

Abstract
Fragile X-associated tremor ataxia syndrome (FXTAS) is a neurodegenerative disorder caused by a CGG trinucleotide repeat expansion in the 5′ UTR of the Fragile X gene, FMR1. FXTAS is thought to arise primarily from an RNA gain-of-function toxicity mechanism. However, recent studies demonstrate that the repeat also elicits production of a toxic polyglycine protein, FMRpolyG, via repeat-associated non-AUG (RAN)-initiated translation. Pathologically, FXTAS is characterized by ubiquitin-positive intranuclear neuronal inclusions, raising the possibility that failure of protein quality control pathways could contribute to disease pathogenesis. To test this hypothesis, we used Drosophila- and cell-based models of CGG-repeat-associated toxicity. In Drosophila, ubiquitin proteasome system (UPS) impairment led to enhancement of CGG-repeat-induced degeneration, whereas overexpression of the chaperone protein HSP70 suppressed this toxicity. In transfected mammalian cells, CGG repeat expression triggered accumulation of a UPS reporter in a length-dependent fashion. To delineate the contributions from CGG repeats as RNA from RAN translation-associated toxicity, we enhanced or impaired the production of FMRpolyG in these models. Driving expression of FMRpolyG enhanced induction of UPS impairment in cell models, while prevention of RAN translation attenuated UPS impairment in cells and suppressed the genetic interaction with UPS manipulation in Drosophila. Taken together, these findings suggest that CGG repeats induce UPS impairment at least in part through activation of RAN translation.
Introduction
Fragile X-associated tremor ataxia syndrome (FXTAS) is an under-recognized, inherited, neurodegenerative disorder that affects upward of 1 in 3000 males and a lower percentage of females over the age of 50 years (1,2). It is characterized clinically by intention tremors, gait ataxia, dementia and peripheral neuropathy (3). FXTAS results from a CGG trinucleotide repeat expansion in the 5′ UTR of the fragile X gene, FMR1 (4). Normally, this gene consists of <45 CGG repeats. Large intergenerational expansion to >200 CGGs results in transcriptional silencing of the FMR1 locus with subsequent loss of the fragile X protein, FMRP (5–9). This causes fragile X syndrome, a common inherited form of intellectual disability and autism (10). However, intermediate expansions to 55–200 CGGs can lead to FXTAS. Unlike in fragile X syndrome, this ‘premutation’-sized repeat remains unmethylated, resulting in enhanced FMR1 transcription and the accumulation of CGG-repeat-containing mRNA (11–15).
Work to date by numerous groups suggests that the CGG repeat can elicit toxicity as RNA, presumably by binding to and sequestering specific RNA-binding proteins (13,16–21). This RNA gain-of-function mechanism has been best established in Myotonic Dystrophy Type I, where a CUG repeat expansion in the 3′ UTR of DMPK binds to and sequesters the Muscleblind (MBNL) family of RNA-splicing factors (22–24). This results in the dysregulated splicing of MBNL target transcripts, which can be reversed by supplying additional MBNL in animal models of the disease (25,26). In FXTAS, CGG repeats have been shown to avidly bind a large number of proteins, including hnRNP A2, Pur α, SAM-68 and the miRNA biogenesis complex Drosha/DGCR8 (18,19,21,27). However, studies demonstrating causality of these interactions in disease pathogenesis remain incomplete.
Recently, our group explored an alternative mechanism by which CGG repeats might trigger neurodegeneration (28). We discovered that CGG repeats within the 5′ UTR of FMR1 trigger a process known as repeat-associated non-AUG (RAN)-initiated translation that leads to production of a polyglycine protein from translation through the repeat itself (28,29). This polyglycine protein, FMRpolyG, accumulates in cellular-, fly- and mouse-based disease models and is found in ubiquitinated neuronal intranuclear inclusions in FXTAS patient brains (28). Moreover, sequence changes that either enhance or suppress FMRpolyG production lead to significant modulation of CGG-repeat-associated phenotypes in transfected cells and in Drosophila (28). RAN translation has also been shown to occur in other disorders, including at CAG repeats in Spinocerebellar Ataxia Type 8 and at GGGGCC and CCCCGG hexanucleotide repeat expansions associated with ALS and frontotemporal dementia (C9ALS/FTD) (29–34).
The discovery of RAN translation at CGG repeats and the early findings in Drosophila disease models suggest that FMRpolyG production may contribute to FXTAS pathogenesis (35). If this is true, then defining how FMRpolyG elicits toxicity would provide important insights. One possible mechanism underlying FMRpolyG-associated toxicity involves alterations in protein quality control pathways (17,28,36–39). Proteostasis has been widely implicated in numerous neurodegenerative disorders, including Alzheimer's disease, Parkinson's disease and polyglutamine disorders such as spinal bulbar muscular atrophy and Huntington's disease (40–60). Although it remains unknown what, if any, functional domains FMRpolyG might possess, it is aggregation prone in multiple model systems when fused to either FLAG or green fluorescent protein (GFP) (28). What functional consequence this propensity to aggregation has on cellular homeostasis, however, remains unknown.
Here, we demonstrate that CGG-repeat-elicited toxicity in Drosophila models is modifiable by alterations in some, but not all, protein quality control pathways. The sensitivity to modulators of protein quality control is dependent on the relative production of FMRpolyG rather than on the expression of CGG repeat RNA, per se, and modulating ubiquitin proteasome system (UPS) function leads to alterations in the number and distribution of FMRpolyG-GFP inclusions. In cell-based reporter assays, expression of CGG repeats leads to induction of UPS impairment, and this induction is largely dependent on RAN translation. Together, these data suggest that RAN translation of FMRpolyG at CGG repeats triggers induction of UPS impairment and alterations in neuronal proteostasis that contribute to pathogenesis in FXTAS.
Results
Ubiquitin proteasome system modulation alters CGG-repeat-associated toxicity in Drosophila
Protein quality control pathways are crucial in ridding cells of toxic or misfolded proteins. Two major protein quality control pathways, the UPS and the autophagy system, play important roles in numerous neurodegenerative disorders (40,44). To evaluate the role of UPS function in CGG-repeat-associated toxicity, we first utilized a series of temperature-sensitive, dominant, negative mutations in the β-2 (DTS7) or β-6 (DTS5) subunit of the proteasome to impair the UPS (61). Expression of either DTS5 or DTS7 alone elicited mild ommatidial degeneration at 28°C, which was greater when two copies of the DTS7 subunit (DTS72C) were expressed (Fig. 1Aii,iv,vi and C; Supplementary Material, Fig. S1) (41). Similarly, expression of 100 CGG repeats in the 5′ UTR of GFP resulted in a moderate rough-eye phenotype (Fig. 1Ai), as described (17,28). When this CGG100GFP line was crossed to DTS5 or DTS7 mutant lines, the resulting flies demonstrated significantly enhanced toxicity compared with either line alone (Fig. 1Aiii, v, vii, C). This finding was reproducible in multiple fly lines generated by different groups using different parent sequences with either 90 or 100 CGG repeats (Supplementary Material, Fig. S1) (18,28). In contrast, overexpression of the chaperone protein HSP70, which enhances UPS-based clearance of proteins, led to suppression of CGG100GFP-induced neurodegeneration (Fig. 1B and C), consistent with published results (17). These data suggest that CGG-repeat-elicited toxicity in Drosophila is modifiable by alterations in UPS activity.
Figure 1.

Impairment of the UPS enhances CGG-repeat-elicited eye degeneration in Drosophila. (A) Representative eye images from Drosophila of the indicated genotypes crossed to gmr-GAL4 at 1–2 days post-eclosion. CGG100GFP flies contain the 5′ UTR of FMR1 with 100 CGG repeats in front of eGFP. Both DTS5 and DTS7 are temperature-sensitive mutations in proteasomal subunits that enhance CGG-repeat-associated toxicity. DTS72C represents two copies of the mutant subunit, DTS7. (B) Representative eye images from Drosophila demonstrating suppression of the CGG100GFP rough-eye phenotype by co-expression of HSP70. (C) Quantitation of observed eye phenotypes. Scale is 0–10, with 0 being normal and 9 being frank necrosis with loss of ommatidial structure and bristle patterning over 50% of the eye. N > 50/genotype from at least two independent experiments. *P < 0.05, Mann–Whitney U-test compared with gmr-GAL4; CGG100GFP alone.
To determine whether other protein quality control pathways were similarly capable of modulating CGG-repeat-elicited toxicity, we utilized a series of Drosophila lines in which expression of either ATG6 or ATG12, critical components of the autophagy pathway, was suppressed by RNAi. When co-expressed with polyglutamine, these RNAi lines trigger a significant worsening of rough-eye phenotypes (41). Knocking down expression of either ATG6 or ATG12 had no significant impact on eye phenotypes in isolation (Supplementary Material, Fig. S2A). Co-expression of either ATG6 or ATG12 RNAi lines with CGG90GFP had no impact on the observed rough-eye phenotype (Supplementary Material, Fig. S2A) (14,62). To evaluate whether enhanced activation of the autophagy pathway was protective, we utilized the drug rapamycin. Rapamycin is known to, among other things, activate the autophagy pathway, and it has been proposed that this activation contributes to its neuroprotective effects in other neurodegenerative disease models (41,63). We reared CGG90GFP flies on food containing 1 and 2 mm of rapamycin or dimethyl sulfoxide (DMSO) as a vehicle control. Rapamycin-reared flies showed no improvement and perhaps a dose-dependent toxicity in the observed eye phenotype compared with controls (Supplementary Material, Fig. S2B and C), consistent with published results (62). Similarly, rapamycin treatment did not suppress repeat-dependent lethality when the transgene was expressed either ubiquitously or throughout the entire nervous system (Supplementary Material, Fig. S2D) (62). It did, however, suppress lethality associated with ubiquitous expression of two copies of the mutated proteasome subunit, DTS7 (Supplementary Material, Fig. S2D) (41). These results suggest that manipulation of the autophagy pathway does not modify CGG-repeat-associated toxicity (14,62).
Repeat-associated non-AUG (RAN) translation leads to inclusion formation in a Drosophila model of CGG repeat toxicity
We recently demonstrated that repeat-associated non-AUG (RAN)-initiated translation triggers inclusion formation and elicits toxicity in FXTAS models (28). Based on the way the transgene construct was generated, we expect two different GFP-containing protein products to be translated (Fig. 2A). First, GFP is generated from an ATG start codon in a Kozak context. Second, RAN translation in the polyglycine (GGC) reading frame in the inserted FMR1 5′ UTR will generate a polyglycine-GFP fusion protein (Fig. 2A). In Drosophila, expression of GFP alone in ommatidia leads to a diffuse pattern of staining within the eye. In contrast, including the 5′ UTR of FMR1 with a CGG repeat expansion leads to inclusions that are detectible by epifluorescence even after fixation and permeabilization of the tissue (Fig. 2B). These aggregates co-localize with HSP70 and ubiquitin (28). To delineate whether modulating UPS activity influences the rates of inclusion formation, we crossed CGG100GFP flies with flies expressing DTS7 or HSP70. GFP cryosections show that UPS-impaired flies produce more inclusions compared with CGG100GFP flies alone and that the burden of inclusions is lessened by overexpression of HSP70 (Fig. 2C and D).
Figure 2.

Altering UPS function modulates RAN translation-elicited inclusion number in CGG-repeat-expressing Drosophila. (A) Schematic of the CGG-repeat transgene. Both FMRpolyG-GFP (via RAN translation) and GFP (via AUG initiated translation) are generated in flies expressing this transcript. Red is human-derived sequence. (B) Flies expressing CGG100GFP, but not GFP alone, produce large GFP-positive aggregates in the ommatidia when crossed to gmr-GAL4. (C) Representative transverse sections through fixed and permeabilized Drosophila eyes of the indicated genotypes. (D) Quantitation of the number of GFP-positive inclusions in flies expressing CGG100GFP alone or with DTS7 or HSP70. *P < 0.05 on Student's t-test compared with gmr-Gal4; CGG100GFP alone.
The sensitivity of CGG-expressing flies to UPS impairment is determined by their ability to produce the CGG RAN translation product, FMRpolyG
We next determined whether the genetic epistasis of CGG repeat phenotypes by UPS modulation was a result of the CGG repeat as RNA or via production of the CGG RAN translation product, FMRpolyG. To accomplish this, we utilized a series of previously described Drosophila lines, which have either enhanced or decreased FMRpolyG production despite similar CGG RNA expression (28). As described earlier, the rough-eye phenotype elicited by ommatidial expression of CGG repeats is enhanced by co-expression of DTS7 (Fig. 3A and B). To evaluate whether the repeats as RNA in the absence of FMRpolyG can interact genetically with UPS impairment lines, we utilized a Drosophila line expressing CGG repeats preceded by a stop codon just 5′ to the repeat. Inclusion of this stop codon inhibits generation of FMRpolyG (28). However, the expression of the CGG repeat as RNA is maintained. This line itself has only a mild rough-eye phenotype (Fig. 3A). When this StopCGG100 line is co-expressed with DTS7, there is a diminished genetic interaction compared with expression of the repeat without the stop codon (Fig. 3A and B).
Figure 3.
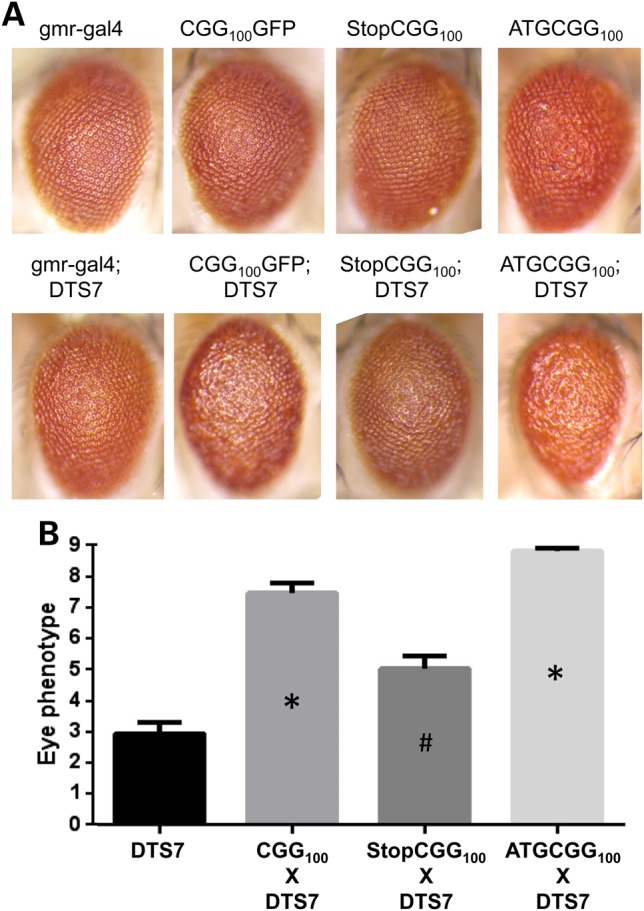
RAN translation is required for modulation of CGG-repeat-mediated toxicity by UPS impairment. (A) Top row: representative images of indicated genotypes. StopCGG100 includes a stop codon just before the repeat that precludes translation of FMRpolyG. ATGCGG100 includes a start codon just before the repeat that enhances FMRpolyG translation. Bottom row: same lines crossed to uas-DTS7, which impairs the proteasome. (B) Quantitation of eye phenotypes from flies expressing the indicated genotypes at 28°C. *P < 0.05 on Mann–Whitney U-test compared with DTS7 alone. # indicates significantly different from DTS7 × CGG100 and DTS7 × ATGCGG100.
To evaluate whether this interaction was influenced by FMRpolyG overproduction, we next utilized a CGG-expressing line in which expression of the RAN-translated product, FMRpolyG, is enhanced by the inclusion of an AUG start codon just 5′ to the repeat (28). These flies exhibit increased FMRpolyG production, decreased CGG RNA expression and a mildly enhanced basal phenotype compared with CGG repeats alone (Fig. 3A and B). When crossed to flies expressing DTS7, however, there was a synergistic enhancement of toxicity, such that all of the ommatidia are fused and there is necrosis of the eye (Fig. 3A and B). Thus, there is an enhanced sensitivity to genetic modulation of UPS function in the setting of the RAN translation product, FMRpolyG.
CGG repeats induce impairment of the ubiquitin proteasome system
The data presented earlier suggest that modulating UPS function can suppress or enhance CGG-repeat-mediated toxicity through clearance of the CGG RAN translation product, FMRpolyG. However, it is unclear whether CGG repeats are capable of eliciting UPS impairment, either directly or indirectly. To determine whether CGG repeats—either as RNA or RAN-translated protein—can induce UPS failure, we utilized a GFP-UPS reporter system in HeLa cells (64). These HeLa cells are stably transfected with a covalently bound G76V ubiquitin-tagged GFP reporter. The GFP protein is constantly turned over using the UPS, resulting in little to no green fluorescent signal (Fig. 4A). Treating these cells with lactacystin, a pharmacological inhibitor of the UPS, led to an accumulation of GFP signal (Fig. 4A). Similarly, transfecting HeLa cells with a truncated Ataxin 3 construct containing an expanded tract of 80 glutamines (Q80) elicited an enhanced GFP signal (Fig. 4A), consistent with published studies (41,64).
Figure 4.

CGG repeats elicit UPS impairment via FMRpolyG translation. (A) Representative fluorescent (top) and DIC (bottom) images from HeLa cells that stably express the UPS reporter G76V-Ub-GFP. Cells were transfected with an empty vector or a vector expressing Ataxin 3 with 80 glutamines, Q80, or were treated for 24 h with 10 µm lactacystin, an inhibitor of the UPS. (B) Schematic of cell-based constructs. (C) Representative images of G76V-Ub-GFP HeLa cells transfected with the indicated constructs and imaged by confocal microscopy. (D) Quantitation of the percentage of red cells (indicating transfection) where green fluorescence was present at 48 h post -transfection. *P < 0.05 compared with mCherry alone. **P < 0.05 compared with the indicated genotype. (E) qRT-PCR quantifying RNA expression from each of the constructs. (F) Western blot to mCherry in cells either treated with lactacystin or transfected with the indicated constructs. MCherry runs at ∼25 kD (indicated by *). Higher-molecular-weight bands represent RAN translation product through the CGG repeat (indicated by <) or AUG-driven production of FMRpolyG (indicated by #).
To evaluate whether CGG repeats can modulate UPS function, we generated a series of new constructs where the 5′ UTR of FMR1 was placed upstream of mCherry, such that RAN translation would elicit an FMRpolyG-mCherry fusion protein (Fig. 4B). Expression of mCherry alone elicited little change in GFP signal (Fig. 4C). However, cells transfected with constructs containing short (30) or long (65) CGG repeats showed an increase in GFP signal that was more prominent at larger repeat sizes (Fig. 4C and D).
To determine whether this induction of UPS impairment results from FMRpolyG production or from the expression of CGG-repeat-containing RNA, we inserted a stop codon 5′ to the long CGG repeats to block RAN-initiated translation (Fig. 4B). This demonstrated significantly reduced levels of GFP compared with the long CGG repeat construct, despite comparable RNA expression levels (Fig. 4C and E). We also introduced an ATG start codon 5′ to the CGG repeats to drive production of the RAN-initiated translation product. Driving expression of FMRpolyG further induced UPS impairment as measured by accumulation of G76V-Ub GFP (Fig. 4C and D). The accumulation of GFP in this assay was not associated with the total CGG RNA generated [as measured by quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR)] but was associated with RAN translation product generation as assessed by western blot (Fig. 4E and F).
In cell-based models, a second RAN translation product, FMRpolyA, is produced from expanded CGG repeats in the alanine (GCG) reading frame (28). Given that blocking FMRpolyG production reduced, but did not eliminate, UPS impairment, we sought to determine whether enhancing FMRpolyA production might impact UPS function. We, therefore, placed an ATG start codon just above the repeat in the alanine (GCG) reading frame and shifted the mCherry reading frame so that it would detect the polyalanine fusion protein (Fig. 4B). Expression of ATG-FMRpolyA-mCherry elicited UPS impairment that was comparable with that caused by CGG75 alone and was less than that observed for ATG-FMRpolyG-mCherry (Fig. 4D, Supplementary Material, Fig. S3). These data suggest that FMRpolyA production is not a major driver of CGG-repeat-elicited UPS impairment.
Discussion
Our studies demonstrate that the CGG repeat expansion responsible for the neurodegenerative disorder FXTAS is capable of interacting genetically with modulators of UPS function in Drosophila and of inducing UPS impairment in human cell lines. These processes are largely dependent on RAN translation of the CGG repeat into the polyglycine-containing protein, FMRpolyG. Taken together with previous studies (17,28,37,38), these findings suggest a model where CGG repeat expansions in FMR1 elicit neuronal proteostasis via RAN translation and contribute to FXTAS pathogenesis.
Impairment of protein quality control pathways is a common feature among almost all neurodegenerative conditions, including Alzheimer's disease, Parkinson's disease, polyglutamine diseases and ALS/frontotemporal dementia (40–60). Together, these conditions have been referred to as neurodegenerative proteinopathies, implying that failures in protein quality control pathways are a proximal shared event in disease pathogenesis (66) and thus a potentially shared route to effective therapeutics. The presence of proteinaceous intranuclear inclusions in FXTAS brains at autopsy suggests that it may similarly be subject to age-dependent decrements in protein quality control and thus share aspects of disease pathogenesis with more common neurodegenerative disorders. However, it was initially unclear how RNA repeats could trigger such a cascade of UPS failure and aggregate formation. Our discovery that CGG repeats support RAN translation to produce homopolymeric proteins containing polyglycine and (in cell culture models) polyalanine suggested one possible mechanism by which this might occur (28). Yet, the exact role that these proteins play in UPS impairment and disease pathogenesis was unclear.
This study provides two important additions to the field. First, we find that not only are models of FXTAS sensitive to alterations in proteostasis pathways but also that CGG repeat expression is capable of inducing UPS impairment. Second, both the genetic interaction with UPS pathways and the induction of UPS impairment are largely dependent on production of a specific CGG RAN translation product, FMRpolyG. These findings imply that a component of disease pathogenesis that is shared with other neurodegenerative disorders is specifically dependent on RAN translation, implying that future therapeutic development may need to take these products and their toxic consequences into account.
While the placement of a stop codon just 5′ to the repeat significantly impaired both the genetic interaction of CGG repeat expression with UPS impairment and the induction of UPS impairment in cell culture, it should be noted that it did not completely eliminate the effects of the repeat. This could be the result of at least two possible mechanisms. First, the CGG repeat as RNA may have a direct impact on UPS function (16,17,36). This idea is supported by work in Drosophila on CAG repeats, where placement of these repeats in the 3′ UTR (where its translation should be minimal) elicits phenotypes that are reversible by co-expression of the heat-shock protein HSP-70 (56). Alternatively, the introduced stop codon does not preclude RAN translation in other reading frames. Thus, production of a polyalanine RAN product from the CGG repeats, FMRpolyA, may serve as an alternative trigger for this genetic interaction and UPS impairment (28). Our data in cell culture studies make this less likely, as driving expression of FMRpolyA causes less induction of UPS failure than similar constructs expressing FMRpolyG. Future studies will be needed to determine the potential roles of both FMRpolyA and the CGG repeat as RNA in UPS impairment and FXTAS pathogenesis.
In animal models of many neurodegenerative disorders, treatment with agents that augment protein quality control pathways can ameliorate neuronal toxicity and behavioral phenotypes, suggesting a role for targeting this pathway in disease therapeutics. Here, we have tried to treat a Drosophila model of FXTAS with rapamycin, a drug that augments autophagy and suppresses polyglutamine toxicity in multiple model systems (41,67–70). Surprisingly, we find that rapamycin has no beneficial effects and in fact has a trend toward enhancing toxicity. Moreover, we see no genetic interaction with autophagy impairment. These results are consistent with previously published studies (14,62). This suggests that the CGG RAN-translated products interact specifically with the UPS, making them somewhat separate from other toxic proteins. However, it may be that other effects of mTOR impairment explain the lack of suppression in CGG repeat models compared with polyglutamine disorders that explain this finding. Indeed, activation of mTOR can suppress some CGG repeat phenotypes in Drosophila (62).
Recent work by other groups demonstrates that RAN translation contributes significantly to toxicity elicited by the GGGGCC repeat expansion in cell- and Drosophila-based models (71–74). Our data are generally consistent with these findings. However, it remains unclear whether the translational initiation mechanisms and behavior of FMRpolyG are directly comparable with the dipeptide repeat proteins generated from hexanucleotide repeat expansions. Early results suggest that each RAN translation product is likely to elicit both non-specific repeat-mediated proteotoxicity coupled with repeat-specific toxicity mediated through protein–protein interactions and perhaps sequestration of specific factors (71,74). However, at least one recent study demonstrates that application of drugs which bind to both CGG and GGGGCC repeat mRNAs impair RAN translation in cellular models (75). Evaluating these types of compounds in vivo should assist us in evaluating the broader impact of RAN translation on disease pathogenesis and provide a model for therapeutic development.
In summary, we provide evidence that RAN translation of FMRpolyG underlies the genetic interaction between protein quality control pathways and CGG-repeat-mediated toxicity and that these products are capable of impairing UPS function. These studies suggest that interventions that either impede RAN translation or enhance UPS function are likely to have beneficial effects in FXTAS and other repeat-mediated disorders.
Materials and Methods
Drosophila fly stocks/eye images
Drosophila lines used in this study have been previously described (17,28,61). Unless stated otherwise, all crosses were done and maintained on standard food at 28°C. For macroscopic eye phenotypes, 1- to 2-day-old anesthetized flies were evaluated with a Leica MZ APO stereomicroscope and photographed with a Leica DFC320 digital camera at 80× as previously described (14,28). For each genotype and condition, at least 50 flies over multiple crosses were evaluated. For whole eye GFP fluorescent images of Drosophila, the flies were anesthetized with CO2, decapitated and immediately imaged on a glass coverslip by epifluorescence on an inverted Olympus IX71 microscope. All images were taken at the same exposure. Images were processed using Slidebook 4.0 software and subtracted for background auto-fluorescence based on images of non-transgenic flies.
Eye phenotype severity was quantified using a validated scale (41). Briefly, over 50 eyes from each genotype were scored on a nine-point scale based on the following criteria: supernumerary inter-ommatidial bristles, abnormal bristle orientation, ommatidium fusion, ommatidium pitting, disorganization of ommatidial array and retinal collapse/necrosis. The presence of each feature is given one point, and additional two points were given if more than 5% of the eyes were affected or four points if more than 50% of eyes were affected. Higher score means that the eyes are more degenerated. For rapamycin drug treatments, flies were maintained on standard food with DMSO or 1 and 2 mm of rapamycin at 28°C.
Drosophila histology/confocal imaging
Transverse sections of Drosophila eyes were prepared as previously described (28). Briefly, 1- to 2-day-old flies were anesthetized, decapitated and immediately frozen in OCT media and cryosectioned at 12 μm. Sections were fixed for 15 min in 4% electron microscopy grade paraformaldehyde in phosphate-buffered saline (PBS) and mounted in Prolong Gold with DAPI. Antibodies were not used to visualize GFP. Fluorescent imaging of histological sections was performed on an Olympus FV1000 confocal microscope. For inclusion counts, five identical-sized boxes were drawn on top of each transverse-sectioned confocal image across samples using Adobe Photoshop CS2. At least five different transverse sections for each sample were used in counting. The same exposure and laser settings were used for all directly compared samples. All post hoc image alterations were applied across the entire image and were applied identically to directly compared samples.
Plasmid constructs
mCherry reporter vectors were derived from previously described GFP plasmids (28,65) by using Xho1 and Not1 endonuclease restriction digestion to remove GFP and insert an mCherry reporter tag. All constructs were verified by Sanger sequencing.
Cell culture and transient transfections
HeLa cells were maintained at 37°C in 5% CO2 incubators. Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 1% non-essential amino acids, 1% G418 and 1% Pen-Strep was used as culture media. Cells were transfected using Lipofectamine LTX with Plus Reagent from Invitrogen using its protocol.
UPS impairment assay
G76V-Ub-GFP HeLa cell lines were previously described (64). Cells were transfected with either empty vector or the indicated constructs. Forty-eight hours post-transfection, cells were fixed for 15 min in EM-grade 4% paraformaldehyde in PBS, permeabilized with 0.1% Triton X for 5 min and then blocked in 5% normal goat serum prior to incubation with primary antibodies to GFP (Roche, Mouse, 1:1000) and mCherry (Invitrogen, rat, 1:1000) overnight. After washing, Alexa-488 (anti-mouse) and Alexa 555 (anti-rat) secondary antibodies were used. Cells were mounted in Prolong Gold with DAPI and imaged using confocal microscopy as described earlier.
Cells were analyzed blind to genotype. For each construct, 100 red cells/well were identified, and then the percent of these cells that appeared green above a background level defined by the empty vector-transfected cells was identified. The data are derived from at least three independent experiments and at least two separate wells per plasmid per experiment. Similar results were obtained by flow cytometry-based separation of green and red cells (not shown).
Statistical analysis
For images of fly eyes, quantitated morphology scores were analyzed using a non-parametric Mann–Whitney U-test with the Bonferroni correction for multiple comparisons. For quantitation of GFP aggregates, Student's t-test was utilized. For comparison of the percentage of red cells that were green, we utilized chi-squared correlation. Error bars represent the standard error of the mean except for proportion numbers, where the error bars represent the 95% confidence interval. *P < 0.05 on a two-sided Student's t-test unless otherwise noted.
Supplementary Material
Funding
Funding for this project was derived from a National Ataxia Foundation grant to F.H. and an AAN CRTF fellowship, NIH R01NS08681001, NIH K08NS069809 and the Department of Veterans Affairs BLRD 1I21BX001841 to P.K.T.
Supplementary Material
Acknowledgements
We thank Peng Jin for generously sharing fly lines. Nico Dantuma originally generated the G76V-Ub GFP stable HeLa cell line. Paul Hagerman provided parent vectors used to derive mCherry constructs. We thank Nicolas Charlet and Chantal Sellier for helpful discussions. We thank Michael Sutton and the MBNI at Michigan for use of their confocal microscope.
Conflict of Interest statement: None declared.
References
- 1.Hagerman R.J., Leehey M., Heinrichs W., Tassone F., Wilson R., Hills J., Grigsby J., Gage B., Hagerman P.J. (2001) Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology, 57, 127–130. [DOI] [PubMed] [Google Scholar]
- 2.Jacquemont S., Hagerman R.J., Leehey M.A., Hall D.A., Levine R.A., Brunberg J.A., Zhang L., Jardini T., Gane L.W., Harris S.W., et al. (2004) Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA, 291, 460–469. [DOI] [PubMed] [Google Scholar]
- 3.Berry-Kravis E., Abrams L., Coffey S.M., Hall D.A., Greco C., Gane L.W., Grigsby J., Bourgeois J.A., Finucane B., Jacquemont S., et al. (2007) Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov. Disord., 22, 2018–2030, quiz 2140. [DOI] [PubMed] [Google Scholar]
- 4.Jacquemont S., Hagerman R.J., Leehey M., Grigsby J., Zhang L., Brunberg J.A., Greco C., Des Portes V., Jardini T., Levine R., et al. (2003) Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am. J. Hum. Genet., 72, 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pieretti M., Zhang F.P., Fu Y.H., Warren S.T., Oostra B.A., Caskey C.T., Nelson D.L. (1991) Absence of expression of the FMR-1 gene in fragile X syndrome. Cell, 66, 817–822. [DOI] [PubMed] [Google Scholar]
- 6.Kremer E.J., Pritchard M., Lynch M., Yu S., Holman K., Baker E., Warren S.T., Schlessinger D., Sutherland G.R., Richards R.I. (1991) Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science, 252, 1711–1714. [DOI] [PubMed] [Google Scholar]
- 7.Verkerk A.J., Pieretti M., Sutcliffe J.S., Fu Y.H., Kuhl D.P., Pizzuti A., Reiner O., Richards S., Victoria M.F., Zhang F.P., et al. (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell, 65, 905–914. [DOI] [PubMed] [Google Scholar]
- 8.Vincent A., Heitz D., Petit C., Kretz C., Oberle I., Mandel J.L. (1991) Abnormal pattern detected in fragile-X patients by pulsed-field gel electrophoresis. Nature, 349, 624–626. [DOI] [PubMed] [Google Scholar]
- 9.Yu S., Pritchard M., Kremer E., Lynch M., Nancarrow J., Baker E., Holman K., Mulley J., Warren S., Schlessinger D., et al. (1991) Fragile X genotype characterized by an unstable region of DNA. Science, 252, 1179–1181. [DOI] [PubMed] [Google Scholar]
- 10.Krueger D.D., Bear M.F. (2011) Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annu. Rev. Med., 62, 411–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tassone F., Hagerman R.J., Taylor A.K., Gane L.W., Godfrey T.E., Hagerman P.J. (2000) Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet., 66, 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tassone F., Beilina A., Carosi C., Albertosi S., Bagni C., Li L., Glover K., Bentley D., Hagerman P.J. (2007) Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA, 13, 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tassone F., Iwahashi C., Hagerman P.J. (2004) FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS). RNA Biol., 1, 103–105. [DOI] [PubMed] [Google Scholar]
- 14.Todd P.K., Oh S.Y., Krans A., Pandey U.B., Di Prospero N.A., Min K.T., Taylor J.P., Paulson H.L. (2010) Histone deacetylases suppress CGG repeat-induced neurodegeneration via transcriptional silencing in models of fragile X tremor ataxia syndrome. PLoS Genet., 6, e1001240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Usdin K., Hayward B.E., Kumari D., Lokanga R.A., Sciascia N., Zhao X.N. (2014) Repeat-mediated genetic and epigenetic changes at the FMR1 locus in the Fragile X-related disorders. Front. Genet., 5, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hagerman P. (2013) Fragile X-associated tremor/ataxia syndrome (FXTAS): pathology and mechanisms. Acta Neuropathol., 126, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin P., Zarnescu D.C., Zhang F., Pearson C.E., Lucchesi J.C., Moses K., Warren S.T. (2003) RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron, 39, 739–747. [DOI] [PubMed] [Google Scholar]
- 18.Jin P., Duan R., Qurashi A., Qin Y., Tian D., Rosser T.C., Liu H., Feng Y., Warren S.T. (2007) Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron, 55, 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sofola O.A., Jin P., Qin Y., Duan R., Liu H., de Haro M., Nelson D.L., Botas J. (2007) RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron, 55, 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sellier C., Usdin K., Pastori C., Peschansky V.J., Tassone F., Charlet-Berguerand N. (2014) The multiple molecular facets of fragile X-associated tremor/ataxia syndrome. J. Neurodev. Disord., 6, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sellier C., Rau F., Liu Y., Tassone F., Hukema R.K., Gattoni R., Schneider A., Richard S., Willemsen R., Elliott D.J., et al. (2010) Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J., 29, 1248–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thornton C.A. (2014) Myotonic dystrophy. Neurol. Clin., 32, 705–719, viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mohan A., Goodwin M., Swanson M.S. (2014) RNA-protein interactions in unstable microsatellite diseases. Brain. Res., 1584, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper T.A., Wan L., Dreyfuss G. (2009) RNA and disease. Cell, 136, 777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanadia R.N., Shin J., Yuan Y., Beattie S.G., Wheeler T.M., Thornton C.A., Swanson M.S. (2006) Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl Acad. Sci. USA, 103, 11748–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Haro M., Al-Ramahi I., De Gouyon B., Ukani L., Rosa A., Faustino N.A., Ashizawa T., Cooper T.A., Botas J. (2006) MBNL1 and CUGBP1 modify expanded CUG-induced toxicity in a Drosophila model of myotonic dystrophy type 1. Hum. Mol. Genet., 15, 2138–2145. [DOI] [PubMed] [Google Scholar]
- 27.Sellier C., Freyermuth F., Tabet R., Tran T., He F., Ruffenach F., Alunni V., Moine H., Thibault C., Page A., et al. (2013) Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep., 3, 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Todd P.K., Oh S.Y., Krans A., He F., Sellier C., Frazer M., Renoux A.J., Chen K.C., Scaglione K.M., Basrur V., et al. (2013) CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron, 78, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zu T., Gibbens B., Doty N.S., Gomes-Pereira M., Huguet A., Stone M.D., Margolis J., Peterson M., Markowski T.W., Ingram M.A., et al. (2010) Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl Acad. Sci. USA, 108, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ash P.E., Bieniek K.F., Gendron T.F., Caulfield T., Lin W.L., Dejesus-Hernandez M., van Blitterswijk M.M., Jansen-West K., Paul J.W., III, Rademakers R., et al. (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron, 77, 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mori K., Weng S.M., Arzberger T., May S., Rentzsch K., Kremmer E., Schmid B., Kretzschmar H.A., Cruts M., Van Broeckhoven C., et al. (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science, 339, 1335–1338. [DOI] [PubMed] [Google Scholar]
- 32.Mori K., Arzberger T., Grasser F.A., Gijselinck I., May S., Rentzsch K., Weng S.M., Schludi M.H., van der Zee J., Cruts M., et al. (2013) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol., 126, 881–893. [DOI] [PubMed] [Google Scholar]
- 33.Zu T., Liu Y., Banez-Coronel M., Reid T., Pletnikova O., Lewis J., Miller T.M., Harms M.B., Falchook A.E., Subramony S.H., et al. (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl Acad. Sci. USA, 110, E4968–E4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gendron T.F., Bieniek K.F., Zhang Y.J., Jansen-West K., Ash P.E., Caulfield T., Daughrity L., Dunmore J.H., Castanedes-Casey M., Chew J., et al. (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol., 126, 829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kearse M.G., Todd P.K. (2014) Repeat-associated non-AUG translation and its impact in neurodegenerative disease. Neurotherapeutics, 11, 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Todd P.K., Paulson H.L. (2010) RNA-mediated neurodegeneration in repeat expansion disorders. Ann. Neurol., 67, 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Handa V., Goldwater D., Stiles D., Cam M., Poy G., Kumari D., Usdin K. (2005) Long CGG-repeat tracts are toxic to human cells: implications for carriers of Fragile X premutation alleles. FEBS Lett., 579, 2702–2708. [DOI] [PubMed] [Google Scholar]
- 38.Iwahashi C.K., Yasui D.H., An H.J., Greco C.M., Tassone F., Nannen K., Babineau B., Lebrilla C.B., Hagerman R.J., Hagerman P.J. (2006) Protein composition of the intranuclear inclusions of FXTAS. Brain, 129, 256–271. [DOI] [PubMed] [Google Scholar]
- 39.Garcia-Arocena D., Yang J.E., Brouwer J.R., Tassone F., Iwahashi C., Berry-Kravis E.M., Goetz C.G., Sumis A.M., Zhou L., Nguyen D.V., et al. (2010) Fibroblast phenotype in male carriers of FMR1 premutation alleles. Hum. Mol. Genet., 19, 299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nedelsky N.B., Todd P.K., Taylor J.P. (2008) Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim. Biophys. Acta., 1782, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pandey U.B., Nie Z., Batlevi Y., McCray B.A., Ritson G.P., Nedelsky N.B., Schwartz S.L., DiProspero N.A., Knight M.A., Schuldiner O., et al. (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature, 447, 859–863. [DOI] [PubMed] [Google Scholar]
- 42.Pickford F., Masliah E., Britschgi M., Lucin K., Narasimhan R., Jaeger P.A., Small S., Spencer B., Rockenstein E., Levine B., et al. (2008) The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Invest., 118, 2190–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ross C.A., Pickart C.M. (2004) The ubiquitin-proteasome pathway in Parkinson's disease and other neurodegenerative diseases. Trends Cell. Biol., 14, 703–711. [DOI] [PubMed] [Google Scholar]
- 44.Sontag E.M., Vonk W.I., Frydman J. (2014) Sorting out the trash: the spatial nature of eukaryotic protein quality control. Curr. Opin. Cell. Biol., 26, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Warrick J.M., Chan H.Y., Gray-Board G.L., Chai Y., Paulson H.L., Bonini N.M. (1999) Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet., 23, 425–428. [DOI] [PubMed] [Google Scholar]
- 46.Auluck P.K., Chan H.Y., Trojanowski J.Q., Lee V.M., Bonini N.M. (2002) Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science, 295, 865–868. [DOI] [PubMed] [Google Scholar]
- 47.Chan H.Y., Warrick J.M., Andriola I., Merry D., Bonini N.M. (2002) Genetic modulation of polyglutamine toxicity by protein conjugation pathways in Drosophila. Hum. Mol. Genet., 11, 2895–2904. [DOI] [PubMed] [Google Scholar]
- 48.Chartier A., Benoit B., Simonelig M. (2006) A Drosophila model of oculopharyngeal muscular dystrophy reveals intrinsic toxicity of PABPN1. EMBO J., 25, 2253–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chan W.M., Tsoi H., Wu C.C., Wong C.H., Cheng T.C., Li H.Y., Lau K.F., Shaw P.C., Perrimon N., Chan H.Y. (2011) Expanded polyglutamine domain possesses nuclear export activity which modulates subcellular localization and toxicity of polyQ disease protein via exportin-1. Hum. Mol. Genet., 20, 1738–1750. [DOI] [PubMed] [Google Scholar]
- 50.Du G., Liu X., Chen X., Song M., Yan Y., Jiao R., Wang C.C. (2010) Drosophila histone deacetylase 6 protects dopaminergic neurons against {alpha}-synuclein toxicity by promoting inclusion formation. Mol. Biol. Cell, 21, 2128–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Estes P.S., Boehringer A., Zwick R., Tang J.E., Grigsby B., Zarnescu D.C. (2011) Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum. Mol. Genet., 20, 2308–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fernandez-Funez P., Nino-Rosales M.L., de Gouyon B., She W.C., Luchak J.M., Martinez P., Turiegano E., Benito J., Capovilla M., Skinner P.J., et al. (2000) Identification of genes that modify ataxin-1-induced neurodegeneration. Nature, 408, 101–106. [DOI] [PubMed] [Google Scholar]
- 53.Ghosh S., Feany M.B. (2004) Comparison of pathways controlling toxicity in the eye and brain in Drosophila models of human neurodegenerative diseases. Hum. Mol. Genet., 13, 2011–2018. [DOI] [PubMed] [Google Scholar]
- 54.Mallik M., Lakhotia S.C. (2010) Improved activities of CREB binding protein, heterogeneous nuclear ribonucleoproteins and proteasome following downregulation of noncoding hsromega transcripts help suppress poly(Q) pathogenesis in fly models. Genetics, 184, 927–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miguel L., Avequin T., Delarue M., Feuillette S., Frebourg T., Campion D., Lecourtois M. (2012) Accumulation of insoluble forms of FUS protein correlates with toxicity in Drosophila. Neurobiol. Aging, 33, 1008 e1001–1015. [DOI] [PubMed] [Google Scholar]
- 56.Shieh S.Y., Bonini N.M. (2011) Genes and pathways affected by CAG-repeat RNA-based toxicity in Drosophila. Hum. Mol. Genet., 20, 4810–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vo S.H., Butzlaff M., Pru S.K., Ni Charthaigh R.A., Karsten P., Lankes A., Hamm S., Simons M., Adryan B., Schulz J.B., et al. (2012) Large-scale screen for modifiers of ataxin-3-derived polyglutamine-induced toxicity in Drosophila. PLoS One, 7, e47452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shulman J.M., Feany M.B. (2003) Genetic modifiers of tauopathy in Drosophila. Genetics, 165, 1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Warrick J.M., Morabito L.M., Bilen J., Gordesky-Gold B., Faust L.Z., Paulson H.L., Bonini N.M. (2005) Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol. Cell, 18, 37–48. [DOI] [PubMed] [Google Scholar]
- 60.Ziviani E., Tao R.N., Whitworth A.J. (2010) Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl Acad. Sci. USA, 107, 5018–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Covi J.A., Belote J.M., Mykles D.L. (1999) Subunit compositions and catalytic properties of proteasomes from developmental temperature- sensitive mutants of Drosophila melanogaster. Arch. Biochem. Biophys., 368, 85–97. [DOI] [PubMed] [Google Scholar]
- 62.Lin Y., Tang C., He H., Duan R. (2013) Activation of mTOR ameliorates fragile X premutation rCGG repeat-mediated neurodegeneration. PLoS One, 8, e62572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berger Z., Ravikumar B., Menzies F.M., Oroz L.G., Underwood B.R., Pangalos M.N., Schmitt I., Wullner U., Evert B.O., O'Kane C.J., et al. (2006) Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet., 15, 433–442. [DOI] [PubMed] [Google Scholar]
- 64.Dantuma N.P., Lindsten K., Glas R., Jellne M., Masucci M.G. (2000) Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol., 18, 538–543. [DOI] [PubMed] [Google Scholar]
- 65.Arocena D.G., Iwahashi C.K., Won N., Beilina A., Ludwig A.L., Tassone F., Schwartz P.H., Hagerman P.J. (2005) Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum. Mol. Genet., 14, 3661–3671. [DOI] [PubMed] [Google Scholar]
- 66.Williams A.J., Paulson H.L. (2008) Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci., 31, 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cuervo A.M., Stefanis L., Fredenburg R., Lansbury P.T., Sulzer D. (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science, 305, 1292–1295. [DOI] [PubMed] [Google Scholar]
- 68.Ravikumar B., Duden R., Rubinsztein D.C. (2002) Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet., 11, 1107–1117. [DOI] [PubMed] [Google Scholar]
- 69.Ravikumar B., Stewart A., Kita H., Kato K., Duden R., Rubinsztein D.C. (2003) Raised intracellular glucose concentrations reduce aggregation and cell death caused by mutant huntingtin exon 1 by decreasing mTOR phosphorylation and inducing autophagy. Hum. Mol. Genet., 12, 985–994. [DOI] [PubMed] [Google Scholar]
- 70.Sarkar S., Davies J.E., Huang Z., Tunnacliffe A., Rubinsztein D.C. (2007) Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem., 282, 5641–5652. [DOI] [PubMed] [Google Scholar]
- 71.May S., Hornburg D., Schludi M.H., Arzberger T., Rentzsch K., Schwenk B.M., Grasser F.A., Mori K., Kremmer E., Banzhaf-Strathmann J., et al. (2014) C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol., 128, 485–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mizielinska S., Gronke S., Niccoli T., Ridler C.E., Clayton E.L., Devoy A., Moens T., Norona F.E., Woollacott I.O., Pietrzyk J., et al. (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science, 345, 1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Y.J., Jansen-West K., Xu Y.F., Gendron T.F., Bieniek K.F., Lin W.L., Sasaguri H., Caulfield T., Hubbard J., Daughrity L., et al. (2014) Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol., 128, 505–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kwon I., Xiang S., Kato M., Wu L., Theodoropoulos P., Wang T., Kim J., Yun J., Xie Y., McKnight S.L. (2014) Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science, 345, 1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Su Z., Zhang Y., Gendron T.F., Bauer P.O., Chew J., Yang W.Y., Fostvedt E., Jansen-West K., Belzil V.V., Desaro P., et al. (2014) Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron, 83, 1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.