L.L., Ansorge O., Wade-Martins R. (2009) LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet., 18, 4022–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piccoli G., Condliffe S.B., Bauer M., Giesert F., Boldt K., De Astis S., Meixner A., Sarioglu H., Vogt-Weisenhorn D.M., Wurst W., et al. (2011) LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J. Neurosci., 31, 2225–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luster A.D., Alon R., von Andrian U.H. (2005) Immune cell migration in inflammation: present and future therapeutic targets. Nat. Immunol., 6, 1182–1190. [DOI] [PubMed] [Google Scholar]
- 53.Honda S., Sasaki Y., Ohsawa K., Imai Y., Nakamura Y., Inoue K., Kohsaka S. (2001) Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J. Neurosci., 21, 1975–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choi H.G., Zhang J., Deng X., Hatcher J.M., Patricelli M.P., Zhao Z., Alessi D.R., Gray N.S. (2012) Brain penetrant LRRK2 inhibitor. ACS Med. Chem. Lett., 3, 658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gallily R., Warwick A., Bang F.B. (1964) Effect of cortisone of genetic resistance to mouse heptatitis virus in vivo and in vitro. Proc. Natl Acad. Sci. USA, 51, 1158–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barski G., Messore G., Lepine P. (1955) Artificial peritoneal exudate, source of cells for virus culture in vitro. Ann. Inst. Pasteur. (Paris), 89, 366–371. [PubMed] [Google Scholar]
- 57.Ghosn E.E., Cassado A.A., Govoni G.R., Fukuhara T., Yang Y., Monack D.M., Bortoluci K.R., Almeida S.R., Herzenberg L.A., Herzenberg L.A. (2010) Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc. Natl Acad. Sci. USA, 107, 2568–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schleicher U., Hesse A., Bogdan C. (2005) Minute numbers of contaminant CD8+ T cells or CD11b+CD11c+ NK cells are the source of IFN-gamma in IL-12/IL-18-stimulated mouse macrophage populations. Blood, 105, 1319–1328. [DOI] [PubMed] [Google Scholar]
- 59.Vodovotz Y., Bogdan C., Paik J., Xie Q.W., Nathan C. (1993) Mechanisms of suppression of macrophage nitric oxide release by transforming growth factor beta. J. Exp. Med., 178, 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ness D., Ren Z., Gardai S., Sharpnack D., Johnson V.J., Brennan R.J., Brigham E.F., Olaharski A.J. (2013) Leucine-rich repeat kinase 2 (LRRK2)-deficient rats exhibit renal tubule injury and perturbations in metabolic and immunological homeostasis. PLoS One, 8, e66164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kigerl K.A., Gensel J.C., Ankeny D.P., Alexander J.K., Donnelly D.J., Popovich P.G. (2009) Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci., 29, 13435–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mikita J., Dubourdieu-Cassagno N., Deloire M.S., Vekris A., Biran M., Raffard G., Brochet B., Canron M.H., Franconi J.M., Boiziau C., et al. (2011) Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult. Scler., 17, 2–15. [DOI] [PubMed] [Google Scholar]
- 63.Mege J.L., Mehraj V., Capo C. (2011) Macrophage polarization and bacterial infections. Curr. Opin. Infect. Dis., 24, 230–234. [DOI] [PubMed] [Google Scholar]
- 64.Kuziel W.A., Morgan S.J., Dawson T.C., Griffin S., Smithies O., Ley K., Maeda N. (1997) Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc. Natl Acad. Sci. USA, 94, 12053–12058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mack M., Cihak J., Simonis C., Luckow B., Proudfoot A.E., Plachy J., Bruhl H., Frink M., Anders H.J., Vielhauer V., et al. (2001) Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J. Immunol., 166, 4697–4704. [DOI] [PubMed] [Google Scholar]
- 66.Mildner A., Mack M., Schmidt H., Brück W., Djukic M., Zabel M.D., Hille A., Priller J., Prinz M. (2009) CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain, 132, 2487–2500. [DOI] [PubMed] [Google Scholar]
- 67.Kuziel W.A., Dawson T.C., Quinones M., Garavito E., Chenaux G., Ahuja S.S., Reddick R.L., Maeda N. (2003) CCR5 deficiency is not protective in the early stages of atherogenesis in apoE knockout mice. Atherosclerosis, 167, 25–32. [DOI] [PubMed] [Google Scholar]
- 68.Meixner A., Boldt K., Van Troys M., Askenazi M., Gloeckner C.J., Bauer M., Marto J.A., Ampe C., Kinkl N., Ueffing M. (2011) A QUICK screen for Lrrk2 interaction partners--leucine-rich repeat kinase 2 is involved in actin cytoskeleton dynamics. Mol. Cell Proteomics, 10, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Caesar M., Felk S., Aasly J.O., Gillardon F. (2015) Changes in actin dynamics and F-actin structure both in synaptoneurosomes of LRRK2(R1441G) mutant mice and in primary human fibroblasts of LRRK2(G2019S) mutation carriers. Neuroscience, 284, 311–324. [DOI] [PubMed] [Google Scholar]
- 70.Parisiadou L., Xie C., Cho H.J., Lin X., Gu X.L., Long C.X., Lobbestael E., Baekelandt V., Taymans J.M., Sun L., et al. (2009) Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J. Neurosci., 29, 13971–13980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mellacheruvu D., Wright Z., Couzens A.L., Lambert J.P., St-Denis N.A., Li T., Miteva Y.V., Hauri S., Sardiu M.E., Low T.Y., et al. (2013) The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods, 10, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Henderson J.L., Kormos B.L., Hayward M.M., Coffman K.J., Jasti J., Kurumbail R.G., Wager T.T., Verhoest P.R., Noell G.S., Chen Y., et al. (2015) Discovery and preclinical profiling of 3-[4-(morpholin-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl]benzonitrile (PF-06447475), a highly potent, selective, brain penetrant, and in vivo active LRRK2 kinase inhibitor. J. Med. Chem., 58, 419–432. [DOI] [PubMed] [Google Scholar]
- 73.Jones G.E. (2000) Cellular signaling in macrophage migration and chemotaxis. J. Leukocyte Biol., 68, 593–602. [PubMed] [Google Scholar]
- 74.Mukai Y., Iwaya K., Ogawa H., Mukai K. (2005) Involvement of Arp2/3 complex in MCP-1-induced chemotaxis. Biochem. Biophys. Res. Commun., 334, 395–402. [DOI] [PubMed] [Google Scholar]
- 75.Baumann K. (2012) Cell migration: Chemotaxis without ARP2/3. Nat. Rev. Mol. Cell Biol., 13, 211–211. [Google Scholar]
- 76.Choi C.-H., Thomason P.A., Zaki M., Insall R.H., Barber D.L. (2012) Phosphorylation of actin-related protein 2 (Arp2) is required for normal development and cAMP chemotaxis in Dictyostelium. J. Biol. Chem., 4, 2464–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cavnar P.J., Mogen K., Berthier E., Beebe D.J., Huttenlocher A. (2012) The actin regulatory protein HS1 interacts with Arp2/3 and mediates efficient neutrophil chemotaxis. J. Biol. Chem., 287, 25466–25477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu C., Asokan S.B., Berginski M.E., Haynes E.M., Sharpless N.E., Griffith J.D., Gomez S.M., Bear J.E. (2012) Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell, 148, 973–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Asokan S.B., Johnson H.E., Rahman A., King S.J., Rotty J.D., Lebedeva I.P., Haugh J.M., Bear J.E. (2014) Mesenchymal chemotaxis requires selective inactivation of myosin II at the leading edge via a noncanonical PLCγ/PKCα pathway. Dev. Cell, 31, 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rey M., Vicente-Manzanares M., Viedma F., Yanez-Mo M., Urzainqui A., Barreiro O., Vazquez J., Sanchez-Madrid F. (2002) Cutting edge: association of the motor protein nonmuscle myosin heavy chain-IIA with the C terminus of the chemokine receptor CXCR4 in T lymphocytes. J. Immunol., 169, 5410–5414. [DOI] [PubMed] [Google Scholar]
- 81.Sandquist J.C., Swenson K.I., Demali K.A., Burridge K., Means A.R. (2006) Rho kinase differentially regulates phosphorylation of nonmuscle myosin II isoforms A and B during cell rounding and migration. J. Biol. Chem., 281, 35873–35883. [DOI] [PubMed] [Google Scholar]
- 82.Kim S.V., Mehal W.Z., Dong X., Heinrich V., Pypaert M., Mellman I., Dembo M., Mooseker M.S., Wu D., Flavell R.A. (2006) Modulation of cell adhesion and motility in the immune system by Myo1f. Science, 314, 136–139. [DOI] [PubMed] [Google Scholar]
- 83.Even-Ram S., Doyle A.D., Conti M.A., Matsumoto K., Adelstein R.S., Yamada K.M. (2007) Myosin IIA regulates cell motility and actomyosin-microtubule crosstalk. Nat. Cell Biol., 9, 299–309. [DOI] [PubMed] [Google Scholar]
- 84.Dulyaninova N.G., House R.P., Betapudi V., Bresnick A.R. (2007) Myosin-IIA heavy-chain phosphorylation regulates the motility of MDA-MB-231 carcinoma cells. Mol. Biol. Cell, 18, 3144–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vicente-Manzanares M., Zareno J., Whitmore L., Choi C.K., Horwitz A.F. (2007) Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J. Cell Biol., 176, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sandquist J.C., Means A.R. (2008) The C-terminal tail region of nonmuscle myosin II directs isoform-specific distribution in migrating cells. Mol. Biol. Cell, 19, 5156–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen C.L., Wang Y., Sesaki H., Iijima M. (2012) Myosin I links PIP3 signaling to remodeling of the actin cytoskeleton in chemotaxis. Sci. Signal, 5, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jaleel M., Nichols R.J., Deak M., Campbell D.G., Gillardon F., Knebel A., Alessi D.R. (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem. J., 405, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beilina A., Rudenko I.N., Kaganovich A., Civiero L., Chau H., Kalia S.K., Kalia L.V., Lobbestael E., Chia R., Ndukwe K., et al. (2014) Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl Acad. Sci. USA, 111, 2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schreij A.M., Chaineau M., Ruan W., Lin S., Barker P.A., Fon E.A., McPherson P.S. (2015) LRRK2 localizes to endosomes and interacts with clathrin-light chains to limit Rac1 activation. EMBO Rep., 16, 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Habig K., Gellhaar S., Heim B., Djuric V., Giesert F., Wurst W., Walter C., Hentrich T., Riess O., Bonin M. (2013) LRRK2 guides the actin cytoskeleton at growth cones together with ARHGEF7 and tropomyosin 4. Biochim. Biophys. Acta., 1832, 2352–2367. [DOI] [PubMed] [Google Scholar]
- 92.Bosgraaf L., Russcher H., Smith J.L., Wessels D., Soll D.R., Van Haastert P.J. (2002) A novel cGMP signalling pathway mediating myosin phosphorylation and chemotaxis in Dictyostelium. EMBO J., 21, 4560–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rougerie P., Miskolci V., Cox D. (2013) Generation of membrane structures during phagocytosis and chemotaxis of macrophages: role and regulation of the actin cytoskeleton. Immunol. Rev., 256, 222–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pecci A., Bozzi V., Panza E., Barozzi S., Gruppi C., Seri M., Balduini C.L. (2011) Mutations responsible for MYH9-related thrombocytopenia impair SDF-1-driven migration of megakaryoblastic cells. Thromb. Haemost., 106, 693–704. [DOI] [PubMed] [Google Scholar]
- 95.Jacobelli J., Bennett F.C., Pandurangi P., Tooley A.J., Krummel M.F. (2009) Myosin-IIA and ICAM-1 regulate the interchange between two distinct modes of T cell migration. J. Immunol., 182, 2041–2050. [DOI] [PubMed] [Google Scholar]
- 96.Fraser K.B., Moehle M.S., Daher J.P., Webber P.J., Williams J.Y., Stewart C.A., Yacoubian T.A., Cowell R.M., Dokland T., Ye T., et al. (2013) LRRK2 secretion in exosomes is regulated by 14–3–3. Hum. Mol. Genet., 22, 4988–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sartor R.B. (2006) Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol., 3, 390–407. [DOI] [PubMed] [Google Scholar]
- 98.Marks D.J., Segal A.W. (2008) Innate immunity in inflammatory bowel disease: a disease hypothesis. J. Pathol., 214, 260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Marks D.J., Harbord M.W., MacAllister R., Rahman F.Z., Young J., Al-Lazikani B., Lees W., Novelli M., Bloom S., Segal A.W. (2006) Defective acute inflammation in Crohn's disease: a clinical investigation. Lancet, 367, 668–678. [DOI] [PubMed] [Google Scholar]
- 100.Dessein R., Chamaillard M., Danese S. (2008) Innate immunity in Crohn's disease: the reverse side of the medal. J. Clin. Gastroenterol., 42, 144–147. [DOI] [PubMed] [Google Scholar]
- 101.Hagge D.A., Ray N.A., Krahenbuhl J.L., Adams L.B. (2004) An in vitro model for the lepromatous leprosy granuloma: fate of mycobacterium leprae from target macrophages after interaction with normal and activated effector macrophages. J. Immunol., 172, 7771–7779. [DOI] [PubMed] [Google Scholar]
- 102.Turk J.L. (1985) The mononuclear phagocyte system in granulomas. Br. J. Dermatol., 113, 49–54. [DOI] [PubMed] [Google Scholar]
- 103.Adams L.B., Scollard D.M., Ray N.A., Cooper A.M., Frank A.A., Orme I.M., Krahenbuhl J.L. (2002) The study of mycobacterium leprae infection in interferon-γ gene—disrupted mice as a model to explore the immunopathologic spectrum of leprosy. J. Infect. Dis., 185, 1–8. [DOI] [PubMed] [Google Scholar]
- 104.Gerhard A., Pavese N., Hotton G., Turkheimer F., Es M., Hammers A., Eggert K., Oertel W., Banati R.B., Brooks D.J. (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol. Dis., 21, 404–412. [DOI] [PubMed] [Google Scholar]
- 105.McGeer P.L., Itagaki S., Akiyama H., McGeer E.G. (1988) Rate of cell death in parkinsonism indicates active neuropathological process. Ann. Neurol., 24, 574–576. [DOI] [PubMed] [Google Scholar]
- 106.Healy D.G., Falchi M., O'Sullivan S.S., Bonifati V., Durr A., Bressman S., Brice A., Aasly J., Zabetian C.P., Goldwurm S., et al. (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol., 7, 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lesage S., Durr A., Tazir M., Lohmann E., Leutenegger A.L., Janin S., Pollak P., Brice A. (2006) LRRK2 G2019S as a cause of Parkinson's disease in North African Arabs. N. Engl. J. Med., 354, 422–423. [DOI] [PubMed] [Google Scholar]
- 108.Ozelius L.J., Senthil G., Saunders-Pullman R., Ohmann E., Deligtisch A., Tagliati M., Hunt A.L., Klein C., Henick B., Hailpern S.M., et al. (2006) LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews. N. Engl. J. Med., 354, 424–425. [DOI] [PubMed] [Google Scholar]
- 109.Bar-Shira A., Hutter C.M., Giladi N., Zabetian C.P., Orr-Urtreger A. (2009) Ashkenazi Parkinson's disease patients with the LRRK2 G2019S mutation share a common founder dating from the second to fifth centuries. Neurogenetics, 10, 355–358. [DOI] [PubMed] [Google Scholar]
- 110.Alves-Filho J.C., de Freitas A., Spiller F., Souto F.O., Cunha F.Q. (2008) The role of neutrophils in severe sepsis. Shock, 30, 3–9. [DOI] [PubMed] [Google Scholar]
- 111.Paula-Neto H.A., Alves-Filho J.C., Souto F.O., Spiller F., Amendola R.S., Freitas A., Cunha F.Q., Barja-Fidalgo C. (2011) Inhibition of guanylyl cyclase restores neutrophil migration and maintains bactericidal activity increasing survival in sepsis. Shock, 35, 17–27. [DOI] [PubMed] [Google Scholar]
- 112.Park D.W., Jiang S., Tadie J.M., Stigler W.S., Gao Y., Deshane J., Abraham E., Zmijewski J.W. (2013) Activation of AMPK enhances neutrophil chemotaxis and bacterial killing. Mol. Med., 19, 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kurihara T., Jones C.N., Yu Y.M., Fischman A.J., Watada S., Tompkins R.G., Fagan S.P., Irimia D. (2013) Resolvin D2 restores neutrophil directionality and improves survival after burns. FASEB J., 27, 2270–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li X., Patel J.C., Wang J., Avshalumov M.V., Nicholson C., Buxbaum J.D., Elder G.A., Rice M.E., Yue Z. (2010) Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J. Neurosci., 30, 1788–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sepulveda B., Mesias R., Li X., Yue Z., Benson D.L. (2013) Short- and long-term effects of LRRK2 on axon and dendrite growth. PLoS One, 8, e61986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hunter R.L., Liu M., Choi D.Y., Cass W.A., Bing G. (2008) Inflammation and age-related iron accumulation in F344 rats. Curr. Aging Sci., 1, 112–121. [DOI] [PubMed] [Google Scholar]
- 117.Bai L., Zhang X., Li X., Liu N., Lou F., Ma H., Luo X., Ren Y. (2015) Somatostatin prevents lipopolysaccharide-induced neurodegeneration in the rat substantia nigra by inhibiting the activation of microglia. Mol. Med. Rep., 1, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fu S.P., Wang J.F., Xue W.J., Liu H.M., Liu B.R., Zeng Y.L., Li S.N., Huang B.X., Lv Q.K., Wang W., et al. (2015) Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson inverted question marks disease models are mediated by GPR109A-dependent mechanisms. J. Neuroinflammation, 12, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hritcu L., Gorgan L.D. (2014) Intranigral lipopolysaccharide induced anxiety and depression by altered BDNF mRNA expression in rat hippocampus. Prog. Neuropsychopharmacol. Biol. Psychiatry, 51, 126–132. [DOI] [PubMed] [Google Scholar]
- 120.Wu X.L., Wang P., Liu Y.H., Xue Y.X. (2014) Effects of poly (ADP-ribose) polymerase inhibitor 3-aminobenzamide on blood-brain barrier and dopaminergic neurons of rats with lipopolysaccharide-induced Parkinson's disease. J. Mol. Neurosci., 53, 1–9. [DOI] [PubMed] [Google Scholar]
- 121.Zhang Z., Hou L., Song J.L., Song N., Sun Y.J., Lin X., Wang X.L., Zhang F.Z., Ge Y.L. (2014) Pro-inflammatory cytokine-mediated ferroportin down-regulation contributes to the nigral iron accumulation in lipopolysaccharide-induced Parkinsonian models. Neuroscience, 257, 20–30. [DOI] [PubMed] [Google Scholar]
- 122.Ailane S., Long P., Jenner P., Rose S. (2013) Expression of integrin and CD44 receptors recognising osteopontin in the normal and LPS-lesioned rat substantia nigra. Eur. J. Neurosci., 38, 2468–2476. [DOI] [PubMed] [Google Scholar]
- 123.Golembiowska K., Wardas J., Noworyta-Sokolowska K., Kaminska K., Gorska A. (2013) Effects of adenosine receptor antagonists on the in vivo LPS-induced inflammation model of Parkinson's disease. Neurotox. Res., 24, 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hoban D.B., Connaughton E., Connaughton C., Hogan G., Thornton C., Mulcahy P., Moloney T.C., Dowd E. (2013) Further characterisation of the LPS model of Parkinson's disease: a comparison of intra-nigral and intra-striatal lipopolysaccharide administration on motor function, microgliosis and nigrostriatal neurodegeneration in the rat. Brain Behav. Immun., 27, 91–100. [DOI] [PubMed] [Google Scholar]
- 125.Chung E.S., Bok E., Chung Y.C., Baik H.H., Jin B.K. (2012) Cannabinoids prevent lipopolysaccharide-induced neurodegeneration in the rat substantia nigra in vivo through inhibition of microglial activation and NADPH oxidase. Brain Res., 1451, 110–116. [DOI] [PubMed] [Google Scholar]
- 126.Ji K.A., Eu M.Y., Kang S.H., Gwag B.J., Jou I., Joe E.H. (2008) Differential neutrophil infiltration contributes to regional differences in brain inflammation in the substantia nigra pars compacta and cortex. Glia, 56, 1039–1047. [DOI] [PubMed] [Google Scholar]
- 127.Weatherly D.B., Atwood J.A., 3rd, Minning T.A., Cavola C., Tarleton R.L., Orlando R. (2005) A Heuristic method for assigning a false-discovery rate for protein identifications from Mascot database search results. Mol. Cell Proteomics, 4, 762–772. [DOI] [PubMed] [Google Scholar]
- 128.Nesvizhskii A.I., Keller A., Kolker E., Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem., 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- 129.Keller A., Nesvizhskii A.I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem., 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- 130.Liu H., Sadygov R.G., Yates J.R., 3rd (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem., 76, 4193–4201. [DOI] [PubMed] [Google Scholar]
- 131.Old W.M., Meyer-Arendt K., Aveline-Wolf L., Pierce K.G., Mendoza A., Sevinsky J.R., Resing K.A., Ahn N.G. (2005) Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell Proteomics, 4, 1487–1502. [DOI] [PubMed] [Google Scholar]
- 132.Beissbarth T., Hyde L., Smyth G.K., Job C., Boon W.M., Tan S.S., Scott H.S., Speed T.P. (2004) Statistical modeling of sequencing errors in SAGE libraries. Bioinformatics, 20, 31–39. [DOI] [PubMed] [Google Scholar]
- 133.Webber P.J., Smith A.D., Sen S., Renfrow M.B., Mobley J.A., West A.B. (2011) Autophosphorylation in the leucine-rich repeat kinase 2 (LRRK2) GTPase domain modifies kinase and GTP-binding activities. J. Mol. Biol., 412, 94–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.