

Abstract
Epidermal growth factor-like domain 7 (Egfl7) is a gene that encodes a partially secreted protein and whose expression is largely restricted to the endothelia. We recently reported that EGFL7 is also expressed by trophoblast cells in mouse and human placentas. Here, we investigated the molecular pathways that are regulated by EGFL7 in trophoblast cells. Stable EGFL7 overexpression in a Jeg3 human choriocarcinoma cell line resulted in significantly increased cell migration and invasiveness, while cell proliferation was unaffected. Analysis of mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways showed that EGFL7 promotes Jeg3 cell motility by activating both pathways. We show that EGFL7 activates the epidermal growth factor receptor (EGFR) in Jeg3 cells, resulting in downstream activation of extracellular regulated kinases (ERKs). In addition, we provide evidence that EGFL7-triggered migration of Jeg3 cells involves activation of NOTCH signaling. EGFL7 and NOTCH1 are co-expressed in Jeg3 cells, and blocking of NOTCH activation abrogates enhanced migration of Jeg3 cells overexpressing EGFL7. We also demonstrate that signaling through EGFR and NOTCH converged to mediate EGFL7 effects. Reduction of endogenous EGFL7 expression in Jeg3 cells significantly decreased cell migration. We further confirmed that EGFL7 stimulates cell migration by using primary human first trimester trophoblast (PTB) cells overexpressing EGFL7. In conclusion, our data suggest that in trophoblast cells, EGFL7 regulates cell migration and invasion by activating multiple signaling pathways. Our results provide a possible explanation for the correlation between reduced expression of EGFL7 and inadequate trophoblast invasion observed in placentopathies.
Keywords: EGFL7, MAPK, NOTCH signaling, PI3K, trophoblast migration
Introduction
The placenta is one of the first structures to form during embryonic development and is essential for the survival of the embryo because it facilitates the exchange of gases, nutrients and waste at the feto-maternal interface. Placental development is a process that is fine-tuned by a variety of factors critical for a successful pregnancy. An imbalance of these factors can lead to placentopathies, eventually resulting in embryo-fetal loss and/or maternal death. A comprehensive understanding of the molecular mechanisms underlying placental development is essential for the treatment of placental disorders.
The invasion of trophoblasts into the maternal tissue and vessels is a crucial process during human pregnancy (Pijnenborg et al., 1981). Research has focused on the molecular mechanisms regulating trophoblast invasion and, over the years, an increasing number of factors, cytokines and signal transduction pathways controlling trophoblast migration have been identified (Fitzgerald et al., 2005; Knöfler, 2010). Various growth factors control trophoblast migration through activation of mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways. Epidermal growth factor (EGF), which is abundant at the fetal–maternal interface, activates MAPK as well as PI3K signaling, leading to increased trophoblast migration (Qiu et al., 2004a). Similarly, activation of the MAPK signaling pathway and consequent increased trophoblast motility have been observed in the presence of endothelin, prostaglandin E2, insulin-like growth factor (IGF)-II, insulin-like growth factor binding protein (IGFBP)-1, and chorionic gonadotrophin (hCG) (Gleeson et al., 2001; McKinnon et al., 2001; Chakraborty et al., 2003; Nicola et al., 2008; Prast et al., 2008). hCG and EGF induced activation of MAPKs and PI3K resulted in the induction of matrix metalloproteinase (MMP)-2 and -9, respectively, suggesting that these proteinases are crucial for trophoblast invasion (Qiu et al., 2004b; Prast et al., 2008). Hunkapiller et al. recently demonstrated that Notch signaling is also involved in trophoblast endovascular invasion. Invasion of primary cytotrophoblats and their expression of the arterial marker ephrin B2 (EFNB2) were significantly decreased by an inhibitor of the γ-secretase pathway, which is commonly used to block Notch activity. In addition, Notch2 down-regulation led to reduced arterial invasion, decreased size of maternal blood vessels and reduced placental perfusion (Hunkapiller et al., 2011).
Recent studies suggested that the secreted factor epidermal growth factor-like domain 7 (EGFL7) may also play a role in this context (Lacko et al., 2014). Egfl7 was originally identified as an endothelial-restricted gene. High levels of EGFL7 expression are observed in actively proliferating endothelial cells (ECs), both during embryogenesis and during physiologic and pathologic angiogenesis (Soncin et al., 2003; Fitch et al., 2004; Parker et al., 2004; Campagnolo et al., 2005; Bambino et al., 2014). During embryonic development, Egfl7 is expressed as early as in pre- and peri-implantation embryos, starting from the 8-cell stage, and in embryonic stem cells that are derived from the inner cell mass (ICM) of the blastocyst (Fitch et al., 2004; Campagnolo et al., 2008). In the mouse, Egfl7 controls embryonic survival and vascular development (Schmidt et al., 2007; Nichol et al., 2010) by promoting EC proliferation, migration, sprouting and invasion (Campagnolo et al., 2005; Schmidt et al., 2007; Durrans and Stuhlmann 2010; Nikolic et al., 2013). In addition to the endothelium, more recently novel sources of EGFL7 have been discovered, including primordial germ cells, adult neural stem cells and cancer cells of various human tumors (Campagnolo et al., 2008; Diaz et al. 2007; Schmidt et al., 2009; Wu et al., 2009; Huang et al., 2010; Delfortrie et al., 2011). In addition, we have recently shown that trophoblast cells in the placenta are a novel source of EGFL7. Importantly, EGFL7 expression is dramatically reduced in both the endothelial and the trophoblastic compartments of pre-eclamptic (PE) placentas (Lacko et al., 2014). PE is a pregnancy-specific disease that originates in the placenta and it is a leading cause of morbidity and mortality for both the mother and fetus. Hallmarks of PE include inadequate invasion of trophoblast cells into the maternal decidua, dysregulated growth of fetal blood vessels, disruption of maternal–fetal vascular connections, and an imbalance of pro- and anti-angiogenic growth factors (Young et al., 2010).
The present study aims to clarify the role of EGFL7 and identify EGFL7-dependent molecular pathways in trophoblast cells by using genetic manipulation of the choriocarcinoma cell line Jeg3 and primary trophoblast cells isolated from first trimester human placentas.
Materials and Methods
Reagents and antibodies
The following inhibitors were used in this study: mitogen-activated protein/extracellular signal-regulated kinase kinase (MEK1/2)-inhibitor U0126 (5 and 10 µM; Calbiochem, La Jolla, CA, USA), PI3K-inhibitor Wortmannin (100 nM; Sigma-Aldrich, Saint Louis, MO, USA), epidermal growth factor receptor (EGFR)-inhibitor AG1478 (1 µM), and γ-secretase inhibitor dual anti-platelet therapy (DAPT; 10 and 25 µM; Sigma-Aldrich). Mouse culture grade EGF (10 ng/ml; Collaborative Research, Waltham, MA, USA) was used to activate EGFR.
The following primary antibodies were used: two goat anti-human EGFL7 (for immunofluorescence and western blot experiments we used anti-EGFL7 from R&D Systems, Wiesbaden, Germany, at 2 and 0.2 µg/ml, respectively; for western blot we also used anti-EGFL7 from Santa Cruz Biotechnology, Santa Cruz, CA, USA, at 0.2 µg/ml), mouse anti-green fluorescent protein (GFP) (1:2500; Millipore, Billerica, MA, USA), rabbit anti-cow Cytokeratin (CK) (53.5 µg/ml, total immunoglobulin fraction; DakoCytomation, Glostrup, Denmark), mouse anti-5-bromo-2-deoxyuridine (BrdU) (1:2.5; Becton Dickinson, San Jose, CA, USA), rabbit anti-phospho- extracellular regulated kinase 1 and 2 (pERK1/2) (1:1000; Cell Signaling Technologies, Danvers, MA, USA), rabbit anti-Erk1/2 (1:1000; Cell Signaling Technologies), rabbit anti-phospho-protein kinase B (pAKT) (1:1000; Cell Signaling Technologies), rabbit anti-Akt (0.2 µg/ml; Santa Cruz Biotechnology), rabbit anti-phospho-EGFR (1:1000; Cell Signaling Technologies), rabbit anti-human EGFR (1:100 for immunofluorescence experiments and 1:1000 for western blot analysis; Cell Signaling Technologies), rabbit anti-human NOTCH1 (2 µg/ml; Abcam, Cambridge, UK), rabbit anti-human NOTCH4 (2 µg/ml; Santa Cruz Biotechnology) and mouse anti-α-tubulin (1 µg/ml; Sigma-Aldrich). Secondary antibodies were as follows: Cy3-donkey-anti-goat (4 µg/ml; Jackson ImmunoResearch, West Grove, PA, USA), Alexa Fluor568 goat-anti-mouse (4 µg/ml; Invitrogen in Life Technologies, Monza, Italy), Alexa Fluor488 donkey-anti-goat (4 µg/ml; Invitrogen), Cy3-donkey-anti-rabbit (4 µg/ml; Millipore) and FITC-donkey-anti-rabbit (4 µg/ml; Millipore). Secondary anti-goat, anti-rabbit or anti-mouse antibodies conjugated to horseradish peroxidase were purchased from Amersham Biosciences (Piscataway, NJ, USA).
Cell culture
The human choriocarcinoma cell line Jeg3 was purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA). Jeg3 cells were cultured in Eagle Minimum Essential Medium (EMEM with l-Glutamine; Lonza, Milan, Italy), supplemented with 10% (v/v) fetal bovine serum (FBS; Lonza), 2 mM l-glutamine (Lonza), 50 U/ml penicillin and 50 μg/ml streptomycin (Lonza), and 1 mM sodium pyruvate (Sigma-Aldrich), in 5% CO2 at 37°C.
Human Embryonic Kidney 293 (HEK293) cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM with 25 mM d-glucose; Lonza), supplemented with 10% (v/v) FBS, 20 mM N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid (HEPES; Lonza), 2 mM l-glutamine, 50 U/ml penicillin, and 50 µg/ml streptomycin, at 37°C in a humidified atmosphere of 5% CO2 in air.
Human Umbilical Vein Endothelial Cells (HUVECs) were purchased from Lonza and cultured in EGM-2 Bullet kit (Lonza) at 37°C in a humidified atmosphere of 5% CO2 in air.
Primary human trophoblast (PTB) cells were isolated from first trimester villous samples according to the protocol of Hunkapiller and Fisher (2008). Samples were obtained from patients undergoing prenatal diagnosis, after obtaining informed consent. All procedures for the collection and manipulation of human samples comply with the principles reported in the Declaration of Helsinki and have been approved by the Bioethical Committee of the University of Tor Vergata. Purity of PTB was ∼90–93%, when assessed by flow cytometry analysis as previously reported (Spitalieri et al., 2009), and confirmed by immunostaining using anti-cytokeratin antibodies. PTB cells were cultured on Matrigel-coated dishes in Dulbecco's Modified Eagle's Medium (DMEM with 25 mM d-glucose, Lonza), supplemented with 10 mM HEPES, 4 mM l-glutamine, 50 U/ml penicillin, 50 µg/ml streptomycin, 50 μg/ml Gentamycin (Lonza), and 2% (v/v) Nutridoma (Roche Diagnostics GmbH, Mannheim, Germany). The cells were kept at 37°C in a humidified atmosphere of 5% CO2 in air.
Lentiviral transduction
A human EGFL7 cDNA was engineered into the pCCLIRESGFP vector provided by Dr. Stefano Rivella at Weill Cornell Medical College. MISSION shRNA lentiviral plasmid TRCN0000053661 (pLKO61), and the control pLKO1 were purchased from Sigma-Aldrich. HEK293 cells in subconfluent culture condition (70–80% confluency) were transfected with a third-generation lentiviral vector system, consisting of the packaging plasmids (pMDL, pREV and pVSVG) and the transfer vector containing either the EGFL7 and GFP transgene (pCCL-EGFL7) or GFP only (pCCL-GFP) using polyethylenimine (2 mg/ml; Polysciences, Warrington, PA, USA). The same protocol was followed for transfection using the shRNA plasmid pLKO61 or the control pLKO1. After 2 days, cell supernatant was collected, filtered and centrifuged at 16 000 g for 2 h at 4°C to concentrate viral particles to half of the initial volume. Concentrated viral suspension was used to infect subconfluent cultures of Jeg3 or PTB. Polybrene (8 µg/ml; Sigma-Aldrich) was used to neutralize cell surface charge and increase the efficiency of infection. Cells over-expressing or knocked-down for EGFL7 were named as follow:
JegGFP: Jeg3 cells infected with empty vector containing GFP;
JegE7: Jeg3 overexpressing EGFL7;
JegKDSCR: Jeg3 cells infected with scrambled vector pLKO1;
JegKDE7: Jeg3 infected with shRNA plasmid pLKO.61;
PTBGFP: PTB cells infected with empty vector containing GFP;
PTBE7: PTB cells overexpressing EGFL7;
EGFL7 expression was determined by quantitative reverse-transcribed polymerase chain reaction (qRT–PCR) and western blot analysis.
RNA isolation and quantitative RT–PCR
RNA from cell cultures was prepared using the TRIZOL Reagent (Roche Diagnostics GmbH) according to the manufacturer's protocol. Possible DNA contamination was removed by DNAse treatment. RNA quality was examined on agarose gels. mRNA was reverse transcribed using random primers and the Superscript First Strand Synthesis System (Invitrogen) following the manufacturer's specifications. Gene expression was measured using Real Master Mix SYBR ROX (Eppendorf, Hamburg, Germany). qRT–PCR was performed using an Applied Biosystems 7300 Real Time PCR System (Applied Biosystems). Differences among gene expression were quantified using the ΔΔCt method with normalization to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Specific primers for EGFL7, NOTCH1, hairy and enhancer of split-related protein 2 (HEY2), hairy and enhancer of split-related protein 1 (HEY1) and hairy and enhancer of split-1 (HES1) were designed using Primer Express software (Applied Biosystems in Life Technologies, Monza, Italy); sequences are listed below:
EGFL7:5′-TCGTGCAGCGTGTGTACCAG-3′
5′-GCGGTAGGCGGTCCTATAGATG-3′
NOTCH1: 5′- GCGGGATCCACTGTGAGAA -3′
5′- CCGTTGAAGCAGGAGCTCTCT -3′;
HEY2: 5′-CGACCTCCGAGAGCGACAT-3′
5′-CTTTGCCCCGAGTAATTGTTCT-3′
HEY1: 5′-CATCGAGGTGGAGAAGGAGAGT-3′
5′-GACATGGAACCTAGAGCCGAACT-3′
HES1: 5′-AAAGATAGCTCGCGGCATTC-3′
5′-AGGTGCTTCACTGTCATTTCCA-3′
CK7: 5′-AAGGTGGATGCCCTGAATGA-3′
5′-CAGCTCTGTCAACTCCGTCTCA-3′
GAPDH: 5′-TCGGAGTCAACGGATTTGGT-3′
5′-GAATTTGCCATGGGTGGAAT-3′
To evaluate the blockage of NOTCH activity in JegE7, cells were treated with 10 µM U0126 or/and 25 µM DAPT in serum-free culture medium for 90 min, followed by RNA extraction as described above.
To analyze miR-126 expression, total RNA from cell cultures was extracted using mirVana miRNA Isolation Kit (Ambion in Life Technologies, Monza, Italy) following the manufacturer's protocol to ensure the recovery of small RNA. MicroRNA was reverse transcribed using microRNA-specific primers and the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems). miR-126 expression was analyzed using the Taqman MicroRNA Assay (Applied Biosystems) and normalized to that of U18. Changes were quantified using the ΔΔCT method.
Immunofluorescence analysis
Cells cultured at subconfluent conditions were fixed in methanol for 5 min at −20°C; non-specific antibody binding was blocked by incubation in 10% (v/v) donkey serum/phosphate buffered saline (PBS) for 1 h at room temperature. Antibodies used for immunofluorescence (IF) analysis and Hoechst 33342 dye (0.5 µg/ml; Sigma-Aldrich) for cell nuclei counterstaining, were diluted in 0.1% (w/v) bovine serum albumin (BSA; Sigma-Aldrich) in PBS (Lonza) and incubated overnight at 4°C. Cells were mounted with Möwiol (Sigma-Aldrich). All images were acquired using an Axioplan 2 imaging microscope (Zeiss, Thornwood, NY, USA).
Western blot analysis
JegGFP and JegE7 cells were seeded in 6-well plates and cultured in serum-supplemented EMEM as reported above. After three washes in PBS, cells were homogenized in lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.5% NP-40, 5 mM ethylenediaminetetraacetic acid (EDTA), 0.5% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride, 20 mM β-glycerophosphate, 1 mM sodium orthovanadate) containing EDTA-free protease inhibitor cocktail (Roche, Penzberg, Germany). To evaluate EGFL7-mediated activation of the EGFR pathway, 80% confluent cultures were serum-starved for 18 h, prior to protein extraction. In parallel experiments, serum-starved JegGFP cells were treated with 10 ng/ml EGF for 30 min. To block activation of EGFR, cells were treated with 1 µM AG1478 in EMEM for 30 min. To assess the possible convergence of the EGFR and Notch pathways, JegE7 and JegGFP cells were treated with 25 µM DAPT and/or 10 µM U0126 in serum-free culture medium for 60 min. Protein concentration was determined using a Bradford assay. Protein samples (40 µg) were separated by electrophoresis on either 10% (v/v) or 8% (v/v) sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) gels and transferred to polyvinylidene difluoride (PVDF) Transfer Membrane Hybond™ (Amersham Biosciences). Membranes were blocked with 5% (w/v) non-fat dry milk in Tris-buffered saline (TBS) containing 0.1% (v/v) Tween 20 (TBS/T) for 1 h at room temperature and incubated overnight at 4°C with the following primary antibodies: goat anti-EGFL7, mouse anti-GFP, rabbit anti-phospho-Erk1/2, rabbit anti-phospho-Akt, and rabbit anti-phospho-EGFR (all diluted 1:1000 in TBS/T with 5% (w/v) BSA). Secondary antibodies, conjugated to horseradish peroxidase (Amersham Biosciences), were incubated for 1 h at room temperature at 1:10 000 dilution in 5% (w/v) non-fat dry milk containing TBS/T. Immunoreactive bands were detected by LiteAblot Plus and LiteAblot Turbo chemiluminescent substrate (Euroclone, Pero, Italy) according to the manufacturer's protocol. Densitometric analysis of the bands was performed using ImageJ software. For normalization of protein expression, membranes were stripped using the Restore Western Blot Stripping Buffer (Thermo Scientific, Rockford, IL, USA) following the manufacturer's protocol, and re-probed with mouse anti-α-tubulin, rabbit anti-Erk1/2, rabbit anti-Akt, or rabbit anti-EGFR.
Cell proliferation
JegGFP and JegE7 cells were plated at a density of 5 × 104 cells/well in 24-well plates in triplicate; cumulative cell numbers were counted every day up to 4 days of culture using a Neubauer chamber (Blaubrand, Wertheim, Germany). In parallel, cell number was evaluated using the 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-1) (Cell Proliferation Reagent WST-1, Roche Diagnostics, Indianapolis, IN, USA) and the BrdU (Amersham) assay. For WST-1, JegGFP and JegE7 cells were plated at a density of 3 × 103 cells/well in 96-well plates and proliferation of the cells was evaluated every day up to 4 days using the WST-1 assay following the manufacturer's specifications. For BrdU incorporation, JegGFP and JegE7 cells were plated at a density of 5 × 104 cells/well in 24-well plates. At 24 h from the plating, BrdU labeling reagent (1:1000; Amersham) was added to the cultures. After 1 h of incubation at 37°C, cells were fixed in 70% (v/v) ethanol, incubated 2 min in 0.07N NaOH and rinsed with PBS to equilibrate the pH. Following the manufacturer specification, anti-BrdU primary antibody, diluted in 0.5% (v/v) Tween 20 in PBS, was incubated for 30 min at room temperature. After several washings in PBS, cells were incubated with a goat anti-mouse Alexa Fluor568 secondary antibody for 1 h at room temperature, counterstained with Hoechst 33342 (0.5 µg/ml; Sigma-Aldrich), and mounted with Möwiol (Sigma-Aldrich). Images were acquired using an Axioplan 2 imaging microscope (Carl Zeiss).
Wounding assay
JegGFP, JegE7, JegKDSCR and JegKDE7 were plated at a density of 2 × 105 cells/well in 24-well plates and incubated for 24 h in normal culture medium. After reaching confluency, a thin scratch was performed on the monolayer using a 0.1–10 µl pipette tip. To block MAPK, PI3K, EGFR and NOTCH pathways, cells were incubated with 5 µM U0126, 100 nM Wortmannin, 1 µM AG1478 or 10 µM DAPT, respectively. The stripped region was photographed at 0, 8, 24 and 32 h, using a Leitz Diavert microscope connected to a Nikon DS-Fi1 camera. The extension of the wounded area was measured at different time points using the ImageJ software (Schneider et al., 2012). Values, expressed in pixels, were used to calculate the area filled according to the formula: area of the scratch at time 0 − area of the scratch at time X.
Transwell migration and invasion assay
Transwell assays were performed using BD Falcon™ cell culture inserts (BD Biosciences, Franklin Lakes, NJ, USA) with pore sizes of 8-µm. Inserts were used either uncoated to evaluate cell migration, or coated with 30 µl of growth-factor-reduced-Matrigel (BD Biosciences) to study cell invasion. JegGFP, JegE7, PTBGFP and PTBE7 (1 × 106 cells/ml) were seeded in 100 µl serum-free medium in the upper chambers of the inserts located in 24-well plates, and serum-supplemented culture medium was placed into the lower compartment as a chemoattractant. Migration and invasion were evaluated after 24 and 48 h incubation, respectively, at 37°C in a humidified atmosphere of 5% CO2 in air. Cells in the upper chamber were carefully removed from the transwell membrane using cotton swabs; efficient removal was checked under the microscope. Transwells were fixed in 10% (w/v) trichloroacetic acid and stained using KaryoMAX Giemsa stain solution (Gibco by Life Technologies, Monza, Italy) diluted 1:17 in Gurr phosphate buffer (Gurr buffer tablets, Gibco). Insert membranes were air dried, excised from the transwell cast and positioned on a glass slide with the lower side facing up, and mounted with Möwiol (Sigma-Aldrich). Using a light microscope, the number of migrated cells was evaluated by counting six random fields at 10× magnification for each assay, and the extent of migration was expressed as an average number of cells per microscopic field.
Statistical analysis
All experiments were performed at least three times. Data were expressed as mean ± standard error (SE) and analyzed using the one-way analysis of variance (ANOVA) test. Asterisks indicate the level of statistical significance (*P < 0.05; **P < 0.001).
Results
EGFL7 promotes Jeg3 cell migration and invasion
In order to investigate the role of EGFL7 in trophoblast cells, we stably overexpressed EGFL7 in the human choriocarcinoma cell line Jeg3. qRT–PCR indicated that endogenous EGFL7 expression was readily detectable in such cells (Supplementary Fig. S1A and B); however, transcript levels were 200-fold lower in Jeg3 when compared with HUVEC cells (Supplementary Fig. S1B), known to express high levels of EGFL7 (Fitch et al., 2004). Even lower transcript levels were observed for the EGFL7-associated miR-126 (Supplementary Fig. S1C). To stably overexpress EGFL7, we generated a lentiviral vector containing the human EGFL7 cDNA and a GFP reporter gene expressed from a bicistronic mRNA under the control of the PGK promoter (Fig. 1A), and infected Jeg3 cells with the lentivirus (JegE7, Fig. 1C). Jeg3 cells infected with a lentivirus vector containing the GFP reporter but no EGFL7 cDNA were used as a control (JegGFP, Fig. 1B). Efficiency of infection was estimated to be higher than 90%, as judged by the fraction of GFP-positive cells (Fig. 1B and C). qRT–PCR demonstrated ∼700-fold increase in EGFL7 expression in JegE7 compared with JegGFP cells (Fig. 1D), while miR-126 expression level was not affected (Fig. 1E). Western blot analysis confirmed that expression of EGFL7 in JegE7 cells was also increased at the protein level (Fig. 1F–G).
Figure 1.
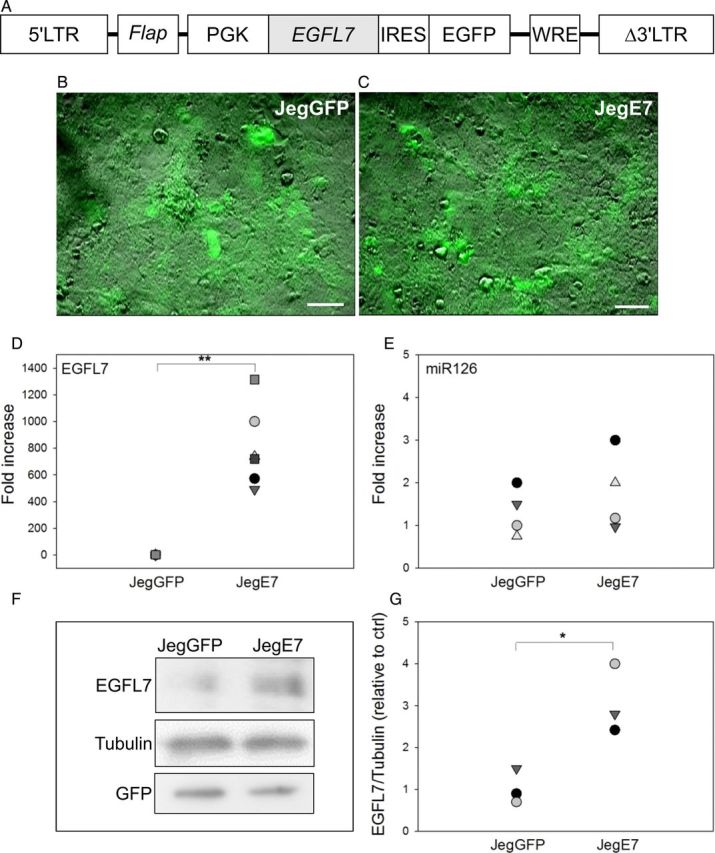
Generation of Jeg3 cells overexpressing epidermal growth factor-like domain 7 (EGFL7). (A) Schematic representation of lentiviral vector containing the human EGFL7 cDNA, an internal ribosome entry site (IRES) and a green fluorescent protein (GFP) reporter gene under the control of a phosphoglycerate kinase 1 (PGK) promoter. (B and C) GFP-positive Jeg3 cells transduced with the empty vector control (JegGFP) (B) and containing an EGFL7 cDNA (JegE7) (C). (D and E) Quantitative RT–PCR data for EGFL7 (D) and miR126 (E) in JegGFP and JegE7 cells. *P < 0.05; **P < 0.001. (F–G) Western blot analysis for EGFL7 (F) and its densitometric analysis (G). Scale bars = 50 µm.
To investigate if EGFL7 is involved in the migration and invasion of trophoblast cells, we used both wounding and transwell assays. In the wounding assay, migration of Jeg3 cells into the wounded area was monitored at 0, 8 and 24 h of culture (Fig. 2A and B). Overexpression of EGFL7 resulted in significantly increased cell migration capability by 8 h, when JegE7 cells had covered a surface of the wound that was about double of that covered by JegGFP cells (Fig. 2A and B). The higher migration activity of JegE7 cells in comparison to JegGFP was maintained also after 24 h (Fig. 2A and B) and 32 h (data not shown). To examine whether the effects of EGFL7 overexpression on Jeg3 cell migration and invasion can be ascribed to an increased proliferation rate, cell counts (Supplementary Fig. S2A) and the WST-1 assay (Supplementary Fig. S2B) to evaluate cell proliferation as metabolic activity were performed. In addition, a BrdU assay was used to evaluate cell proliferation as DNA replication (Supplementary Fig. S2C and D). However, neither assay revealed any differences between JegE7 and JegGFP cells (Supplementary Fig. S2), suggesting that EGFL7 promotes Jeg3 cell migration and invasion, rather than cell proliferation. To confirm further, the stimulating effect of EGFL7 overexpression on Jeg3 cell migration, transwell migration assays were performed. After 24 h of culture, the number of cells moving through the filters toward the lower chamber containing the culture medium supplemented with FBS was counted (Fig. 2C–E). EGFL7 overexpression significantly increased the number of migrating Jeg3 cells by 4-fold (Fig. 2C–E). JegE7 cells also invaded transwell chambers coated with a thick Matrigel layer more efficiently than JegGFP after 48 h of culture (Fig. 2F–H).
Figure 2.

Epidermal growth factor-like domain 7 (EGFL7) stimulates migration and invasion of Jeg3 cells. (A) Wounding assay at 0, 8 and 24 h. Dotted lines indicate the edges of the monolayer. (B) Quantification of Jeg3 migration in wounding assay; the dot plots represent the area filled after 8 and 24 h. (C–E) JegGFP (control) (C) and JegE7 (overexpressing EGFL7) (D) cells migrated under the filter of a transwell chamber after 24 h of culture. Quantification of cell migration is shown in the dot plot graph (E). (F–H) JegGFP (F) and JegE7 (G) that invaded into the Matrigel and migrated under the filter of the transwell chamber after 48 h of culture. Quantification of cell invasion is shown in the dot plot graph (H). All data are represented as cells counted in each of at least six different fields for each experiment. *P < 0.05, **P < 0.001. Scale bars (A) = 120 µm; (C;D;F;G) = 60 µm.
EGFL7 knockdown reduces migration of Jeg3 cells
To further investigate the involvement of EGFL7 in Jeg3 cell migration, we used lentivirus-mediated knockdown of endogenous EGFL7 expression.
qRT–PCR analysis demonstrated a 60% reduction of EGFL7 expression in Jeg3 cells infected with the lentivirus knockdown vector for EGFL7 (JegKDE7) compared with the scrambled infected cells (JegKDSCR) (Fig. 3A). Using the wounding assay, we observed that at 8 h post-wounding, JegKDE7 cells had covered an area that was about five times less than that covered by JegKDSCR cells (Fig. 3B and C). Reduced cell migration of JegKDE7 compared with JegKDSCR was confirmed at 24 (Fig. 3B and C) and 32 h (data not shown).
Figure 3.

Epidermal growth factor-like domain 7 (EGFL7) knockdown reduces migration of Jeg3 cells. (A) Quantitative RT–PCR data for EGFL7 in scrambled control (JegKDSCR) and EGFL7 knockdown Jeg3 cells (JegKDE7). (B) Wounding assay at 0, 8 and 24 h. Dotted lines indicate the edges of the monolayer. (C) Quantification of Jeg3 migration in wounding assay, represented as dot plot showing the area filled after at 8 and 24 h. *P < 0.05, **P < 0.001. Scale bars = 120 µm.
EGFL7 promotes migration of primary human first trimester trophoblast cells
To support our findings obtained in the Jeg3 cell line, we isolated primary trophoblast (PTB) cells from first trimester human chorionic villus samples. Isolated PTB expressed the trophoblast marker CK by IF (Supplementary Fig. S3A) and the mRNA for CK7, which is a specific marker of the trophoblast (Potgens et al., 2001) (Supplementary Fig. S3B). When EGFL7 was overexpressed in PTB cells using lentivirus vectors (PTBE7), by qRT–PCR we observed an estimated 400-fold increase of EGFL7 mRNA levels in PTBE7 compared with PTBGFP cells (Fig. 4A). Similarly to what was observed in JegE7, miR-126 levels were not affected (data not shown).
Figure 4.
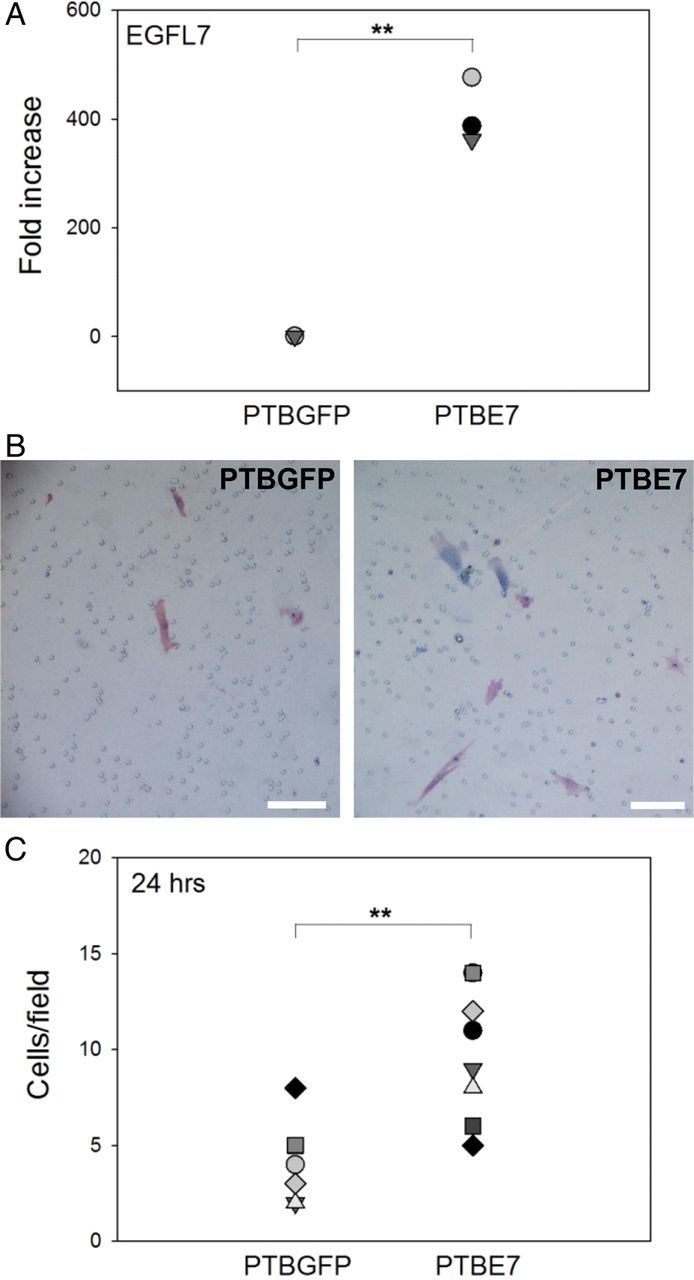
Generation of long-term primary human first trimester trophoblast (PTB) cells overexpressing epidermal growth factor-like domain 7 (PTBE7). EGFL7 stimulates migration of PTB cells. (A) Quantitative RT–PCR data for EGFL7. (C and B) PTBGFP (control) and PTBE7 cells migrated under the filter of transwell chamber after 24 h of culture. Quantification of cell migration is shown in the dot plot graph (F). **P < 0.001. Scale bar (A-B) = 50 µm; (D-E) = 100 µm.
To examine whether EGFL7 promotes cell migration also in PTB cells, we performed transwell assays using PTBE7 and PTBGFP cells. Overall, PTB cells appeared to migrate slower than Jeg3 cells. However, similar to our results using Jeg3 cells, the number of cells moving toward the lower side of the filter was significantly higher in PTBE7 than in PTBGFP cells after 24 h (Fig. 4B and C).
EGFL7 stimulates Jeg3 cell migration by activating MAPK and PI3K signaling pathways
Activation of the MAPK and PI3K signaling pathways plays a major role in trophoblast migration (Qiu et al., 2004a; Prast et al., 2008). We therefore investigated whether overexpression of EGFL7 induced activation of these pathways. Protein extracts from serum-starved JegGFP and JegE7 were analyzed by western blot for levels of ERK and AKT phosphorylation. Approximately 2-fold higher levels of the phosphorylated forms of ERKs (Fig. 5A and B) and AKT (Fig. 5C and D) were observed in extracts from JegE7 compared with JegGFP cells. To test whether inhibition of MAPK and PI3K signaling could abolish increased migration of JegE7 cells, we performed wounding assays in the presence of U0126 (Fig. 6A and B) and Wortmannin (Fig. 6C and D), inhibitors of ERK and AKT pathways, respectively. Either compound was able to significantly decrease JegE7 cell migration, while it did not affect migration of the control JegGFP cells (Fig. 6A–D).
Figure 5.

Epidermal growth factor-like domain 7 (EGFL7) activates mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways. Western blot analysis of cell extracts from JegGFP (control) and JegE7 (overexpressing EGFL7) cells for phosphorylated extracellular regulated kinase 1 and 2 (pERK1/2) (A) and phosphorylated protein kinase B (pAKT) (C) in JegGFP and JegE7, with Tubulin as a loading control. The respective densitometric analysis (B and D). Data are presented as percentage of control. *P < 0.05.
Figure 6.

Epidermal growth factor-like domain 7 (EGFL7) promotes Jeg3 cell migration through mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) signaling pathways. Dotted lines highlight the edges of the monolayer. (A) Wounding assay at 0 and 24 h with or without addition of the mitogen-activated protein/extracellular signal-regulated kinase kinase (MEK 1/2)-inhibitor U0126 (5 µM). (B) Quantification of Jeg3 migration in wounding assay, represented as dot plot, showing the total area filled at 24 h. (C) Wounding assay at 0 and 24 h with or without addition of the PI3K-inhibitor Wortmannin (100 nM). (D) Quantification of Jeg3 migration in wounding assay, represented as dot plot showing the total area filled at 24 h. Different letters are to be considered within each culture time-point and indicate statistically significant differences. P < 0.001 is indicated by **. Scale bars = 120 µm. WM = Wortmannin.
The effects of EGFL7 on MAPK and PI3K activation and Jeg3 cell motility are mediated by EGFR
Both MAPK and PI3K signaling pathways are activated in EGF-induced trophoblast migration (Qiu et al., 2004a, b). Since secreted EGFL7 contains two internal EGF-like domains, we hypothesized that EGFL7 could activate MAPK and PI3K through its interaction with EGFR. To test this, we first investigated if EGFL7 possesses homology at positions of amino acid known to be involved in binding of EGF to EGFR (Van Zoelen et al., 2000). Alignment of the amino acid sequences of EGFL7 and EGF shows that, in addition to the cysteine and glycine residues that are required for the three-dimensional folding of these proteins (Van Zoelen et al., 2000), most of the amino acids involved in binding to EGFR are also conserved between EGF and EGFL7 (Fig. 7A). On this premise, we next investigated the effects of EGFL7 overexpression on EGFR tyrosine phosphorylation. For this, Jeg3 cells were serum starved overnight and the activation of EGFR evaluated by western blot analysis. An about 2-fold increase in pEGFR was observed in JegE7 cells compared with control cells (Fig. 7B and C), suggesting that EGFL7 overexpression induces EGFR activation. This was further supported by our observation that EGFL7 and EGFR co-localize in Jeg3 cells, as determined by IF (Fig. 7D).
Figure 7.
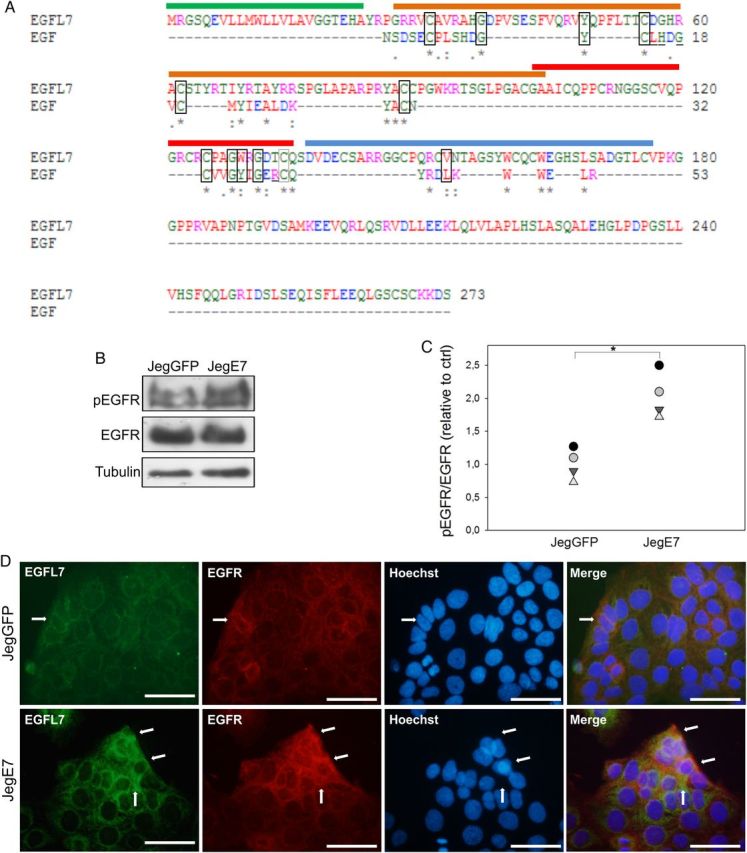
Epidermal growth factor-like domain 7 (EGFL7) interacts with and activates epidermal growth factor receptor (EGFR) in Jeg3 cells. (A) Alignment of the amino acid (aa) sequences of EGFL7 and epidermal growth factor (EGF). The signal peptide of EGFL7 is marked with a green line (aa 1–23), the EMI domain with an orange line (aa 27–104), the EGF-like domain with a red line (aa 103–135), the Ca2+ EGF-like domain with a blue line (aa 137–177). Amino acids involved in EGFR binding that are conserved between EGF and EGFL7 are depicted by rectangles. Amino acids that are involved in EGFR binding but are not conserved between EGF and EGFL7 are underlined (Van Zoelen et al., 2000). Asterisks = positions which have a single, fully conserved residue; colons = conservation between groups of strongly similar properties; periods = conservation between groups of weakly similar properties. Legend for the colors: Red = AVFPMILW, small, hydrophobic; Blue = DE, acidic; Magenta = RK, basic; Green = STYHCNGQ, hydroxyl, sulfhydryl, amine; Grey = others, unusual aa. (B) Western blot analysis of pEGFR in JegGFP (control) and JegE7 (overexpressing EGFL7), with densitometric analysis (C). (D) Double immunofluorescent staining of JegGFP and JegE7 cells for EGFL7 (green), EGFR (red), and Hoechst (blue). Arrows indicate co-localization of EGFL7 and EGFR. *P < 0.05. Scale bars = 50 µm.
Next, we tested whether EGFL7-induced up-regulation of ERKs would be mediated by EGFR activation. For this, AG1478 (1 µM), a specific inhibitor of EGFR kinase activity, was added to transduced Jeg3 cells in serum-free culture medium in the absence or presence of EGF (10 ng/ml), and phosphorylation of EGFR and ERKs was evaluated by western blot analysis. When JegGFP cells were stimulated with EGF, we detected, as expected, increased phosphorylation of EGFR (Fig. 8A and B), concomitant with an increase in ERK phosphorylation (Fig. 8C and D). Activation of EGFR and MAPKs was partly abolished by the treatment with AG1478 (Fig. 8A–D). Similarly, overexpression of EGFL7 triggered increased EGFR and MAPK phosphorylation in JegE7 cells; importantly, phosphorylation levels were also reduced when AG1478 was added to the culture medium (Fig. 8A–D).
Figure 8.
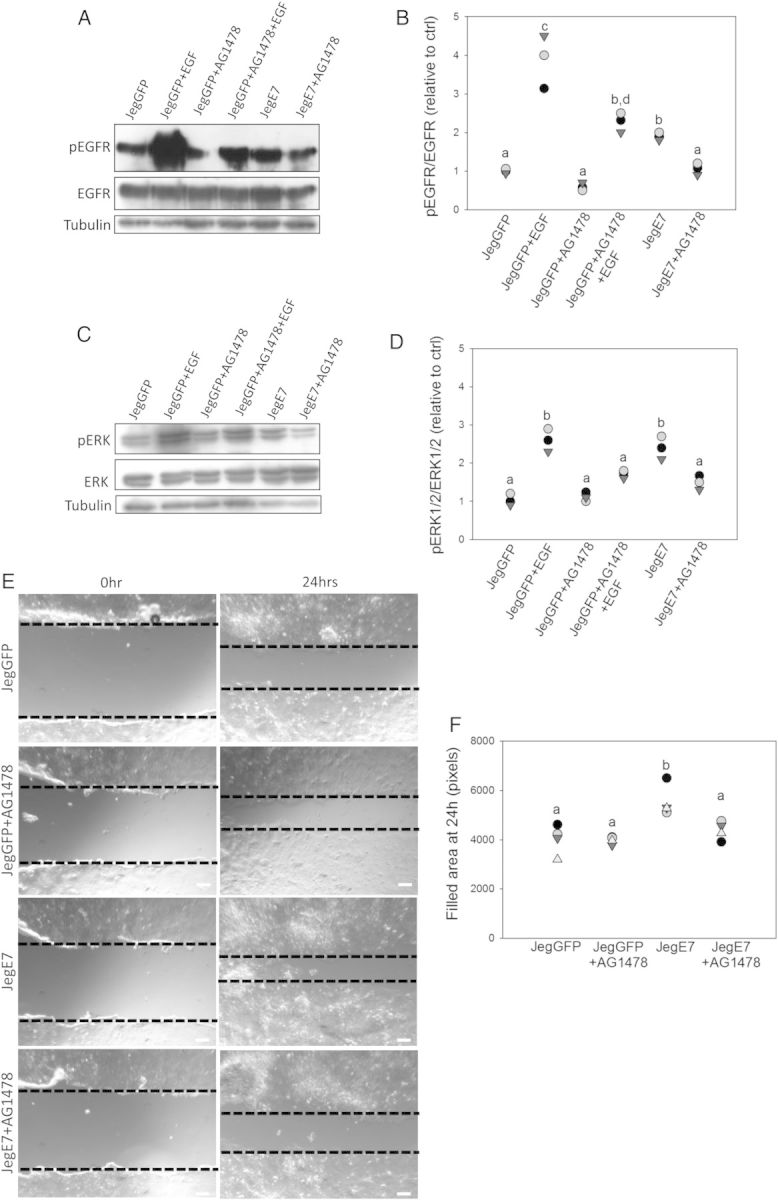
Epidermal growth factor-like domain 7 (EGFL7) activates the mitogen-activated protein kinase (MAPK) signaling pathway and promotes cell migration through epidermal growth factor receptor (EGFR). Jeg3 cells were treated with the EGFR-inhibitor AG1478 (1 µM) in serum-free culture medium for 30 min and/or stimulated with 10 ng/ml epidermal growth factor (EGF) for 30 min consecutively. Phosphorylation levels of EGFR (A) and extracellular regulated kinase 1 and 2 (ERK1/2) (C) were analyzed by western blot analysis. Respective densitometric analysis for (A and C) is shown in (B and D). (E) Wounding assay at 0 and 24 h in the presence of AG1478 (1 µM). Dotted lines highlight the edges of the monolayer. (F) Quantification of Jeg3 migration in the wounding assay, represented as dot plot showing the area filled at 24 h. Different letters are to be considered within each culture time-point and indicate statistically significant differences (P < 0.05). Scale bars = 120 µm.
To assess the role of EGFR activation in migration capability of JegE7 cells, we performed wounding assays in the absence or presence of AG1478. The results showed that addition of AG1478 abrogated the increased migratory ability of JegE7 at 24 (Fig. 8E and F) and 32 h (not shown).
NOTCH signaling is involved in EGFL7-mediated Jeg3 migration
Previous studies showed that EGFL7 is able to interact with all four NOTCH receptors and to function, at least in part, by modulating NOTCH signaling (Schmidt et al., 2009; Nichol et al., 2010). We therefore assessed the expression of the NOTCH family proteins in Jeg3 cells. Using antibodies specific for NOTCH1 and NOTCH4 for IF analysis, we detected expression of NOTCH1 in both JegGFP and JegE7 (Fig. 9A). NOTCH1 appeared to partially co-localize with EGFL7 (Fig. 9A). In contrast, no staining was observed using NOTCH4 antibodies, indicating very low or no expression of this receptor. This conclusion was confirmed by qRT–PCR analysis (data not shown). We next examined if EGFL7 overexpression correlated with changes in the levels of NOTCH1 and its target gene transcripts by performing qRT–PCR analysis. A significant increase of NOTCH1, HEY2, HEY1 and HES1 expression was observed in JegE7 cells compared with the JegGFP control cells (Fig. 9B–E).
Figure 9.
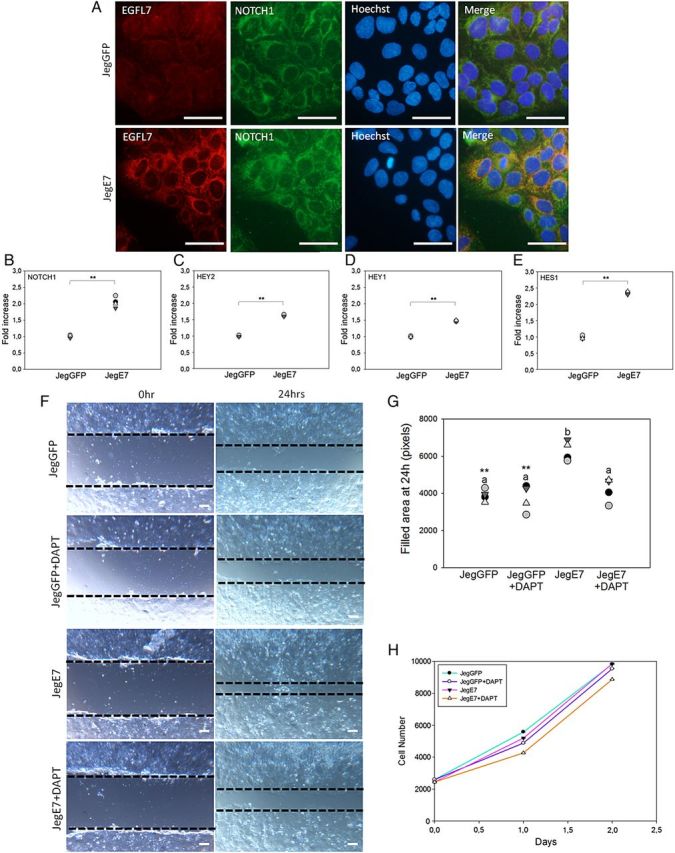
Epidermal growth factor-like domain 7 (EGFL7) promotes Jeg3 cell migration also through NOTCH signaling pathway. (A) Double immunofluorescent staining of JegGFP (control) and JegE7 (overexpressing EGFL7) cells for EGFL7 (red), NOTCH1 (green), and Hoechst (blue). (B–E) Quantitative RT–PCR data for transcript level of NOTCH1 (B), hairy and enhancer of split-related protein 2 (HEY-2) (C), hairy and enhancer of split-related protein 1 (HEY-1) (D), and hairy and enhancer of split-1 (HES-1) (E) in JegGFP and JegE7 cells. (F) Wounding assay at 0 and 24 h in presence of the γ-secretase inhibitor dual anti-platelet therapy (DAPT) (10 µM). Dotted lines highlight the edges of the monolayer. (G) Quantification of Jeg3 migration in wounding assay, represented as dot plot showing the area filled after 24 h. (H) JegGFP and JegE7 proliferation in the presence of DAPT (10 µM) was measured over 2 consecutive days by using the colorimetric 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-1) assay; n = 4. Different letters are to be considered within each culture time-point and indicate statistically significant differences (P < 0.05); **P < 0.001. Scale bars (A) = 50 µm; (F) = 120 µm.
To test whether the NOTCH1 signaling pathway is also involved in mediating the migratory activity of JegE7, we performed wounding assay in the presence of DAPT, a specific inhibitor of NOTCH activation. DAPT significantly abolished the EGFL7-induced migration of Jeg3 cells capability, while it did not affect control cell migration (Fig. 9F–G).
NOTCH signaling has been reported to be involved in the regulation of Jeg3 cell proliferation (Wagener et al., 2013). We therefore investigated if activation of NOTCH could affect the proliferation rate of JegGFP and JegE7 cells. Using the WST-1 colorimetric assay in the presence of DAPT, no significant differences in terms of metabolic activity were observed between JegE7 and JegGFP either in the presence or absence of DAPT (Fig. 9H). Together, these results suggest that EGFL7 mediated activation of NOTCH1 does not affect proliferation.
Convergence of MAPK and NOTCH signaling pathways
To evaluate if EGFR and NOTCH1 signaling pathways were cooperatively regulating JegE7 migration, we tested whether inhibition of the MAPK pathway could also inhibit NOTCH1 signaling and vice versa. To inhibit ERK phosphorylation and NOTCH1 activation, we treated serum starved JegE7 cells with 10 µM U0126 or 25 µM DAPT, respectively. A significant decrease in HES1 gene expression level was detected at 90 min after the addition of either U0126 or DAPT (Fig. 10A) and persisted up to 120 min (data not shown). To examine if the effect of MEK inhibition was NOTCH-dependent, we treated the cells with both U0126 and DAPT. Interestingly, we did not observe an additive effect in the reduction of HES1 expression (Fig. 10A), suggesting that the effect of MAPK on HES1 expression was indeed NOTCH-dependent.
Figure 10.

Inhibition of extracellular regulated kinase 1 and 2 (ERK1/2) activation reduces NOTCH signaling and blockage of NOTCH activity inhibits mitogen-activated protein kinase (MAPK) signaling pathway. (A) JegE7 (overexpressing EGFL7) cells were treated with the mitogen-activated protein/extracellular signal-regulated kinase kinase (MEK 1/2)-inhibitor U0126 (10 µM) or/and the γ-secretase inhibitor dual anti-platelet therapy DAPT (25 µM) in serum-free culture medium for 90 min. Transcript level of hairy and enhancer of split-1 (HES1) was analyzed by quantitative RT–PCR. (B and C) JegE7 cells were treated with DAPT (25 µM) or/and U0126 (10 µM) in serum-free culture medium for 60 min. Phosphorylation levels of ERK1/2 (B) were analyzed by western blot analysis. Respective densitometric analysis is shown in (C). *P < 0.05, **P < 0.001.
To further confirm the convergence of the two pathways, we treated serum starved JegE7 cells with 25 µM DAPT for 60 min. Phosphorylation of ERKs was evaluated by western blot analysis. Untreated JegE7 showed high levels of phosphorylated ERK1/2, and treatment with DAPT led to a significant reduction of ERK1/2 activation (Fig. 10B and C). Treatment of JegE7 cells with U0126 resulted in a more robust inhibition of ERK phosphorylation; however, no additional inhibition was observed when cells were treated with both U0126 and DAPT (Fig. 10B and C). Similar results were obtained in JegGFP cells (not shown).
Discussion
We recently reported that EGFL7 is expressed by trophoblast cells in the human placenta (Lacko et al., 2014). Others and we also demonstrated that EGFL7 regulates the proper spatial organization of endothelial cells during angiogenic sprouting, and influences their collective movement during physiological and pathological angiogenesis (Campagnolo et al., 2005; Schmidt et al., 2007). When Egfl7 is deleted in HUVECs, cell proliferation, migration and sprouting are notably impaired (Nichol et al., 2010). Based on these observations, we considered the possibility that EGFL7 may regulate trophoblast cell migration during uterine tissue invasion, by acting in an autocrine or paracrine manner, as demonstrated in other cell systems (Nichol and Stuhlmann, 2012). In the present study, we have tested this hypothesis by performing gain- and loss-of-function studies in vitro using the human choriocarcinoma cell line Jeg3 overexpressing EGFL7 (JegE7). Our results demonstrate that JegE7 cells possess a higher ability to cover the wounded areas in wounding assays and invade Matrigel thick layers more efficiently than control cells. Conversely, EGFL7 knockdown resulted in impaired migration of Jeg3 cells, consistent with a role for EGFL7 in regulating trophoblast migration/invasion.
One limitation with using long-term cultures of lentivirus transduced cells is that the precision with which signaling pathways are measured could be affected. To overcome this limitation, we have used primary trophoblast cultures and showed that the results obtained in the Jeg3 choriocarcinoma cell line were consistent with results in PTB isolated from human samples. Although manipulation and analysis of PTB cells is challenging when compared with established cell lines, in part due to their slow proliferation and migration, we show here that overexpression of EGFL7 stimulates PTB cell migration in a transwell assay.
Several signaling pathways that are activated at the feto-maternal interface are involved in trophoblast motility and are triggered by various cytokines and growth factors (MacPhee et al., 2001; Shiokawa et al., 2002; Qiu et al., 2004a, b; Fitzgerald et al., 2005; Knöfler, 2010; Hunkapiller et al., 2011). Among these, EGF and EGFR are known to be important players during physiological development of the placenta; EGFR homozygous mutant mice die at midgestation due to placental defects (Threadgill et al., 1995; Faxén et al., 1998; Dackor et al., 2009). In the present work, we show that EGFL7, which contains two internal EGF-like domains (Soncin et al., 2003; Fitch et al., 2004), is able to activate the EGFR pathway. Using double IF analysis, we observed partial co-localization of EGFL7 and EGFR in both JegGFP and JegE7 cells, mainly at the periphery of the cell patches where cell are more prone to migration. Increased phosphorylation of EGFR in JegE7 cells was accompanied by significantly increased activation of MAPK and PI3K signaling, both of which are known to be important players of placental development (Knöfler, 2010). We confirmed the specificity of these responses by using specific inhibitors for EGFR, and MAPK and PI3K pathways. When cells were cultured in the presence of the EGFR specific inhibitor AG1478, increased migration of Jeg3 cells overexpressing EGFL7 was attenuated, while surprisingly no effect was observed in control cells. This latter observation is in line with previous data showing that AG1478 has no effect on unstimulated extra villous trophoblast cell migration (Liu et al., 2009). Together, our results indicate that EGFL7 stimulates migration of trophoblast cells through activation of EGFR. It is possible that activation of EGFR is directly induced by EGFL7; alternatively, EGFL7 might trans-activate EGFR through other receptors. These possibilities need to be further investigated.
While preparing this manuscript for publication, a study was published showing that EGFL7-mediated activation of the EGFR-AKT signaling pathway in gastric cancer cell lines enhances their invasive and migratory capacity (Luo et al., 2014), suggesting that EGFL7 is an important player of these processes in various cell types.
A direct role for EGFL7 in the migration and invasion of trophoblast cells provides a possible explanation for its reduced expression levels that we observed in pre-eclamptic placentas (Lacko et al., 2014). In preeclampsia, inadequate invasion of trophoblast cells into the maternal decidua contributes to the onset of the pathology (Young et al., 2010). We also reported that in placentas of women affected by early onset PE, decreased expression of EGFL7 parallels an alteration of the NOTCH signaling pathway (Lacko et al., 2014), which in turn is known to be associated with impaired trophoblast invasion (Cobellis et al., 2007; Gasperowicz and Otto, 2008; Hunkapiller et al., 2011). In addition, data from the literature report that Jeg3 proliferation and migration/invasion properties are in part controlled by the NOTCH pathway (Wagener et al., 2013). In line with what has been reported in other cell systems (Schmidt et al., 2009; Nichol et al., 2010), we found that EGFL7 and NOTCH1 co-localize in JegE7 cells, and that overexpression of EGFL7 was associated with a concomitant increase in the expression of NOTCH1 and its target genes. In addition, inhibition of NOTCH1 activation by the γ-secretase inhibitor DAPT was able to significantly reduce JegE7 cell migration. We therefore speculated that EGFL7 mediated activation of NOTCH might influence migration by modulating cell proliferation. However, our results showed that inhibition of the NOTCH1 pathway did not result in any change in the proliferation rate of JegE7 and JegGFP cells, suggesting that activation of NOTCH1 was directly involved in migration.
A number of studies in other cell types demonstrated a crosstalk between EGFR and NOTCH pathways (Weijzen et al., 2002; Sundaram, 2005; Izrailit et al., 2013; Tremblay et al., 2013). We investigated whether MAPK and NOTCH signaling pathways act alone or together to mediate the effects of EGFL7 on trophoblast cell migration and invasion. By inhibiting the MAPK pathway, we observed a reduced expression of the NOTCH target gene HES1. Similarly, inhibition of NOTCH activity resulted in reduced activation of ERKs, thus suggesting that the two pathways co-operate. In conclusion, the present study identifies EGFL7 as a novel player involved in the regulation of trophoblast migration and invasion. Our data suggest that EGFL7, through the activation of EGFR and NOTCH signaling pathways (Fig. 11), might play a pivotal role in stimulating and sustaining trophoblast migration and decidua invasion. These observations will be crucial for the better understanding of specific placentopathies characterized by inadequate invasion of the maternal decidua, including PE.
Figure 11.

Schematic representation of cell signaling pathways involved in epidermal growth factor-like domain 7 (EGFL7)-induced trophoblast migration and invasion. Based on our findings, we propose that EGFL7 regulates trophoblast migration/invasion through the activation of the epidermal growth factor receptor (EGFR) and NOTCH pathways. The two pathways converge to mediate EGFL7 effects, as represented by the dashed lines. Question marks indicate possible other molecules mediating the interaction between EGFL7 and either EGFR or NOTCH1.
Supplementary data
Supplementary data are available at http://molehr.oxfordjournals.org/.
Authors' roles
M.M. performed the experiments, prepared the figures and tables and contributed to writing the manuscript; L.V. and D.P. helped performing part of the experiments; P.S. isolated the human trophoblast cells. F.A., S.S and S.F contributed to designing the experiments and to interpreting the data. H.S. hosted and trained M.M. in lentiviral transduction for 3 months in her laboratory, contributed to the study design, analysis of the results and preparation of the manuscript; L.C. designed the study, analyzed and interpreted the results, and wrote the manuscript.
Funding
This work was partly supported by the March of Dimes Foundation #6-FY14-411 (L.C. and H.S.), the NIH grant #R01 HL082098 (H.S.) and the ASM, Associazione Studio Malformazioni onlus (L.C.).
Conflict of interest
None declared.
Acknowledgements
We would like to thank Dr Stefano Rivella (Weill Cornell Medical College) for providing the pCCLIRESGFP vector, Prof. Antonietta Salustri for providing the kinase inhibitors (U0126 and Wortmannin) and helping with the interpretation of part of the data, and Prof. Claudio Sette for providing the EGFR inhibitor. We would also like to thank Prof. Massimo De Felici for critical reading of the manuscript.
References
- Bambino K, Lacko LA, Hajjar KA, Stuhlmann H. Epidermal growth factor-like domain 7 is a marker of the endothelial lineage and active angiogenesis. Genesis 2014;52:657–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagnolo L, Leahy A, Chitnis S, Koschnick S, Fitch MJ, Fallon JT, Loskutoff D, Taubman MB, Stuhlmann H. EGFL7 is a chemoattractant for endothelial cells and is up-regulated in angiogenesis and arterial injury. Am J Pathol 2005;167:275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagnolo L, Moscatelli I, Pellegrini M, Siracusa G, Stuhlmann H. Expression of EGFL7 in primordial germ cells and in adult ovaries and testes. Gene Expr Patterns 2008;8:389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty C, Barbin YP, Chakrabarti S, Chidiac P, Dixon SJ, Lala PK. Endothelin-1 promotes migration and induces elevation of [ca2+]i and phosphorylation of map kinase of a human extravillous trophoblast cell line. Mol Cell Endocrinol 2003;201:63–73. [DOI] [PubMed] [Google Scholar]
- Cobellis L, Mastrogiacomo A, Federico E, Schettino MT, De Falco M, Manente L, Coppola G, Torella M, Colacurci N, De Luca A. Distribution of Notch protein members in normal and preeclampsia-complicated placentas. Cell Tissue Res 2007;330:527–534. [DOI] [PubMed] [Google Scholar]
- Dackor J, Caron KM, Threadgill DW. Placental and embryonic growth restriction in mice with reduced function epidermal growth factor receptor alleles. Genetics 2009;183:207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delfortrie S, Pinte S, Mattot V, Samson C, Villain G, Caetano B, Lauridant-Philippin G, Baranzelli MC, Bonneterre J, Trottein F, et al. Egfl7 promotes tumor escape from immunity by repressing endothelial cell activation. Cancer Res 2011;71:7176–7186. [DOI] [PubMed] [Google Scholar]
- Diaz LE, Chuan YC, Lewitt M, Fernandez-Perez L, Carrasco-Rodríguez S, Sanchez-Gomez M, Flores-Morales A. IGF-II regulates metastatic properties of choriocarcinoma cells through the activation of the insulin receptor. Mol Hum Reprod 2007;13:567–576. [DOI] [PubMed] [Google Scholar]
- Durrans A, Stuhlmann H. A role for Egfl7 during endothelial organization in the embryoid body model system. J Angiogenes Res 2010;2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faxén M, Nasiell J, Blanck A, Nisell H, Lunell NO. Altered mRNA expression pattern of placental epidermal growth factor receptor (EGFR) in pregnancies complicated by preeclampsia and/or intrauterine growth retardation. Am J Perinatol 1998;15:9–13. [DOI] [PubMed] [Google Scholar]
- Fitch MJ, Campagnolo L, Kuhnert F, Stuhlmann H. Egfl7, a novel epidermal growth factor-domain gene expressed in endothelial cells. Dev Dyn 2004;230:316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald JS, Busch S, Wengenmayer T, Foerster K, de la Motte T, Poehlmann TG, Markert UR. Signal transduction in trophoblast invasion. Chem Immunol Allergy 2005;88:181–199. [DOI] [PubMed] [Google Scholar]
- Gasperowicz M, Otto F. The Notch signaling pathway in the development of the mouse placenta. Placenta 2008;29:651–659. [DOI] [PubMed] [Google Scholar]
- Gleeson LM, Chakraborty C, McKinnon T, Lala PK. Insulin-like growth factor-binding protein 1 stimulates human trophoblast migration by signaling through alpha 5 beta 1 integrin via mitogen-activated protein kinase pathway. J Clin Endocrinol Metab 2001;86:2484–2493. [DOI] [PubMed] [Google Scholar]
- Huang CH, Li XJ, Zhou YZ, Luo Y, Li C, Yuan XR. Expression and clinical significance of EGFL7 in malignant glioma. J Cancer Res Clin Oncol 2010;136:1737–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunkapiller NM, Fisher SJ. Placental remodeling of the uterine vasculature. Methods Enzymol 2008;445:281–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunkapiller NM, Gasperowicz M, Kapidzic M, Plaks V, Maltepe E, Kitajewski J, Cross JC, Fisher S. A role for Notch signaling in trophoblast endovascular invasion and in the pathogenesis of pre-eclampsia. Development 2011;138:2987–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izrailit J, Berman HK, Datti A, Wrana JL, Reedijk M. High throughput kinase inhibitor screens reveal TRB3 and MAPK-ERK/ TGFbeta pathways as fundamental Notch regulators in breast cancer. Proc Natl Acad Sci USA 2013;110:1714–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöfler M. Critical growth factors and signaling pathways controlling human trophoblast invasion. Int J Dev Biol 2010;54:269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacko LA, Massimiani M, Sones JL, Hurtado R, Salvi S, Ferrazzani S, Davisson RL, Campagnolo L, Stuhlmann H. Novel expression of EGFL7 in placental trophoblast and endothelial cells and its implication in preeclampsia. Mech Dev 2014;133:163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, MacCalman CD, Wang YI, Leung PCK. Promotion of human trophoblasts invasion by gonadotropin-releasing hormone (GnRH) I and GnRH II via distinct signaling pathways. Mol Endocrinol 2009;23:1014–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo BH, Xiong F, Wang JP, Li JH, Zhong M, Liu QL, Luo GQ, Yang XJ, Xiao N, Xie B, et al. Epidermal growth factor-like domain-containing protein 7 (EGFL7) enhances EGF receptor-AKT signaling, epithelial-mesenchymal transition, and metastasis of gastric cancer cells. PLoS One 2014;9:e99922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macphee DJ, Mostachfi H, Han R, Lye SJ, Post M, Caniggia I. Focal adhesion kinase is a key mediator of human trophoblast development. Lab Invest 2001;81:1469–1483. [DOI] [PubMed] [Google Scholar]
- McKinnon T, Chakraborty C, Gleeson LM, Chidiac P, Lala PK. Stimulation of human extravillous trophoblast migration by IGF-II is mediated by IGF type 2 receptor involving inhibitory g protein(s) and phosphorylation of MAPK. J Clin Endocrinol Metab 2001;86:3665–3674. [DOI] [PubMed] [Google Scholar]
- Nichol D, Stuhlmann H. EGFL7: a unique angiogenic signaling factor in vascular development and disease. Blood 2012;119:1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichol D, Shawber C, Fitch MJ, Bambino K, Sharma A, Kitajewski J, Stuhlmann H. Impaired angiogenesis and altered Notch signaling in mice overexpressing endothelial Egfl7. Blood 2010;116:6133–6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola C, Lala PK, Chakraborty C. Prostaglandin e2-mediated migration of human trophoblast requires rac1 and cdc42. Biol Reprod 2008;78:976–982. [DOI] [PubMed] [Google Scholar]
- Nikolic I, Stankovic ND, Bicker F, Meister J, Braun H, Awwad K, Baumgart J, Simon K, Thal SC, Patra C, et al. EGFL7 ligates αvβ3 integrin to enhance vessel formation. Blood 2013;121:3041–3050. [DOI] [PubMed] [Google Scholar]
- Parker LH, Schmidt M, Jin SW, Gray AM, Beis D, Pham T, Frantz G, Palmieri S, Hillan K, Stainier DY, et al. The endothelial-cell-derived-secreted factor Egfl7 regulates vascular tube formation. Nature 2004;428:754–758. [DOI] [PubMed] [Google Scholar]
- Pijnenborg R, Bland JM, Robertson WB, Dixon G, Brosens I. The pattern of interstitial trophoblastic invasion of the myometrium in early human pregnancy. Placenta 1981;2:303–316. [DOI] [PubMed] [Google Scholar]
- Potgens AJ, Gaus G, Frank HG, Kaufmann P. Characterization of trophoblast cell isolations by a modified flow cytometry assay. Placenta 2001;22:251–255. [DOI] [PubMed] [Google Scholar]
- Prast J, Saleh L, Husslein H, Sonderegger S, Helmer H, Knofler M. Human chorionic gonadotropin stimulates trophoblast invasion through extracellularly regulated kinase and AKT signaling. Endocrinology 2008;149:979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Q, Yang M, Tsang BK, Gruslin A. Both mitogen-activated protein kinase and phosphatidylinositol 3-kinase signalling are required in epidermal growth factor-induced human trophoblast migration. Mol Hum Reprod 2004a;10:677–684. [DOI] [PubMed] [Google Scholar]
- Qiu Q, Yang M, Tsang BK, Gruslin A. EGF-induced trophoblast secretion of MMP-9 and TIMP-1 involves activation of both PI3K and MAPK signalling pathways. Reproduction 2004b;128:355–363. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Paes K, De Maziere A, Smyczek T, Yang S, Gray A, French D, Kasman I, Klumperman J, Rice DS, et al. EGFL7 regulates the collective migration of endothelial cells by restricting their spatial distribution. Development 2007;134:2913–2923. [DOI] [PubMed] [Google Scholar]
- Schmidt MH, Bicker F, Nikolic I, Meister J, Babuke T, Picuric S, Müller-Esterl W, Plate KH, Dikic I. Epidermal growth factor-like domain 7 (EGFL7) modulates Notch signalling and affects neural stem cell renewal. Nat Cell Biol 2009;11:873–880. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiokawa S, Iwashita M, Akimoto Y, Nagamatsu S, Sakai K, Hanashi H, Kabir-Salmani M, Nakamura Y, Uehata M, Yoshimura Y. Small guanosine triphosphatase rhoa and rho-associated kinase as regulators of trophoblast migration. J Clin Endocrinol Metab 2002;87:5808–5816. [DOI] [PubMed] [Google Scholar]
- Soncin F, Mattot V, Lionneton F, Spruyt N, Lepretre F, Begue A, Stehelin D. VE-statin, an endothelial repressor of smooth muscle cell migration. EMBO J 2003;22:5700–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitalieri P, Cortese G, Pietropolli A, Filareto A, Dolci S, Klinger FG, Giardina E, Di Cesare S, Bernardini L, Lauro D, et al. Identification of multipotent cytotrophoblast cells from human first trimester chorionic villi. Cloning Stem Cells 2009;11:535–556. [DOI] [PubMed] [Google Scholar]
- Sundaram MV. The love-hate relationship between Ras and Notch. Genes Dev 2005;19:1825–1839. [DOI] [PubMed] [Google Scholar]
- Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science 1995;269:230–234. [DOI] [PubMed] [Google Scholar]
- Tremblay I, Paré E, Arsenault D, Douziech M, Boucher MJ. The MEK/ERK pathway promotes NOTCH signalling in pancreatic cancer cells. PLoS One 2013;8:e85502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zoelen EJ, Stortelers C, Lenferink AE, Van de Poll ML. The EGF domain: requirements for binding to receptors of the ErbB family. Vitam Horm 2000;59:99–131. [DOI] [PubMed] [Google Scholar]
- Wagener J, Yang W, Kazuschke K, Winterhager E, Gellhaus A. CCN3 regulates proliferation and migration properties in Jeg3 trophoblast cells via ERK1/2, Akt and Notch signalling. Mol Hum Reprod 2013;19:237–249. [DOI] [PubMed] [Google Scholar]
- Weijzen S, Rizzo P, Braid M, Vaishnav R, Jonkheer SM, Zlobin A, Osborne BA, Gottipati S, Aster JC, Hahn WC, et al. Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat Med 2002;8:979–986. [DOI] [PubMed] [Google Scholar]
- Wu F, Yang LY, Li YF, Ou DP, Chen DP, Fan C. Novel role for epidermal growth factor-like domain 7 in metastasis of human hepatocellular carcinoma. Hepatology 2009;50:1839–1850. [DOI] [PubMed] [Google Scholar]
- Young BC, Levine RJ, Karumanchi SA. Pathogenesis of preeclampsia. Annu Rev Pathol 2010;5:173–192. [DOI] [PubMed] [Google Scholar]
- Zhao HB, Wang C, Li RX, Tang CL, Li MQ, Du MR, Hou XF, Li DJ. E-cadherin, as a negative regulator of invasive behavior of human trophoblast cells, is down-regulated by cyclosporin A via epidermal growth factor/extracellular signal-regulated protein kinase signaling pathway. Biol Reprod 2010;83:370–376. [DOI] [PubMed] [Google Scholar]