

Abstract
Human RPE65 mutations cause a spectrum of blinding retinal dystrophies from severe early-onset disease to milder manifestations. The RPE65 P25L missense mutation, though having <10% of wild-type (WT) activity, causes relatively mild retinal degeneration. To better understand these mild forms of RPE65-related retinal degeneration, and their effect on cone photoreceptor survival, we generated an Rpe65/P25L knock-in (KI/KI) mouse model. We found that, when subject to the low-light regime (∼100 lux) of regular mouse housing, homozygous Rpe65/P25L KI/KI mice are morphologically and functionally very similar to WT siblings. While mutant protein expression is decreased by over 80%, KI/KI mice retinae retain comparable 11-cis-retinal levels with WT. Consistently, the scotopic and photopic electroretinographic (ERG) responses to single-flash stimuli also show no difference between KI/KI and WT mice. However, the recovery of a-wave response following moderate visual pigment bleach is delayed in KI/KI mice. Importantly, KI/KI mice show significantly increased resistance to high-intensity (20 000 lux for 30 min) light-induced retinal damage (LIRD) as compared with WT, indicating impaired rhodopsin regeneration in KI/KI. Taken together, the Rpe65/P25L mutant produces sufficient chromophore under normal conditions to keep opsins replete and thus manifests a minimal phenotype. Only when exposed to intensive light is this hypomorphic mutation manifested physiologically, as its reduced expression and catalytic activity protects against the successive cycles of opsin regeneration underlying LIRD. These data also help define minimal requirements of chromophore for photoreceptor survival in vivo and may be useful in assessing a beneficial therapeutic dose for RPE65 gene therapy in humans.
Introduction
In the initial step in the visual process, the visual pigment, retinal opsin, with covalently bound 11-cis-retinal chromophore, undergoes photoactivation upon photon absorption and subsequently bleaches to opsin and all-trans-retinal. To regenerate rhodopsin and maintain normal visual sensitivity, the all-trans isomer must be released, metabolized and reisomerized to 11-cis-retinal in the retinal pigment epithelium (RPE) in a process called the visual (or retinoid) cycle (1), involving movement of retinoid between the photoreceptor and RPE. RPE65 is the key isomerase in this process, converting all-trans-retinyl ester to 11-cis-retinol (2–4). Evolutionarily conserved, RPE65 was the first RPE-specific gene identified as associated with disease, in patients with early-onset severe retinal dystrophy (EOSRD) or Leber congenital amaurosis 2 (LCA2) (5,6). In humans, >100 pathogenic mutations of RPE65 have been identified, spread over all 14 exons of the gene and their boundaries, about half of which are missense mutations (7) (see also http://www.retina-international.org/files/sci-news/rpe65mut.htm, and OMIM 204100). These mutations are associated with a spectrum of retinal dystrophies ranging from early-onset, severe blindness to later-onset, milder retinal degeneration (8,9). In less severe forms resembling retinitis pigmentosa, patients often have well-preserved visual function early in their lives and a slower progression of the disease (10,11).
Transgenic mouse models have been employed to understand inherited retinal degeneration caused by RPE65 mutations. The Rpe65 knockout (KO) mouse showed that RPE65 is necessary for 11-cis retinoid production in the visual cycle (12). With no RPE65 expression, the KO mice over-accumulate the all-trans-retinyl ester substrate in the RPE, while 11-cis-retinoids are absent. Consequently, these mice have a slow retinal degeneration but abolished light sensitivity owing to lack of visual chromophore in retinae. The Rpe65 KO model has been of great utility in understanding retinal physiology and biochemistry, and their alterations caused by or related to variations in chromophore status (13–20). Rd12, a spontaneous null mutation in Rpe65 (21), and an Rpe65/R91W knock-in (KI) mouse mutant have also been reported (22). With synthesis of very low levels of 11-cis-retinal by the R91W RPE65 mutant protein, these mice show better cone functions at young age than the KO mice, but their rod system is severely desensitized. Though more slowly than the KO, the KI retinae do degenerate, consistent with the phenotype of RPE65/R91W patients diagnosed with EOSRD (23).
Based on these observations in both human patients and transgenic mouse models, it appears that the level of impairment of the isomerase activity caused by RPE65 mutations correlates with severity of retinal dysfunction. Therefore, studies on less severe forms of RPE65-related retinal dystrophy may help us not only understand the full spectrum of RPE65-related pathology but also in assessing a beneficial therapeutic dose for RPE65 gene therapy. Recently, a homozygous P25L missense mutation of RPE65 has been associated with mild retinal pathology in a young patient (11). With a greatly reduced isomerase activity [∼8% of wild type (WT) in vitro], though less severe than R91W, mutant RPE65/P25L sustained near-normal visual acuity in the affected patient at the age of 6 years. The retinal structure was relatively well preserved with no obvious increase in fundus autofluorescence. However, rod function was greatly impaired (night blindness), and short-wavelength cones appeared more impaired than long-wavelength cones.
To better understand the pathogenic pathway of the P25L mutation, we generated an Rpe65/P25L KI transgenic mouse model to study mild forms of RPE65-related retinal degeneration. Surprisingly, the phenotype manifested under normal light regime was quite similar to WT controls, but under damaging light intensity, like Rpe65 KO and unlike WT, it was protected against light-induced retinal damage (LIRD).
Results
Normal transcription of Rpe65/P25L gene in Rpe65P25L/P25L knock-in mice
To gain further insights into the pathology of RPE65/P25L missense mutation in human disease, we generated Rpe65P25L/P25L KI mice carrying the missense mutation to leucine at proline 25 (Fig. 1A). Specifically, a targeting construct for mouse Rpe65 changing codon 25 in exon 2, from Pro (CCA) to Leu (CTA) and incorporating a neomycin-resistance cassette flanked by loxP sites (Fig. 1A), was introduced into 129/Sv-derived R1 mouse embryonic stem (ES) cells by electroporation. ES clones with an accurate site-specific recombination were selected by Southern blot (Fig. 1B) and long-range polymerase chain reaction (PCR) analysis (data not shown). Two correctly recombined R1 ES clones were injected into C57BL/6 mouse blastocysts to obtain chimeric mice, which were then backcrossed with WT to obtain F1 mice with germ-line transmission of the KI allele containing a neo cassette (Fig. 1C). Heterozygous F1 mice (HET) were crossed with a female germ-line cell-specific cre line, Zp3-cre (24), to eliminate the neo-selection cassette. A single PCR assay with primers (see Supplementary Material, Table S1 for sequences used) flanking the excised loxP site was performed to detect the presence of the WT allele (225 bp) and the KI allele (KI, 277 bp) (Fig. 1D). The KI mice were then backcrossed five times with 129/Sv mice to obtain mutated mice in a pure congenic background. Consequently, the WT allele is the normal variant of mouse RPE65 encoding the Leu residue at codon 450 (25,26). The resultant heterozygous KI (wt/KI) mice were intercrossed to generate homozygous KI (KI/KI) mice and WT littermate controls. Homozygous KI/KI mice are viable and fertile. Inheritance of the P25L mutation was confirmed by sequencing the exon 2 (Fig. 1D). Quantitative reverse transcription–polymerase chain reaction (RT–PCR) analyses revealed that Rpe65 mRNA levels in the homozygous mouse eyecup were comparable with those found in the WT and heterozygous siblings (Fig. 1E), demonstrating that the necessary genetic manipulations for KI generation did not affect the transcription efficiency of the Rpe65 gene.
Figure 1.

Generation of a mouse model with Rpe65/P25L mutation. (A) Strategy for targeted insertion of the P25L point mutation into the murine Rpe65 locus to generate a KI mouse model. The targeting vector carried a C to T mutation at codon 25 in exon 2 of Rpe65 (star) and a loxP-flanked neo cassette and included the 1.5-kb flanking sequence from Rpe65 genomic DNA as the 5′ left homology arm (LA) and 2.9 kb as the 3′ right homology arm (RA). The neo cassette was removed by Cre-loxP recombination to generate Rpe65/P25L KI mice. This mouse retained an extra 52-bp sequence with added restriction sites at the loxP site for the convenience of genotyping in intron 2. (B) Southern blot analysis of ES clones after homologous recombination: ES clone genomic DNA was digested with NcoI, followed by hybridization with a 5′ probe outside the LA (Fig. 1A); genomic DNA from the wt allele only shows a 5-kb hybridization signal in WT, whereas homologous recombination gives rise to an additional 3.5-kb fragment owing to the presence of an NcoI restriction site within the neo cassette (Fig. 1A) that is seen in heterozygous animals carrying one copy of the KI+neo allele (HET). (C) Germ-line transmission of the KI+neo allele. Following blastocyst injection, germ-line transmission was identified by long-range PCR of both the LA and the RA (left panel, Fig. 1A and Table 1), and then by simple amplification of a 303-bp amplimer in the HET offspring (right panel) along with a WT 143-bp amplimer, the only one seen in WT (see Supplementary Material, Table S1 for primer sequences). (D) Removal of the neo cassette. The neo cassette was removed via Cre recombinase by crossing F1 heterozygous mice with Zp3-Cre mice carrying a germ-line Cre transgene (24) in the C57B6/J background. PCR primers P1 and P3 were designed to amplify the intron 2 region flanking the 52-bp loxP sequence. The wt allele and KI allele produced 225- and 277-bp PCR products, respectively (top panel; see Supplementary Material, Table S1 for primer sequences). Genomic sequencing confirmed the presence of the C to T mutation at codon P25 in the KI/KI mice (bottom panels). (E) Relative levels of Rpe65 mRNA expressed in eyecups of WT, KI/KI (homozygous) and wt/KI (heterozygous) mice at 16–20 weeks of age as determined by Q-PCR. Expression levels were normalized to two reference genes (Gapdh and Hprt). Error bars indicate SDs from biological triplicates within the experiment.
Reduced level of RPE65/P25L protein expression in the Rpe65P25L/P25L mouse
While transcription of Rpe65 was not altered in the KI/KI mice, western blot analysis of eyecups revealed that RPE65 protein levels of Rpe65P25L/P25L mutants were lowered to ∼18% of WT levels (Fig. 2A and B). Interestingly, the level of expression in the heterozygote wt/KI animals was lower at ∼43% than the expected 60%. It is possible that the mutant P25L protein can destabilize dimers (27) that it forms with WT protein, thus reducing apparent expression in heterozygotes. Immunofluorescence staining of eye cross-sections from KI/KI mice showed that the mutant protein was correctly located in RPE (Fig. 2C), albeit at lower expression level. The expression of the mutant protein did not alter as the transgenic animal grew older but remained <20% of the WT control from the age of 6 weeks (Fig. 2A) to 52 weeks (data not shown). The reduced protein level in the KI/KI mouse eyecup is consistent with our previous observation in vitro (11), suggesting that the Rpe65/P25L acts as a hypomorphic mutant wherein reduced expression levels result in reduced isomerase activity. Despite reduced level of the mutant RPE65 protein, expression levels of other visual cycle components, such as RLBP1 and RDH5, remained at steady levels in the eyecup of the KI/KI mice, comparable with those found in controls (Supplementary Material, Fig. S1).
Figure 2.

Reduced mutant protein levels in the KI/KI mice. (A) RPE65 protein expression in eyecups was determined by western blot analysis in WT, heterozygous wt/KI and homozygous KI/KI (KI) mice. GAPDH protein expression was used as loading control. Representative blot shown; (B) quantification of RPE65 expression after normalization with GAPDH. Error bars indicate SDs from biological triplicates within the experiment; (C) immunofluorescence of eyeball sections from WT and KI mice with antibody against RPE65 (red), and nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI; blue). The upper panels cover the full span of the retina, whereas the lower panels are higher magnifications of the RPE. INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium.
Retinal structure and opsin expression are normal in the Rpe65P25L/P25L mouse
Retinal integrity of the KI/KI mice was continuously monitored by both in vivo optical coherence tomography (OCT) imaging and toluidine staining of the eyeball sections from 6 weeks up to 1 year old. Figure 3B and D shows typical retinal structure of a KI/KI mouse at the age of 7 months, revealing no detectable abnormalities; comparable results were seen at older ages also (see Supplementary Material, Fig. S2 for representative 14- and 20-month sections). The length and compactness of the outer segments (OS) were similar to the WT control, and the thickness of the outer nuclear layer (ONL) was comparable with the WT. The well-preserved retinal structure in the Rpe65P25L/P25L KI mice is in contrast with the progressive retinal degeneration observed in Rpe65 KO mice (12) and R91W mice (22) at young age.
Figure 3.

Retinal histology of Rpe65P25L/P25L KI mice is similar to WT. Retinal integrity at the age of 7 months was monitored by both in vivo OCT imaging (A and B) and toluidine staining of the eyeball sections (C and D). There are no detectable differences in retinal structure between KI/KI (B and D) and its WT siblings (A and C) when raised under normal dim light conditions. Scale bar = 50 µm.
It has been well established that the expression of the opsins, particularly the cone opsins, is sensitive to RPE65 expression levels (16). In the Rpe65 KO mice, drastic reductions in the expression of cone opsins were detected at Postnatal Day 25, and a significant portion of the remaining opsins were mislocalized in the cell membrane of the inner segment, cell body, axon and synaptic pedicle (data not shown) (16). In contrast, both cone opsins were correctly localized to the OS in the 7-month-old KI/KI retina as in the WT control (Fig. 4A). Further immunoblot analyses revealed that the total expression levels of the rod and cone opsins were comparable with those found in the WT retinae (Fig. 4B). The normal rod and cone opsin expression level is consistent with survival of the unaffected rod and cone photoreceptors in KI/KI mice.
Figure 4.
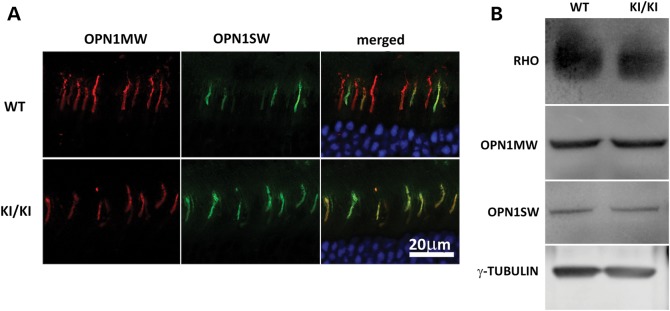
Expression and localization of opsins. (A) Retinal sections of WT (upper) and Rpe65P25L/P25L (lower) mice were stained with antibodies against short-wave-sensitive opsin 1 (OPN1SW) and medium-wave-sensitive opsin 1 (OPN1MW). Both were correctly localized to the OS in the KI/KI mice. (B) Western blotting of opsin proteins from 7-month-old WT or Rpe65P25L/P25L mice. Similar levels of rod (RHO) or cone opsins were detected in retinae of mutant mice as in the WT mice.
In vivo isomerase activity of the RPE65P25L/P25L mutant is sufficient to keep opsins replete
As RPE65 is indispensable for the synthesis of 11-cis-retinal and consequent regeneration of visual pigment (12,22), we examined the retinoid composition in the 4-month-old Rpe65P25L/P25L KI/KI mice. HPLC retinoid analyses of the retinae extracts showed that homozygous KI/KI mice generate a comparable amount of 11-cis-retinal sufficient to keep photoreceptor visual pigments as replete as the WT controls (Fig. 5A), despite the greatly reduced expression levels of the mutant protein (Fig. 2). On the other hand, there was a significant excess accumulation of retinyl esters in the RPE layer of the KI/KI mice (Fig. 5B). This was an ∼3-fold increase compared with the retinyl ester content in the WT littermates at 4 months. These levels, however, are not as high as the ≥20-fold increase in RPE retinyl ester level observed in Rpe65 KO mice (12) and Rpe65R91W/R91W KI/KI mice (22) at comparable ages.
Figure 5.

Analysis of visual cycle retinoids from Rpe65P25L/P25L KI mice at the age of 4 months. (A) Quantitation of HPLC peak (lower panel, representative example of chromatogram) of 11-cis-retinyloxime from dissected retinae shows comparable amount of 11-cis-retinal present in the KI/KI mice as in the WT controls. (B) Quantitation of HPLC peak (lower panel, representative example of chromatogram) of total retinyl esters extracted from dissected eyecup samples showed ester accumulation in RPE. Error bars indicate SDs from biological triplicates within the experiment; *P<0.05.
Visual function of Rpe65P25L/P25L KI mice is similar to WT
To evaluate rod- and cone-mediated light responses, we performed in vivo electroretinogram (ERG) recordings on KI/KI mice. Under dark-adapted (scotopic) conditions, a- and b-wave amplitudes of the KI/KI mouse retinae were similar to those observed in WT and heterozygous wt/KI retinae (Fig. 6A). Similarly, the Rpe65/P25L KI/KI mice showed no significant changes in either a- or b-wave amplitudes under light-adapted (photopic) conditions (Fig. 6B). To further isolate the sensitivities of the UV-cone- and M-cone-specific b-wave responses in the KI mice, we exposed mice to flash light stimuli of particular wavelength. As shown in Figure 6C, we found that KI/KI mice exhibited cone b-wave amplitudes similar to those in the WT controls in response to both UV (triangles) and green (circles) flash stimuli. Thus, the Rpe65/P25L mutation does not have a significant effect on the sensitivity of rod or cone responses to light stimuli when raised under the subdued light regime in standard mouse husbandry.
Figure 6.

Rpe65P25L/P25L KI mouse has full-field scotopic (rod), photopic (cone) ERG responses similar to WT but has delayed a-wave recovery after moderate visual pigment bleach. Dark-adapted (scotopic) ERG (A) and light-adapted (photopic) ERG (B) b-wave responses were obtained from 3-month-old homozygous KI/KI (open circle), heterozygous wt/KI (half-filled circle) and the littermate WT (filled circle) controls. Each point represents the average of six mice. Error bars show ±SD. (C) Cone signal isolation in Rpe65P25L/P25L KI mouse. B-wave amplitudes in response to a series of flashes varying intensity of UV (triangles) and green light (circles) stimuli were measured for 6-month-old homozygous KI/KI mice, heterozygous wt/KI and the littermate WT controls. Each point represents the average of four mice. Error bars show ±SD. (D) Representative a-wave recovery after moderate visual pigment bleach in KI/KI (open circle), WT (filled circle) and wt/KI (half-filled circle) mice. Continuous curves are plotted from Equation (1) fitted to the data with a nonlinear regression.
We next characterized the recovery of rod photoreceptors function following moderate visual pigment bleach in the KI/KI mouse retinae. After being exposed to a bleaching background light (1000 cd/m2) for 30 s [eliciting a fractional bleach of ∼9.5%, as calculated from data in Pawar et al. (28)], ERG responses to a low-intensity flash stimulus (10 cd.s/m2) were continuously recorded for 1 h at 2 min intervals. The a-wave amplitude recovery was plotted against post-bleaching time; Figure 6D shows representative recovery curves for KI/KI, wt/KI and WT mouse. Each plotted recovery curve was fitted with a nonlinear regression using the published equation (see Supplementary Materials and Methods) (29). The three values Amax, ca and τa were calculated for each plot, and the constant values between different genotype groups were analyzed using a multiple t-test. As shown in Table 1, there were no significant differences observed among groups in the value of maximal a-wave amplitude following bleaching (Amax) and the degree of amplitude reduction right after bleaching (ca). However, the time constant for recovery (τa) is significantly longer in the KI/KI mice compared with the WT and heterozygous mice (P < 0.01), suggesting a delayed recovery rate in the KI/KI mice following a moderate visual pigment bleach.
Table 1.
Average values of the parameters used in fitting recovery of a-wave responses after strong light bleaching
| Amax | ca | τa | |
|---|---|---|---|
| WT (n = 20) | 283.48 ± 81.60 | 17.22 ± 10.00 | 6.60 ± 1.95 |
| wt/KI (n = 8) | 297.1875 ± 62.15 | 13.32 ± 5.44 | 6.59 ± 1.08 |
| KI/KI (n = 22) | 285.44 ± 127 | 12.00 ± 6.45 | 9.10 ± 2.43a |
aThe calculated τa values for KI/KI mice are significantly larger than those for the WT mice and the wt/KI mice (P < 0.01). There are no significant differences in the values of Amax or ca between the KI/KI group and the WT controls; n indicates the number of eyes subject to the test of recovery responses in each genotype group.
Rpe65P25L/P25L KI mice are protected from LIRD
As a critical component of the visual cycle, RPE65 modulates retinal susceptibility to intensive light damage via affecting the kinetics of rhodopsin regeneration (14). It has been demonstrated that impaired chromophore synthesis owing to RPE65 mutations increases retinal resistance to intensive light damage (26,30). We examined retinal susceptibility to light damage in the Rpe65P25L/P25L KI mice using OCT and light microscopy. With dilated pupils, mouse retinae were exposed to 20 000 lux white light for 30 min. The structural integrity of the retinae was examined via OCT imaging and histology 2 weeks after the light treatment. The ONL of the WT mice was reduced, and rod outer segments were greatly shortened (Fig. 7B and C), whereas much less severe degeneration was observed in the same area of the KI/KI mice (Fig. 7F and G). Measurement of both the total retinal thickness (Fig. 7I) and the photoreceptor span (Fig. 7J) showed that their thicknesses after light damage were significantly reduced in the WT mice compared with the KI/KI mice, whereas the thicknesses before light damage treatment were comparable between the two genotype groups (Fig. 7I and J). Consistent with decreased sensitivity to LIRD, retinae of the KI/KI mice did not display significant cell death by apoptosis after intensive light treatment (Fig. 7H), nor did they show accumulation of A2E (Supplementary Material, Fig. S2), a derivative of free all-trans-retinal (31,32), as found in the light-treated WT retinae.
Figure 7.

Rpe65P25L/P25L KI mice are protected from light damage. Eyeballs from WT (A–D) and KI/KI (E–H) mice were treated with high-intensity light (20 000 lux) for 30 min following pupil dilation. Retinal integrity was monitored by OCT before (A and E) and 2 weeks after light damage (B and F). Fine retinal structures after light exposure were examined by light microscopic analysis of sections of retinal tissues of WT mice (C) and KI/KI mice (G). Cell apoptosis after light treatment was surveyed via TUNEL staining of frozen sections of retinae of WT mice (D) and KI/KI mice (H). Thickness of the total retina (I) and the photoreceptor layer (J) before and after light damage was measured on 10 WT and 15 KI/KI mice. Error bars show ±SD; *P < 0.05.
We hypothesized that a suppressed visual cycle was the underlying mechanism of protection from intense light damage in the KI/KI mice. Therefore, we examined the regeneration of 11-cis-retinal following the exposure to 20 000 lux white light for 30 min. We found that the 11-cis-retinal pool has already been fully replenished in the WT retinae 1 h after acute light treatment (Fig. 8A). In contrast, the level of 11-cis-retinal in KI/KI mice was much lower, with an observed 5-fold less 11-cis-retinal content 1 h after light treatment than WT, and it was still 3-fold less 2 h later. On the other hand, the amount of retinyl esters in the KI/KI RPE was 4-fold more than that in the WT RPE at 1 h post-light treatment, and at 2 h post-light was about 10-fold more than the completely recovered WT (Fig. 8B). As a further measure of the slowed recycling of visual cycle retinoids, we found that all-trans-retinal levels were also significantly lower in the KI/KI retinae than that in WT, both at 1 and at 2 h after light treatment (Supplementary Material, Fig. S4). These measures of retinoid product and substrate levels indicate that the reduced expression and catalytic activity of the hypomorphic RPE65/P25L enzyme dramatically slow the synthesis of visual chromophore, thereby protecting the KI/KI mice subjected to acute light stress from the successive cycles of opsin regeneration that drive the light damage outcome seen in WT animals.
Figure 8.

Recovery of 11-cis-retinal levels in retinae and reduction in retinyl esters in RPE are slower in KI/KI mice. Dark-adapted mice were exposed to a light of 20 000 lux for 30 min and returned to the dark until retinae, and eyecups were collected for HPLC retinoid analysis at different time points. (A) Levels of 11-cis-retinal in retinae were measured at 1 and 2 h time points AFTER light exposure. (B) Retinyl ester levels in RPE layer were measured at 1 and 2 h time points after light exposure. Error bars indicate the SD of the mean (n = 4 for WT and n = 3 for KI/KI); *P < 0.05; **P < 0.001; LE: light exposure.
Discussion
Herein, we present the somewhat paradoxical phenotype of the Rpe65P25L/P25L KI mouse we have generated. Paradoxical in that KI/KI mice are phenotypically very similar to WT under regular husbandry conditions but manifest an Rpe65 KO-like aspect of complete resistance to LIRD. The Rpe65P25L/P25L KI mouse phenotype parallels, in general terms, the mild phenotype of the human RPE65 homozygous P25L proband (11) for which it is a model but may differ in species-specific details. This model complements null (12,21) and near-null (22) Rpe65 models previously studied. As maintenance of an adequate visual cycle is critical for normal retinal development and function, and overwhelming evidence implicates RPE65 as playing the major role in setting the dynamic range of visual pigment regeneration, an adequate chromophore level may be crucial in preventing or limiting degeneration in patients with mutations of RPE65, or in RPE65 gene therapy, and preserving cone function is a key concern in both cases. A recent study by Jacobson et al. (33) finds that current RPE65 gene therapy fails to stop an apparently ongoing program of retinal degeneration in treated patients. Our experiments may have translational significance for the management of RPE65 gene therapy by providing a minimal estimate for an effective replacement level of chromophore that might prevent progressive photoreceptor loss.
RPE65/P25L leads to mild phenotype in KI model but is protective against LIRD
In line with the mild phenotype in a human patient homozygous for P25L (11), KI/KI mice display intact retinae with normal expression of cone and rod opsins. The P25L mutation has minimal effects on visual functions under standard mouse husbandry. Only when subject to increased light intensities was a retinal phenotype manifested. When raised under the normal light intensity of ∼100 lux, KI/KI mice can maintain chromophore levels comparable with WT and therefore manifest normal photopic and scotopic ERG parameters. It is noticeable that, while sufficient chromophore is generated, there is still significant retinyl ester over-accumulation in the RPE of KI/KI mice compared with WT. It seems that under dim light where minimal top-up of chromophore levels is required, the slower isomerization rate does not affect the rate-limiting step in the visual cycle and thus does not disrupt regeneration. However, this slower rate may not be able to keep up with the continuous influx of all-trans-retinol from both the circulation (34,35) and the retina, resulting in ester accumulation in the RPE. Consistent with this, the reduced isomerase activity in KI/KI mice does not replenish depletions of the 11-cis-retinal pool following bleaches as rapidly as WT, resulting in a significant delay in the recovery of rod photoresponses.
While a sustained level of chromophore is required for photoreceptor survival (12,21,36,37), it has been suggested that a burst influx of 11-cis-retinal following intense light actually triggers massive apoptosis and subsequent retinal degeneration by inducing the rapid release of all-trans-retinal from meta II opsin (38,39). Therefore, while Rpe65 KO mice display a progressive retinal degeneration and total lack of visual sensitivity (12), their lack of an active visual cycle actually protects them from acute LIRD (14,30). Just as in the Rpe65 KO mice, when exposed to acute white light (∼20 000 lux for 30 min), KI/KI mice retinae are protected from LIRD. It is remarkable that while the level of chromophore regeneration supported by P25L RPE65 is sufficient to support near WT phenotype at normal light rearing, that, with as much as 8% WT activity (11), P25L is also protective against LIRD. While the overall mechanism of LIRD is far from clear, it involves formation of stressors that may include rhodopsin photo-intermediates, retinoid intermediates, generation of oxidative species including lipid products and calcium influx owing to excessive activation of the phototransduction cascade (40). The stressor most germane to the present experiments involves the release of all-trans-retinal from meta II opsin and the influx of 11-cis-retinal to replace it. It is likely that WT photoreceptors are bathed in toxic retinals (41) during the 30 min of light damage that are rapidly recycled back to 11-cis-retinal for repeated iterations of this process, whereas inefficient isomerization in the KI/KI short-circuits this adverse cycling and is protective. The protection from LIRD, together with delayed dark adaptation, demonstrates a reduced isomerase activity of the mutant RPE65/P25L, resulting in impaired visual cycle machinery (slower accumulation of 11-cis-retinal/slower decrease of retinyl esters/slower cycling of retinoids under bright light conditions).
Phenotypic similarities between P25L KI model and human hypomorphic patient
In the present study, we have demonstrated that the RPE65/P25L mutant has greatly reduced RPE65 protein levels in the transgenic mouse model, less than one-fifth of the WT RPE65. As the mutant mRNA level is comparable with that of WT, the decreased protein expression may be caused by either an inefficient mRNA translation, or more likely, destabilization of the mutant protein, which may in turn lead to lower isomerase activity in the RPE of KI/KI mice. With respect to the lower than the expected level of expression in the heterozygote wt/KI, a possible explanation is that the mutant P25L protein can destabilize dimers (27) formed with WT protein. This apparently reduced expression, however, does not appear to have a detectable physiological effect on ERGs. This is of additional interest because an apparently dominant-acting RPE65 mutation has been described in an Irish pedigree (42). However, the parents of the described P25L patient did not manifest any ophthalmologic symptoms (11).
The degree of severity of RPE65 retinal phenotypes in both humans and mouse models is correlated with the expression levels and isomerase activities in the various mutants. While null/functional null RPE65 mutations lead to early-onset blindness in humans and light-insensitive mice in the KO and null models (12), the R91W missense mutation with residual protein expression (5% of WT) and isomerase activity (<1%) gives better early preservation of retinal structures and visual functions than null/functional null, in both human (23) and in mouse models (22). Nonetheless, severe retinal dystrophy at young ages and a progressive retinal degeneration are observed in both the null and R91W point mutation. The Rpe65/P25L mutation also causes greatly impaired isomerase activity (∼8% of WT in vitro) (11). However, in vivo, this residual activity is enough to mitigate the extent of retinal dystrophy in the homozygous human patient (11) and can successfully preserve normal retinal structures and visual functions in Rpe65P25L/P25L KI mice raised under relatively dim ‘normal’ light conditions. Similar to the RPE65/P25L, other missense mutations of RPE65, including L22P, E95Q and Y79H, display reduced protein expression/isomerase activity (11) and a variable degree of mild retinal dystrophy (7,10). Further studies in different RPE65 mutants with diverse degrees of retinal dystrophy will help us appreciate how RPE65 activity sustains retinal functions.
Phenotypic differences between mouse knock-in models and human patients
Conversely, Rpe65P25L/P25L KI mice fail to recapitulate certain features of the human pathology, including that of night blindness, though we argue that the delayed recovery in KI/KI mice is a manifestation of this, and impaired sensitivity of blue cones. In respect of the latter, differences in cone abundance and distribution between primates and rodents may confound correlation of given aspects of the phenotype. However, the features in common may be more important than those that are not. The fact that cone survival and function in both the human P25L individual (11) and its mouse model are close to their respective WT situations suggests a common lower limit for survival in respect of chromophore supply. While the retinal visual cycle (43,44) supplements cone chromophore requirements in cone, supply from the ‘canonical’ RPE visual cycle is nonetheless crucial to cone survival.
However, consistent with our observations, a number of mouse models with disruption/mutations of genes involved in the retinoid cycle exhibit less severe phenotypes than the clinical retinopathies in human patients. Such mouse models include mutations in Rdh11 (45), Rdh12 (46), Rlbp1 (47) and Rgr (48). Similar to the Rpe65P25L/P25L KI model, phenotypic manifestation of these mutations may be largely dependent on the light intensities to which the animals are subject. Many of these mouse models have delayed dark adaptation kinetics upon bright light bleaching but regular scotopic and photopic ERG when raised under dim light conditions. Therefore, it appears that phenotypic discrepancies between human and mouse may reflect at least in part, the widely divergent photic environments occupied by these species. Indeed, the general lighting condition in vivaria is ∼100 lux, whereas the light level of a sunny midday can reach 20 000 lux in shaded areas. The mildly impaired visual cycle that can sustain the visual system in the mouse models will most likely more adversely affect visual functions in humans with the orthologous mutations, as chromophore requirements are greater. It will be interesting to raise these mice under conditions of higher ambient light levels to investigate the chronic effects of the missense mutation on retinal structures and function. By in vivo titration of chromophore turnover, it might be possible to further determine effects on retinal physiology and cone survival/function and to determine a minimum level of chromophore turnover to maintain cone viability and function. In addition to better understanding of cone physiology, this may have further translational significance for the application and management of RPE65 gene therapy.
Materials and Methods
Generation of Rpe65P25L/P25L knock-in mice
Through RecE/RecT recombination as described (49), a BAC clone encompassing 5′ flanking sequence and exons 1–3 of mouse Rpe65 was used to construct a targeting vector in which a single nucleotide replacement from C to T was introduced into exon 2, changing codon 25 from proline to leucine. The targeting vector contained a PGK-driven DTA (modified diphtheria toxin A) cassette as the negative selection marker (50) for screening of the BAC clones containing the correct mutation (51). The selected construct was linearized and electroporated into ES cells (SV129). G418-resistant clones were picked, expanded and screened for homologous recombination by both long-range genomic PCR (see Supplementary Material, Table S1 for primer sequences) and Southern blotting with a 5′ probe outside of the targeting vector after NcoI digestion (Fig. 1A). Correctly targeted clones were further analyzed by karyotyping. ES cell clones carrying the targeted allele were isolated, and two clones were injected into blastocysts. The selectable marker (loxP-Neo-loxP) was removed by mating to Zp3-Cre (24) mice (Jackson Laboratories, Bar Harbor, ME, USA) expressing Cre recombinase. Successful excision of the selectable marker cassette was confirmed by PCR of a region spanning the loxP insertion site (see Supplementary Material, Table S1 for primer sequences).
Quantitative RT–PCR (Q-PCR)
Total RNA was isolated from the adult mouse eyecup using RNeasy Plus Mini Kit (Qiagen, Valencia, CA, USA); 500 ng RNA was reverse-transcribed with a cDNA synthesis kit (Life Technologies, Grand Island, NY, USA). Q-PCR was performed with synthesized cDNA as a template and Taqman probe for mouse Rpe65 gene (Life Technologies). Gapdh and Hprt were used as reference genes for normalization. Q-PCR analysis was performed in biological triplicates for each genotype.
Animals
All procedures concerning animals were in accordance with institutional regulations and with the statement of the Association for Research in Vision and Ophthalmology for the use of animals in research and were carried out under an institutional Animal Study Protocol approved by the National Eye Institute (NEI), NIH Animal Care and Use Committee. The mice were raised in cyclic light (∼100 lux, 12:12 h).
Immunoblotting
Mouse retinae or eyecups were dissected and solubilized in phosphate-buffered saline (PBS) supplemented with 0.15 mg/ml dodecyl maltoside and Complete protease inhibitor (Roche). Ten to twenty micrograms of protein extracts from mouse eyecup were analyzed by western blot using antibodies against RPE65 (PETLET, 1:2000), RDH5 (1:1000), RLBP1 (1:10 000; gift of J. Saari, University of Washington) and GAPDH (Cell Signaling, 1:1000). Five to ten micrograms of protein extracts from mouse retinae were analyzed by western blot using antibodies against RHODOPSIN (1D5, 1:10 000), OPN1MW (1:5000, gift of T. Li, NEI), OPN1SW (1:5000, gift of T. Li, NEI) and γ-Tubulin (Cell Signaling, 1:1000). Relative expression levels were quantified using the Image Studio Lite software package (LI-COR Biotechnology, Lincoln, NE, USA) on gel files imported from a Typhoon 9410 imaging system (GE Healthcare).
Histology and immunofluorescence microscopy
Enucleated eyeballs were fixed in 2% paraformaldehyde and 2.5% glutaraldehyde and embedded in methacrylate. Serial vertical sections were cut through the pupillary-optic nerve plane, stained with toluidine and analyzed by light microscopy. For immunofluorescence, eyes were enucleated and fixed in 4% formaldehyde for 1–2 h. The anterior segments and lenses were removed. The fixed tissues were equilibrated in 30% sucrose/PBS for 3 h to overnight, snap-frozen and sectioned along the superior–inferior meridian at 10 µm thickness. Immunolabeling on frozen sections was performed using rabbit anti-RPE65 (1:500), goat anti-OPN1SW (1:100, Santa Cruz) and rabbit anti-OPN1MW (1:500, Millipore). Stained sections were imaged by confocal laser scanning microscopy (model TCS SP2; Leica, Wetzlar, Germany). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) for nuclei undergoing apoptosis was performed following the manufacturers' instructions on frozen sections (prepared as above) using the ApopTag® Red In Situ Apoptosis Detection Kit (EMD Millipore, Billerica, MA, USA).
Retinoid analysis
All procedures involving retinoids were conducted under dim red light. For analysis of neural retinas, a single mouse retina was homogenized in 0.5 ml of freshly made hydroxylamine buffer (50 mm MOPS, 10 mm NH2OH, pH 6.5) using a disposable micro tissue homogenizer (BioMasher II, Warrington, PA, USA). The homogenate was transferred to a 15-ml polypropylene screw-cap tube, and an additional 0.5 ml of hydroxylamine buffer was added along with 1 ml ethanol. Samples were incubated for 30 min in the dark at room temperature. Following this, retinoids were extracted twice by addition of 4 ml hexane, vortexing and centrifugation (2000g, 8 min). The upper hexane phases were collected and pooled, and solvent was evaporated under argon at 37°C. The dried samples were redissolved in 100 µl hexane for analysis. Retinaloxime standards were prepared following Garwin and Saari (52). Standards and samples were separated on a LiChrospher Si-60 (5 µm; Merck, Darmstadt, Germany) normal-phase column using a 11.2% ethyl acetate, 2% dioxane and 1.4% octanol (v/v/v) in hexane mobile phase at a flow rate of 0.7 ml/min. Absorbance was monitored at 350 nm, and peak areas for all-trans and 11-cis-retinaloximes were integrated and quantified using external calibration curves. Data were analyzed using Empower 3 software (Waters Corp., Milford, MA, USA).
To measure retinyl esters in RPE, individual eyecups were homogenized in 1 ml ethanol using all-glass tissue grinders (Kontes Duall 21). Retinoids were extracted twice by addition of 5 ml hexane, vortex mixing and centrifugation. The upper hexane phases were combined, solvent evaporated and the remaining residue redissolved in 100 µl hexane. Retinoids were separated as mentioned earlier. Absorbance was monitored at 325 nm, and peak areas for retinyl ester were integrated and quantified using external calibration curves. Data were analyzed as mentioned earlier.
Electroretinograms
After overnight dark adaptation, the eyes of anesthetized mice were dilated with a drop of tropicamide and phenylephrine. Tetracaine (0.5%) drops were applied for local anesthesia of cornea. Body temperature was maintained at 37°C with a heating pad. Electroretinograms (ERGs) were recorded from both eyes using gold wire loop electrodes connected to an Espion e2 Visual Electrophysiology System (Diagnosys, Lowell, MA, USA). A gold wire loop placed in the mouth was used as a reference electrode. Dark-adapted ERG was performed using flashes with intensities ranging from 0.0001 to 10 sc cd.s/m2. Light adaptation was performed with white light at 20 sc cd/m2 for 2 min, and the ERG response was recorded using flashes with intensities ranging from 0.3 to 100 sc cd.s/m2. A UV colordome connected to an Espion e2 system was used to provide Ganzfeld UV (365 nm) flashes, and electroretinogram recordings were performed on at least six mice per genotype.
For bleaching experiments, mice were dark-adapted overnight and then subjected to a moderate visual pigment bleach with the background light of a Ganzfeld chamber (1000 cd/m2) for 30 s. After the light was turned off, a single-flash ERG at 10 cd.s/m2 was used to monitor recovery of a-wave amplitude every 2 min for 60 min. To analyze the data, the recovery of a-wave amplitude [A(t)] following bleaching is plotted against post-bleach time (t). The following equation was fitted to the raw data using Prism 6 (GraphPad Software, San Diego, CA, USA) (29):
| (1) |
where Amax denotes the fully recovered a-wave amplitude, ca represents the degree of reduction immediately after the bleach and τa is the time constant of recovery. These three parameters were calculated for each plot, and statistical significance among WT, heterozygous and homozygous KI mice was analyzed using one-way ANOVA.
Optical coherence tomography (OCT) imaging
Mice were anesthetized and their pupils dilated as described earlier. Artificial tears (Systane Ultra, Alcon, Fort Worth, TX, USA) were used to maintain corneal moisture and clarity. OCT images were obtained using the Bioptigen Spectral Domain Ophthalmic Imaging System (SDOIS; Bioptigen Envisu R2200, Bioptigen, Morrisville, NC, USA). Image acquisition software was provided by vendor. Blue light-induced fundus autofluorescence was recorded with Spectralis HRA-OCT (Heidelberg Engineering, Heidelberg, Germany).
Induction of retinal light damage
Mice were dark-adapted for 7 days before exposure to bright light. Acute retinal damage was induced by exposing WT and KI/KI animals with dilated pupils (as described earlier) to 20 000 lux of diffuse white fluorescent light for 30 min. Eyes were collected following euthanization 1 or 2 h after exposure for retinoid analyses, or, subsequent to OCT imaging, 2 weeks after exposure for histology or TUNEL staining.
Supplementary Material
Funding
This research was supported by the Intramural Research Program of the National Eye Institute, National Institutes of Health (US Department of Health and Human Services).
Supplementary Material
Acknowledgements
We acknowledge the advice of Dr Robert N. Fariss and assistance of Dr Maria M. Campos (Biological Imaging Core, NEI), Dr Jianguo Fan (Molecular Structure and Functional Genomics Section, NEI) and of Dr Baerbel Rohrer (Medical University of South Carolina, Charleston, SC, USA). E.G. and Y.C.-P. were supported by the Diversity in Vision Research and Ophthalmology summer program of the NEI Office of the Director.
Conflict of Interest statement. None declared.
References
- 1.Kiser P.D., Golczak M., Maeda A., Palczewski K. (2012) Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim. Biophys. Acta., 1821, 137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin M., Li S., Moghrabi W.N., Sun H., Travis G.H. (2005) Rpe65 is the retinoid isomerase in bovine retinal pigment epithelium. Cell, 122, 449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moiseyev G., Chen Y., Takahashi Y., Wu B.X., Ma J.X. (2005) RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc. Natl Acad. Sci. USA, 102, 12413–12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Redmond T.M., Poliakov E., Yu S., Tsai J.Y., Lu Z., Gentleman S. (2005) Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc. Natl Acad. Sci. USA, 102, 13658–13663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gu S.M., Thompson D.A., Srikumari C.R., Lorenz B., Finckh U., Nicoletti A., Murthy K.R., Rathmann M., Kumaramanickavel G., Denton M.J., et al. (1997) Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat. Genet., 17, 194–197. [DOI] [PubMed] [Google Scholar]
- 6.Marlhens F., Bareil C., Griffoin J.M., Zrenner E., Amalric P., Eliaou C., Liu S.Y., Harris E., Redmond T.M., Arnaud B., et al. (1997) Mutations in RPE65 cause Leber's congenital amaurosis. Nat. Genet., 17, 139–141. [DOI] [PubMed] [Google Scholar]
- 7.Thompson D.A., Gyurus P., Fleischer L.L., Bingham E.L., McHenry C.L., Apfelstedt-Sylla E., Zrenner E., Lorenz B., Richards J.E., Jacobson S.G., et al. (2000) Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Invest. Ophthalmol. Vis. Sci., 41, 4293–4299. [PubMed] [Google Scholar]
- 8.Lorenz B., Gyurus P., Preising M., Bremser D., Gu S., Andrassi M., Gerth C., Gal A. (2000) Early-onset severe rod-cone dystrophy in young children with RPE65 mutations. Invest. Ophthalmol. Vis. Sci., 41, 2735–2742. [PubMed] [Google Scholar]
- 9.Yzer S., van den Born L.I., Schuil J., Kroes H.Y., van Genderen M.M., Boonstra F.N., van den Helm B., Brunner H.G., Koenekoop R.K., Cremers F.P. (2003) A Tyr368His RPE65 founder mutation is associated with variable expression and progression of early onset retinal dystrophy in 10 families of a genetically isolated population. J. Med. Genet., 40, 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marlhens F., Griffoin J.M., Bareil C., Arnaud B., Claustres M., Hamel C.P. (1998) Autosomal recessive retinal dystrophy associated with two novel mutations in the RPE65 gene. Eur. J. Hum. Genet., 6, 527–531. [DOI] [PubMed] [Google Scholar]
- 11.Lorenz B., Poliakov E., Schambeck M., Friedburg C., Preising M.N., Redmond T.M. (2008) A comprehensive clinical and biochemical functional study of a novel RPE65 hypomorphic mutation. Invest. Ophthalmol. Vis. Sci., 49, 5235–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Redmond T.M., Yu S., Lee E., Bok D., Hamasaki D., Chen N., Goletz P., Ma J.X., Crouch R.K., Pfeifer K. (1998) Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat. Genet., 20, 344–351. [DOI] [PubMed] [Google Scholar]
- 13.Doyle S.E., Castrucci A.M., McCall M., Provencio I., Menaker M. (2006) Nonvisual light responses in the Rpe65 knockout mouse: rod loss restores sensitivity to the melanopsin system. Proc. Natl Acad. Sci. USA, 103, 10432–10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grimm C., Wenzel A., Hafezi F., Yu S., Redmond T.M., Reme C.E. (2000) Protection of Rpe65-deficient mice identifies rhodopsin as a mediator of light-induced retinal degeneration. Nat. Genet., 25, 63–66. [DOI] [PubMed] [Google Scholar]
- 15.Lyubarsky A.L., Savchenko A.B., Morocco S.B., Daniele L.L., Redmond T.M., Pugh E.N., Jr. (2005) Mole quantity of RPE65 and its productivity in the generation of 11-cis-retinal from retinyl esters in the living mouse eye. Biochemistry, 44, 9880–9888. [DOI] [PubMed] [Google Scholar]
- 16.Rohrer B., Lohr H.R., Humphries P., Redmond T.M., Seeliger M.W., Crouch R.K. (2005) Cone opsin mislocalization in Rpe65-/- mice: a defect that can be corrected by 11-cis retinal. Invest. Ophthalmol. Vis. Sci., 46, 3876–3882. [DOI] [PubMed] [Google Scholar]
- 17.Seeliger M.W., Grimm C., Stahlberg F., Friedburg C., Jaissle G., Zrenner E., Guo H., Reme C.E., Humphries P., Hofmann F., et al. (2001) New views on RPE65 deficiency: the rod system is the source of vision in a mouse model of Leber congenital amaurosis. Nat. Genet., 29, 70–74. [DOI] [PubMed] [Google Scholar]
- 18.Tu D.C., Owens L.A., Anderson L., Golczak M., Doyle S.E., McCall M., Menaker M., Palczewski K., Van Gelder R.N. (2006) Inner retinal photoreception independent of the visual retinoid cycle. Proc. Natl Acad. Sci. USA, 103, 10426–10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Hooser J.P., Aleman T.S., He Y.G., Cideciyan A.V., Kuksa V., Pittler S.J., Stone E.M., Jacobson S.G., Palczewski K. (2000) Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc. Natl Acad. Sci. USA, 97, 8623–8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodruff M.L., Wang Z., Chung H.Y., Redmond T.M., Fain G.L., Lem J. (2003) Spontaneous activity of opsin apoprotein is a cause of Leber congenital amaurosis. Nat. Genet., 35, 158–164. [DOI] [PubMed] [Google Scholar]
- 21.Pang J.J., Chang B., Hawes N.L., Hurd R.E., Davisson M.T., Li J., Noorwez S.M., Malhotra R., McDowell J.H., Kaushal S., et al. (2005) Retinal degeneration 12 (rd12): a new, spontaneously arising mouse model for human Leber congenital amaurosis (LCA). Mol. Vis., 11, 152–162. [PubMed] [Google Scholar]
- 22.Samardzija M., von Lintig J., Tanimoto N., Oberhauser V., Thiersch M., Reme C.E., Seeliger M., Grimm C., Wenzel A. (2008) R91W mutation in Rpe65 leads to milder early-onset retinal dystrophy due to the generation of low levels of 11-cis-retinal. Hum. Mol. Genet., 17, 281–292. [DOI] [PubMed] [Google Scholar]
- 23.El Matri L., Ambresin A., Schorderet D.F., Kawasaki A., Seeliger M.W., Wenzel A., Arsenijevic Y., Borruat F.X., Munier F.L. (2006) Phenotype of three consanguineous Tunisian families with early-onset retinal degeneration caused by an R91W homozygous mutation in the RPE65 gene. Graefes Arch. Clin. Exp. Ophthalmol., 244, 1104–1112. [DOI] [PubMed] [Google Scholar]
- 24.Lewandoski M., Wassarman K.M., Martin G.R. (1997) Zp3-cre, a transgenic mouse line for the activation or inactivation of loxP-flanked target genes specifically in the female germ line. Curr. Biol., 7, 148–151. [DOI] [PubMed] [Google Scholar]
- 25.Danciger M., Matthes M.T., Yasamura D., Akhmedov N.B., Rickabaugh T., Gentleman S., Redmond T.M., La Vail M.M., Farber D.B. (2000) A QTL on distal chromosome 3 that influences the severity of light-induced damage to mouse photoreceptors. Mamm. Genom., 11, 422–427. [DOI] [PubMed] [Google Scholar]
- 26.Wenzel A., Reme C.E., Williams T.P., Hafezi F., Grimm C. (2001) The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J. Neurosci., 21, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiser P.D., Golczak M., Lodowski D.T., Chance M.R., Palczewski K. (2009) Crystal structure of native RPE65, the retinoid isomerase of the visual cycle. Proc. Natl Acad. Sci. USA, 106, 17325–17330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pawar A.S., Qtaishat N.M., Little D.M., Pepperberg D.R. (2008) Recovery of rod photoresponses in ABCR-deficient mice. Invest. Ophthalmol. Vis. Sci., 49, 2743–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas M.M., Lamb T.D. (1999) Light adaptation and dark adaptation of human rod photoreceptors measured from the a-wave of the electroretinogram. J. Physiol., 518 (Pt 2), 479–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samardzija M., Wenzel A., Naash M., Reme C.E., Grimm C. (2006) Rpe65 as a modifier gene for inherited retinal degeneration. Eur. J. Neurosci., 23, 1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poliakov E., Strunnikova N.V., Jiang J.K., Martinez B., Parikh T., Lakkaraju A., Thomas C., Brooks B.P., Redmond T.M. (2014) Multiple A2E treatments lead to melanization of rod outer segment-challenged ARPE-19 cells. Mol. Vis., 20, 285–300. [PMC free article] [PubMed] [Google Scholar]
- 32.Sparrow J.R., Wu Y., Kim C.Y., Zhou J. (2010) Phospholipid meets all-trans-retinal: the making of RPE bisretinoids. J. Lipid Res., 51, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cideciyan A.V., Jacobson S.G., Beltran W.A., Sumaroka A., Swider M., Iwabe S., Roman A.J., Olivares M.B., Schwartz S.B., Komaromy A.M., et al. (2013) Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc. Natl Acad. Sci. USA, 110, E517–E525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maeda A., Palczewski K. (2013) Retinal degeneration in animal models with a defective visual cycle. Drug Discov. Today Dis. Models, 10, e163–e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qtaishat N.M., Redmond T.M., Pepperberg D.R. (2003) Acute radiolabeling of retinoids in eye tissues of normal and rpe65-deficient mice. Invest. Ophthalmol. Vis. Sci., 44, 1435–1446. [DOI] [PubMed] [Google Scholar]
- 36.Batten M.L., Imanishi Y., Maeda T., Tu D.C., Moise A.R., Bronson D., Possin D., Van Gelder R.N., Baehr W., Palczewski K. (2004) Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J. Biol. Chem., 279, 10422–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu L., Gudas L.J. (2005) Disruption of the lecithin:retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency. J. Biol. Chem., 280, 40226–40234. [DOI] [PubMed] [Google Scholar]
- 38.Rozanowska M., Sarna T. (2005) Light-induced damage to the retina: role of rhodopsin chromophore revisited. Photochem. Photobiol., 81, 1305–1330. [DOI] [PubMed] [Google Scholar]
- 39.Rozanowska M.B. (2012) Light-induced damage to the retina: current understanding of the mechanisms and unresolved questions: a symposium-in-print. Photochem. Photobiol., 88, 1303–1308. [DOI] [PubMed] [Google Scholar]
- 40.Organisciak D.T., Vaughan D.K. (2010) Retinal light damage: mechanisms and protection. Prog. Retin. Eye Res., 29, 113–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y., Okano K., Maeda T., Chauhan V., Golczak M., Maeda A., Palczewski K. (2012) Mechanism of all-trans-retinal toxicity with implications for stargardt disease and age-related macular degeneration. J. Biol. Chem., 287, 5059–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bowne S.J., Humphries M.M., Sullivan L.S., Kenna P.F., Tam L.C., Kiang A.S., Campbell M., Weinstock G.M., Koboldt D.C., Ding L., et al. (2011) A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur. J. Hum. Genet., 19, 1074–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang J.S., Kefalov V.J. (2011) The cone-specific visual cycle. Progr. Retin. Eye Res., 30, 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xue Y., Shen S.Q., Jui J., Rupp A.C., Byrne L.C., Hattar S., Flannery J.G., Corbo J.C., Kefalov V.J. (2015) CRALBP supports the mammalian retinal visual cycle and cone vision. J. Clin. Invest., 125, 727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim T.S., Maeda A., Maeda T., Heinlein C., Kedishvili N., Palczewski K., Nelson P.S. (2005) Delayed dark adaptation in 11-cis-retinol dehydrogenase-deficient mice: a role of RDH11 in visual processes in vivo. J. Biol. Chem., 280, 8694–8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chrispell J.D., Feathers K.L., Kane M.A., Kim C.Y., Brooks M., Khanna R., Kurth I., Hubner C.A., Gal A., Mears A.J., et al. (2009) Rdh12 activity and effects on retinoid processing in the murine retina. J. Biol. Chem., 284, 21468–21477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saari J.C., Nawrot M., Kennedy B.N., Garwin G.G., Hurley J.B., Huang J., Possin D.E., Crabb J.W. (2001) Visual cycle impairment in cellular retinaldehyde binding protein (CRALBP) knockout mice results in delayed dark adaptation. Neuron, 29, 739–748. [DOI] [PubMed] [Google Scholar]
- 48.Chen P., Hao W., Rife L., Wang X.P., Shen D., Chen J., Ogden T., Van Boemel G.B., Wu L., Yang M., et al. (2001) A photic visual cycle of rhodopsin regeneration is dependent on Rgr. Nat. Genet., 28, 256–260. [DOI] [PubMed] [Google Scholar]
- 49.Muyrers J.P., Zhang Y., Benes V., Testa G., Rientjes J.M., Stewart A.F. (2004) ET recombination: DNA engineering using homologous recombination in E. coli. Methods Mol. Biol., 256, 107–121. [DOI] [PubMed] [Google Scholar]
- 50.Yagi T., Nada S., Watanabe N., Tamemoto H., Kohmura N., Ikawa Y., Aizawa S. (1993) A novel negative selection for homologous recombinants using diphtheria toxin A fragment gene. Anal. Biochem., 214, 77–86. [DOI] [PubMed] [Google Scholar]
- 51.Thomas K.R., Capecchi M.R. (1987) Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell, 51, 503–512. [DOI] [PubMed] [Google Scholar]
- 52.Garwin G.G., Saari J.C. (2000) High-performance liquid chromatography analysis of visual cycle retinoids. Meth. Enzymol., 316, 313–324. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.