

Abstract
Spinocerebellar ataxia type 6 (SCA6) belongs to the family of CAG/polyglutamine (polyQ)-dependent neurodegenerative disorders. SCA6 is caused by abnormal expansion in a CAG trinucleotide repeat within exon 47 of CACNA1A, a bicistronic gene that encodes α1A, a P/Q-type calcium channel subunit and a C-terminal protein, termed α1ACT. Expansion of the CAG/polyQ region of CACNA1A occurs within α1ACT and leads to ataxia. There are few animal models of SCA6. Here, we describe the generation and characterization of the first Drosophila melanogaster models of SCA6, which express the entire human α1ACT protein with a normal or expanded polyQ. The polyQ-expanded version of α1ACT recapitulates the progressively degenerative nature of SCA6 when expressed in various fly tissues and the presence of densely staining aggregates. Additional studies identify the co-chaperone DnaJ-1 as a potential therapeutic target for SCA6. Expression of DnaJ-1 potently suppresses α1ACT-dependent degeneration and lethality, concomitant with decreased aggregation and reduced nuclear localization of the pathogenic protein. Mutating the nuclear importer karyopherin α3 also leads to reduced toxicity from pathogenic α1ACT. Little is known about the steps leading to degeneration in SCA6 and the means to protect neurons in this disease are lacking. Invertebrate animal models of SCA6 can expand our understanding of molecular sequelae related to degeneration in this disorder and lead to the rapid identification of cellular components that can be targeted to treat it.
Introduction
Spinocerebellar ataxia type 6 (SCA6) is one of the nine currently incurable diseases that comprise the polyglutamine (polyQ) family of age-related neurodegenerative disorders, including Huntington's disease (HD), dentatorubral-pallidoluysian atrophy, spinobulbar muscular atrophy and six SCAs (1, 2, 3, 6, 7 and 17) (1,2). PolyQ diseases are rooted in anomalous CAG triplet repeat expansion in the genes that encode nine otherwise-unrelated proteins. SCA6 is a dominantly inherited ataxia that manifests later in life as a ‘pure’ gait ataxia accompanied by dysarthria, visual symptoms, episodic vertigo and tremors. The CAG repeat that is linked to SCA6 resides in the gene CACNA1A (3,4), which is bicistronic (5). The normal CAG repeat is between 4 and 18, whereas the SCA6-causing allele shows an expansion between 19 and 33 repeats (4). CACNA1A encodes two proteins: the α1A P/Q type calcium channel subunit and the transcription factor α1ACT. α1ACT contains the polyQ region (Fig. 1A). Recently, it was demonstrated that polyQ expansion within α1ACT is sufficient to cause SCA6-like degeneration in mice (5).
Figure 1.

New transgenic fly lines that express human α1ACT. (A) Diagrammatic representation of the α1A protein, including its C-terminal portion (α1ACT) where the polyQ region resides. α1ACT is expressed independently of α1A through an internal ribosomal entry site in the gene CACNA1A. For our new fly lines, we cloned the complete α1ACT into the pWalium expression vector. (B) Diagram of α1ACT with native start and stop codons that was cloned into pWalium10(moe) and inserted into the site attP2 on the third chromosome of Drosophila (Supplementary Material, Fig. S1) for Gal4-UAS-dependent expression. (C) Western blots from dissected fly heads homogenized in boiling SDS lysis buffer. Each lane represents an independent transformant line. Driver: gmr-Gal4 (eye-specific). The (no α1ACT) lane was homozygous for both gmr-Gal4 and the empty targeting vector inserted into attP2. All other flies were also homozygous for all transgenes. Asterisks: non-specific bands. (D) Subcellular fractionation of dissected fly heads expressing the noted versions of α1ACT, or its empty targeting vector (Ctrl), driven by gmr-Gal4. Flies were heterozygous for all transgenes. Dotted line indicates where the lower boundary of the expanded version of α1ACT would run in the cytoplasmic fraction. Oblong smear underneath the dotted line on the cytoplasmic portion of the membrane for Q70 is a blotting artifact.
As with all of the other polyQ diseases, SCA6 is incurable and symptomatic treatments are not very effective. There are presently few animal models for SCA6 (5–7), and none employs the potency and flexibility of Drosophila genetics. The fruit fly has proved to be a powerful tool to model various polyQ diseases and to identify potential therapeutic targets for them (8–11). We reasoned that fruit fly models of SCA6 that are based on the targeted expression of the complete, human α1ACT protein with a normal or expanded polyQ repeat would serve as versatile platforms for investigating mechanisms of neuroprotection for SCA6. Here, we describe the generation and biochemical, histological and physiological characterization of new Drosophila models of SCA6, and their use to identify suppressor genes.
Compared with the expression of α1ACT with a normal polyQ repeat, expression of a polyQ-expanded version of this protein causes developmental lethality when it is expressed throughout the organism, or reduced motility and longevity when it is expressed in neuronal, glial or muscle cells. Expression of a pathogenic version of α1ACT in fly eyes causes progressive degeneration and the presence of densely staining aggregates. We subsequently used a panel of candidate suppressors to demonstrate the utility of these new lines for the identification of potential therapeutic targets of SCA6. We found that the expression of the co-chaperone DnaJ-1, and mutation or knockdown of the nuclear importin karyopherin α3, suppress degeneration caused by pathogenic α1ACT without eliminating the toxic protein, but by reducing aggregation and nuclear localization. Our studies highlight the versatility of Drosophila as a useful model to identify and characterize therapeutic strategies for α1ACT-dependent degeneration.
Results
New fly models of SCA6
We generated new transgenic fly lines that express the entire human sequence of α1ACT through the Gal4-UAS system (12,13) (Fig. 1B and Supplementary Material, Fig. S1) using the phiC31 targeted integration system that allows unidirectional, site-specific, single construct integration (14). This method enables homogeneous expression among lines because constructs are integrated at precisely the same chromosomal site. In our case, this site is attP2 on the third chromosome. AttP2 allows high Gal4-induced expression with minimal background (15).
The α1ACT construct that we cloned into the targeting vector [pWalium10(moe)] comprises the complete mRNA sequence of human α1ACT with its native start and stop codons and without any exogenous tags. We were concerned that the introduction of non-native sequences could perturb the localization and handling of α1ACT in vivo. For the non-pathogenic, wild-type version, we utilized a polyQ of 11 repeats, whereas for the polyQ-expanded version we utilized a repeat of 70Q. We selected a hyper-expanded repeat for the pathogenic version, even though it is outside of what is observed in SCA6 patients. Our reasoning was that this line would likely prove severely and consistently pathogenic, and that it would enable us to identify strong suppressors of the disease. Studies conducted in mice have highlighted the utility of a hyper-expanded CAG repeat within CACNA1A as a suitable choice to model degeneration in animals (6,7).
As shown in Supplementary Material, Figure S1, we obtained several founders for both Q11 and Q70 transgenic lines, all of which were integrated into the attP2 site and in the correct orientation. In Figure 1C, we show that α1ACT with 11Q or 70Q is expressed in Drosophila at the expected molecular weight. These western blots were from dissected fly heads where α1ACT was expressed in the compound eye. As highlighted, with Q11 we observe a single prominent band, whereas with Q70 we observe a band as well as a smear in the upper portion of the gel, to which we refer as SDS-soluble and SDS-resistant species, respectively (Fig. 1C). We have described similar SDS-soluble and -resistant species with other polyQ peptides that have toxic properties in Drosophila (16). The homogenization protocol that we utilized for Figure 1C and in similar panels in later figures was chosen based on its ability to provide us with the most prominent SDS-soluble species (16). All of the studies described subsequently in this report were conducted with both the lines shown in Figure 1C for Q11 and Q70 versions, with similar results within both groups.
In mice, α1ACT is a transcriptional protein with a nuclear presence (5). Consequently, we examined whether exogenously expressed α1ACT localizes in nuclei in Drosophila. We expressed α1ACT in fly eyes and dissected intact fly heads soon after adults emerged from the pupal case. Subcellular fractionation showed that both the normal and polyQ-expanded α1ACT localize in nuclei (Fig. 1D). The pathogenic version of α1ACT is predominantly found in nuclear fractions rather than cytoplasmic ones, compared with α1ACT(Q11) (Fig. 1D). In some cases, we observe no α1ACT(Q70) in the cytoplasm (Fig. 1D), and in others we notice a signal from this species outside of the nucleus (Fig. 7C shows an example). Ultimately, both species can be found in the nucleus and the cytoplasm, and the polyQ-expanded version of α1ACT is predominantly in the nucleus.
Figure 7.

DnaJ-1 expression reduces aggregation and nuclear localization of α1ACT(Q70). (A) Western blots from dissected fly heads expressing the noted transgenes, driven by gmr-Gal4. Fifteen heads per group were homogenized in boiling SDS lysis buffer. Flies were heterozygous for all transgenes (UAS-α1ACT(Q70), gmr-Gal4, UAS-DnaJ-1) or mutations (kap-α3[D93]). Ctrl: isogenic control for modifiers. (B) Quantification of signal from blots on the left and other similar, independent experiments. Shown are means of SDS-soluble and SDS-resistant α1ACT(Q70) species ± standard deviations. Asterisks: P < 0.05 from ANOVA with Tukey's post-hoc correction comparing middle and right histograms to the one on the left of each graph. α1ACT signal was normalized to its respective tubulin loading control. After normalization, signal from α1ACT in the (Ctrl) lane in each individual experimental repeat was set to 100%. (C) Subcellular fractionation of dissected fly heads expressing α1ACT(Q70) in the absence or presence of UAS-DnaJ-1 or kap-α3[D93], driven by gmr-Gal4. Flies were heterozygous for driver, transgenes and/or mutations. Fifteen dissected fly heads were utilized per group. Ctrl: isogenic control for modifiers. (D) Quantification of signal from blots on the left and other independent experiments. Shown are means of nuclear and cytoplasmic α1ACT species ± standard deviations. Asterisks: P < 0.05 from ANOVA with Tukey's post-hoc correction comparing middle and right histograms to the one on the left of each graph. α1ACT signal was normalized to its respective loading control from anti-tubulin or anti-lamin blots. Normalized α1ACT signal from the (Ctrl) lane in each individual experimental repeat was set to 100%.
Expression of polyQ-expanded α1ACT causes toxicity in Drosophila
To investigate the toxicity of α1ACT in different cell types, we expressed α1ACT(Q11) and α1ACT(Q70) using a series of Gal4 drivers: ubiquitous, pan-neuronal, muscle cell-specific and glial cell-specific (17). As summarized in Figure 2A, widespread expression of α1ACT(Q70) is lethal in late pharate stages and during the eclosion of adults from the pupal case; no adults eclose successfully.
Figure 2.

Effect of α1ACT expression in various fly tissues. (A) Summary of effects observed when α1ACT with Q11 or Q70 is expressed in the noted tissues. Flies were heterozygous for all transgenes. Controls consisted of the noted Gal4 driver in trans with the empty targeting vector inserted into attP2. (B, D and F) Longevity data from at least 200 flies per group. Flies were heterozygous for all transgenes. (C and E) Negative geotaxis results from 50 or more flies per group, from experiments repeated at least three times. Flies were heterozygous for all transgenes. P-values are from ANOVA with Tukey's post-hoc correction comparing no α1ACT empty vector controls with α1ACT-expressing lines. Shown are means ± standard deviations.
Expression of α1ACT(Q70) pan-neuronally leads to earlier adult lethality (Fig. 2A and B) and progressively reduced motility (Fig. 2C). Similarly, expression of α1ACT(Q70) in glial cells leads to earlier adult death and progressive loss of climbing ability (Fig. 2A, D and E). Finally, when α1ACT(Q70) is expressed only in muscle cells, adults die soon after eclosion (Fig. 2A and F).
Overall, expression of α1ACT(Q11) ubiquitously or in the select tissues mentioned above does not lead to deleterious effects, compared with fly lines that contain the targeting expression vector (pWalium), but without including α1ACT (Fig. 2). Based on these collective results, we conclude that α1ACT(Q70) is progressively toxic in Drosophila.
Retinal degeneration caused by α1ACT
Drosophila eyes have been used extensively to model neurodegeneration caused by various toxic proteins (11,18–23) and to identify genes that modify toxicity from polyQ diseases such as HD, SCA1 and SCA3 (8,18,24,25). As summarized in Figure 3A, expression of either form of α1ACT does not cause clear anomalies in the external portion of the retina early on in adults. By 4 weeks of age, we notice very light pigmentary aberration of the external retina when α1ACT(Q70) is expressed. Dyspigmentation is sufficiently mild that it is difficult to image; a representative example is shown in the bottom row of Figure 3A.
Figure 3.

Effect of α1ACT expression in fly eyes. External retinal photos (A) and histological sections (B) of fly eyes expressing the empty targeting vector (Ctrl) or α1ACT in fly eyes, driven by gmr-Gal4. Flies were heterozygous for all transgenes. In (B), red boxes highlight some densely staining punctate structures, white bracketed lines indicate retinal depth, empty arrowheads highlight separation of retinal structures and red bracketed line demarcates ommatidial boundaries.
Conversely, histological sections of adult retinae show degeneration of the ommatidial array as flies age into 4 weeks, highlighted by separation of retinal structures (arrowheads in Fig. 3B), the disappearance of the ommatidial boundaries in the presence of α1ACT(Q70) (red bracketed line in Fig. 3B) and, in some cases, a restriction of the retinal depth (white bracketed lines, Fig. 3B). Also present with age are densely staining punctate structures when α1ACT(Q70) is expressed in fly eyes (red boxes in Fig. 3B, and in Fig. 6C and D). Aggregates have been observed in SCA6 patients and in mouse models of this disease (7,26,27). Because our α1ACT antibody reagents are only useful for immunoblotting, and not for immunohistochemistry, we do not know the precise subcellular localization of these densely staining structures in the adult retina. Qualitatively, expression of α1ACT(Q11) does not lead to clear anomalies in the external or internal portions of the retina, compared with controls that do not express α1ACT (Fig. 3).
Figure 6.

DnaJ-1 and kap-α3 modulate degeneration caused by α1ACT(Q70). (A) Membrane-targeted GFP fluorescence photos from flies of the specified ages, expressing α1ACT(Q70), driven by gmr-Gal4. Ctrl: isogenic control for modifiers. Flies were heterozygous for all transgenes or mutations. All photos were taken using the same exposure and collection parameters. α1ACT(Q70), DnaJ-1, ataxin-3 and RNAi constructs are UAS-based. kap-α3[D93] is a loss of function allele. (B) Quantification of data from (A) and other images. Asterisks: P < 0.05 from ANOVA with Tukey's post-hoc correction comparing the α1ACT(Q70) + Ctrl group to others. Shown are means ± standard deviations. (C and D) Histological sections from adult fly retinae. Flies were heterozygous for transgenes (UAS-α1ACT(Q70), gmr-Gal4, UAS-DnaJ-1, UAS-DnaJ-1-RNAi), or mutations (kap-α3[D93]). Ctrl: isogenic control for modifiers. Red boxes: some of the densely staining punctate structures; red bracketed lines: ommatidial boundaries; open arrowheads: separation of retinal structures.
We recently reported a rapid and highly sensitive method for identifying modulators of degeneration in Drosophila eyes in vivo (28). We reasoned that this platform, which is GFP-based and reports eye integrity reliably and quantitatively, could be utilized to identify suppressors of α1ACT-dependent degeneration as potential therapeutic targets for SCA6. Consequently, we assayed retinal degeneration in α1ACT-expressing eyes by examining the fluorescence from membrane-targeted CD8-GFP. Healthy fly eyes show uniform GFP fluorescence, whereas diseased eyes show progressive loss of GFP fluorescence and increased mosaicism as retinal cells degenerate (Fig. 4A (Ctrl)) (28).
Figure 4.

Progressive loss of GFP fluorescence when α1ACT is expressed in fly eyes. (A) Membrane-targeted GFP fluorescence photos from flies of the specified ages expressing, or not, α1ACT driven by gmr-Gal4. Ctrl: gmr-Gal4 in trans to empty targeting vector inserted into attP2. Flies were heterozygous for all transgenes (including gmr-Gal4, UAS-α1ACT, UAS-CD8-GFP, empty targeting vector control). All photos were taken using the same exposure and collection parameters, which were chosen to enable viewing of minimal fluorescence [e.g. 14-day α1ACT(Q70)] as well as maximal signal (e.g. Ctrl) while avoiding saturation. (B) Quantification of data from (A) and other images. Asterisks: P < 0.05, from ANOVA with Tukey's post-hoc correction comparing no α1ACT empty vector controls with α1ACT-expressing lines. Shown are means ± standard deviations.
As shown in photos and quantitative measurements in Figure 4A and B, expression of both α1ACT(Q11) and α1ACT(Q70) leads to a reduction of mean GFP fluorescence, a phenomenon that is particularly noticeable and progressive for α1ACT(Q70). With the expanded version of α1ACT, GFP fluorescence is barely above background by 2 weeks of age. It is somewhat surprising that expression of α1ACT(Q11) also leads to some loss of GFP fluorescence, especially since we did not notice anomalies from this form of α1ACT when it was expressed in other tissues or in histological sections from fly eyes. However, the GFP-based method is more sensitive than retinal histology (28). In other models of polyQ SCAs, overexpression of the wild-type form of various SCA proteins has led to toxicity (9,29). Our results that α1ACT(Q11) is also mildly toxic to fly eyes should serve as a reminder of its importance to normal physiological processes, and suggest that α1ACT-dependent toxicity may stem, at least in part, from gain-of-function processes, as observed with other polyQ SCAs (1,2).
Collectively, the data in Figures 3 and 4 demonstrate toxicity caused by α1ACT in fly eyes and highlight the use of membrane-targeted GFP as a potential investigative tool to identify suppressors of α1ACT-dependent phenotypes.
α1ACT(Q11) does not suppress toxicity from the Q70 version
We examined whether wild-type α1ACT modulates toxicity caused by the expanded counterpart. We reasoned that if α1ACT(Q70) is toxic due to a loss-of-function effect in the fruit fly, expression of the Q11 form of this protein may alleviate toxicity. Co-expression of α1ACT(Q11) together with Q70 does not affect the stage at which the polyQ-expanded allele causes lethality; whether Q11 was co-expressed or not, expression of α1ACT(Q70) throughout developing flies is lethal at pharate stages and during eclosion from the pupal case (Supplementary Material, Fig. S2A). Similarly, based on the membrane-bound GFP reporter, α1ACT(Q11) does not appear to affect toxicity by the Q70 version in fly eyes (Fig. 5). Together, these data suggest a toxic gain-of-function aspect for α1ACT(Q70)-related degeneration in Drosophila.
Figure 5.
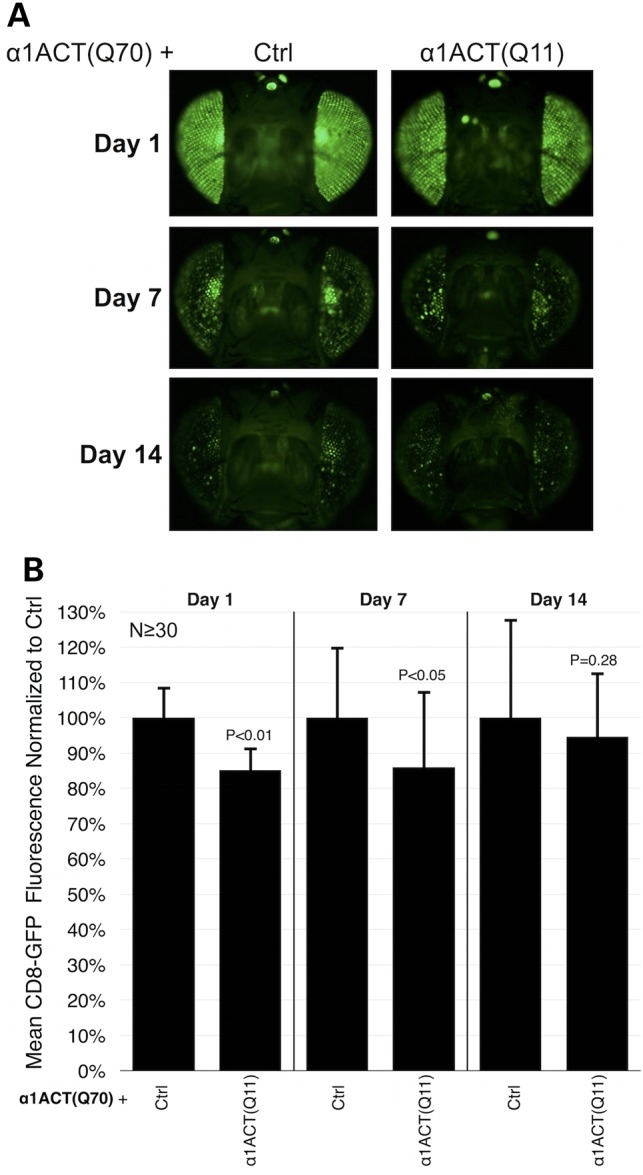
α1ACT(Q70)-dependent toxicity is not suppressed by co-expressing α1ACT(Q11). (A) GFP fluorescence photos of fly eyes expressing α1ACT(Q70) without or with α1ACT(Q11), driven by gmr-Gal4. Flies were heterozygous for all transgenes. Ctrl: isogenic control for α1ACT(Q11). (B) Quantification of data from (A) and other images. Overall means of fluorescence readings from (Q70 + Q11) flies were normalized to readings from the (Q70 + Ctrl) group in each day cohort. P-values are based on Student's T-tests with two tails comparing (Q70 + Q11) flies to (Q70 + Ctrl) flies in each day cohort. Shown are means -/+ standard deviations.
DnaJ-1 and karyopherin α3 suppress α1ACT-dependent toxicity
A primary goal for the generation of the α1ACT transgenic lines is their utilization to isolate modulators of SCA6 in vivo. Consequently, we examined the ability of specific proteins to suppress α1ACT(Q70)-dependent loss of GFP fluorescence in fly eyes. We chose the following proteins: DnaJ-1, which is a co-chaperone of the HSP40 family, shown to suppress polyQ-dependent toxicity in an HD model in Drosophila (18); ataxin-3, a polyQ protein (SCA3) and deubiquitinase that suppresses toxicity in fly models of HD, SCA1 and SCA3 (16,28,30); karyopherin α3 (kap-α3), a protein involved in nuclear shuttling, which we selected based on previous work showing that nuclear localization of α1ACT is toxic in mammalian cell culture (29,31); and the deubiquitinases, trbd and the fly orthologue to OTUB1 (CG4968 in flies) (17), which suppress toxicity from expression of a nearly pure polyQ tract in flies (Tsou and Todi, unpublished data).
Results are summarized in photos and quantitative data in Figure 6. We find that expression of DnaJ-1, a mutation in kap-α3 or RNAi targeting its gene have a marked suppressive effect on α1ACT(Q70) throughout the period of observations (Fig. 6A and B). Whereas membrane-targeted GFP fluorescence is lost when α1ACT(Q70) is present in fly eyes, co-expression of DnaJ-1, or a mutation or RNAi targeting kap-α3, significantly suppresses this phenotype. Additionally, as shown in Figure 6C, co-expression of DnaJ-1, or a mutation in kap-α3, together with α1ACT(Q70) leads to improved ommatidial morphology in histological sections: the boundaries among retinal units are clear and the separation of the ommatidial array from the underlying layer is no longer present. The importance of DnaJ-1 in protecting against retinal degeneration caused by α1ACT(Q70) is also supported by a substantial increase in toxicity from the polyQ-expanded version of this protein when DnaJ-1 is knocked down through RNAi (Fig. 6D). Knockdown of DnaJ-1 leads to marked α1ACT(Q70)-dependent ommatidial degeneration and retinal structure separation in adults as young as 2 weeks old—a time point when we do not observe clear structural anomalies by histology—when compared with α1ACT(Q70) expression by itself (Figs 3B and 6D).
We then examined whether these two suppressors can also protect against lethality caused by α1ACT(Q70) when it is expressed in all fly tissues. Expression of DnaJ-1 is even able to suppress lethality in the new model of SCA6, whereas mutating kap-α3, even though it has a protective role in fly eyes, is unable to rescue lethality (Supplementary Material, Fig. S2B and C).
Some of the other potential modifiers have suppressive effects, or exacerbate α1ACT(Q70)-dependent loss of GFP fluorescence (Fig. 6A and B). Knockdown of the fly orthologue of OTUB1 (CG4968) by RNAi leads to increased GFP signal. Knockdown of trbd seems to have a suppressive role on Day 7, but this effect is no longer statistically significant by 14 days (Fig. 6A and B). Expression of ataxin-3, which has been shown to reduce retinal toxicity caused by other polyQ proteins (16,30) does not seem to repress the decline of GFP signal in the presence of α1ACT(Q70). Indeed, exogenous expression of this deubiquitinase appears to exacerbate the phenotype (Fig. 6A and B). This finding suggests that the new SCA6 model recapitulates aspects of this disease that are specific to α1ACT and that are not necessarily shared with other polyQ proteins.
Modifiers reduce α1ACT(Q70) nuclear localization in fly eyes
Based on the suppressive effect of DnaJ-1 and kap-α3 against α1ACT(Q70)-dependent toxicity, we sought to gain some molecular insight into the protective role of these two proteins. We first examined whether the co-chaperone or the importin affect overall protein levels of α1ACT(Q70). As shown in Figure 7A, co-expression of DnaJ-1 together with α1ACT(Q70) does not eliminate the protein. Instead, we notice a larger amount of SDS-soluble α1ACT(Q70) in the presence of the co-chaperone, concomitant with a reduction of the SDS-resistant portion of the SCA6 protein (Fig. 7B). When α1ACT(Q70) is expressed in the background of mutated kap-α3, we do not observe significant differences in SDS-soluble or -resistant species of α1ACT(Q70) (Fig. 7A and B).
We next investigated the effect of DnaJ-1 and kap-α3 on the subcellular distribution of the toxic protein. We conducted nuclear/cytoplasmic fractionation of dissected fly heads and examined the sub-compartmentalization of α1ACT(Q70) when it is expressed selectively in fly eyes. We noticed a marked increase in the cytoplasmic fraction of the SCA6 protein when DnaJ-1 is present, accompanied by a reduction of the nuclear fraction, compared with the control group (Fig. 7C and D). Mutating kap-α3 has a milder, though still statistically significant, effect on the nuclear and cytoplasmic distribution of this polyQ protein (Fig. 7C and D). Collectively, these data indicate that DnaJ-1 suppresses degeneration caused by α1ACT(Q70) not by eliminating this protein, but by decreasing its aggregating propensity and by limiting its nuclear localization; they also suggest kap-α3 as a potential nuclear shuttling factor for the SCA6 protein.
Discussion
Several animal models of SCA6 have been generated and have provided critical insight into the biology of this disease (5–7). We add to this collection a Drosophila system with which to rapidly investigate potential therapeutic options for this neurodegenerative disorder. We report the generation of a fly model of SCA6 that utilizes tissue-specific expression of the disease protein α1ACT with a wild-type or expanded polyQ repeat, and the discovery of suppressors of α1ACT-dependent toxicity, which provides critical clues into protective pathways that can be utilized for SCA6 therapy. We find that widespread expression of polyQ-expanded α1ACT throughout the fly or in select neuronal and non-neuronal tissues is lethal during development, or causes reduced motility and premature death in adults. The phenotypes that we observed are consistently progressive. We also find a degenerative phenotype when α1ACT(Q70) is expressed in fly eyes, characterized by retinal disruption and the presence of densely staining structures that are reminiscent of aggregates reported in SCA6 post-mortem tissue (26,27,32).
Through a membrane-targeted GFP reporter, we identified genes/proteins with a suppressive effect on α1ACT(Q70). Especially notable is the co-chaperone DnaJ-1, which markedly protects against α1ACT(Q70) when this protein is expressed only in eyes, or throughout the fly. DnaJ-1 is a member of the family of J/HSP40 proteins that provide specificity for HSP70/Hsc70 chaperones (33,34). DnaJ-1 has been previously reported to suppress toxicity caused by a polyQ-expanded fragment of the HD protein, huntingtin, in fly eyes (18). We confirmed this same protective effect from DnaJ-1 on degeneration caused by a generic model of polyQ diseases in Drosophila eyes that express an isolated polyQ tract of 78 repeats (Tsou and Todi, manuscript in preparation). As in those other fly models of polyQ diseases, expression of DnaJ-1 leads to strong suppression of the phenotype caused by α1ACT(Q70). Repressed toxicity from the pathogenic version of the SCA6 protein is accompanied by reduced SDS-resistant species based on western blots, absence of aggregated structures in fly eyes based on histological sections and by increased cytoplasmic localization of the toxic polyQ protein according to fractionation protocols. These findings suggest that DnaJ-1 protects from α1ACT(Q70)-dependent degeneration by decreasing its aggregation, preventing its nuclear accumulation, or both. Collectively, these data highlight DnaJ-1 and its human paralogs (DNAJB1, B4 and B5, based on BLASTp results) as potential therapeutic options for SCA6. The precise molecular mechanism by which DnaJ-1 protects against α1ACT(Q70)-dependent toxicity in flies, the chaperone(s) with which it functions, and how this protein folding component prevents the nuclear localization and aggregation of α1ACT(Q70) remain to be investigated.
Another suppressor was kap-α3, a nuclear shuttle protein. Mutating the gene that encodes this protein suppresses degeneration caused by α1ACT(Q70) in fly eyes, although it does not have a noticeable effect on lethality when the SCA6 protein is expressed throughout the organism. This variation in the protective role from mutating kap-α3 could be due to insufficient inhibition of the nuclear localization of α1ACT(Q70) when the toxic protein is expressed everywhere. The finding that reduced nuclear localization of pathogenic α1ACT ameliorates toxicity is perhaps not entirely surprising. Restriction of nuclear localization of polyQ proteins leads to significantly milder toxicity in cultured cells and in mouse models of polyQ diseases, including SCA6, whereas nuclear localization can be strongly toxic (29,31,35–37). It deserves mention that a polyQ-expanded C-terminal fragment of the α1A protein that localized to the cytoplasm was found to be toxic in cultured HEK-293 and PC12 cells (38). We could surmise that benefits from subcellular restriction of the SCA6 protein are context- or cell-type-specific, thus leading to reduced degeneration of retinal cells in the background of kap-α3 mutation, but not sufficiently helpful when α1ACT(Q70) is expressed in other tissues in the same background. The locations where the SCA6 protein exerts its pathogenic effect are controversial. Two studies detected the C-terminal portion of α1A in the nucleus and the cytoplasm of neuronal cells (32,39), and another observed toxicity with this protein localized to lysosomes in vivo (7). Our findings support the notion of nuclear-related toxicity from α1ACT, open the possibility that kap-α3 is directly involved with the nuclear shuttling of this protein, and suggest the potential of this transposase as a therapeutic target for SCA6.
As assessed by membrane-targeted GFP, a previously reported suppressor of polyQ toxicity in Drosophila, ataxin-3, does not appear to protect against α1ACT(Q70) in fly eyes. This deubiquitinase protected against degeneration caused by an essentially generic form of polyQ diseases, consisting largely of a Q78 repeat (20), as well as polyQ-expanded versions of huntingtin (HD) and ataxin-1 (SCA1) (16,30). Since ataxin-3 did not prevent the progressive loss of GFP fluorescence caused by α1ACT(Q70) in the present study, we reason that toxicity caused by the SCA6 protein occurs through mechanisms that are at least somewhat different from those in other polyQ models mentioned earlier.
Our results also offer some clues into α1ACT(Q70)-dependent degeneration. Co-expression of α1ACT(Q11) together with α1ACT(Q70) does not rescue pharate lethality from the polyQ-expanded version when the transgenes are expressed throughout the fly, or the loss of GFP fluorescence when they are co-expressed in fly eyes. These findings argue against loss-of-function and support the notion of a toxic-gain-of function mechanism for α1ACT-related toxicity in Drosophila. Such an interpretation is consistent with results from cell and mouse models of SCA6 (5), and has also been ascribed to other polyQ disorders (1,2,40–42).
With our most sensitive reporter of retinal integrity, membrane-targeted GFP, we observed some toxicity in fly eyes when the normal allele of α1ACT was expressed in this organ. When present in other tissues, the wild-type allele of α1ACT does not cause detectable anomalies, based on longevity and motility; it is possible that these latter assays are not as sensitive reporters of tissue health as membrane-targeted GFP. This discrepancy regarding a potentially toxic effect from α1ACT(Q11) in different Drosophila tissues highlights the sensitivity of the GFP-based technique, and might also be due to stronger expression of α1ACT in fly eyes, driven by gmr-Gal4, compared with its expression by other Gal4 drivers. As also observed with wild-type alleles in other polyQ SCA models (9,29), overexpression of α1ACT(Q11) could have some potentially toxic effects and may be of use in the future as yet another means through which to investigate the molecular basis of SCA6, especially in relation to loss- or gain-of-function mechanisms.
The work that we described here establishes Drosophila as a flexible and sensitive organism with which to study degeneration caused by α1ACT, with a particular focus on mechanisms of protection against this disease protein. Our findings highlight two effective suppressors of α1ACT-dependent degeneration and suggest cytoplasmic retention of the disease protein as a potential route for SCA6 therapy. Drosophila does not contain a structure that resembles the cerebellum, the major target of SCA6. Thus, the utility of this organism in investigating the precise steps of SCA6 etiology is somewhat restricted to shared molecular components and cellular pathways between this invertebrate and mammals. Nonetheless, as shown by numerous studies (24,43,44), this model organism can provide invaluable insight into basic cellular pathways important for neurodegenerative disorders, and can identify potential therapeutic targets for the treatment of these diseases in humans.
Materials and Methods
Antibodies
Mouse, monoclonal anti-Tubulin (1:10 000; Sigma-Aldrich); mouse, monoclonal anti-Lamin (ADL84.12; 1:200; Developmental Hybridoma Bank, Iowa City, IA, USA); rabbit, polyclonal anti-α1A(1:500; described before) (5); rabbit, monoclonal anti-α1A ab181371 (1:1000; AbCam); 1:5000 peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch).
Fly stocks and cloning and generation of new Drosophila transgenics
Commonly used stocks were purchased from Bloomington Drosophila Stock Center (BDSC). Wild-type ataxin-3-expressing line was described before (16), and sqh-Gal4 was also described before (45–47). UAS-DnaJ-1 [w[*]; P{w[+mC] = UAS-DnaJ-1.K}3] is stock #30533 from BDSC, karyopherin mutation [w[*]; Kap-alpha3[D93]/TM6B, Tb[1]] is stock #25397 from BDSC, RNAi to karyopherin [y[1] v[1]; P{y[+t7.7] v[+t1.8] = TRiP.JF02686}attP2] is stock number 27535 from BDSC, RNAi to OTUB1 (CG4968) [w[1118]; P{GD11489}v21978] is stock number 21978 from Vienna Drosophila RNAi Center (VDRC); RNAi to trbd [w[1118]; P{GD14218}v24030], is stock number 24030 from VDRC. Drosophila husbandry was conducted at 25°C and ∼60% humidity in regulated diurnal environments. A complete list of fly lines used in this report and their provenance is included in Supplementary Material, Table S1.
To generate transgenic flies that express α1ACT through the Gal4-UAS system, DNA sequences encompassing exon 40 to the 3′ end of the human CACNA1A gene were PCR-amplified with the forward primer: 5′-ATGATCATGGAGTACTACCGGCAGA-3′ and the reverse primer: 5′-TTAGCACCAATCATCGTCACTCTCG-3′, and inserted into the EcoRI/BglII site of the pWalium10-moe plasmid (generated by the Perrimon lab, procured from the DNA Resource Core at Harvard Medical School). Injections of pWalium-α1ACT constructs and empty pWalium were conducted by the Duke University Model System Injection Service into the y, w1118; +; attP2 line. For transgene insertion verification, we extracted genomic DNA from different founder lines and performed PCR using primers white-end-F: 5′-TTCAATGATATCCAGTGCAGTAAAA-3′ and A2-3L-R: 5′-CTCTTTGCAAGGCATTACATCTG-3′. All lines used here were placed into w1118 background.
Nuclear/cytoplasmic isolation, lysis protocols and western blotting
Proteins were extracted using the ReadyPrep Protein Extraction Kit (Bio-Rad) per the manufacturer's protocol. Fifteen dissected adult Drosophila heads per group were homogenized in 250 µl of cytoplasmic protein extraction buffer and incubated on ice for 2 min. After a quick spin, supernatant was transferred to a clean tube and centrifuged at 1000g for 10 min at 4°C. The supernatant containing cytoplasmic proteins was transferred and the nuclear pellet was washed thrice with 100 µl cytoplasmic protein extraction buffer. The nuclear pellet was then resuspended in 100 µl complete protein solubilization buffer and incubated on ice for 5 min. Protein loading buffer was added to each sample, boiled, and loaded onto SDS–PAGE gels. For western blotting of whole head lysates, 15 fly heads per group were homogenized in boiling SDS lysis buffer (50 mm Tris pH 6.8, 2% SDS, 10% glycerol, 100 mm dithiothreitol). The choice of this specific buffer was determined in an earlier report, where we established that this particular protocol provided us with the highest quantity of SDS-soluble and SDS-resistant species of polyQ species (16). Heads were mechanically disrupted by using a pestle and then sonicated for 15 s, boiled for 10 min, centrifuged at top speed at room temperature for 10 min, and loaded onto SDS–PAGE gels. Western blots were developed using a CCD-equipped VersaDoc 5000MP system (Bio-Rad) or PXi (Syngene), as described previously (16,48).
Longevity and motility assays
A total of 200–400 adults were collected and aged in conventional cornmeal fly media. Flies were transferred to fresh vials every 3–4 days, until the day when all adults died. For motility assays, 10 adults per vial were tapped on the bench and the number of adults that reached the top was recorded at 15 and at 30 s. Flies that reached the top and ventured downwards to subsequently climb upwards again were counted only once.
Fluorescence measurements
All membrane-targeted GFP (UAS-CD8-GFP) fluorescence images were taken using an Olympus BX53 microscope and CellSens software. The same camera and image settings were used for all images in each specific experiment. Image capture, quantification and statistical analyses were conducted by separate investigators. The overall mean of the (Ctrl) groups in each figure panel was set to 100%. Image capture settings for each collective figure panel were set to show high and low intensities without reaching saturation.
Histology
Adult flies, whose proboscises and wings were removed, were fixed overnight in 2% glutaraldehyde/2% paraformaldehyde in Tris-buffered saline with 0.1% Triton X-100. Fixed flies were then dehydrated in a series of 30, 50, 75 and 100% ethanol and propylene oxide, embedded in Poly/Bed812 (Polysciences) and sectioned at 5 µm. Sections were stained with toluidine blue.
Quantitative RT–PCR
Total RNA was extracted from pupae using TRIzol reagent (Life Technologies). Extracted RNA was treated with TURBO DNAse (Ambion) to eliminate contaminating DNA. Reverse transcription was performed with the High-Capacity cDNA Reverse Transcription Kit (ABI). Messenger RNA levels were quantified by using the StepOnePlus Real-Time PCR System with Fast SYBR Green Master Mix (ABI). rp49 was used as an internal control. Primers:
α1ACT-F: 5′-CTAACTCTCAGTCCGTGGAGATG-3′,
α1ACT-R: 5′-GTCTGAGATGGTACTGAGGTTATTCC-3′,
rp49-F: 5′-AGATCGTGAAGAAGCGCACCAAG-3′,
rp49-R: 5′-CACCAGGAACTTCTTGAATCCGG-3′.
Statistical analyses
Student's T-test and ANOVA with Tukey's post-hoc correction were used to assess the statistical significance of the results shown in this work. ANOVA was used for Figures 2C and E, 4B, 6B, 7B and D, comparing the various experimental groups to their specific internal controls. Two-tailed Student's T-tests were used for Figure 5B comparing (Q70 + Q11) groups to their day-specific (Q70 + ctrl) ones.
Supplementary Material
Funding
This work was funded by R01NS33202 to C.M.G. from NINDS and by R01NS086778 to S.V.T. from NINDS.
Supplementary Material
Acknowledgements
We thank the Bloomington Drosophila Stock Center (funded by the National Institutes of Health grant P40OD018537), the Vienna Drosophila RNAi Center and the TRiP at Harvard Medical School (funded by National Institutes of Health grant R01GM084947) for providing transgenic fly stocks and plasmid vectors used in this study. We also thank Ms. Candace Savonen for her assistance with statistical work.
Conflict of Interest statement. None declared.
References
- 1.Orr H.T., Zoghbi H.Y. (2007) Trinucleotide repeat disorders. Annu. Rev. Neurosci., 30, 575–621. [DOI] [PubMed] [Google Scholar]
- 2.Todi S.V., Williams A., Paulson H. (2007) In Waxman S.G. (ed.), Molecular Neurology. Academic Press, London, pp. 257–276. [Google Scholar]
- 3.Matsuyama Z., Kawakami H., Maruyama H., Izumi Y., Komure O., Udaka F., Kameyama M., Nishio T., Kuroda Y., Nishimura M., et al. (1997) Molecular features of the CAG repeats of spinocerebellar ataxia 6 (SCA6). Hum. Mol. Genet., 6, 1283–1287. [DOI] [PubMed] [Google Scholar]
- 4.Zhuchenko O., Bailey J., Bonnen P., Ashizawa T., Stockton D.W., Amos C., Dobyns W.B., Subramony S.H., Zoghbi H.Y., Lee C.C. (1997) Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet., 15, 62–69. [DOI] [PubMed] [Google Scholar]
- 5.Du X., Wang J., Zhu H., Rinaldo L., Lamar K.M., Palmenberg A.C., Hansel C., Gomez C.M. (2013) Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell, 154, 118–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watase K., Barrett C.F., Miyazaki T., Ishiguro T., Ishikawa K., Hu Y., Unno T., Sun Y., Kasai S., Watanabe M., et al. (2008) Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels. Proc. Natl Acad. Sci. USA, 105, 11987–11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Unno T., Wakamori M., Koike M., Uchiyama Y., Ishikawa K., Kubota H., Yoshida T., Sasakawa H., Peters C., Mizusawa H., et al. (2012) Development of Purkinje cell degeneration in a knockin mouse model reveals lysosomal involvement in the pathogenesis of SCA6. Proc. Natl Acad. Sci. USA, 109, 17693–17698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park J., Al-Ramahi I., Tan Q., Mollema N., Diaz-Garcia J.R., Gallego-Flores T., Lu H.C., Lagalwar S., Duvick L., Kang H., et al. (2013) RAS-MAPK-MSK1 pathway modulates ataxin 1 protein levels and toxicity in SCA1. Nature, 498, 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez-Funez P., Nino-Rosales M.L., de Gouyon B., She W.C., Luchak J.M., Martinez P., Turiegano E., Benito J., Capovilla M., Skinner P.J., et al. (2000) Identification of genes that modify ataxin-1-induced neurodegeneration. Nature, 408, 101–106. [DOI] [PubMed] [Google Scholar]
- 10.Romero E., Cha G.H., Verstreken P., Ly C.V., Hughes R.E., Bellen H.J., Botas J. (2008) Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron, 57, 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pandey U.B., Nie Z., Batlevi Y., McCray B.A., Ritson G.P., Nedelsky N.B., Schwartz S.L., DiProspero N.A., Knight M.A., Schuldiner O., et al. (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature, 447, 859–863. [DOI] [PubMed] [Google Scholar]
- 12.Brand A.H., Manoukian A.S., Perrimon N. (1994) Ectopic expression in Drosophila. Methods Cell Biol., 44, 635–654. [DOI] [PubMed] [Google Scholar]
- 13.Brand A.H., Perrimon N. (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development, 118, 401–415. [DOI] [PubMed] [Google Scholar]
- 14.Groth A.C., Fish M., Nusse R., Calos M.P. (2004) Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics, 166, 1775–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Markstein M., Pitsouli C., Villalta C., Celniker S.E., Perrimon N. (2008) Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat. Genet., 40, 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsou W.L., Burr A.A., Ouyang M., Blount J.R., Scaglione K.M., Todi S.V. (2013) Ubiquitination regulates the neuroprotective function of the deubiquitinase ataxin-3 in vivo. J. Biol. Chem., 288, 34460–34469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsou W.-L., Sheedlo M.J., Morrow M.E., Blount J.R., McGregor K.M., Das C., Todi S.V. (2012) Systematic analysis of the physiological importance of deubiquitinating enzymes. PLoS One, 7, e43112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kazemi-Esfarjani P., Benzer S. (2000) Genetic suppression of polyglutamine toxicity in Drosophila. Science, 287, 1837–1840. [DOI] [PubMed] [Google Scholar]
- 19.Jackson G.R., Salecker I., Dong X., Yao X., Arnheim N., Faber P.W., MacDonald M.E., Zipursky S.L. (1998) Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron, 21, 633–642. [DOI] [PubMed] [Google Scholar]
- 20.Warrick J.M., Paulson H.L., Gray-Board G.L., Bui Q.T., Fischbeck K.H., Pittman R.N., Bonini N.M. (1998) Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell, 93, 939–949. [DOI] [PubMed] [Google Scholar]
- 21.Lanson N.A., Jr, Maltare A., King H., Smith R., Kim J.H., Taylor J.P., Lloyd T.E., Pandey U.B. (2011) A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum. Mol. Genet., 20, 2510–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feany M.B., Bender W.W. (2000) A Drosophila model of Parkinson's disease. Nature, 404, 394–398. [DOI] [PubMed] [Google Scholar]
- 23.Bonner J.M., Boulianne G.L. (2011) Drosophila as a model to study age-related neurodegenerative disorders: Alzheimer's disease. Exp. Gerontol., 46, 335–339. [DOI] [PubMed] [Google Scholar]
- 24.Bonini N.M., Fortini M.E. (2003) Human neurodegenerative disease modeling using Drosophila. Annu. Rev. Neurosci., 26, 627–656. [DOI] [PubMed] [Google Scholar]
- 25.Bilen J., Bonini N.M. (2007) Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet., 3, 1950–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Q., Hashizume Y., Yoshida M., Wang Y., Goto Y., Mitsuma N., Ishikawa K., Mizusawa H. (2000) Morphological Purkinje cell changes in spinocerebellar ataxia type 6. Acta Neuropathol., 100, 371–376. [DOI] [PubMed] [Google Scholar]
- 27.Ishikawa K., Fujigasaki H., Saegusa H., Ohwada K., Fujita T., Iwamoto H., Komatsuzaki Y., Toru S., Toriyama H., Watanabe M., et al. (1999) Abundant expression and cytoplasmic aggregations of [alpha]1A voltage-dependent calcium channel protein associated with neurodegeneration in spinocerebellar ataxia type 6. Hum. Mol. Genet., 8, 1185–1193. [DOI] [PubMed] [Google Scholar]
- 28.Burr A.A., Tsou W.L., Ristic G., Todi S.V. (2014) Using membrane-targeted green fluorescent protein to monitor neurotoxic protein-dependent degeneration of Drosophila eyes. J. Neurosci. Res., 92, 1100–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kordasiewicz H.B., Thompson R.M., Clark H.B., Gomez C.M. (2006) Carboxyl termini of P/Q-type Ca2+ channel {alpha}1A subunits translocate to nuclei and promote polyglutmine-mediated toxicity. Hum. Mol. Genet., 15, 1587–1599. [DOI] [PubMed] [Google Scholar]
- 30.Warrick J.M., Morabito L.M., Bilen J., Gordesky-Gold B., Faust L.Z., Paulson H.L., Bonini N.M. (2005) Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol. Cell, 18, 37–48. [DOI] [PubMed] [Google Scholar]
- 31.Li L., Saegusa H., Tanabe T. (2009) Deficit of heat shock transcription factor 1-heat shock 70 kDa protein 1A axis determines the cell death vulnerability in a model of spinocerebellar ataxia type 6. Genes Cells, 14, 1253–1269. [DOI] [PubMed] [Google Scholar]
- 32.Ishikawa K., Owada K., Ishida K., Fujigasaki H., Shun Li M., Tsunemi T., Ohkoshi N., Toru S., Mizutani T., Hayashi M., et al. (2001) Cytoplasmic and nuclear polyglutamine aggregates in SCA6 Purkinje cells. Neurology, 56, 1753–1756. [DOI] [PubMed] [Google Scholar]
- 33.Kampinga H.H., Craig E.A. (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol., 11, 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koutras C., Braun J.E. (2014) J protein mutations and resulting proteostasis collapse. Front. Cell Neurosci., 8, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bichelmeier U., Schmidt T., Hubener J., Boy J., Ruttiger L., Habig K., Poths S., Bonin M., Knipper M., Schmidt W.J., et al. (2007) Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: in vivo evidence. J. Neurosci., 27, 7418–7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klement I.A., Skinner P.J., Kaytor M.D., Yi H., Hersch S.M., Clark H.B., Zoghbi H.Y., Orr H.T. (1998) Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell, 95, 41–53. [DOI] [PubMed] [Google Scholar]
- 37.Peters M.F., Nucifora F.C., Jr, Kushi J., Seaman H.C., Cooper J.K., Herring W.J., Dawson V.L., Dawson T.M., Ross C.A. (1999) Nuclear targeting of mutant Huntingtin increases toxicity. Mol. Cell Neurosci., 14, 121–128. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi M., Obayashi M., Ishiguro T., Sato N., Niimi Y., Ozaki K., Mogushi K., Mahmut Y., Tanaka H., Tsuruta F., et al. (2013) Cytoplasmic location of alpha1A voltage-gated calcium channel C-terminal fragment (Cav2.1-CTF) aggregate is sufficient to cause cell death. PLoS One, 8, e50121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishiguro T., Ishikawa K., Takahashi M., Obayashi M., Amino T., Sato N., Sakamoto M., Fujigasaki H., Tsuruta F., Dolmetsch R., et al. (2010) The carboxy-terminal fragment of alpha(1A) calcium channel preferentially aggregates in the cytoplasm of human spinocerebellar ataxia type 6 Purkinje cells. Acta Neuropathol., 119, 447–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.La Spada A.R., Wilson E.M., Lubahn D.B., Harding A.E., Fischbeck K.H. (1991) Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature, 352, 77–79. [DOI] [PubMed] [Google Scholar]
- 41.Palhan V.B., Chen S., Peng G.H., Tjernberg A., Gamper A.M., Fan Y., Chait B.T., La Spada A.R., Roeder R.G. (2005) Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc. Natl Acad. Sci. USA, 102, 8472–8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sopher B.L., Thomas P.S., Jr, LaFevre-Bernt M.A., Holm I.E., Wilke S.A., Ware C.B., Jin L.W., Libby R.T., Ellerby L.M., La Spada A.R. (2004) Androgen receptor YAC transgenic mice recapitulate SBMA motor neuronopathy and implicate VEGF164 in the motor neuron degeneration. Neuron, 41, 687–699. [DOI] [PubMed] [Google Scholar]
- 43.Jaiswal M., Sandoval H., Zhang K., Bayat V., Bellen H.J. (2012) Probing mechanisms that underlie human neurodegenerative diseases in Drosophila. Annu. Rev. Genet., 46, 371–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Casci I., Pandey U.B. (2015) A fruitful endeavor: Modeling ALS in the fruit fly. Brain Res., 1607, 47–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Franke J.D., Boury A.L., Gerald N.J., Kiehart D.P. (2006) Native nonmuscle myosin II stability and light chain binding in Drosophila melanogaster. Cell Motil. Cytoskeleton, 63, 604–622. [DOI] [PubMed] [Google Scholar]
- 46.Franke J.D., Montague R.A., Kiehart D.P. (2005) Nonmuscle myosin II generates forces that transmit tension and drive contraction in multiple tissues during dorsal closure. Curr. Biol., 15, 2208–2221. [DOI] [PubMed] [Google Scholar]
- 47.Kiehart D.P., Franke J.D., Chee M.K., Montague R.A., Chen T.L., Roote J., Ashburner M. (2004) Drosophila crinkled, mutations of which disrupt morphogenesis and cause lethality, encodes fly myosin VIIA. Genetics, 168, 1337–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blount J.R., Tsou W.L., Ristic G., Burr A.A., Ouyang M., Galante H., Scaglione K.M., Todi S.V. (2014) Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23. Nat. Commun., 5, 4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.