

Abstract
Cannabinoid receptor 1 (CB1) antagonists are potentially useful for the treatment of several diseases. However, clinical development of several CB1 antagonists was halted due to central nervous system (CNS)-related side effects including depression and suicidal ideation in some users. Recently, studies have indicated that selective regulation of CB1 receptors in the periphery is a viable strategy for treating several important disorders. Past efforts to develop peripherally selective antagonists of CB1 have largely targeted rimonabant, an inverse agonist of CB1. Reported here are our efforts toward developing a peripherally selective CB1 antagonist based on the otenabant scaffold. Even though otenabant penetrates the CNS, it is unique among CB1 antagonists that have been clinically tested because it has properties that are normally associated with peripherally selective compounds. Our efforts have resulted in an orally absorbed compound that is a potent and selective CB1 antagonist with limited penetration into the CNS.
INTRODUCTION
Cannabinoid receptors belong to the endocannabinoid (EC) system, which consists of receptors, transporters, endocannabinoids, and the enzymes involved in synthesis and degradation of endocannabinoids. To date, two receptors have been identified — CB1 and CB2.1 Both CB1 and CB2 are G protein-coupled receptors (GPCRs) primarily activating inhibitory G proteins (Gi/o).2 Of the two, CB1 is prominently expressed in the central nervous system (CNS).1 However, it is also expressed peripherally in a number of peripheral tissues. The CB2 receptor is nominally expressed in the brain. However, it is highly expressed in cells of the immune system.3 The CB1 receptor is also a prominent target of drugs of abuse including (−)-Δ9-tetrahydrocannabinol (THC), the main psychoactive constituent of marijuana.4
In recent years, CB1 antagonists have received attention in the treatment of disorders that have a central nervous system (CNS)-related craving component, including alcoholism.5 Further, CB1 is a validated drug target to treat obesity, metabolic syndrome, liver disease, diabetes, and dyslipidemias through both CNS (anorexic) and peripheral (metabolic) effects.6 Despite the therapeutic promise of CB1 antagonists, this drug class has been limited by adverse effects including anxiety and depression, and the first clinically approved CB1 antagonist (inverse agonist) for weight loss, rimonabant (1, SR141716A) (Figure 1), was withdrawn from Europe.7 Consequently, development or clinical trials of several CB1 antagonists, such as taranabant, otenabant (2), and ibipinabant, were halted due to regulatory concerns generated by 1.8
Figure 1.
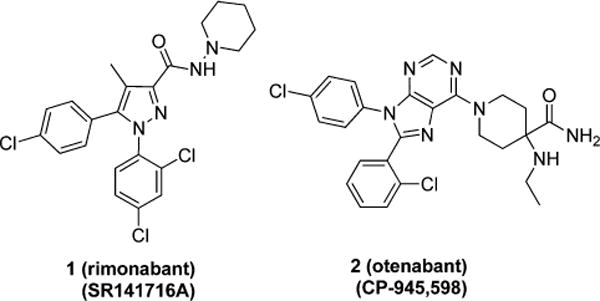
Examples of CB1 antagonists.
An alternative strategy to target this receptor is to develop antagonists that are peripherally restricted by virtue of not being able to cross the blood–brain barrier (BBB) and thus avoiding CNS-mediated adverse effects noted with nontissue selective compounds. A similar strategy has been successful in the development of peripherally selective opioids.9 Several groups are currently pursuing this strategy.10 A few peripherally selective CB1 antagonists have been reported (Figure 2), and their further validation and characterization are underway. It should be noted almost all reported efforts (e.g., 3–7) at designing peripherally restricted antagonists of CB1 to date have focused on compounds that closely resemble 1.
Figure 2.
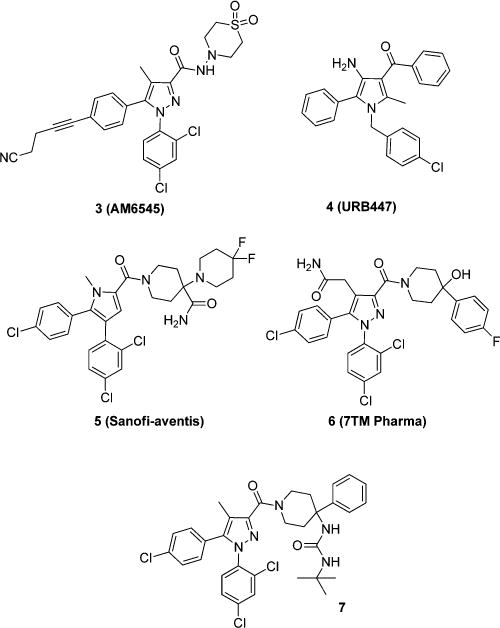
CB1 antagonists that are reported to be selective for the periphery.
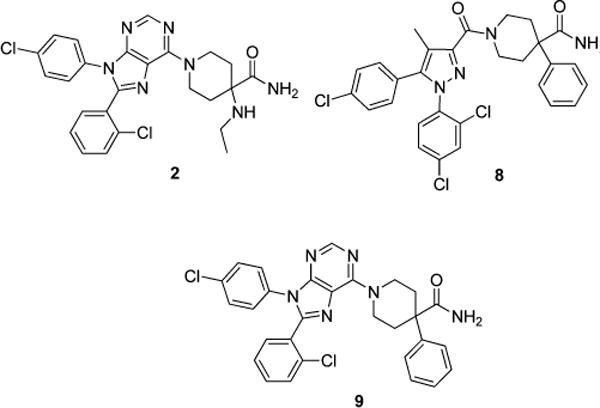
Our group has been involved in the development of peripherally selective CB1 antagonists based on amide variations of compound 1 using two different approaches. First, charged compounds were produced based on the observation that such compounds cross the BBB only when transported by a specific transporter.11 However, the compounds synthesized to date using this approach have low potency as reported in our previous publication.11b The second strategy involved generation of compounds with high topological polar surface areas (TPSAs). Past studies indicate that compounds with high TPSA, usually >100, have lower permeability into the CNS.12 This strategy was more successful and led to the identification of 7 that we have previously reported.13 While this compound was promising, having a brain to plasma ratio of less than 4% in a simple pharmacokinetic experiment, oral absorption of this compound was less than optimal.13
In this paper, we report our efforts toward the synthesis and characterization of peripherally selective CB1 antagonists based on 2. This compound is a highly selective CB1 receptor antagonist developed by Pfizer that was later abandoned due to CNS-related adverse effects during phase 3 clinical trials. This compound is unique among CB1 antagonists that have been clinically tested because it has properties that would be normally associated with a peripherally selective compound, including a TPSA of 102, three hydrogen bond donors, and formula weight >500.14 However, 2 is CNS permeable. It was suggested that the intramolecular H-bonding observed in the X-ray structure between the primary amide and the ethyl amine portion of the molecule effectively lowered the polarity of this compound, allowing for penetration into the CNS.14 Thus upon examination, this scaffold appeared to be more favorable for peripheralization as synthesis of compounds based on 2 but incapable of forming intramolecular H-bonding would be expected to be peripherally selective. This innovative approach to ligand design has now provided us with several promising compounds for further refinement.
RESULTS
Approaches to Ligand Design and Testing
Synthesis, purification, and characterization of compounds are described in the Experimental Section. Antagonism of each compound toward CB1 was quantitatively tested using an in vitro calcium mobilization assay for CB1 as has been previously described,13,15 and this antagonism is expressed as an apparent dissociation affinity constant (Ke). Compounds that demonstrated Ke < 100 nM were further characterized using competitive radioligand displacement of [3H]CP55940 at CB1 and CB2 overexpressing membrane preparations as described previously.13 Displacement data were quantified and expressed as an equilibrium dissociation constant (Ki) value at each receptor. Potent and selective compounds were progressed into an in vitro model of brain penetration in monolayers of MDCK-mdr1cells as described previously.11b,13 Certain compounds were also characterized for metabolic stability in human hepatic microsomal fractions (S9). Finally, in vivo testing was conducted in Sprague–Dawley (SD) rats to measure CNS permeability of these compounds.
Compound Development Strategy and Results
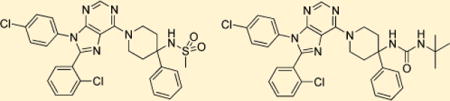
During our pursuit of peripherally selective CB1 antagonists, 8 (Figure 3) was synthesized and found to be highly potent, selective, and to have relatively low permeability in the MDCK-mdr1 model.13 These results along with our interest in CB1 antagonists that possess a purine core, like 2, moved us to make purine 9. Purine 9 is a potent (Ke = 9 nM) and selective (CB2/CB1 selectivity ratio ∼ 3077) hybrid of 2 and 8 with a TPSA of 90. Despite having a TPSA that is lower than that of 2, 9 cannot form intramolecular hydrogen bonds, and therefore, was predicted to be more peripherally selective than 2. This hypothesis was tested and confirmed (Table 1) using the MDCK-mdr1 permeability assay,13 where 9 was only 6% permeable (A–B), while 2 was ∼40% permeable (apical to basal, A–B) in the same assay. These positive results warranted further SAR exploration around 9.
Figure 3.
Design of compound 9; a hybrid of compounds 2 and 8.
Table 1.
Functional Assessment of Compound 9
| compd | TPSA | Ke CB1 (nM) | Ki CB1 (nM) [3H] CP55940 | Ki CB2 (nM) [3H] CP55940 | CB2/CB1 Ki ratio | MDCK-mdr1 A to B (%)a |
|---|---|---|---|---|---|---|
| 2 | 102 | 8.7 | 40 | |||
| 8 | 81 | 0.45 | 3.44 | 5504 | 1600 | 8 |
| 9 | 90 | 9 | 1.79 | 5507 | 3077 | 6 |
Compound’s permeability was measured from apical to basal (A to B) sides of the membrane.
To further improve upon 9, the amide was changed to more polar functionalities such as sulfonamide and urea (Figure 4, Table 2). Carbamate 10 and amine 11 were first synthesized, the latter of which also serves as a common synthetic intermediate for other analogues. Interestingly, carbamate 10 was very potent (Ke = 3.9 nM), selective (CB2/CB1 = 1959), and poorly permeable in the MDCK-mdr1 model (A to B <1%). Similarly, amine 11 was also very potent (Ke = 15.9 nM), selective (CB2:CB1 = 151), and poorly permeable in the MDCK-mdr1 model (A to B <1%). With amine 11 in hand, sulfonamide 12 and a series of ureas 13a–g were synthesized. Sulfonamide 12 was especially interesting because it has high potency (Ke = 2.9 nM), selectivity (CB2/CB1 = 153), poor permeability in the MDCK-mdr1 model (A to B is <1%), and a high TPSA (101), similar to 2 (TPSA = 102); it also lacks the ability to hydrogen bond in an intramolecular fashion. Two members of structural series 13 were found to have interesting pharmacological profiles. Urea 13a was potent (Ke = 19.2 nM), selective (CB2/CB1 = 20000), and poorly permeable in the MDCK-mdr1assay (A to B is <1%). Urea 13e was also found to be potent (Ke = 62 nM) and selective (CB2/CB1 = 74) as well.
Figure 4.
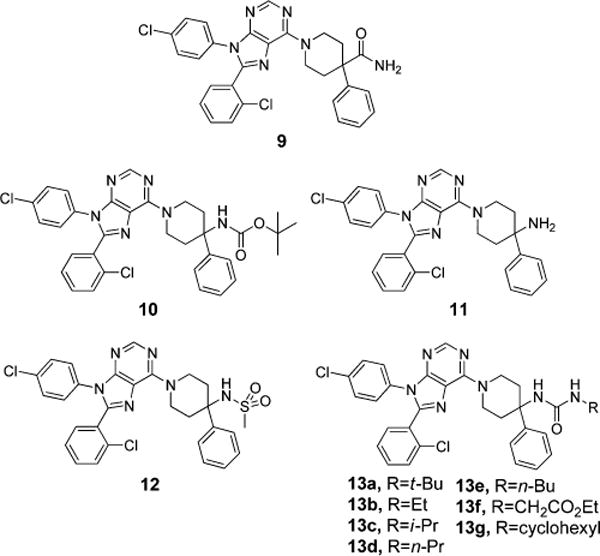
Ligand design around compound 9, replacement of the amide functionality.
Table 2.
Functional Assessment of Analogues of Compound 9
| compd | TPSA | Ke CB1 (nM) | Ki CB1 (nM) [3H] CP55940 | Ki CB2 (nM) [3H] CP55940 | CB2/CB1 Ki ratio | MDCK-mdr1 A to B (%)a |
|---|---|---|---|---|---|---|
| 10 | 85 | 3.9 | 7.1 | 13947 | 1959 | <1 |
| 11 | 73 | 15.9 | 11 | 1657 | 151 | <1 |
| 12 | 101 | 2.9 | 6.2 | 948 | 153 | <1 |
| 13a | 88 | 19.2 | 25.5 | 20000 | 784 | <1 |
| 13b | 88 | 443 | 195 | 4645 | 24 | |
| 13c | 88 | 46 | 337 | 8946 | 27 | |
| 13d | 88 | 99 | 220 | 7779 | 35 | |
| 13e | 88 | 62 | 269 | >20000 | >74 | |
| 13f | 114 | 171 | ||||
| 13g | 88 | 199 |
Compound’s permeability was measured from apical to basal (A to B) sides of the membrane.
The positive result observed with compounds 10–13g led to further exploration of the SAR around purines (Figure 5). The 4-amino-4-phenylpiperidine linker of compounds 10–13g was replaced with 4- and 3-aminopiperidine linkers to test the importance of the juxtaposition of the terminal polar groups of compounds 10–13g. These changes also had the benefit of reducing the molecular weight of analogues that were synthesized by removing the phenyl group present in 10–13g. Structural series 14, 15, 16, and 17 were all made and evaluated pharmacologically. The data (Table 3) for these series show that the 4-aminopiperidine linker of structural series 17 was superior to the linkers in structural series 14, 15, and 16 for all direct comparisons of the R group. However, in almost every case the 4-amino-4-phenylpiperidine linker of 10–13g was more potent than its direct analogues in structural series 14–17.
Figure 5.
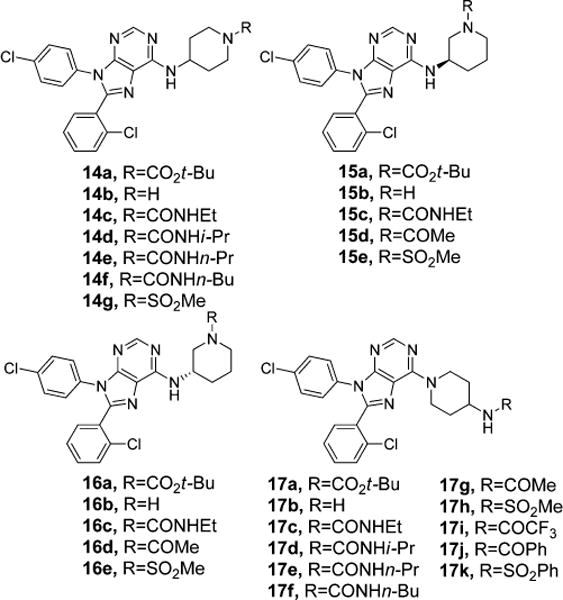
Exploration of different spacers.
Table 3.
Functional Assessment of Different Spacers and Functional Groups
| compd | TPSA | Ke CB1 (nM) | Ki CB1 (nM) [3H] CP55940 | Ki CB2 (nM) [3H] CP55940 | CB2/CB1 Ki ratio |
|---|---|---|---|---|---|
| 14a | 85 | 92 | 24 | 14000 | 568 |
| 14b | 68 | 5706 | |||
| 14c | 88 | 1148 | |||
| 14d | 88 | 495 | |||
| 14e | 88 | 1964 | |||
| 14f | 88 | 71 | 3343 | >20000 | >5 |
| 14g | 101 | 1645 | |||
| 15a | 85 | 326 | |||
| 15b | 68 | 5546 | |||
| 15c | 88 | 1500 | |||
| 15d | 76 | 920 | |||
| 15e | 101 | 346 | |||
| 16a | 85 | 3927 | |||
| 16b | 68 | 8406 | |||
| 16c | 88 | 1153 | |||
| 16d | 76 | 4449 | |||
| 16e | 101 | 6044 | |||
| 17a | 85 | 11 | 1.5 | 1713 | 1142 |
| 17c | 88 | 149 | |||
| 17d | 88 | 133 | 14 | 2453 | 175 |
| 17e | 88 | 132 | 28 | 153 | 5 |
| 17f | 88 | 49 | 25 | 1548 | 62 |
| 17g | 76 | 631 | |||
| 17h | 101 | 71 | 29 | 1073 | 37 |
| 17i | 76 | 3.2 | |||
| 17j | 76 | 0.7 | |||
| 17k | 101 | 0.3 | 1.2 | 5865 | 4888 |
However, several interesting compounds were identified in structural series 14–17. The tert-butoxycarbonyl (BOC) analogue 14a was potent (Ke = 92 nM) and selective (CB2/CB1 = 568). In general, compounds from structural series 15 and 16 were weakly active against CB1 in the calcium mobilization assay and the compounds were not characterized further. Several analogues in structural series 17 were very interesting. The BOC analogue 17a was very potent (Ke = 11 nM) and selective (CB2/CB1 = 1142). The urea analogues 17c–f all showed good activity (Ke ranging from 49 to 149 nM). Selectivity (CB2/CB1) for ureas 17c–f ranged from 5 to 175 fold. Amides and sulfonamides 17g–k were generally active with 3 of the 5 analogues having Ke < 10 nM. One of these compounds, 17k, was found to be highly selective (CB2/CB1 = 4888).
In Vitro Metabolic Stability and In Vivo Evaluation of Brain Penetration
A small set of potent and selective compounds were progressed into in vitro models of metabolic stability (Table 4). Stability in human plasma and human hepatic S9 fractions was measured to gauge potential metabolic liabilities of these compounds. All compounds were stable in plasma up to 1 h. The stabilities in S9 fractions for these compounds were more variable. All compounds displayed acceptable metabolic stabilities; however, 13a did show significant metabolism in S9 fraction and was deemed unsuitable for further development.
Table 4.
Functional Assessment of Select Compounds in for in Vitro Metabolic Stability
|
in vitro metabolic stability
|
||||||
|---|---|---|---|---|---|---|
| compd | TPSA | Ke CB1 (nM) | CB2/CB1 Ki CP55940 | MDCKmdr1 A to B (%) | S9% remaining 120 min | plasma % remaining 60 min |
| 9 | 90 | 0.45 | 3077 | 6 | >90 | >90 |
| 12 | 101 | 2.9 | 153 | <1 | >90 | >90 |
| 13a | 88 | 19.2 | 784 | <1 | 43 | >90 |
Several compounds showed excellent in vitro properties, including compounds 17a–k. Because of limited resources only sulfonamide 12 was advanced into an in vivo experiment to evaluate its brain penetration in SD rats upon oral dosing. Future studies are planned with additional compounds. Sulfonamide 12 was dosed orally in SD rats at 10 mg/kg, and both plasma and brain levels were analyzed out to 8 h post dose (Table 5). The compound showed slow absorption and did not reach a maximum concentration until 240 min post dose. Sulfonamide 12 also showed diminished brain penetration with the brain to plasma ratio ranging from 0.05 to 0.11. It should be noted that unperfused brain tissues, which are expected to contain 2–4% blood, were examined.16 Thus, it is concluded that 12 has limited brain penetration. Furthermore, 12 demonstrated excellent oral absorption and favorable clearance upon oral dosing in rats. Thus, this compound may be suitable for further development.
Table 5.
Pharmacokinetic Analyses of 12
| compd | dose mg/kg oral | sacrifice time (min) | plasma conc. (ng/mL) | brain conc. (ng/mL) | brain/plasma ratio |
|---|---|---|---|---|---|
| 12 | 10 | 30 | 752 | 38 | 0.05 |
| 60 | 767 | 58 | 0.08 | ||
| 120 | 1188 | 122 | 0.10 | ||
| 240 | 1653 | 184 | 0.11 | ||
| 480 | 914 | 89 | 0.10 |
Synthesis
Compound 9 (Scheme 1) was prepared from commercially available piperidine 19 and the readily available chloro-purine 18.14 Chloro-purine 18, piperidine 19, and triethylamine were heated in ethanol at 55 °C for 3 days yielding 9 in 63% yield. In a similar manner chloro-purine 18 was reacted with piperidine 20 and triethylamine in ethanol at 80 °C for 16 h to yield 10 in 84% yield (Scheme 2). The BOC group of 10 was removed using 20% trifluoroacetic acid (TFA) in dichloromethane to yield the primary amine 11 in 99%. Sulfonamide 12 was prepared by reacting 11, methanesulfonyl chloride, and triethylamine in tetrahydrofuran (THF) in 82% yield. The ureas 13a–g were made by reacting amine 11, triethylamine, and the appropriate isocyanate in THF in yields ranging from 69 to 92%.
Scheme 1. Synthesis of Compound 9a.
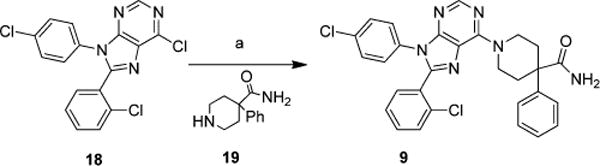
aReagents and conditions: (a) 19, triethylamine, ethanol, 55 °C, 3 days.
Scheme 2. Synthesis of Analogues of Compound 9a.
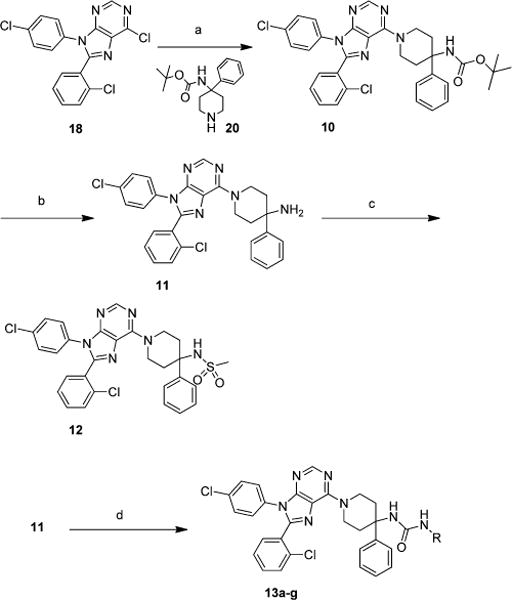
aReagents and conditions: (a) 20, triethylamine, ethanol, 80 °C, 16 h; (b) 20% TFA in dichloromethane; (c) methanesulfonyl chloride, triethylamine, THF; (d) appropriate isocyanate, triethylamine, THF.
Chloro-purine 18 was converted to 14a, 15a, and 16a in yields ranging from 86 to 95% by reaction with the appropriate amine and triethylamine in ethanol at 80 °C for 16 h (Scheme 3). The BOC groups were removed from 14a, 15a, and 16a to form amines 14b, 15b, and 16b using 30% TFA in dichloromethane in yields ranging from 65 to 88%. Amines 14b, 15b, and 16b were reacted with the appropriate isocyanate and triethylamine in THF to generate ureas 14c–f, 15c, and 16c in yields ranging from 65 to 95%. Amines 15b and 16b were reacted with acetic anhydride in pyridine to yield amides 15d and 16d in 87% and 81% respectively. Sulfonamides 14g, 15e, and 16e were made from 14b, 15b, and 16b by reaction with methanesulfonyl chloride and triethylamine in THF in yields ranging from 51 to 91%.
Scheme 3. Synthesis of Compounds with Different Spacers and Functional Groupsa.
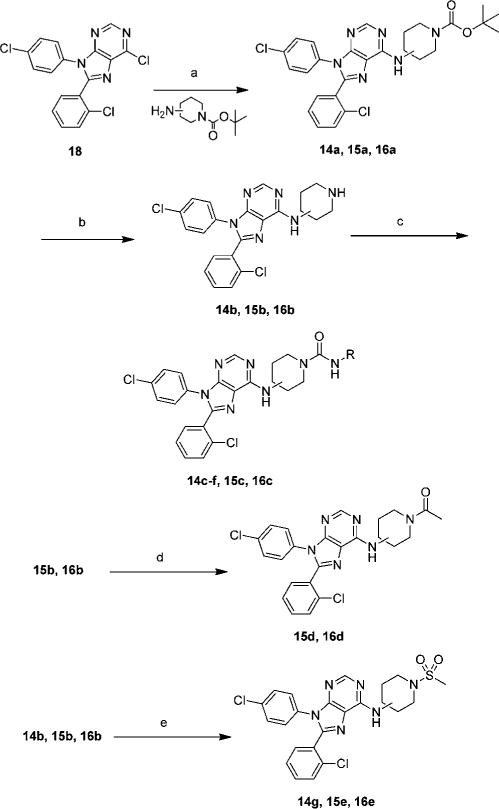
aReagents and conditions: (a) triethylamine, ethanol, 80 °C, 16 h; (b) 30% TFA in dichloromethane; (c) appropriate isocyanate, triethylamine, THF, rt; (d) acetic anhydride, pyridine; (e) methanesulfonyl chloride, triethylamine, THF.
Chloro-purine 18 and piperidine 21 were heated in ethanol at 80 °C for 16 h yielding 17a in 99% yield (Scheme 4). The remaining chemistry, to generate 17b–k, was performed in a similar manner to reactions discussed above.
Scheme 4. Synthesis of Compounds with Different Spacers and Functional Groups Continueda.
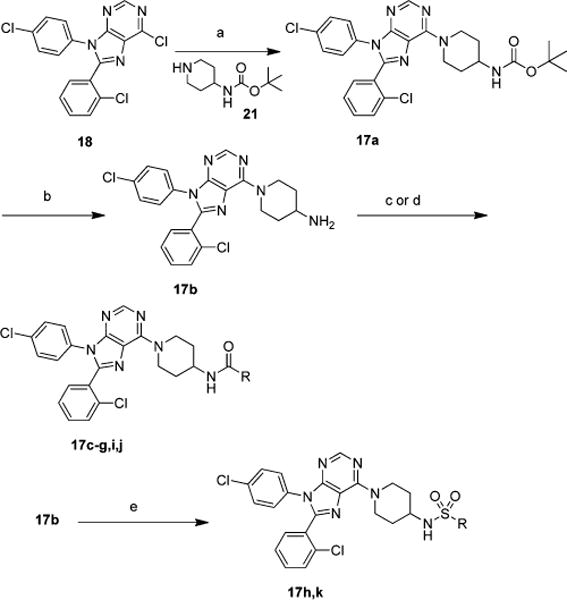
aReagents and conditions: (a) 21, ethanol, 80 °C, 16 h; (b) 30% TFA in dichloromethane; (c) appropriate isocyanate, triethylamine, THF; (d) appropriate anhydride, triethylamine, THF; (e) appropriate sulfonyl chloride, triethylamine, THF.
DISCUSSION
CB1 receptor antagonism is a validated strategy to treat several important disorders including diabetes, obesity, metabolic syndromes, liver diseases, and dyslipidemias. However, non-tissue selective antagonists of this receptor are not clinically useful due to adverse CNS effects. CB1 receptors are also expressed in peripheral organs and targeting of these peripheral receptors has produced promising data in experimental models of important diseases. Thus, several groups are trying to develop peripherally selective antagonists of CB1 for therapeutic use. While some progress has been made in this direction, currently disclosed CB1 antagonists that are peripherally selective need further improvement and validation prior to clinical use. Furthermore, most of these compounds are closely related to 1. In this report, we disclose our efforts to develop peripherally selective antagonists of CB1 based on 2, which we believe is a promising scaffold for optimization. This is because 2 has several chemical properties that would indicate poor brain penetration. However, intramolecular hydrogen bonding may effectively lower the polarity of 2 and this has been suggested by Griffith and co-workers.14 Thus, we hypothesized that optimized design and synthesis of compounds based on 2 that do not have the ability to form intramolecular hydrogen bonds could lead to CB1 antagonists with limited brain penetration.
Several potent and selective CB1 antagonists have been reported in this paper including nine compounds that have Ke < 100 nM and selectivity >100-fold versus CB2. Potency at CB1 has been obtained with a variety of functional groups on the terminus of the aminopiperidine in structures 10–17k. Carbamates, amines, sulfonamides, ureas, and amides have all been well tolerated at this position. The 4-amino-4-phenylpiperidine linker present in compounds 10–13g was the most potent linker noted in this study. 4-Aminopiperidine derivatives were more potent that 3-aminopiperidine analogues. However, of the 1–3 aminopiperidine compounds in structural series 15 and 16, the R enantiomers (15) were generally more potent at CB1 (four out of five examples) than the S enantiomers (16) in head to head comparisons. Of the 4-aminopiperidine linker compounds, lacking a phenyl group on the piperidine ring, in structural series 14 and 17, the analogues where the piperidine nitrogen was bonded to the purine core (structural series 17) were favored over the isomers where the 4-amino group was bonded to the core (structural series 14). Compounds with branched terminal R groups were found to be more selective for CB1 over CB2 than compounds with less sterically demanding R groups. This trend was especially prevalent in ureas 13a–d and 17c–f. In both of these series, 13a–d and 17c–f, the compounds with the most branched R group have the highest selectivity ratio for CB2/CB1. This selectivity is achieved not by gains in CB1 binding affinity, but by lowering the binding affinity for CB2. While 2 is an inverse agonist at CB1, it is currently not known if the compounds described in this publication act as inverse agonist or neutral antagonists at the present time. Those studies will be performed in the future.
The removal of the intramolecular hydrogen bond found in 2 allowed us to synthesize compounds with significantly lower permeability in the MDCK-mdr1 assay. This can be demonstrated by comparing 2 and 12. While 2 and 12 have almost identical TPSAs (102 vs 101), 12 which lacks the ability to hydrogen bond in an intramolecular fashion has vastly reduced permeability in the MDCK-mdr1 assay. Thus, confirming Griffith and co-worker’s hypothesis.14
Compounds 9, 12, and 13a were identified for further evaluation. All of these compounds showed reasonable stability to human plasma. Both 9 and 12 had good stability in human S9 fraction with over 90% of compound remaining after 120 min of incubation. The stability of 13a to S9 fraction was acceptable, but it did show significant metabolism in the assay. Compound 12 was advanced into in vivo models to assess its brain penetration. In a rat PK assay, 12 showed brain to plasma ratios ranging from 0.05 to 0.11, indicating limited brain penetration. Generally, compounds that have <10% brain penetration are considered peripherally selective. This is also a significant reduction in brain penetration in comparison to 2 in SD rats, which has ∼33% brain exposure at 2–3 h postdose.17
In conclusion, through our studies, we have discovered novel analogues of 2 with vastly reduced brain penetration. Of the compounds reported, 12 had excellent potency, selectivity, and oral absorption in rats. This compound has low levels of brain penetration as well. This compound will be evaluated in proof of concept studies for diseases such as obesity, liver fibroses, and diabetes in the future. Furthermore, sulfonamide 12 will also serve as a starting point for refinement to further lower the brain penetration of future compounds, if necessary. However, any such endeavor must be dictated by the proposed use, and an acceptable balance between therapeutic benefits and undesirable effects must be achieved. For example, antiobesity drugs should have a very favorable profile as far as adverse effects are concerned. By contrast, in other diseases such as liver disease or diabetes, a more permissive profile of adverse effects could be tolerated.
EXPERIMENTAL SECTION
Compound Synthesis and Characterization
Chemistry General
Purity and characterization of compounds were established by a combination of HPLC, TLC, and NMR analytical techniques described below. 1H and 13CNMR spectra were recorded on a Bruker Avance DPX-300 (300 MHz) spectrometer and were determined in CHCl3-d or MeOH-d4 with tetramethylsilane (TMS) (0.00 ppm) or solvent peaks as the internal reference unless otherwise noted. Chemical shifts are reported in ppm relative to the solvent signal, and coupling constant (J) values are reported in hertz (Hz). Thin-layer chromatography (TLC) was performed on EMD precoated silica gel 60 F254 plates, and spots were visualized with UV light or I2 detection. Low-resolution mass spectra were obtained using a Waters Alliance HT/Micromass ZQ system (ESI). All test compounds were greater than 95% pure as determined by HPLC on an Agilent 1100 system using an Agilent Zorbax SB-Phenyl, 2.1 × 150 mm, 5 μm column with gradient elution using the mobile phases (A) H2O containing 0.05% CF3COOH and (B) methanol. A flow rate of 1.0 mL/min was used. All compounds were isolated as white or off-white solids.
1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-4-phe-nylpiperidine-4-carboxamide (9)
To a solution of 6-chloro-8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purine (9) (23.1 mg, 0.062 mmol, 1 equiv) in 2 mL of ethanol was added 4-carbamoyl-4-phenylpiperidin-1-ium trifluoroacetate (19) (25 mg, 0.123 mmol, 2 equiv) and triethylamine (0.03 mL, 0.19 mmol, 3 equiv). The reaction was heated to 55 °C for 3 days. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield 21 mg (63%) of desired product. 1H NMR (300 MHz, methanol-d4) δ ppm 2.02–2.21 (m, 2 H) 2.62 (d, J = 13.37 Hz, 2 H) 3.82 (br. s., 2 H) 4.98 (d, J = 15.54 Hz, 2 H) 7.07–7.67 (m, 13 H) 8.02–8.29 (m, 1 H), [M + H]+ 543.6.
tert-Butyl N-{1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-4-phenylpiperidin-4-yl}carbamate (10)
To a solution of 18 (50 mg, 0.133 mmol, 1 equiv) in 2 mL of ethanol was added tert-butyl N-(4-phenylpiperidin-4-yl)carbamate (20) (44 mg, 0.16 mmol, 1.2 equiv) and triethylamine (0.03 mL, 0.20 mmol, 1.5 equiv). The reaction was heated to 80 °C for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield 69 mg (84%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.31–1.49 (m, 9 H) 2.09–2.28 (m, 2 H) 2.43 (br. s., 2 H) 3.62 (br. s., 2 H) 4.96 (s, 1 H) 5.30 (br. s., 2 H) 7.03–7.61 (m, 13 H) 8.40 (s, 1 H), [M + H]+ 615.4.
1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-4-phe-nylpiperidin-4-amine (11)
A solution of 10 (2.65 g, 4.3 mmol) was stirred in dichloromethane (32 mL) and trifluoroacetic acid (8 mL) for 1.5 h. The reaction was concentrated in vacuo. The crude product was dissolved in ethyl acetate and washed with 3.8 M NaOH. The aqueous layer was extracted twice with ethyl acetate. The combined organic layer was washed with brine and dried with MgSO4 to yield 2.21 g (99%) of pure desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.87 (d, J = 13.56 Hz, 2 H) 2.15–2.39 (m, 2 H) 3.82–4.11 (m, 2 H) 5.10 (br. s., 2 H) 7.11–7.62 (m, 13 H) 8.40 (s, 1 H), [M + H]+ 515.8.
N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-4-phenylpiperidin-4-yl}methanesulfonamide (12)
To a solution of 11 (8.4 mg, 0.016 mmol, 1 equiv) in 2 mL of THF was added methanesulfonyl chloride (0.01 mL) and triethylamine (0.02 mL). The mixture was stirred for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield 8 mg (82%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 2.22 (s, 3 H) 2.30–2.44 (m, 2 H) 2.47–2.66 (m, 2 H) 4.20 (br. s., 2 H) 4.75 (s, 2 H) 7.08–7.64 (m, 13 H) 8.38 (s, 1 H), [M + H]+ 593.3.
General Procedure for Making Ureas from 1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-4-phenylpiperidin-4-amine (11)
To a solution of 11 (19 mg, 0.036 mmol, 1 equiv) in 2 mL of THF was added triethylamine (0.015 mL, 0.108 mmol, 3 equiv) and the appropriate isocyanate (1.5 equiv). The reaction was stirred for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield pure compound.
3-tert-Butyl-1-{1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-4-phenylpiperidin-4-yl}urea (13a)
Reaction proceeded in 81% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 1.12 (s, 9 H) 2.13–2.27 (m, 2 H) 2.27–2.43 (m, 2 H) 3.67 (br. s., 2 H) 3.96 (br. s., 1 H) 4.78 (br. s., 1 H) 5.10–5.68 (m, 2 H) 7.07–7.63 (m, 13 H) 8.40 (s, 1 H), [M + H]+ 614.7.
1-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-4-phenylpiperidin-4-yl}-3-ethylurea (13b)
Reaction proceeded in 81% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 0.85–1.01 (m, 3 H) 2.11–2.29 (m, 2 H) 2.30–2.47 (m, 2 H) 2.96–3.20 (m, 2 H) 3.64 (br. s., 2 H) 4.10–4.24 (m, 1 H) 5.00 (s, 1 H) 5.32 (d, J = 14.41 Hz, 2 H) 7.10–7.60 (m, 13 H) 8.40 (s, 1 H), [M + H]+ 586.8.
1-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-4-phenylpiperidin-4-yl}-3-(propan-2-yl)urea (13c)
Reaction proceeded in 79% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 0.94 (d, J = 6.50 Hz, 6 H) 2.13–2.29 (m, 2 H) 2.29–2.42 (m, 2 H) 3.51–3.76 (m, 2 H) 3.95 (d, J = 7.82 Hz, 1 H) 4.93 (s, 1 H) 5.32 (d, J = 13.56 Hz, 2 H) 7.07–7.63 (m, 13 H) 8.40 (s, 1 H), [M + H]+ 600.7.
1-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-4-phenylpiperidin-4-yl}-3-propylurea (13d)
Reaction proceeded in 88% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 0.64–0.77 (m, 3 H) 1.19–1.37 (m, 2 H) 2.10–2.27 (m, 2 H) 2.29–2.45 (m, 2 H) 2.87–3.08 (m, 2 H) 3.63 (br. s., 2 H) 4.28 (br. s., 1 H) 5.11 (s, 1 H) 5.36 (br. s., 2 H) 7.10–7.60 (m, 13 H) 8.40 (s, 1 H), [M + H]+ 600.5.
3-Butyl-1-{1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-4-phenylpiperidin-4-yl}urea (13e)
Reaction proceeded in 82% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 0.74–0.86 (m, 3 H) 1.11 (dq, J = 14.79, 7.22 Hz, 2 H) 1.20–1.31 (m, 2 H) 2.11 – 2.27 (m, 2 H) 2.29–2.41 (m, 2 H) 3.05 (q, J = 6.59 Hz, 2 H) 3.63 (br. s., 2 H) 4.22 (t, J = 5.13 Hz, 1 H) 5.07 (s, 1 H) 5.34 (br. s., 2 H) 7.12–7.56 (m, 13 H) 8.40 (s, 1 H), [M + H]+ 614.7.
Ethyl 2-[({1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-4-phenylpiperidin-4-yl}carbamoyl)amino]acetate (13f)
Reaction proceeded in 92% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 1.18–1.28 (m, 3 H) 2.11–2.29 (m, 2 H) 2.33–2.52 (m, 2 H) 3.64 (br. s., 2 H) 3.82–3.92 (m, 2 H) 4.09–4.24 (m, 2 H) 5.13 (br. s., 1 H) 5.23–5.52 (m, 2 H) 5.60 (s, 1 H) 7.05–7.59 (m, 13 H) 8.39 (s, 1 H), [M + H]+ 644.4.
3-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-4-phenylpiperidin-4-yl}-1-cyclohexylurea (13g)
Reaction proceeded in 69% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 0.77–1.13 (m, 3 H) 1.15–1.36 (m, 3 H) 1.36–1.54 (m, 3 H) 1.56–1.76 (m, 1 H) 2.12–2.28 (m, 2 H) 2.29–2.42 (m, 2 H) 3.34–3.55 (m, 1 H) 3.65 (br. s., 2 H) 3.96–4.20 (m, 1 H) 4.96 (s, 1 H) 5.34 (br. s., 2 H) 7.06–7.61 (m, 13 H) 8.40 (s, 1 H), [M + H]+ 640.5.
tert-Butyl 4-{[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]amino}piperidine-1-carboxylate (14a)
To a solution of 18 (151 mg, 0.403 mmol, 1 equiv) in 4 mL of ethanol was added tert-butyl 4-aminopiperidine-1-carboxylate (96 mg, 0.483 mmol, 1.2 equiv) and triethylamine (0.08 mL, 0.6 mmol, 1.5 equiv). The reaction was heated to 80 °C for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield 203 mg (94%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.38–1.55 (m, 11 H) 2.14 (d, J = 12.34 Hz, 2 H) 2.98 (t, J = 12.10 Hz, 2 H) 4.00–4.21 (m, 2 H) 4.28–4.52 (m, 1 H) 5.83 (d, J = 7.63 Hz, 1 H) 7.17–7.27 (m, 2 H) 7.31–7.44 (m, 5 H) 7.50 (d, J = 6.88 Hz, 1 H) 8.44 (s, 1 H), [M + H]+ 539.4.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-N-(piperidin-4-yl)-9H-purin-6-amine (14b)
A solution of 14a (180 mg, 0.33 mmol) was stirred in dichloromethane (9 mL) and trifluoroacetic acid (4 mL) for 16 h. The reaction was concentrated in vacuo. The crude product was dissolved in ethyl acetate and washed with saturated NaHCO3. The aqueous layer was extracted twice with ethyl acetate. The combined organic layer was washed with brine and dried with MgSO4. The crude material was purified by silica gel column chromatography using 0–100% CMA 80 (80% chloroform, 18% methanol, and 2% ammonium hydroxide)/ethyl acetate to yield 129 mg (88%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.45 (qd, J = 11.66, 3.81 Hz, 2 H) 2.10 (d, J = 11.87 Hz, 2 H) 2.64–2.88 (m, 2 H) 3.10 (d, J = 12.62 Hz, 2 H) 4.27 (br. s., 1 H) 5.80 (d, J = 6.31 Hz, 1 H) 7.01–7.55 (m, 8 H) 8.37 (s, 1 H), [M + H]+ 439.6.
General Procedure for Making Ureas from 8-(2-chlorophenyl)-9-(4-chlorophenyl)-N-(piperidin-4-yl)-9H-purin-6-amine (14b)
To a solution of 14b (18.4 mg, 0.042 mmol, 1 equiv) in 2 mL of THF was added triethylamine (0.02 mL, 0.126 mmol, 3 equiv) and the appropriate isocyanate (1.5 equiv). The reaction is stirred for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% CMA 80/ethyl acetate to yield pure compound.
4-{[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]amino}-N-ethylpiperidine-1-carboxamide (14c)
Reaction proceeded in 86% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 1.09 (t, J = 7.21 Hz, 3 H) 1.34–1.60 (m, 2 H) 2.11 (d, J = 12.34 Hz, 2 H) 2.83–3.06 (m, 2 H) 3.13–3.32 (m, 2 H) 3.92 (d, J = 13.37 Hz, 2 H) 4.40 (d, J = 4.90 Hz, 2 H) 5.80 (d, J = 7.82 Hz, 1 H) 7.04–7.51 (m, 8 H) 8.37 (s, 1 H), [M + H]+ 510.4.
4-{[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]amino}-N-(propan-2-yl)piperidine-1-carboxamide (14d)
Reaction proceeded in 91% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 1.18 (d, J = 6.50 Hz, 6 H) 1.46–1.68 (m, 2 H) 2.19 (d, J = 10.55 Hz, 2 H) 3.03 (t, J = 11.59 Hz, 2 H) 3.89–4.10 (m, 3 H) 4.29 (d, J = 7.16 Hz, 1 H) 4.42 (br. s., 1 H) 5.88 (d, J = 7.82 Hz, 1 H) 7.13–7.64 (m, 8 H) 8.45 (s, 1 H), [M + H]+ 524.7.
4-{[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]amino}-N-propylpiperidine-1-carboxamide (14e)
Reaction proceeded in 95% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 0.86–1.01 (m, 3 H) 1.44–1.67 (m, 4 H) 2.19 (d, J = 10.64 Hz, 2 H) 3.05 (t, J = 11.73 Hz, 2 H) 3.22 (q, J = 6.59 Hz, 2 H) 3.92–4.08 (m, 2 H) 4.43 (br. s., 1 H) 4.54 (t, J = 5.04 Hz, 1 H) 5.88 (d, J = 7.72 Hz, 1 H) 7.09–7.58 (m, 8 H) 8.45 (s, 1 H), [M + H]+ 524.8.
N-Butyl-4-{[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]amino}piperidine-1-carboxamide (14f)
Reaction proceeded in 91% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 0.79–0.92 (m, 3 H) 1.29 (dq, J = 14.75, 7.20 Hz, 2 H) 1.36–1.58 (m, 4 H) 2.11 (d, J = 10.64 Hz, 2 H) 2.96 (t, J = 11.73 Hz, 2 H) 3.17 (q, J = 6.75 Hz, 2 H) 3.81–4.01 (m, 2 H) 4.22–4.51 (m, 2 H) 5.79 (d, J = 7.54 Hz, 1 H) 7.01–7.53 (m, 8 H) 8.37 (s, 1 H), [M + H]+ 538.4.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-N-(1-methanesulfonylpiperidin-4-yl)-9H-purin-6-amine (14g)
To a solution of 14b (18.4 mg, 0.042 mmol, 1 equiv) in 2 mL of THF was added methanesulfonyl chloride (0.005 mL, 0.063 mmol, 1.5 equiv) and triethylamine (0.02 mL, 0.126 mmol, 3 equiv). The mixture was stirred for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield 20 mg (91%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.58–1.76 (m, 2 H) 2.21 (d, J = 10.64 Hz, 2 H) 2.76 (s, 3 H) 2.90 (t, J = 10.93 Hz, 2 H) 3.77 (d, J = 12.15 Hz, 2 H) 4.33 (br. s., 1 H) 5.84 (br. s., 1 H) 7.04–7.51 (m, 8 H) 8.36 (s, 1 H), [M – H]− 515.7.
tert-Butyl (3R)-3-{[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]amino}piperidine-1-carboxylate (15a)
To a solution of 18 (100 mg, 0.267 mmol, 1 equiv) in 3 mL of ethanol was added tert-butyl (3R)-3-aminopiperidine-1-carboxylate (64 mg, 0.32 mmol, 1.2 equiv) and triethylamine (0.06 mL, 0.4 mmol, 1.5 equiv). The reaction was heated to 80 °C for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield 124 mg (86%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.33 (br. s., 9 H) 1.49–1.67 (m, 2 H) 1.74 (d, J = 6.97 Hz, 1 H) 2.00 (d, J = 2.83 Hz, 1 H) 3.20 (br. s., 2 H) 3.50 (br. s., 1 H) 3.88 (br. s., 1 H) 4.32 (br. s., 1 H) 5.87 (d, J = 7.44 Hz, 1 H) 7.02–7.54 (m, 8 H) 8.38 (s, 1 H), [M + H]+ 539.3.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-N-[(3R)-piperidin-3-yl]-9H-purin-6-amine (15b)
A solution of tert-butyl 15a (109 mg, 0.202 mmol) was stirred in dichloromethane (7 mL) and trifluoroacetic acid (3 mL) for 16 h. The reaction was concentrated in vacuo. The crude product was dissolved in ethyl acetate and washed with saturated NaHCO3. The aqueous layer was extracted twice with ethyl acetate. The combined organic layer was washed with brine and dried with MgSO4. The crude material was purified by silica gel column chromatography using 0–100% CMA 80/ethyl acetate to yield 70 mg (79%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.43–1.82 (m, 4 H) 1.91–2.03 (m, 1 H) 2.67 (dt, J = 11.68, 7.16 Hz, 2 H) 2.76–2.94 (m, 1 H) 3.21 (dd, J = 11.82, 2.87 Hz, 1 H) 4.28 (br. s., 1 H) 6.11 (d, J = 6.12 Hz, 1 H) 6.98–7.57 (m, 8 H) 8.36 (s, 1 H), [M + H]+ 439.6.
(3R)-3-{[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-amino}-N-ethylpiperidine-1-carboxamide (15c)
To a solution of 15b (20 mg, 0.046 mmol, 1 equiv) in 2 mL of THF was added triethylamine (0.02 mL, 0.137 mmol, 3 equiv) and ethyl isocyanate (0.005 mL, 0.068 mmol, 1.5 equiv). The reaction is stirred for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield 15 mg (65%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.03–1.34 (m, 3 H) 1.56–1.96 (m, 4 H) 2.18 (br. s., 1 H) 3.07 (br. s., 2 H) 3.32 (br. s., 1 H) 3.73–4.42 (m, 3 H) 5.05 (br. s., 1 H) 6.02 (d, J = 5.84 Hz, 1 H) 7.09–7.63 (m, 8 H) 8.45 (br. s., 1 H), [M + H]+ 510.3.
1-[(3R)-3-{[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-amino}piperidin-1-yl]ethan-1-one (15d)
A solution of 15b (20 mg, 0.046 mmol, 1 equiv) in 1 mL of acetic anhydride and 1 mL of pyridine was stirred for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield 19 mg (87%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.53–1.92 (m, 3 H) 2.07 (br. s., 4 H) 2.84–3.37 (m, 2 H) 3.86–4.59 (m, 3 H) 5.88 (br. s., 1 H) 7.04–7.60 (m, 8 H) 8.37 (br. s., 1 H), [M + H]+ 481.3.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-N-[(3R)-1-methanesulfonylpiperidin-3-yl]-9H-purin-6-amine (15e)
To a solution of 15b (20 mg, 0.046 mmol, 1eq.) in 2 mL of dichloromethane was added methanesulfonyl chloride (0.007 mL, 0.091 mmol, 2 equiv) and triethylamine (0.02 mL, 0.137 mmol, 3 equiv). The mixture was stirred for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield 12 mg (51%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.81–2.18 (m, 4 H) 2.77–2.94 (m, 3 H) 3.07–3.29 (m, 2 H) 3.42 (br. s., 1 H) 3.82 (d, J = 9.89 Hz, 1 H) 4.68 (br. s., 1 H) 6.13 (br. s., 1 H) 7.12–7.65 (m, 8 H) 8.48 (s, 1 H), [M + H]+ 517.8.
tert-Butyl (3S)-3-{[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]amino}piperidine-1-carboxylate (16a)
To a solution of 18 (100 mg, 0.267 mmol, 1 equiv) in 3 mL of ethanol was added tert-butyl (3S)-3-aminopiperidine-1-carboxylate (64 mg, 0.32 mmol, 1.2 equiv) and triethylamine (0.06 mL, 0.4 mmol, 1.5 equiv). The reaction was heated to 80 °C for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield 137 mg (95%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.43 (br. s., 9 H) 1.60–1.76 (m, 2 H) 1.77–1.93 (m, 1 H) 2.08–2.22 (m, 1 H) 3.30 (br. s., 2 H) 3.60 (br. s., 1 H) 3.98 (br. s., 1 H) 4.42 (br. s., 1 H) 5.96 (d, J = 7.54 Hz, 1 H) 7.14–7.68 (m, 8 H) 8.48 (s, 1 H), [M + H]+ 539.4.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-N-[(3S)-piperidin-3-yl]-9H-purin-6-amine (16b)
A solution of 16a (120 mg, 0.22 mmol) was stirred in dichloromethane (7 mL) and trifluoroacetic acid (3 mL) for 16 h. The reaction was concentrated in vacuo. The crude product was dissolved in ethyl acetate and washed with saturated NaHCO3. The aqueous layer was extracted twice with ethyl acetate. The combined organic layer was washed with brine and dried with MgSO4. The crude material was purified by silica gel column chromatography using 0–100% CMA 80/ethyl acetate to yield 64 mg (65%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.55–1.86 (m, 4 H) 2.01–2.14 (m, 1 H) 2.66–2.85 (m, 2 H) 2.87–3.04 (m, 1 H) 3.32 (dd, J = 11.77, 2.35 Hz, 1 H) 4.39 (br. s., 1 H) 6.20 (d, J = 5.84 Hz, 1 H) 7.12–7.66 (m, 8 H) 8.47 (s, 1 H), [M + H]+ 439.6.
(3S)-3-{[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-amino}-N-ethylpiperidine-1-carboxamide (16c)
To a solution of 16b (16 mg, 0.036 mmol, 1 equiv) in 2 mL of THF was added triethylamine (0.015 mL, 0.109 mmol, 3 equiv) and ethyl isocyanate (0.004 mL, 0.055 mmol, 1.5 equiv). The reaction is stirred for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield 16 mg (86%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.06–1.34 (m, 3 H) 1.53–1.95 (m, 4 H) 2.12–2.29 (m, 1 H) 3.07 (br. s., 2 H) 3.32 (br. s., 1 H) 3.72–4.41 (m, 3 H) 5.06 (br. s., 1 H) 6.02 (d, J = 6.12 Hz, 1 H) 7.08–7.66 (m, 8 H) 8.45 (br. s., 1 H), [M + H]+ 510.3.
1-[(3S)-3-{[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-amino}piperidin-1-yl]ethan-1-one (16d)
A solution of 16b (15.7 mg, 0.036 mmol, 1 equiv) in 1 mL of acetic anhydride and 1 mL of pyridine was stirred for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield 14 mg (81%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.48–1.91 (m, 3 H) 2.07 (s, 4 H) 2.90–3.39 (m, 2 H) 3.96–4.57 (m, 3 H) 5.83 (d, J = 6.50 Hz, 1 H) 7.04–7.56 (m, 8 H) 8.37 (s, 1 H), [M + H]+ 481.3.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-N-[(3S)-1-methanesulfonylpiperidin-3-yl]-9H-purin-6-amine (16e)
To a solution of 16b (15.3 mg, 0.035 mmol, 1 equiv) in 2 mL of THF was added methanesulfonyl chloride (0.005 mL, 0.07 mmol, 2 equiv) and triethylamine (0.015 mL, 0.11 mmol, 3 equiv). The mixture was stirred for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield 16 mg (89%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.63–2.02 (m, 4 H) 2.77 (s, 3 H) 2.98–3.19 (m, 2 H) 3.31 (br. s., 1 H) 3.71 (d, J = 10.55 Hz, 1 H) 4.56 (br. s., 1 H) 6.01 (d, J = 7.54 Hz, 1 H) 6.97–7.56 (m, 8 H) 8.37 (s, 1 H), [M + H]+ 517.4.
tert-Butyl N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}carbamate (17a)
To a solution of 18 (309 mg, 0.824 mmol, 1 equiv) in 10 mL of ethanol was added tert-butyl N-(piperidin-4-yl)carbamate (21) (329 mg, 1.65 mmol, 2 equiv). The reaction was heated to 80 °C for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexanes to yield 442 mg (99%) of desired product. 1H NMR (300 MHz, chloroform-d) δ ppm 1.40–1.53 (m, 11 H) 2.12 (d, J = 11.40 Hz, 2 H) 3.32 (t, J = 11.63 Hz, 2 H) 3.80 (br. s., 1 H) 4.41–4.68 (m, 1 H) 5.40 (br. s., 2 H) 7.16–7.24 (m, 2 H) 7.26–7.43 (m, 5 H) 7.51 (d, J = 6.59 Hz, 1 H) 8.38 (s, 1 H), [M + H]+ 539.2.
1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-piperidin-4-amine (17b)
A solution of 17a (380 mg, 0.705 mmol) was stirred in dichloromethane (7 mL) and trifluoroacetic acid (3 mL) for 16 h. The reaction was concentrated in vacuo. The crude product was dissolved in ethyl acetate and washed with saturated NaHCO3. The aqueous layer was extracted twice with ethyl acetate. The combined organic layer was washed with brine and dried with MgSO4. The crude material was purified by silica gel column chromatography using 0–100% CMA 80/ethyl acetate to yield 294 mg (95%) of the desired compound. Compound was determined to be 95% pure by 1H NMR and was carried forward into further chemistry. 1H NMR (300 MHz, chloroform-d) δ ppm 1.31–1.54 (m, 2 H) 1.92–2.08 (m, 2 H) 2.92–3.12 (m, 1 H) 3.27 (t, J = 11.87 Hz, 2 H) 5.42 (br. s., 2 H) 7.07–7.61 (m, 8 H) 8.38 (s, 1 H), [M + H]+ 439.4.
General Procedure for Making Ureas from 1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-amine (17b)
To a solution of 17b (20 mg, 0.046 mmol, 1 equiv) in 2 mL of THF was added triethylamine (0.02 mL, 0.136 mmol, 3 equiv) and the appropriate isocyanate (1.5 equiv). The reaction was stirred for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexane to yield pure compound.
1-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-piperidin-4-yl}-3-ethylurea (17c)
Reaction proceeded in 56% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 1.13 (t, J = 7.21 Hz, 3 H) 1.33–1.57 (m, 2 H) 2.04–2.20 (m, 2 H) 3.14–3.25 (m, 2 H) 3.32 (t, J = 12.06 Hz, 2 H) 3.86–4.05 (m, 1 H) 4.23–4.45 (m, 2 H) 5.39 (br. s., 2 H) 7.03–7.59 (m, 8 H) 8.37 (s, 1 H), [M + H]+ 510.2.
1-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-piperidin-4-yl}-3-(propan-2-yl)urea (17d)
Reaction proceeded in 84% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 1.14 (d, J = 6.50 Hz, 6 H) 1.35–1.55 (m, 2 H) 2.12 (d, J = 10.46 Hz, 2 H) 3.32 (t, J = 12.10 Hz, 2 H) 3.84 (dd, J = 13.66, 6.69 Hz, 1 H) 3.89–4.04 (m, 1 H) 4.12–4.36 (m, 2 H) 5.39 (br. s., 2 H) 7.19 (d, J = 8.67 Hz, 2 H) 7.28–7.43 (m, 5 H) 7.51 (d, J = 6.69 Hz, 1 H) 8.37 (s, 1 H), [M + H]+ 524.6.
1-{1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-piperidin-4-yl}-3-propylurea (17e)
Reaction proceeded in 71% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 0.83–0.98 (m, 3 H) 1.37–1.59 (m, 4 H) 2.13 (d, J = 10.27 Hz, 2 H) 3.12 (q, J = 6.66 Hz, 2 H) 3.32 (t, J = 12.01 Hz, 2 H) 3.83–4.07 (m, 1 H) 4.20–4.47 (m, 2 H) 5.40 (br. s., 2 H) 7.13–7.24 (m, 2 H) 7.28–7.44 (m, 5 H) 7.51 (d, J = 6.69 Hz, 1 H) 8.37 (s, 1 H), [M + H]+ 524.1.
3-Butyl-1-{1-[8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-yl}urea (17f)
Reaction proceeded in 69% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 0.84–0.99 (m, 3 H) 1.28–1.55 (m, 6 H) 2.04–2.20 (m, 2 H) 3.15 (q, J = 6.75 Hz, 2 H) 3.32 (t, J = 12.15 Hz, 2 H) 3.83–4.08 (m, 1 H) 4.18–4.39 (m, 2 H) 5.40 (br. s., 2 H) 7.08–7.43 (m, 7 H) 7.51 (d, J = 6.69 Hz, 1 H) 8.37 (s, 1 H), [M + H]+ 538.4.
General Procedure for Making Amides from 1-[8-(2-Chlorophen-yl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-amine (17b)
To a solution of 17b (21 mg, 0.048 mmol, 1 equiv) in 2 mL of THF was added triethylamine (0.02 mL, 0.143 mmol, 3 equiv) and the appropriate anhydride (2 equiv). The reaction is stirred for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexane to yield pure compound.
N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-piperidin-4-yl}acetamide (17g)
Reaction proceeded in 65% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 1.42–1.56 (m, 2 H) 1.99 (s, 3 H) 2.12 (d, J = 10.08 Hz, 2 H) 3.30 (t, J = 12.24 Hz, 2 H) 4.09–4.25 (m, 1 H) 5.41 (d, J = 8.01 Hz, 3 H) 7.13–7.43 (m, 7 H) 7.51 (d, J = 6.78 Hz, 1 H) 8.38 (s, 1 H), [M + H]+ 481.4.
N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-piperidin-4-yl}-2,2,2-trifluoroacetamide (17i)
Reaction proceeded in 39% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 1.62 (qd, J = 11.99, 3.86 Hz, 2 H) 2.18 (d, J = 10.83 Hz, 2 H) 3.31 (t, J = 12.43 Hz, 2 H) 4.11–4.29 (m, 1 H) 5.53 (br. s., 2 H) 6.19 (d, J = 7.06 Hz, 1 H) 7.01–7.57 (m, 8 H) 8.40 (s, 1 H), [M + H]+ 535.4.
N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-piperidin-4-yl}benzamide (17j)
Reaction proceeded in 19% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 1.82–1.97 (m, 2 H) 2.77 (d, J = 12.34 Hz, 2 H) 3.08 (br. s., 2 H) 3.46–3.62 (m, 1 H) 3.66–3.80 (m, 1 H) 4.01–4.13 (m, 1 H) 5.26–5.46 (m, 1 H) 7.12–7.59 (m, 13 H) 8.38 (s, 1 H), [M + H]+ 543.6.
General Procedure for Making Sulfonamides from 1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperidin-4-amine (17b)
To a solution of 17b (21 mg, 0.048 mmol, 1 equiv) in 2 mL of THF was added triethylamine (0.02 mL, 0.143 mmol, 3 equiv) and the appropriate sulfonyl chloride (2 equiv). The reaction is stirred for 16 h. The reaction was concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–100% ethyl acetate/hexane to yield pure compound.
N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-piperidin-4-yl}methanesulfonamide (17h)
Reaction proceeded in 32% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 1.55–1.67 (m, 2 H) 2.18 (d, J = 12.53 Hz, 2 H) 3.03 (s, 3 H) 3.36 (t, J = 12.29 Hz, 2 H) 3.59–3.80 (m, 1 H) 4.31 (d, J = 7.44 Hz, 1 H) 5.43 (d, J = 9.89 Hz, 2 H) 7.13–7.44 (m, 7 H) 7.51 (d, J = 6.78 Hz, 1 H) 8.39 (s, 1 H), [M + H]+ 517.6.
N-{1-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-piperidin-4-yl}benzenesulfonamide (17k)
Reaction proceeded in 53% yield. 1H NMR (300 MHz, chloroform-d) δ ppm 1.43–1.59 (m, 2 H) 1.94 (d, J = 10.46 Hz, 2 H) 3.30 (t, J = 11.96 Hz, 2 H) 3.41–3.66 (m, 1 H) 4.62 (d, J = 7.54 Hz, 1 H) 5.26 (d, J = 10.08 Hz, 2 H) 7.07–7.69 (m, 10 H) 7.92 (d, J = 7.35 Hz, 2 H) 8.35 (s, 1 H), [M + H]+ 579.4.
Calcium Mobilization and Radioligand Displacement Assays
Each compound was pharmacologically characterized using a functional fluorescent CB1 activated Gαq16-coupled intracellular calcium mobilization assay in CHO-K1 cells, as has been described in our previous publications and apparent affinity (Ke) values were determined.13 Briefly, CHO-K1 cells were engineered to coexpress human CB1 and Gαq16. Activation of CB1 by an agonist then leads to generation of inositol phospahatase 3 (IP3) and activation of IP3 receptors, which leads to mobilization of intracellular calcium. Calcium flux was monitored in a 96-well format using the fluorescent dye Calcein-4 a.m. in an automated platereader (Flexstation, Molecular Devices). The antagonism of a test compound was measured by its ability to shift the concentration response curve of the synthetic CB1 agonist CP55940 rightwards using the equation:
Ke = [Ligand]/[DR-1] where DR is the EC50 ratio of CP55940 in the presence or absence of a test agent. Standard errors were between 5 and 25% of mean in most cases and have been left out for clarity.
Further characterization of select compounds was performed using radioligand displacement of [3H]SR141716 and equilibrium dissociation constant (Ki) values were determined as described previously.13 Selectivity of these compounds at CB1 versus CB2 was also determined by obtaining Ki values at either receptor using displacement of [3H]CP55940 in membranes of CHO-K1 cells overexpressing either receptor. Data reported are average values from 3 to 6 measurements. The standard errors for most measurements were between 5 and 25% of mean and have been left out from the tables and figures for clarity.
MDCK-mdr1 Permeability Assays
MDCK-mdr1 cells permeability assays were preformed as previously described.11b,13 MDCK-mdr1 cells obtained from Netherlands Cancer Institute were grown on Transwell type filters (Corning) for 4 days to confluence in DMEM/F12 media containing 10% fetal bovine serum and antibiotics as has been described previously.13 Compounds were added to the apical side at a concentration of 10 μM in a transport buffer comprising of 1× Hank’s balanced salt solution, 25 mM D-glucose and buffered with HEPES to pH 7.4. Samples were incubated for 1 h at 37 °C and carefully collected from both the apical and basal side of the filters. Compounds selected for MDCK-mdr1 cell assays were infused on an Applied Biosystems API-4000 mass spectrometer to optimize for analysis using multiple reaction monitoring (MRM). Flow injection analysis was also conducted to optimize for mass spectrometer parameters. Samples from the apical and basolateral side of the MDCK cell assay were dried under nitrogen on a Turbovap LV. The chromatography was conducted with an Agilent 1100 binary pump with a flow rate of 0.5 mL/min. Mobile phase solvents were A, 0.1% formic acid in water, and B, 0.1% formic acid in methanol. The initial solvent conditions were 10% B for 1 min, then a gradient was used by increasing to 95% B over 5 min, then returning to initial conditions. Data reported are average values from 2 to 3 measurements.
In Vitro Stability Testing
Stability of compounds to plasma and S9 fraction was preformed as previously described.13 In vitro testing for metabolic stability was conducted in pooled samples of mixed gender human plasma from BioChemed Services, Winchester, VA and human mixed gender pooled hepatic S9 fraction supplied by Xenotech, LLC, Lenexa, KS. Identity of the donors was unknown.
For the hepatic S9 metabolism studies, all samples were tested at 10 μM final concentration in a 1 mL volume containing 1 mg/mL S9. Samples were incubated in a buffer containing 50 mM potassium phosphate, pH 7.4 with 3 mM MgCl2 and a NADPH regeration system comprising of NADP (1 mM), glucose-6-phosphate (5 mM) and glucose-6-phosphate dehydrogenase (1 unit/mL). Triplicate samples were incubated for 0, 15, 30, 60, and 120 min. Reactions were terminated by addition of 3 vol of acetonitrile and processed as described for the MDCK-mdr1 assays, but standard curves were prepared in blank matrix for each compound for quantitative assessment.
The plasma stability studies were conducted at 37 °C in a volume of 1 mL plasma per sample. All compounds were tested at 10 μM final concentration at 0, 30, and 60 min after a 5 min preincubation. Reactions were terminated by addition of acetonitrile and analyzed as described above.
Pharmacokinetic Evaluation in Vivo
Male Sprague–Dawley rats aged 8 weeks at time of dosing were acquired from Charles River Laboratories and were dosed orally. Three to four animals were used per time point. Dose was formulated in food-grade corn oil at 10 mg/kg. Plasma, liver, and brain were taken from all rats at 0.5, 1, 2, 4, and 8 h postdose. Samples were prepared and analyzed as follows: Plasma (50 μL) was mixed with 10 μL of 10 μL of acetonitrile, and 300 μL of acetonitrile, vortexed, and centrifuged at 9000g for 5 min. Supernatants were transferred to autosampler vials with low-volume inserts and injected without dilution. Brains were homogenized with 50:50 ethanol/water (3:1, v/v) using a Potter Elvehjem type homogenizer. Homogenate (50 μL) was mixed with 10 μL of acetonitrile, and 300 μL of acetonitrile, vortexed and centrifuged at 9000g for 5 min. Supernatant was transferred to autosampler vials with low-volume inserts and injected without dilution. Standards were prepared as above for each compound in blank plasma, blank liver homogenate, and blank brain homogenate. Standards used were within 15% of nominal; except for 20% at LOQ. Compounds for LC-MS/MS analyses were supplied at 1 mg/mL in methanol. The stock solutions were further diluted to ∼100 ng/mL. The 100 ng/mL solutions were used to optimize the mass spectrometer for MRM transitions and mass spectrometer parameters. Infusion and flow injection optimization were also performed. LC-MS/MS was conducted using an Applied Biosystems API 4000 coupled with an Agilent 1100 HPLC system. Chromatography was performed with a Phenomenex Luna C18 (50 × 2 mm, 5 μm) column. Mobile phases were 0.1% formic acid and 10 mM ammonium formate in water (A), and 0.1% formice acid and 10 mM ammonium formate in methanol. Initial conditions were 10% B and held for one minute, followed by a linear gradient to 90% B over 5 min. 90% B was held for 2 min before returning to initial conditions. Compound 12 was analyzed with multiple reaction monitoring in the positive mode with a transition of 595.28 → 370.0. The following parameters were used, DP = 116, CE = 45, CXP = 36, CAD = 4, CUR = 10, GS1 = 40, GS2 = 60, IS = 4000, and TEM = 650.
Supplementary Material
Acknowledgments
The authors would like to thank Ann Gilliam for performing the binding assays, and Ms. Sherry Black and Ms. Purvi Patel for help with metabolic stability studies. We express our gratitude to the NIDA drug supply program for providing radiolabeled probes and to Dr. Brian Thomas for supplying the CB1 cells. This research was funded by research grants 1R21AA019740-01 and 1R03AA017514-01 to R.M. from NIAAA.
ABBREVIATIONS USED
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- CMA 80
80% chloroform, 18% methanol, and 2% ammonium hydroxide
- CNS
central nervous system
- BBB
blood–brain barrier
- TPSA
topological polar surface area
- ECS
endocannabinoid system
- CBR
cannabinoid receptors
- Ke
apparent affinity constant
- MDCK-mdr1
Madin-Darby canine kidney cells transfected with the human MDR1 gene
- A
apical
- B
basal
- BOP
benzotriazole-1-yl-oxytris(dimethylamino)-phosphonium hexafluorophosphate
- CHO-K1
Chinese hamster ovary cells
- IP3
inositol phospahatase 3
- MRM
multiple reaction monitoring
- LOQ
below limit of quantitation
- NA
not applicable
Footnotes
HPLC and melting point data of target compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Reggio PH. Endocannabinoid binding to the cannabinoid receptors: what is known and what remains unknown. Curr Med Chem. 2010;17:1468–1486. doi: 10.2174/092986710790980005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mouslech Z, Valla V. Endocannabinoid system: An overview of its potential in current medical practice. Neuroendocrinol Lett. 2009;30:153–179. [PubMed] [Google Scholar]
- 4.Demuth DG, Molleman A. Cannabinoid signalling. Life Sci. 2006;78:549–563. doi: 10.1016/j.lfs.2005.05.055. [DOI] [PubMed] [Google Scholar]
- 5.(a) De Vries TJ, Schoffelmeer AN. Cannabinoid CB1 receptors control conditioned drug seeking. Trends Pharmacol Sci. 2005;26:420–426. doi: 10.1016/j.tips.2005.06.002. [DOI] [PubMed] [Google Scholar]; (b) Le Foll B, Goldberg SR. Cannabinoid CB1 receptor antagonists as promising new medications for drug dependence. J Pharmacol Exp Ther. 2005;312:875–883. doi: 10.1124/jpet.104.077974. [DOI] [PubMed] [Google Scholar]; (c) Wierzbicki AS. Rimonabant: endocannabinoid inhibition for the metabolic syndrome. Int J Clin Pract. 2006;60:1697–1706. doi: 10.1111/j.1742-1241.2006.01210.x. [DOI] [PubMed] [Google Scholar]
- 6.Janero DR, Makriyannis A. Cannabinoid receptor antagonists: pharmacological opportunities, clinical experience, and translational prognosis. Expert Opin Emerging Drugs. 2009;14:43–65. doi: 10.1517/14728210902736568. [DOI] [PubMed] [Google Scholar]
- 7.(a) Janero DR, Makriyannis A. Cannabinoid receptor antagonists: pharmacological opportunities, clinical experience, and translational prognosis. Expert Opin Emerging Drugs. 2009;14:43–65. doi: 10.1517/14728210902736568. [DOI] [PubMed] [Google Scholar]; (b) Di Marzo V. CB(1) receptor antagonism: biological basis for metabolic effects. Drug Discovery Today. 2008;13:1026–1041. doi: 10.1016/j.drudis.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 8.Lee HK, Choi EB, Pak CS. The Current Status and Future Perspectives of Studies of Cannabinoid Receptor 1 Antagonists as Anti-Obesity Agents. Curr Top Med Chem. 2009;9:482–503. doi: 10.2174/156802609788897844. [DOI] [PubMed] [Google Scholar]
- 9.Stein C, Schafer M, Machelska H. Attacking pain at its source: new perspectives on opioids. Nat Med. 2003;9:1003–1008. doi: 10.1038/nm908. [DOI] [PubMed] [Google Scholar]
- 10.(a) Tam J, Vemuri VK, Liu J, Batkai S, Mukhopadhyay B, Godlewski G, Osei-Hyiaman D, Ohnuma S, Ambudkar SV, Pickel J, Makriyannis A, Kunos G. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest. 2010;120:2953. doi: 10.1172/JCI42551. Corrigendum: J. Clin. Invest. 2010120, 3735–3735. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tarzia G, LoVerme J, Duranti A, Tontini A, Spadoni G, Mor M, Rivara S, Stella N, Xu C, Piomelli D. Synthesis and characterization of a peripherally restricted CB(1) cannabinoid antagonist, URB447, that reduces feeding and body-weight gain in mice. Bioorg Med Chem Lett. 2009;19:639–643. doi: 10.1016/j.bmcl.2008.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Barth F, Hortala L, Rinaldi-Carmona M, Congy C, Boulu L, Sadoun F, Fabre G, Finance O. Rational design of a novel peripherally-restricted, orally active CB(1) cannabinoid antagonist containing a 2,3-diarylpyrrole motif. Bioorg Med Chem Lett. 2010;20:4573–4577. doi: 10.1016/j.bmcl.2010.06.017. [DOI] [PubMed] [Google Scholar]; (d) Hogberg T, Cooper M, Receveur JM, Bjurling E, Norregaard PK, Nielsen PA, Skold N. Exploring SAR features in diverse library of 4-cyanomethyl-pyrazole-3-carboxamides suitable for further elaborations as CB1 antagonists. Bioorg Med Chem Lett. 2010;20:26–30. doi: 10.1016/j.bmcl.2009.11.047. [DOI] [PubMed] [Google Scholar]; (e) Hogberg T, Receveur JM, Murray A, Linget JM, Norregaard PK, Cooper M, Bjurling E, Nielsen PA. Conversion of 4-cyanomethyl-pyrazole-3-carboxamides into CB1 antagonists with lowered propensity to pass the blood-brain-barrier. Bioorg Med Chem Lett. 2010;20:453–457. doi: 10.1016/j.bmcl.2009.12.003. [DOI] [PubMed] [Google Scholar]; (f) Sasmal PK, Reddy DS, Talwar R, Venkatesham B, Balasubrahmanyam D, Kannan M, Srinivas P, Kumar KS, Devi BN, Jadhav VP, Khan SK, Mohan P, Chaudhury H, Bhuniya D, Iqbal J, Chakrabarti R. Novel pyrazole-3-carboxamide derivatives as cannabinoid-1 (CB1) antagonists: Journey from non-polar to polar amides. Bioorg Med Chem Lett. 2011;21:562–568. doi: 10.1016/j.bmcl.2010.10.055. [DOI] [PubMed] [Google Scholar]
- 11.(a) Chen RZ, Frassetto A, Lao JZ, Huang RRC, Xiao JC, Clements MJ, Walsh TF, Hale JJ, Wang JY, Tong XC, Fong TM. Pharmacological evaluation of LH-21, a newly discovered molecule that binds to cannabinoid CB1 receptor. Eur J Pharmacol. 2008;584:338–342. doi: 10.1016/j.ejphar.2008.02.029. [DOI] [PubMed] [Google Scholar]; (b) Fulp A, Bortoff K, Zhang Y, Seltzman H, Snyder R, Maitra R. Towards rational design of cannabinoid receptor 1 (CB1) antagonists for peripheral selectivity. Bioorg Med Chem Lett. 2011;21:5711–5714. doi: 10.1016/j.bmcl.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark DE, Pickett SD. Computational methods for the prediction of drug-likeness. Drug Discovery Today. 2000;5:49–58. doi: 10.1016/s1359-6446(99)01451-8. [DOI] [PubMed] [Google Scholar]
- 13.Fulp A, Bortoff K, Seltzman H, Zhang Y, Mathews J, Snyder R, Fennell T, Maitra R. Design and synthesis of cannabinoid receptor 1 antagonists for peripheral selectivity. J Med Chem. 2012;55:2820–2834. doi: 10.1021/jm201731z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffith DA, Hadcock JR, Black SC, Iredale PA, Carpino PA, DaSilva-Jardine P, Day R, DiBrino J, Dow RL, Landis MS, O’Connor RE, Scott DO. Discovery of 1-[9-(4-chlorophenyl)-8-(2-chlorophenyl)-9H-purin-6-yl]-4-ethylaminopiperi dine-4-carboxylic acid amide hydrochloride (CP-945,598), a novel, potent, and selective cannabinoid type 1 receptor antagonist. J Med Chem. 2009;52:234–237. doi: 10.1021/jm8012932. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Gilliam A, Maitra R, Damaj MI, Tajuba JM, Seltzman HH, Thomas BF. Synthesis and biological evaluation of bivalent ligands for the cannabinoid 1 receptor. J Med Chem. 2010;53:7048–7060. doi: 10.1021/jm1006676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Doran A, Obach RS, Smith BJ, Hosea NA, Becker S, Callegari E, Chen C, Chen X, Choo E, Cianfrogna J, Cox LM, Gibbs JP, Gibbs MA, Hatch H, Hop CE, Kasman IN, Laperle J, Liu J, Liu X, Logman M, Maclin D, Nedza FM, Nelson F, Olson E, Rahematpura S, Raunig D, Rogers S, Schmidt K, Spracklin DK, Szewc M, Troutman M, Tseng E, Tu M, Van Deusen JW, Venkatakrishnan K, Walens G, Wang EQ, Wong D, Yasgar AS, Zhang C. The impact of P-glycoprotein on the disposition of drugs targeted for indications of the central nervous system: evaluation using the MDR1A/1B knockout mouse model. Drug Metab Dispos. 2005;33:165–174. doi: 10.1124/dmd.104.001230. [DOI] [PubMed] [Google Scholar]; (b) Polli JW, Olson KL, Chism JP, John-Williams LS, Yeager RL, Woodard SM, Otto V, Castellino S, Demby VE. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2- (methylsulfonyl)ethy l]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016) Drug Metab Dispos. 2009;37:439–442. doi: 10.1124/dmd.108.024646. [DOI] [PubMed] [Google Scholar]
- 17.Hadcock JR, Griffith DA, Iredale PA, Carpino PA, Dow RL, Black SC, O’Connor R, Gautreau D, Lizano JS, Ward K, Hargrove DM, Kelly-Sullivan D, Scott DO. In vitro and in vivo pharmacology of CP-945,598, a potent and selective cannabinoid CB(1) receptor antagonist for the management of obesity. Biochem Biophys Res Commun. 2010;394:366–371. doi: 10.1016/j.bbrc.2010.03.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.